

Abstract
To investigate cross-ancestry genetics of complex traits, we conducted a phenome-wide analysis of loci with heterogeneous effects across African, Admixed-American, Central/South Asian, East Asian, European and Middle Eastern participants of the UK Biobank (N = 441 331). Testing 843 phenotypes, we identified 82 independent genomic regions mapping variants showing genome-wide significant (GWS) associations (P < 5 × 10−8) in the trans-ancestry meta-analysis and GWS heterogeneity among the ancestry-specific effects. These included (i) loci with GWS association in one ancestry and concordant but heterogeneous effects among the other ancestries and (ii) loci with a GWS association in one ancestry group and an experiment-wide significant discordant effect (P < 6.1 × 10−4) in at least another ancestry. Since the trans-ancestry GWS associations were mostly driven by the European ancestry sample size, we investigated the differences of the allele frequency (ΔAF) and linkage disequilibrium regulome tagging (ΔLD) between European populations and the other ancestries. Within loci with concordant effects, the degree of heterogeneity was associated with European–Middle Eastern ΔAF (P = 9.04 × 10−6) and ΔLD of European populations with respect to African, Admixed-American and Central/South Asian groups (P = 8.21 × 10−4, P = 7.17 × 10−4 and P = 2.16 × 10−3, respectively). Within loci with discordant effects, ΔAF and ΔLD of European populations with respect to African and Central/South Asian ancestries were associated with the degree of heterogeneity (ΔAF: P = 7.69 × 10−3 and P = 5.31 × 10−3, ΔLD: P = 0.016 and P = 2.65 × 10−4, respectively). Considering the traits associated with cross-ancestry heterogeneous loci, we observed enrichments for blood biomarkers (P = 5.7 × 10−35) and physical appearance (P = 1.38 × 10−4). This suggests that these specific phenotypic classes may present considerable cross-ancestry heterogeneity owing to large allele frequency and LD variation among worldwide populations.
Introduction
Genome-wide association studies (GWAS) are a powerful tool to identify the genetic variants associated with human traits and diseases (1). As of 15 December 2020, 4809 publications and 227 262 associations have been listed in the GWAS Catalog (2). This unprecedented amount of information has revolutionized our understanding of the predisposition to complex phenotypes, demonstrating that a large portion of the heritability of complex traits resides in common genetic variation [i.e. polymorphisms in the human genome that show a minor allele frequency (MAF) greater than 1%] (1). In recent years, the investigations of massive cohorts from 100 000 to more than 1 000 000 participants were possible because of large collaborative projects combining numerous studies (3–6), the availability of biobanks enrolling an unprecedented number of participant (7–9) and collaboration with direct-to-consumer genetic testing companies (10). These large-scale GWAS identifying ever-greater numbers of risk loci with ever-smaller individual effects demonstrated that the genetic architecture of common diseases is highly polygenic and their heritability is likely owing to the contribution of several thousands (or even more) of risk loci across the human genome (11–14). One of the main GWAS promises is that the knowledge gained can be used to develop genetic instruments useful to predict disease risk, treatment response and disease prognosis. Leveraging data generated by large-scale GWAS, a growing number of studies are developing approaches to test the utility of polygenic information with respect to the human phenotypic spectrum (15–18). Although these successful experiments strongly support the movement toward the application of GWAS data to develop new strategies to prevent and treat human diseases, important challenges remain. Among them, one of the most pressing challenges is related to the limited ancestry and ethnic diversity of large-scale GWAS which have created a large gap between the genetic data available for populations of European versus non-European descent (19). Applying GWAS data generated from European ancestry cohorts to non-European individuals raises serious issues, including much lower predictive power than that observed in comparisons between like populations (20,21) and possible biases (e.g. reflecting unaccounted population stratification rather than the phenotype of interest) owing to the genetic variability among human populations (22,23). The most reliable solution to this problem is to conduct large-scale GWAS in populations with non-European ancestry. Ongoing efforts such as the Million Veteran Program (24) and the All of US Research Program (25) are investigating multiple ancestry groups representative of the US population to reduce this gap. Although these projects are expected to reduce the population disparities in human genetic research, this is likely to be a long-term outcome. To date, to contribute to a more comprehensive understanding of human genetic diversity, we can leverage the data available, combining large-scale genome-wide association datasets generated from cohorts, mainly including participants of European descent with reference panels representative of the genetic diversity among worldwide populations (26–29). In the present study, we focused our attention on the UK Biobank (UKB). This large cohort includes more than 500 000 participants, with >90% of them as British individuals of European descent (30). In addition to participants of European descent, the UKB cohort includes individuals of African (AFR), Admixed-American (AMR), Central/South Asian (CSA), East Asian (EAS) and Middle Eastern (MID) ancestral backgrounds. Combining UKB cross-ancestry data with information regarding the inter-population variability in the linkage disequilibrium (LD) tagging of regulatory elements, we investigated the cross-ancestry heterogeneity of loci associated with the human phenotypic spectrum (Supplementary Material, Table S1).
Results
Considering UKB genome-wide association statistics related to 843 phenotypes investigated across the six ancestry groups available, we identified 20 287 associations presenting genome-wide significance (P < 5 × 10−8) in both the trans-ancestry meta-analysis and the heterogeneity test (Supplementary Material, Table S2; 14 708 variants and 93 phenotypes). Based on our positional mapping strategy (described in the Materials and Methods), we identified 82 independent genomic regions mapping the 20 287 genome-wide significant (GWS) trans-ancestry associations with GWS heterogeneity among the ancestry-specific effects. Because our goal was to identify loci with the strongest genetic heterogeneity among the ancestry groups investigated, the GWAS index variants were defined within each of the 82 independent genomic regions as the variant with the strongest statistical evidence of the cross-ancestry heterogeneity (Supplementary Material, Table S3). Among the GWAS index variants, we observed two different scenarios: (i) loci with GWS association in one ancestry and concordant but with heterogeneous effects among the other ancestries (i.e. cross-ancestry differences in the effect size) and (ii) loci with a GWS association in one ancestry group and an experiment-wide significant discordant effect (P < 6.1 × 10−4, i.e. Bonferroni correction accounting for the number of independent genomic regions) in at least one other ancestry. Among the concordant but heterogeneous index variants, the strongest degree of heterogeneity was observed in the association of SLC45A2 rs35390*A with ‘hair color (natural, before graying): black’ (trans-ancestry beta = −0.456, P = 1.95 × 10−78, heterogeneity P = 6.49 × 10−192) that showed a much larger effect size in the UKB EUR participants (EUR beta = −1.928, P = 5.12 × 10−265) than in other ancestries (AMR beta = −0.629, P = 1.03 × 10−5; MID beta = −0.380, P = 1.54 × 10−4). With respect to the ‘discordant-effect’ loci, the association of LUC7L rs7189975*A with mean corpuscular hemoglobin (trans-ancestry beta = 0.105, P = 2.82 × 10−102, heterogeneity P = 6.03 × 10−104) was positive in the UKB EUR participants (EUR beta = 0.134, P = 2.26 × 10−151) and negative in the UKB AFR participants (AFR beta = −0.336, P = 1.81 × 10−58). Owing to the large sample size of the UKB EUR sample, most of the trans-ancestry associations of the GWAS index variants were driven by the EUR-specific analysis. Only three trans-ancestry associations presented the strongest ancestry-specific signals in non-EUR samples: UGT1 family members A3-A10 rs12466997*C (total bilirubin; trans-ancestry beta = −0.107, P = 3.54 × 10−91; AFR beta = −0.554, P = 5.18 × 10−142; heterogeneity P = 1.5 × 10−109), F8 rs782604098*ATG (glycated hemoglobin; trans-ancestry beta = −0.027, P = 2.71 × 10−9; AFR beta = −0.544, P = 1.45 × 10−109; heterogeneity P = 5.59 × 10−103) and ABO rs9411476*A (alkaline phosphatase; trans-ancestry beta = −0.088, P = 2.99 × 10−13; AFR beta = −0.228, P = 3.21 × 10−16; heterogeneity P = 5 × 10−24). The heterogeneous effect of these AFR-driven associations showed concordant directions across the other ancestries. Table 1 reports the details of the associations described before. Supplementary Material, Table S4 shows the negligible LD among the 82 GWAS index variants (R2 < 0.01) identified through our positional mapping strategy.
Table 1.
Loci with most significant concordant and discordant heterogeneity and loci with cross-ancestry associations driven by the UKB AFR ancestry sample
| Heterogeneity | rsID | REF | ALT | Phenotype description | Ancestry | Effect allele frequency | Beta | SE | P | P heterogeneity |
|---|---|---|---|---|---|---|---|---|---|---|
| Concordant | rs35390 | C | A | Hair color (natural, before graying): black | AFR | 0.572/0.620 | -0.111 | 0.056 | 4.64E-02 | 6.49E-192 |
| AMR | 0.622/0.804 | -0.629 | 0.143 | 1.03E-05 | ||||||
| CSA | 0.3885/0.410 | -0.058 | 0.037 | 1.19E-01 | ||||||
| EAS | 0.417/0.456 | 0.013 | 0.075 | 8.62E-01 | ||||||
| EUR | 0.959/0.985 | -1.928 | 0.055 | 5.12E-265 | ||||||
| MID | 0.6306/0.784 | -0.380 | 0.101 | 1.54E-04 | ||||||
| Meta-analysis | 0.9372/0.964 | -0.456 | 0.024 | 1.95E-78 | ||||||
| Discordant | rs7189975 | G | A | Mean corpuscular hemoglobin | AFR | 0.479 | -0.336 | 0.021 | 1.81E-58 | 6.03E-104 |
| AMR | 0.096 | -0.134 | 0.084 | 1.12E-01 | ||||||
| CSA | 0.043 | 0.059 | 0.039 | 1.30E-01 | ||||||
| EAS | 0.006 | 0.197 | 0.210 | 3.47E-01 | ||||||
| EUR | 0.068 | 0.134 | 0.005 | 2.26E-151 | ||||||
| MID | 0.131 | 0.002 | 0.061 | 9.79E-01 | ||||||
| Meta-analysis | 0.074 | 0.105 | 0.005 | 2.82E-102 | ||||||
| AFR-concordant | rs12466997 | T | C | Total bilirubin | AFR | 0.284 | -0.554 | 0.022 | 5.18E-142 | 1.50E-109 |
| AMR | 0.106 | -0.097 | 0.085 | 2.56E-01 | ||||||
| CSA | 0.176 | -0.214 | 0.020 | 6.11E-26 | ||||||
| EAS | 0.202 | 0.081 | 0.033 | 1.53E-02 | ||||||
| EUR | 0.054 | -0.072 | 0.006 | 8.42E-36 | ||||||
| MID | 0.132 | -0.173 | 0.056 | 2.07E-03 | ||||||
| Meta-analysis | 0.061 | -0.107 | 0.005 | 3.54E-91 | ||||||
| rs9411476 | G | A | Alkaline phosphatase | AFR | 0.125 | -0.228 | 0.028 | 3.21E-16 | 5.00E-24 | |
| AMR | 0.017 | -0.203 | 0.181 | 2.63E-01 | ||||||
| CSA | 0.056 | -0.208 | 0.034 | 1.63E-09 | ||||||
| EAS | 0.158 | -0.292 | 0.036 | 6.39E-16 | ||||||
| EUR | 0.007 | 0.026 | 0.016 | 1.14E-01 | ||||||
| MID | 0.031 | -0.045 | 0.115 | 6.97E-01 | ||||||
| Meta-analysis | 0.011 | -0.088 | 0.012 | 2.99E-13 | ||||||
| rs782604098 | A | ATG | Glycated hemoglobin | AFR | 0.251 | -0.544 | 0.024 | 1.45E-109 | 5.59E-103 | |
| AMR | 0.042 | -0.475 | 0.106 | 6.84E-06 | ||||||
| CSA | 0.011 | -0.022 | 0.083 | 7.95E-01 | ||||||
| EAS | 0.015 | 0.155 | 0.133 | 2.44E-01 | ||||||
| EUR | 0.019 | -0.007 | 0.005 | 1.01E-01 | ||||||
| MID | 0.075 | -0.117 | 0.064 | 6.98E-02 | ||||||
| Meta-analysis | 0.022 | -0.027 | 0.004 | 2.71E-09 |
The alternate allele (ALT) is the effect allele. For binary outcomes, we report the effect allele frequency for both cases and controls. REF, reference allele; SE, standard error; P: P-value.
To understand how the genetic variation across ancestries affects the heterogeneity observed among the ancestry-specific associations, we investigated differences with respect to allele frequency and LD. Figure 1 reports Spearman’s correlation among the allele frequencies and LD–regulome tagging score (RTS) of the 82 GWAS index variants among the ancestries investigated. Since the EUR sample was the main driver of most of the trans-ancestry associations investigated (96%), we analyzed the relationship of the degree of heterogeneity among these associations with respect to the ΔAF and ΔLD of the European populations with the other ancestry groups (Table 2). Among the concordant but heterogeneous associations, the median-based linear regression (MBLM) two-dimensional analysis highlighted the effect of four variables on the degree of heterogeneity: the EUR–MID ΔAF (PMBLM = 9.04 × 10−6) and the ΔLD of EUR sample with respect to AFR (PMBLM = 8.21 × 10−3), AMR (PMBLM = 7.17 × 10−4) and CSA (PMBLM = 2.16 × 10−3). The locally estimated scatterplot smoothing (LOESS)–generalized additive model (GAM) multi-dimensional analysis confirmed that the effect of EUR–MID ΔAF (PLOESS–GAM = 3.76 × 10−7), EUR–AFR ΔLD (PLOESS–GAM = 0.026) and EUR–AMR ΔLD (PLOESS–GAM = 3.47 × 10−3) are independent of each other. A different pattern was observed with respect to the GWAS index variants with discordant, heterogeneous effects. Among them, the degree of heterogeneity observed was associated with the differences of EUR populations with AFR and CSA ancestries (EUR–AFR: ΔAF PMBLM = 7.69 × 10−3, ΔLD PMBLM = 0.016; EUR–CSA: ΔAF PMBLM = 5.31 × 10−3, ΔLD PMBLM = 5.31 × 10−3). However, these effects appeared to be driven by EUR–AFR ΔAF (PLOESS–GAM = 0.026). To explore further the effect of ΔAF and ΔLD on cross-ancestry heterogeneity, we analyzed their associations considering all GWAS index variants (Supplementary Material, Table S5). Across loci with concordant and discordant heterogeneous cross-ancestry effects, the degree of heterogeneity was associated independently with EUR–MID ΔAF (PLOESS–GAM = 8 × 10−6), EUR–AFR ΔLD (PLOESS–GAM = 5.1 × 10−3) and EUR–AMR ΔLD (PLOESS–GAM = 4.4 × 10−4). With respect to both concordant and discordant cross-ancestry heterogeneity, the strength of the ΔLD and ΔAF associations was not correlated with the sample size of the non-European ancestry cohorts investigated (P > 0.05; Supplementary Material, Table S6).
Figure 1 .

Correlation (Spearman’s rho) among allele frequencies (upper triangle) and LD–RTS (lower triangle) of the ancestral groups investigated by considering the GWAS index variants tested. The correlations presented survive Bonferroni multiple testing correction accounting for the number of tests performed.
Table 2.
Association of ΔAF and ΔLD with the degree of heterogeneity considering GWAS index variants with concordant and discordant heterogeneity
| Heterogeneity | Variable | MBLM | LOESS–GAM | |||
|---|---|---|---|---|---|---|
| V-value | Pr (>|V|) | F-value | Pr (>|t|) | |||
| Concordant | ΔAF EUR versus | AFR | 997 | 0.714 | — | — |
| AMR | 1122 | 0.206 | — | — | ||
| CSA | 998 | 0.709 | — | — | ||
| EAS | 1113 | 0.23 | — | — | ||
| MID | 1564 | 9.04E-06 | 1564 | 9.04E-06 | ||
| ΔLD EUR versus | AFR | 577 | 8.21E-03 | 577 | 8.21E-03 | |
| AMR | 1417 | 7.17E-04 | 1417 | 7.17E-04 | ||
| CSA | 518 | 2.16E-03 | 518 | 2.16E-03 | ||
| EAS | 1175 | 0.1 | — | — | ||
| Discordant | ΔAF EUR versus | AFR | 145 | 0.00769 | 6.32 | 0.026 |
| AMR | 108 | 0.338 | — | — | ||
| CSA | 21 | 5.31E-03 | 2.48 | 0.140 | ||
| EAS | 50 | 0.127 | — | — | ||
| MID | 69 | 0.486 | — | — | ||
| ΔLD EUR versus | AFR | 31 | 0.0159 | 1.13 | 0.308 | |
| AMR | 46 | 0.089 | — | — | ||
| CSA | 16 | 2.65E-03 | 0.11 | 0.744 | ||
| EAS | 70 | 0.514 | — | — | ||
V-value: the intercept value of the Theil-Sen regression; Pr (>|V|): p-value of the Theil-Sen regression slope; F-value: statistics related to the V-test; Pr(>|F): p-value for the t-test.
The 82 GWAS index variants showed associations with a total of 59 phenotypes (Supplementary Material, Table S2). To investigate the enrichment for specific phenotypic classes, we considered only the phenotypic association with the strongest statistical evidence of cross-ancestry heterogeneity for each GWAS index variant. Among them (N = 40), we observed blood biomarkers (82.5%), physical appearance (10%) and anthropometric traits (7.5%). Accordingly, we observed that the same trait was associated with multiple GWAS index variants located in independent genomic regions. Among blood biomarkers, low-density lipoprotein (LDL) cholesterol adjusted by medication showed five independent associations with loci with cross-ancestry heterogeneous effects (i.e. PCSK9 rs2479413 on chromosome 1, DYNC2LI1 rs4953016 on chromosome 2, ANKRD31 rs55810502 on chromosome 5, TXNL4B rs217181 on chromosome 16 and SPC24 rs79668907 on chromosome 19). Similarly, ‘hair color (natural, before graying): black’ was associated with four independent loci with cross-ancestry heterogeneity (i.e. SLC45A2 rs35390 on chromosome 5, IRF4 rs11308001 on chromosome 6, rs11437447 on chromosome 12 and SLC24A4 rs4904871 on chromosome 14). Other 20 traits in these phenotypic classes showed an association with at least two GWAS index variants. Comparing the distribution of phenotypic classes associated with loci with cross-ancestry heterogeneous effects with that of the 843 traits tested initially, we observed an over-representation for associations with blood biomarkers (enrichment = 11.04, P = 5.7 × 10−35) and traits related to physical appearance (enrichment = 12.04, P = 1.38 × 10−4). We also verified whether the GWAS index variants are enriched for evolutionary signatures. Considering the integrated haplotype score (iHS) (31), the composite of multiple signals (CMS; long haplotypes, differentiated alleles and high frequency derived alleles) (32) and the Neanderthal local ancestry (LA) (33), we observed that only 1.2, 2.4 and 4.9% of the GWAS index variants were in the top 2% of these scores (Supplementary Material, Table S7). These proportions were not significantly different from those observed from randomly selected variants matched for genomic characteristics (Supplementary Material, Table S8).
Discussion
To provide a more comprehensive understanding of the genetics of complex traits across worldwide populations, we investigated heterogeneity among loci identified by a cross-ancestry GWAS meta-analysis. The results obtained provide a comprehensive overview of how genetic differences among human population groups affect the genetic associations of complex traits. Specifically, we observed that loci showing cross-ancestry heterogeneous effects present specific genetic and phenotypic characteristics.
Allele frequency differences among human populations can affect certain genotype–phenotype associations because the number of effect-allele carriers changes the statistical power of the association analysis conducted (34). The Population Architecture using Genomics and Epidemiology study highlighted that the effect sizes of the multi-ancestry joint analyses were significantly weaker than those observed in homogenous cohorts, suggesting truly differential effect sizes between ancestries rather than the comparisons being biased by the ‘winner’s curse’ (34).
The LD structure among human populations also plays a key role in the functional implication of the variants identified by GWAS (35,36). Indeed, it has been proposed that cross-ancestry meta-analyses can be a useful tool to fine map causal loci responsible for GWS loci (37–39). Our study demonstrates that both allele frequency and LD differences among human populations are significant contributors to the cross-ancestry heterogeneity across the human phenotypic spectrum. Additionally, we showed that the cross-ancestry variability in the tagging properties of regulatory elements is linked to both the differences in LD structure and variant density among human populations (Supplementary Material, Figs S1–S4). Considering the population diversity of the UKB cohort, we identified many loci with a cross-ancestry GWS association with a certain trait and GWS heterogeneity among the ancestry-specific effects. The heterogeneity observed was both qualitative and quantitative. Among the GWAS index variants located in independent genomic regions, 16 loci (22%) showed qualitative heterogeneity among the ancestry specific effects, i.e. a GWS association in one ancestry group and an experiment-wide significant discordant effect in at least one another ancestry group. Applying a two-dimensional model (i.e. MBLM approach), the degree of heterogeneity was associated with the genetic differences of EUR ancestry with respect to the AFR and CSA population groups. However, the multi-dimensional LOESS–GAM approach highlighted that the qualitative heterogeneity observed was primarily driven by the EUR–AFR ΔAF. This finding is in line with the human evolutionary history. Because of the African origin of the human species and the subsequent out-of-Africa bottlenecks (40,41), African populations present a much greater genetic variation than any other human group. Previous investigations highlighted how these genetic differences affect the statistical power of genetic association analyses (42). To our knowledge, the present findings represent the first comprehensive assessment of how the genetic diversity of African populations can lead to a discordant effect in the genotype–phenotype associations when compared with other ancestries. Additionally, although UKB includes a limited number of individuals of non-European descent, we identified three independent associations related to the blood biomarkers (i.e. rs12466997, total bilirubin; rs782604098, glycated hemoglobin; rs9411476, alkaline phosphatase) with cross-ancestry heterogeneity driven by the UKB participant of African descent. This strongly supports that investigating diverse populations can improve the gene discovery for complex traits.
We observed a different scenario with respect to the loci with quantitative heterogeneity (i.e. concordant effects with heterogeneous effect size). The degree of heterogeneity among these loci was independently associated with primarily EUR–MID ΔAF and EUR–AMR ΔLD and to a lesser extent with the EUR–AFR ΔLD. This highlights that, while the diversity of AFR populations is still a significant contributor, the cross-ancestry quantitative heterogeneity observed within the UKB cohort was more strongly affected by the differences of EUR ancestry with MID and AMR populations. This is particularly interesting because of the demographic history of these two human groups. Indeed, although they occurred on a different time scale and extent, a certain degree of admixture is present in both. AMR populations present an admixture of Native American, Sub-Saharan African and European ancestries, and the proportions of these three components can vary greatly across the American continent reflecting pre-Colombian civilizations, European colonization and the Atlantic slave trade (43). MID populations present an admixture of Mediterranean African, Southern European and Central Asian ancestries related to the demographic history that occurred over a long period of time in that region (e.g. rise and fall of empires, invasions, migrations and trade) (44). Accordingly, our study highlights that the genetic admixture is likely a key mechanism contributing to the qualitative heterogeneity observed among the ancestry-specific effects.
Beyond their genetic characteristics, the loci identified as heterogeneous among the ancestries investigated showed associations with specific phenotypic classes. We observed that certain traits are enriched for associations with variants presenting strong heterogeneity among the ancestry-specific effects. Additionally, GWAS index variants located in independent genomic regions are associated with the same traits. This strongly highlights how cross-ancestry genetic heterogeneity is widespread in the genetic associations with certain phenotypic classes and how it does not represent a unique or exceptional event. Across the 14 categories identified within the 843 traits tested (Supplementary Material, Table S1), we observed enrichments for cross-ancestry heterogeneous associations with respect to traits related to blood biomarkers and physical appearance. The enrichment for the first categories showed the strongest significance. Out of the 63 blood biomarkers tested (Supplementary Material, Table S1), 33 (52%) showed one or more associations with loci with cross-ancestry heterogeneous effects. These included blood biomarkers related to lipid metabolism (e.g. LDL cholesterol, lipoprotein A and apolipoprotein B), liver function (alanine aminotransferase and alkaline phosphatase), inflammation (C-reactive protein), glucose metabolism (glucose and glycated hemoglobin), immune function (e.g. eosinophil percentage, monocyte count and neutrophil count), hematic parameters (e.g. platelet count and erythrocyte distribution width), hormonal regulation (sex hormone-binding globulin) and vitamin levels (vitamin D). Accordingly, we hypothesize that cross-ancestry genetic heterogeneity is not related to a specific function but is rather pervasive across the genetics of multiple molecular phenotypes. Genetic ancestry has been previously associated with the variability of certain blood biomarkers among worldwide populations (45). In line with these genetic differences, multiple studies highlighted that blood biomarkers related to different health outcomes (e.g. cardiometabolic risk and brain aging) present significant differences among ancestry groups (46,47). These previous data support that reference intervals for blood biomarkers should be tailored to ancestry groups and that the integration of genetic information into clinical practice could help to develop precision medicine protocols among diverse populations with respect to certain health outcomes. Our present findings not only expand this previous evidence but also provide novel insights into the underlying mechanisms responsible for this variability. With respect to ΔAF and ΔLD contribution to the genetic heterogeneity of blood biomarkers, we hypothesize that natural selection may have also contributed in addition to the human demographic history. Indeed, loci associated with certain blood biomarkers (e.g. those related to immune function) present genomic selective signatures owing to the selective pressures of pathogens, diet changes and several other environmental variables that shaped the human evolutionary history (48). With respect to the second phenotypic domain enriched for loci presenting cross-ancestry heterogeneity, we expect that evolutionary pressures had played a much larger role. Indeed, the traits identified were related to hair and skin appearance. ‘Hair color (natural, before graying)’ showed strong heterogeneity among AMR, CSA, EUR and MID groups. In line with the minimal hair color variation within AFR and EAS individuals, no effect was observed with respect to these human groups. Conversely, alleles associated with black and dark brown hair colors showed concordant but heterogeneous effects among the remaining ancestries available in the UKB cohort. Among the seven GWAS index variants located in independent genomic regions (four for black hair color and three for dark brown hair color), the associated allele generally showed the largest effect size with respect to the EUR sample. However, in one case (the association of NPLOC4 rs9895741*G allele with dark brown hair color), we observed a discordant effect between EUR (beta = 0.104, P = 2.32 × 10−75) and CSA (beta = −0.194, P = 1.95 × 10−5). The phenotype related to skin pigmentation (i.e. ‘ease of skin tanning’ ranging from ‘never tan, only burn’ to ‘get very tanned’) showed the same pattern across three associated independent GWAS index variants (TYR rs7941686*A on chromosome 11, SLC24A5 rs1426654*G on chromosome 15 and TCF25 rs577053706*C on chromosome 16): negative GWS association in EUR, consistent effect direction among the other non-AFR samples and positive effect direction in AFR. The heterogeneity observed within these loci is likely to be affected by the evolutionary mechanisms that shaped human skin and hair pigmentation (49). Indeed, the variability of human pigmentation reflects the subsequent adaptive processes from the protective, dark, eumelanin-enriched coloration useful to protect the naked skin in the tropical regions where Homo sapiens originated to the loss of melanin pigmentation that occurred during the dispersal of H. sapiens into non-tropical landmasses (49–51). With respect to this previous knowledge, we expand the understanding of the genetic mechanisms underlying this phenotypic variation, also putting this in the context of the genetics of the human phenotypic spectrum. Although the phenotypic classes identified are known to be related to human evolutionary history, we did not observe enrichments for positive selection measures. This can be owing to the fact that cross-ancestry genetic heterogeneity observed in certain phenotypic domains may be related to polygenic adaptation mechanisms not reflected by the measures tested (52).
Although our investigation provides comprehensive evidence regarding how genetic variation across human populations affects the predisposition to complex traits, there are limitations to acknowledge. Our analysis is based on the UKB cohort, which presents a strong ancestry imbalance (>90% British of European descent). This permitted us to provide information mainly regarding the differences between European populations and other ancestry groups, and the loci and phenotypic classes identified are those with very large heterogeneity. A better representation of human genetic variation would have permitted us to detect heterogeneous signals across non-European populations and to increase our power to detect loci with a lower degree of heterogeneity. For the same reason, we did not attempt to model cross-ancestry heterogeneity in the context of polygenicity. More diverse genome-wide datasets will support the investigation of genetic heterogeneity with respect to how the polygenic architecture of complex traits varies across human populations. Additionally, increased population diversity among functional studies of gene regulation across human tissues and cells will permit to test the mechanisms underlying the cross-ancestry heterogeneity observed in each of the loci identified. Another important limitation is owing to the fact that our analysis was conducted only on the UKB cohort because this was the only large-scale resource publicly available at that time. Although UKB includes mainly British individuals of European descent, a consistent cross-replication between the genetic effects observed in UKB and those derived from independent non-British cohorts of European descent (53–59) has been reported, supporting that UKB findings can be generalized to other populations of European descent. Comparing cross-ancestry heterogeneity generated from independent cohorts will permit us to generalize our findings and to understand how the sample characteristics and recruitment strategies affect the gene discovery of complex traits across worldwide populations. Finally, our positional mapping approach did not permit us to investigate independent loci within the same genomic regions. The modeling of multiple ancestry-specific LD reference panels may increase the ability to uncover independent loci with cross-ancestry heterogeneity within the same genomic regions. To our knowledge, current methods can calculate the ancestry-specific posterior probability with respect to an association identified in a multi-ancestry GWAS (60). However, this analysis should be conducted with respect to each phenotype investigated, complicating the scaling to large-scale multi-ancestry phenome-wide investigations.
In conclusion, this study provided novel evidence regarding the predisposition to complex traits in the context of human genetic variation. We observed that loci with heterogeneous effects across ancestries are enriched for traits shaped by human demographic history and natural selection. Both inter-population differences in allele frequencies and LD tagging of regulatory elements affect the genotype–phenotype associations both qualitatively and quantitatively (effect direction and effect size, respectively). However, we showed how the strongest genetic heterogeneity (i.e. discordant effect direction) was mainly driven by the differences between European and African populations, while loci with heterogeneous but concordant effects were mainly affected by the differences of European ancestry with respect to populations with a complex genetic makeup (e.g. AMR and MID). Finally, although our data contribute to increasing our knowledge regarding how cross-ancestry genetic diversity affects the predisposition to complex traits, they strongly highlight that there is an urgent need for greater population diversity in GWAS and functional studies of gene regulation.
Materials and Methods
UK Biobank
UKB is a large population-based prospective study to explore different life-threatening disorders using information about environmental factors and genes in order to improve diagnosis and treatment (9). A wide variety of phenotypic information, including socio-demographic and lifestyle factors, electronic health records data and physiological conditions, have been collected for more than 500 000 UKB participants (30). UKB genetic data were used to generate genome-wide association datasets that can be employed to explore the genetics of complex traits. In our study, we used the Pan-UKB genome-wide association statistics generated from the analysis of six ancestries (AFR N = 6636; AMR N = 980; CSA N = 8876; EAS N = 2709; EUR N = 429531; MID N = 1599). Pan-UKB data are available at https://pan.ukbb.broadinstitute.org/downloads. A detailed description of the methods used to generate these data is available at https://pan.ukbb.broadinstitute.org/. Briefly, the ancestry assignment of UKB participants was conducted with respect to combined reference data from the 1000 Genomes Project (1KG) (61) and the Human Genome Diversity Project (HGDP) (62) using a two-stage approach: (i) assign continental ancestries and (ii) prune ancestry outliers within continental groups. The top six principal components (PCs) from the reference data were used to train a random forest classifier that was then applied to the UKB PC data. UKB participants were assigned to an ancestry group based on a random forest probability >50%. Individuals with a probability of <50% were excluded from the analysis. The genetic association analysis investigated variants with an imputation INFO score > 0.8 and a minimum allele count of 20. Phenotypes analyzed included binary, ordinal and continuous traits. For binary traits, a minimum of 50 cases were required in each ancestry group with the exception of the European ancestry sample where at least 100 cases were required. To investigate cross-ancestry heterogeneity, we analyzed 843 traits (Supplementary Material, Table S1) that were assessed across all six ancestry groups available in the UKB cohort. Within each ancestry, the genome-wide association analysis was conducted using the Scalable and Accurate Implementation of GEneralized (SAIGE) mixed model (63) and including a kinship matrix as a random effect and covariates as fixed effects. The covariates included age, sex, age × sex, age2, age2 × sex and the top 10 within-ancestry PCs. The ancestry-specific genome-wide associations were meta-analyzed using a fixed-effect inverse variance weighted method, and a Cochran’s Q heterogeneity test of the cross-ancestry meta-analysis was also performed. The code used for the trans-ancestry meta-analysis is available at https://github.com/atgu/ukbb_pan_ancestry.
Positional mapping to identify independent loci
To identify the number of independent signals, we decided to apply a conservative approach based on positional mapping of the variants showing genome-wide significance (P < 5 × 10−8) in both the trans-ancestry meta-analysis and the heterogeneity test. We did not use LD information because the cross-ancestry meta-analysis was generated from six ancestry groups with different LD patterns. Accordingly, we defined independent blocks considering a pairwise distance of 10 Mb. Specifically, we applied a two-variant window and assigned variants to the same block if closer than 10 Mb; a novel block is defined when two variants are more distant than 10 Mb. The 10 Mb window was defined based on previous strategies to identify GWS associations with negligible residual LD (64).
Regulome LD tagging properties across ancestry groups
To investigate how the LD structure across worldwide populations affects the ability of the GWAS index variants to tag functional elements in the surrounding regions, we leveraged 1KG reference superpopulations (61) and information regarding regulatory variants from RegulomeDB (65). We used LD information from the 1KG reference panel instead of the UKB cohort because 1KG data generated from whole-genome sequencing are more informative of LD tagging variability than the UKB data generated from the GWAS array plus imputation. Additionally, UKB genetic data have been imputed using 1000 Genomes phase 3 as one of the reference panels specifically to help with non-European ancestry UKB participants (30). Using LDlink (66,67), we tested the effect of the LD structure variability across ancestry groups on the ability of variants to tag (measured as LD R2) functional variants in the surrounding regions (±500 Kb). The 1KG reference superpopulations include AFR, AMR, EAS, EUR and South Asian. UKB CSA sample was defined by combining 1KG South Asian and HGDP South/Central Asian reference data. Accordingly, we refer to the 1KG South Asian reference panel as CSA hereafter. RegulomeDB (65) was used to score the regulatory effect of the tagged variants on the basis of high-throughput, experimental datasets as well as computational predictions and manual annotations. RegulomeDB scoring scheme ranges from 1 (highest number of known and predicted data regarding regulatory function) to 7 (lowest number of known and predicted data regarding regulatory function). To quantify the ancestry-specific ability of the GWAS index variants to tag regulatory elements in their surrounding regions, we defined an LD–RTS for each GWAS index variant calculated as: 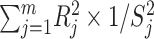 , where j is a genetic polymorphism in m polymorphisms within ±500 Kb of the GWAS index variant, R is the LD correlation coefficient between the polymorphism j and the GWAS index variant, and S is the RegulomeDB score of the polymorphism j. LD–RTS scores were calculated for each GWAS index variants with respect to the LD information available from each of the 1KG reference superpopulations. Owing to the lack of a 1KG MID superpopulation, LD–RTS was not calculated for this group. The ancestry differences of LD–RTS distribution (Supplementary Material, Fig. S1) reflect variant densities (Supplementary Material, Fig. S2) and LD patterns (Supplementary Material, Figs S3 and S4) present in the ancestries investigated. Accordingly, LD–RTS was not normalized with respect to the LD structure and variant density to model fully the differences between the ancestries.
, where j is a genetic polymorphism in m polymorphisms within ±500 Kb of the GWAS index variant, R is the LD correlation coefficient between the polymorphism j and the GWAS index variant, and S is the RegulomeDB score of the polymorphism j. LD–RTS scores were calculated for each GWAS index variants with respect to the LD information available from each of the 1KG reference superpopulations. Owing to the lack of a 1KG MID superpopulation, LD–RTS was not calculated for this group. The ancestry differences of LD–RTS distribution (Supplementary Material, Fig. S1) reflect variant densities (Supplementary Material, Fig. S2) and LD patterns (Supplementary Material, Figs S3 and S4) present in the ancestries investigated. Accordingly, LD–RTS was not normalized with respect to the LD structure and variant density to model fully the differences between the ancestries.
Non-parametric regression analyses
To understand the relationship between the heterogeneity observed in the trans-ancestry meta-analysis and genetic variation among worldwide populations, we conducted non-parametric regression analyses. We decided to apply non-parametric tests to avoid assumptions related to the distribution of the variables investigated and to apply normalization procedures to the variables of interest. Because of the much larger UKB EUR sample, the trans-ancestry GWS associations were mostly driven by EUR-specific results. Accordingly, the heterogeneity observed in the UKB trans-ancestry meta-analysis should be mainly owing to the genetic differences of EUR populations with respect to the other ancestry groups. For this reason, we considered the differences of the allele frequency and LD–RTS (ΔAF and ΔLD, respectively) of European populations with respect to the other ancestries investigated. These variables were entered in the non-parametric regression models by considering the degree of heterogeneity as the outcome of interest. The latter was quantified as the negative of the base-10 logarithm of the P-values obtained from the trans-ancestry meta-analysis heterogeneity test. Initially, we applied the MBLM approach (available at https://cran.r-project.org/web/packages/mblm/index.html) to investigate the relationship of the degree of heterogeneity with each of the ΔAF and ΔLD, e.g. 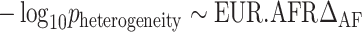 . Then, we entered the significant MBLM variables in a multivariate regression applying the LOESS process (available at https://www.rdocumentation.org/packages/stats/versions/3.6.2/topics/loess) and testing it using the GAM approach (available at https://cran.r-project.org/web/packages/gam/index.html). Similarly to a previous study (68), we applied this two-stage approach to screen the relevant variables to avoid affecting the statistical power of the multivariate regression.
. Then, we entered the significant MBLM variables in a multivariate regression applying the LOESS process (available at https://www.rdocumentation.org/packages/stats/versions/3.6.2/topics/loess) and testing it using the GAM approach (available at https://cran.r-project.org/web/packages/gam/index.html). Similarly to a previous study (68), we applied this two-stage approach to screen the relevant variables to avoid affecting the statistical power of the multivariate regression.
Enrichment analyses
To test whether there is an over-representation of certain phenotypic classes among the associations presenting cross-ancestry heterogeneity, we calculated the significance of the phenotypic enrichment applying the cumulative distribution function of the hyper-geometric distribution to the proportions of the phenotypic classes associated with the GWAS index variants investigated with respect to the proportion of phenotypic classes across the overall phenotypic spectrum investigated (Supplementary Material, Table S1). Additionally, we also verified whether the GWAS index variants were enriched for evolutionary signatures. We selected three evolutionary measures: iHS (31), CMS (32) and Neanderthal LA (33). Similarly to previous studies (68), these were converted to binary annotations as bins of the top 2% of the scores genome-wide. Using the cumulative distribution function of the hyper-geometric distribution, we compared the proportions observed in the GWAS index variants (Supplementary Material, Table S7) with those obtained from the variants matched by MAF (±5%), gene density (±50%), distance to the nearest gene (±50%) and LD independence (R2 = 0.5, ±50%). The matched variants were identified using SNPsnap (69).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank the Pan-UKB investigators for sharing their data and Dr Riccardo Pennacchi for the computational support. Yale investigators acknowledge support from the National Institutes of Health (R21 DA047527, R21 DC018098 and F32 MH122058). The sources of funding had no role in the design of the study and the collection, analysis and interpretation of data and in the writing of the manuscript.
Conflict of Interest statement. None declared.
Contributor Information
Antonella De Lillo, Department of Psychiatry, Yale University School of Medicine, West Haven, CT 06516, USA; Department of Biology, University of Rome Tor Vergata, Rome 00133, Italy.
Salvatore D'Antona, Department of Biology, University of Rome Tor Vergata, Rome 00133, Italy.
Gita A Pathak, Department of Psychiatry, Yale University School of Medicine, West Haven, CT 06516, USA; VA CT Healthcare Center, West Haven, CT 06516, USA.
Frank R Wendt, Department of Psychiatry, Yale University School of Medicine, West Haven, CT 06516, USA; VA CT Healthcare Center, West Haven, CT 06516, USA.
Flavio De Angelis, Department of Psychiatry, Yale University School of Medicine, West Haven, CT 06516, USA; Department of Biology, University of Rome Tor Vergata, Rome 00133, Italy; VA CT Healthcare Center, West Haven, CT 06516, USA.
Maria Fuciarelli, Department of Biology, University of Rome Tor Vergata, Rome 00133, Italy.
Renato Polimanti, Department of Psychiatry, Yale University School of Medicine, West Haven, CT 06516, USA; VA CT Healthcare Center, West Haven, CT 06516, USA.
References
- 1. Visscher, P.M., Wray, N.R., Zhang, Q., Sklar, P., McCarthy, M.I., Brown, M.A. and Yang, J. (2017) 10 years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet., 101, 5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Buniello, A., MacArthur, J.A.L., Cerezo, M., Harris, L.W., Hayhurst, J., Malangone, C., McMahon, A., Morales, J., Mountjoy, E., Sollis, E. et al. (2019) The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res., 47, D1005–D1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sullivan, P.F., Agrawal, A., Bulik, C.M., Andreassen, O.A., Borglum, A.D., Breen, G., Cichon, S., Edenberg, H.J., Faraone, S.V., Gelernter, J. et al. (2018) Psychiatric genomics: an update and an agenda. Am. J. Psychiatry, 175, 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thompson, P.M., Stein, J.L., Medland, S.E., Hibar, D.P., Vasquez, A.A., Renteria, M.E., Toro, R., Jahanshad, N., Schumann, G., Franke, B. et al. (2014) The ENIGMA Consortium: large-scale collaborative analyses of neuroimaging and genetic data. Brain Imaging Behav., 8, 153–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kim, Y., Han, B.G. and Ko, G.E.S.g. (2017) Cohort profile: the Korean genome and epidemiology study (KoGES) consortium. Int. J. Epidemiol., 46, e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Colodro-Conde, L., Cross, S.M., Lind, P.A., Painter, J.N., Gunst, A., Jern, P., Johansson, A., Lund Maegbaek, M., Munk-Olsen, T., Nyholt, D.R. et al. (2017) Cohort profile: nausea and vomiting during pregnancy genetics consortium (NVP Genetics Consortium). Int. J. Epidemiol., 46, e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fan, C.T., Lin, J.C. and Lee, C.H. (2008) Taiwan Biobank: a project aiming to aid Taiwan's transition into a biomedical island. Pharmacogenomics, 9, 235–246. [DOI] [PubMed] [Google Scholar]
- 8. Kubo, M. and Guest, E. (2017) BioBank Japan project: epidemiological study. J. Epidemiol., 27, S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sudlow, C., Gallacher, J., Allen, N., Beral, V., Burton, P., Danesh, J., Downey, P., Elliott, P., Green, J., Landray, M. et al. (2015) UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med., 12, e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Check Hayden, E. (2017) The rise and fall and rise again of 23andMe. Nature, 550, 174–177. [DOI] [PubMed] [Google Scholar]
- 11. Evangelou, E., Warren, H.R., Mosen-Ansorena, D., Mifsud, B., Pazoki, R., Gao, H., Ntritsos, G., Dimou, N., Cabrera, C.P., Karaman, I. et al. (2018) Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat. Genet., 50, 1412–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Timmers, P.R., Mounier, N., Lall, K., Fischer, K., Ning, Z., Feng, X., Bretherick, A.D., Clark, D.W., eQTLGen Consortium, Agbessi, M. et al. (2019) Genomics of 1 million parent lifespans implicates novel pathways and common diseases and distinguishes survival chances. elife, 8, e39856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Karlsson Linner, R., Biroli, P., Kong, E., Meddens, S.F.W., Wedow, R., Fontana, M.A., Lebreton, M., Tino, S.P., Abdellaoui, A., Hammerschlag, A.R. et al. (2019) Genome-wide association analyses of risk tolerance and risky behaviors in over 1 million individuals identify hundreds of loci and shared genetic influences. Nat. Genet., 51, 245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee, J.J., Wedow, R., Okbay, A., Kong, E., Maghzian, O., Zacher, M., Nguyen-Viet, T.A., Bowers, P., Sidorenko, J., Karlsson Linner, R. et al. (2018) Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat. Genet., 50, 1112–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Weigl, K., Chang-Claude, J., Knebel, P., Hsu, L., Hoffmeister, M. and Brenner, H. (2018) Strongly enhanced colorectal cancer risk stratification by combining family history and genetic risk score. Clin. Epidemiol., 10, 143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sparano, J.A., Gray, R.J., Ravdin, P.M., Makower, D.F., Pritchard, K.I., Albain, K.S., Hayes, D.F., Geyer, C.E., Jr., Dees, E.C., Goetz, M.P. et al. (2019) Clinical and genomic risk to guide the use of adjuvant therapy for breast cancer. N. Engl. J. Med., 380, 2395–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Khera, A.V., Chaffin, M., Wade, K.H., Zahid, S., Brancale, J., Xia, R., Distefano, M., Senol-Cosar, O., Haas, M.E., Bick, A. et al. (2019) Polygenic prediction of weight and obesity trajectories from birth to adulthood. Cell, 177, 587–596.e589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Inouye, M., Abraham, G., Nelson, C.P., Wood, A.M., Sweeting, M.J., Dudbridge, F., Lai, F.Y., Kaptoge, S., Brozynska, M., Wang, T. et al. (2018) Genomic risk prediction of coronary artery disease in 480,000 adults: implications for primary prevention. J. Am. Coll. Cardiol., 72, 1883–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sirugo, G., Williams, S.M. and Tishkoff, S.A. (2019) The missing diversity in human genetic studies. Cell, 177, 1080. [DOI] [PubMed] [Google Scholar]
- 20. Martin, A.R., Kanai, M., Kamatani, Y., Okada, Y., Neale, B.M. and Daly, M.J. (2019) Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet., 51, 584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mostafavi, H., Harpak, A., Agarwal, I., Pritchard, J.K., Conley, D. and Przeworski, M. (2020) Variable prediction accuracy of polygenic scores within an ancestry group. elife, 9, e48376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Martin, A.R., Gignoux, C.R., Walters, R.K., Wojcik, G.L., Neale, B.M., Gravel, S., Daly, M.J., Bustamante, C.D. and Kenny, E.E. (2017) Human demographic history impacts genetic risk prediction across diverse populations. Am. J. Hum. Genet., 100, 635–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Duncan, L., Shen, H., Gelaye, B., Meijsen, J., Ressler, K., Feldman, M., Peterson, R. and Domingue, B. (2019) Analysis of polygenic risk score usage and performance in diverse human populations. Nat. Commun., 10, 3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gaziano, J.M., Concato, J., Brophy, M., Fiore, L., Pyarajan, S., Breeling, J., Whitbourne, S., Deen, J., Shannon, C., Humphries, D. et al. (2016) Million Veteran Program: a mega-biobank to study genetic influences on health and disease. J. Clin. Epidemiol., 70, 214–223. [DOI] [PubMed] [Google Scholar]
- 25. Sankar, P.L. and Parker, L.S. (2017) The Precision Medicine Initiative’s All of Us Research Program: an agenda for research on its ethical, legal, and social issues. Genet. Med., 19, 743–750. [DOI] [PubMed] [Google Scholar]
- 26. Daub, J.T., Hofer, T., Cutivet, E., Dupanloup, I., Quintana-Murci, L., Robinson-Rechavi, M. and Excoffier, L. (2013) Evidence for polygenic adaptation to pathogens in the human genome. Mol. Biol. Evol., 30, 1544–1558. [DOI] [PubMed] [Google Scholar]
- 27. Hofer, T., Ray, N., Wegmann, D. and Excoffier, L. (2009) Large allele frequency differences between human continental groups are more likely to have occurred by drift during range expansions than by selection. Ann. Hum. Genet., 73, 95–108. [DOI] [PubMed] [Google Scholar]
- 28. Iorio, A., De Angelis, F., Di Girolamo, M., Luigetti, M., Pradotto, L.G., Mazzeo, A., Frusconi, S., My, F., Manfellotto, D., Fuciarelli, M. et al. (2017) Population diversity of the genetically determined TTR expression in human tissues and its implications in TTR amyloidosis. BMC Genomics, 18, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Polimanti, R., Yang, C., Zhao, H. and Gelernter, J. (2015) Dissecting ancestry genomic background in substance dependence genome-wide association studies. Pharmacogenomics, 16, 1487–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bycroft, C., Freeman, C., Petkova, D., Band, G., Elliott, L.T., Sharp, K., Motyer, A., Vukcevic, D., Delaneau, O., O'Connell, J. et al. (2018) The UK Biobank resource with deep phenotyping and genomic data. Nature, 562, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Voight, B.F., Kudaravalli, S., Wen, X. and Pritchard, J.K. (2006) A map of recent positive selection in the human genome. PLoS Biol., 4, e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Grossman, S.R., Shlyakhter, I., Karlsson, E.K., Byrne, E.H., Morales, S., Frieden, G., Hostetter, E., Angelino, E., Garber, M., Zuk, O. et al. (2010) A composite of multiple signals distinguishes causal variants in regions of positive selection. Science, 327, 883–886. [DOI] [PubMed] [Google Scholar]
- 33. Sankararaman, S., Mallick, S., Dannemann, M., Prufer, K., Kelso, J., Paabo, S., Patterson, N. and Reich, D. (2014) The genomic landscape of Neanderthal ancestry in present-day humans. Nature, 507, 354–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wojcik, G.L., Graff, M., Nishimura, K.K., Tao, R., Haessler, J., Gignoux, C.R., Highland, H.M., Patel, Y.M., Sorokin, E.P., Avery, C.L. et al. (2019) Genetic analyses of diverse populations improves discovery for complex traits. Nature, 570, 514–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Magi, R., Horikoshi, M., Sofer, T., Mahajan, A., Kitajima, H., Franceschini, N., McCarthy, M.I., Cogent-Kidney Consortium, TDGC and Morris, A.P. (2017) Trans-ethnic meta-regression of genome-wide association studies accounting for ancestry increases power for discovery and improves fine-mapping resolution. Hum. Mol. Genet., 26, 3639–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Asimit, J.L., Hatzikotoulas, K., McCarthy, M., Morris, A.P. and Zeggini, E. (2016) Trans-ethnic study design approaches for fine-mapping. Eur. J. Hum. Genet., 24, 1330–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Koyama, S., Ito, K., Terao, C., Akiyama, M., Horikoshi, M., Momozawa, Y., Matsunaga, H., Ieki, H., Ozaki, K., Onouchi, Y. et al. (2020) Population-specific and trans-ancestry genome-wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Nat. Genet., 52, 1169–1177. [DOI] [PubMed] [Google Scholar]
- 38. Gelernter, J., Sun, N., Polimanti, R., Pietrzak, R., Levey, D.F., Bryois, J., Lu, Q., Hu, Y., Li, B., Radhakrishnan, K. et al. (2019) Genome-wide association study of post-traumatic stress disorder reexperiencing symptoms in >165,000 US veterans. Nat. Neurosci., 22, 1394–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mahajan, A., Spracklen, C.N., Zhang, W., Ng, M.C.Y., Petty, L.E., Kitajima, H., Yu, G.Z., Rueger, S., Speidel, L., Kim, Y.J. et al. (2020) Trans-ancestry genetic study of type 2 diabetes highlights the power of diverse populations for discovery and translation. medRxiv, in press, 2020.2009.2022.20198937. doi: 10.1101/2020.09.22.20198937. preprint: not peer reviewed September 23, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Scerri, E.M.L., Chikhi, L. and Thomas, M.G. (2019) Beyond multiregional and simple out-of-Africa models of human evolution. Nat Ecol Evol, 3, 1370–1372. [DOI] [PubMed] [Google Scholar]
- 41. Groucutt, H.S., Petraglia, M.D., Bailey, G., Scerri, E.M., Parton, A., Clark-Balzan, L., Jennings, R.P., Lewis, L., Blinkhorn, J., Drake, N.A. et al. (2015) Rethinking the dispersal of Homo sapiens out of Africa. Evol. Anthropol., 24, 149–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bentley, A.R., Callier, S.L. and Rotimi, C.N. (2020) Evaluating the promise of inclusion of African ancestry populations in genomics. NPJ Genom. Med., 5, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gravel, S., Zakharia, F., Moreno-Estrada, A., Byrnes, J.K., Muzzio, M., Rodriguez-Flores, J.L., Kenny, E.E., Gignoux, C.R., Maples, B.K., Guiblet, W. et al. (2013) Reconstructing Native American migrations from whole-genome and whole-exome data. PLoS Genet., 9, e1004023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Scott, E.M., Halees, A., Itan, Y., Spencer, E.G., He, Y., Azab, M.A., Gabriel, S.B., Belkadi, A., Boisson, B., Abel, L. et al. (2016) Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat. Genet., 48, 1071–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sjaarda, J., Gerstein, H.C., Kutalik, Z., Mohammadi-Shemirani, P., Pigeyre, M., Hess, S. and Pare, G. (2020) Influence of genetic ancestry on human serum proteome. Am. J. Hum. Genet., 106, 303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Morris, J.C., Schindler, S.E., McCue, L.M., Moulder, K.L., Benzinger, T.L.S., Cruchaga, C., Fagan, A.M., Grant, E., Gordon, B.A., Holtzman, D.M. et al. (2019) Assessment of racial disparities in biomarkers for Alzheimer disease. JAMA Neurol., 76, 264–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hackler, E., 3rd, Lew, J., Gore, M.O., Ayers, C.R., Atzler, D., Khera, A., Rohatgi, A., Lewis, A., Neeland, I., Omland, T. et al. (2019) Racial differences in cardiovascular biomarkers in the general population. J. Am. Heart Assoc., 8, e012729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Quintana-Murci, L. (2019) Human immunology through the lens of evolutionary genetics. Cell, 177, 184–199. [DOI] [PubMed] [Google Scholar]
- 49. Pavan, W.J. and Sturm, R.A. (2019) The genetics of human skin and hair pigmentation. Annu. Rev. Genomics Hum. Genet., 20, 41–72. [DOI] [PubMed] [Google Scholar]
- 50. Jablonski, N.G. and Chaplin, G. (2017) The colours of humanity: the evolution of pigmentation in the human lineage. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci., 372, 20160349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wilde, S., Timpson, A., Kirsanow, K., Kaiser, E., Kayser, M., Unterlander, M., Hollfelder, N., Potekhina, I.D., Schier, W., Thomas, M.G. et al. (2014) Direct evidence for positive selection of skin, hair, and eye pigmentation in Europeans during the last 5,000 y. Proc. Natl. Acad. Sci. U. S. A., 111, 4832–4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Barghi, N., Hermisson, J. and Schlotterer, C. (2020) Polygenic adaptation: a unifying framework to understand positive selection. Nat. Rev. Genet., 21, 769–781. [DOI] [PubMed] [Google Scholar]
- 53. Dashti, H.S., Daghlas, I., Lane, J.M., Huang, Y., Udler, M.S., Wang, H., Ollila, H.M., Jones, S.E., Kim, J., Wood, A.R. et al. (2021) Genetic determinants of daytime napping and effects on cardiometabolic health. Nat. Commun., 12, 900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Aguirre, M., Tanigawa, Y., Venkataraman, G.R., Tibshirani, R., Hastie, T. and Rivas, M.A. (2021) Polygenic risk modeling with latent trait-related genetic components. Eur. J. Hum. Genet., doi: 10.1038/s41431-021-00813-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Di Narzo, A., Frades, I., Crane, H.M., Crane, P.K., Hulot, J.S., Kasarskis, A., Hart, A., Argmann, C., Dubinsky, M., Peter, I. et al. (2021) Meta-analysis of sample-level dbGaP data reveals novel shared genetic link between body height and Crohn’s disease. Hum. Genet., doi: 10.1007/s00439-020-02250-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhao, X., Qiao, D., Yang, C., Kasela, S., Kim, W., Ma, Y., Shrine, N., Batini, C., Sofer, T., Taliun, S.A.G. et al. (2020) Whole genome sequence analysis of pulmonary function and COPD in 19,996 multi-ethnic participants. Nat. Commun., 11, 5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Palmer, M.R., Kim, D.S., Crosslin, D.R., Stanaway, I.B., Rosenthal, E.A., Carrell, D.S., Cronkite, D.J., Gordon, A., Du, X., Li, Y.K. et al. (2021) Loci identified by a genome-wide association study of carotid artery stenosis in the eMERGE network. Genet. Epidemiol., 45, 4–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhang, Y.X., Zhang, S.S., Ran, S., Liu, Y., Zhang, H., Yang, X.L., Hai, R., Shen, H., Tian, Q., Deng, H.W. et al. (2021) Three pleiotropic loci associated with bone mineral density and lean body mass. Mol. Gen. Genomics., 296, 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hindy, G., Aragam, K.G., Ng, K., Chaffin, M., Lotta, L.A., Baras, A., Regeneron Genetics, C., Drake, I., Orho-Melander, M., Melander, O. et al. (2020) Genome-wide polygenic score, clinical risk factors, and long-term trajectories of coronary artery disease. Arterioscler. Thromb. Vasc. Biol., 40, 2738–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen, M.H., Raffield, L.M., Mousas, A., Sakaue, S., Huffman, J.E., Moscati, A., Trivedi, B., Jiang, T., Akbari, P., Vuckovic, D. et al. (2020) Trans-ethnic and ancestry-specific blood-cell genetics in 746,667 individuals from 5 global populations. Cell, 182, 1198–1213.e1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. 1000 Genomes Project Consortium, Auton, A., Brooks, L.D., Durbin, R.M., Garrison, E.P., Kang, H.M., Korbel, J.O., Marchini, J.L., McCarthy, S., McVean, G.A. et al. (2015) A global reference for human genetic variation. Nature, 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li, J.Z., Absher, D.M., Tang, H., Southwick, A.M., Casto, A.M., Ramachandran, S., Cann, H.M., Barsh, G.S., Feldman, M., Cavalli-Sforza, L.L. et al. (2008) Worldwide human relationships inferred from genome-wide patterns of variation. Science, 319, 1100–1104. [DOI] [PubMed] [Google Scholar]
- 63. Zhou, W., Nielsen, J.B., Fritsche, L.G., Dey, R., Gabrielsen, M.E., Wolford, B.N., LeFaive, J., VandeHaar, P., Gagliano, S.A., Gifford, A. et al. (2018) Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet., 50, 1335–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hemani, G., Zheng, J., Elsworth, B., Wade, K.H., Haberland, V., Baird, D., Laurin, C., Burgess, S., Bowden, J., Langdon, R. et al. (2018) The MR-Base platform supports systematic causal inference across the human phenome. elife, 7, e34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Boyle, A.P., Hong, E.L., Hariharan, M., Cheng, Y., Schaub, M.A., Kasowski, M., Karczewski, K.J., Park, J., Hitz, B.C., Weng, S. et al. (2012) Annotation of functional variation in personal genomes using RegulomeDB. Genome Res., 22, 1790–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Machiela, M.J. and Chanock, S.J. (2015) LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics, 31, 3555–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Machiela, M.J. and Chanock, S.J. (2018) LDassoc: an online tool for interactively exploring genome-wide association study results and prioritizing variants for functional investigation. Bioinformatics, 34, 887–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wendt, F.R., Pathak, G.A., Overstreet, C., Tylee, D.S., Gelernter, J., Atkinson, E.G. and Polimanti, R. (2021) Characterizing the effect of background selection on the polygenicity of brain-related traits. Genomics, 113, 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Pers, T.H., Timshel, P. and Hirschhorn, J.N. (2015) SNPsnap: a web-based tool for identification and annotation of matched SNPs. Bioinformatics, 31, 418–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.