

Abstract
Multicentric Osteolysis, Nodulosis, and Arthropathy (MONA) syndrome is a rare genetic skeletal dysplasia. Its diagnosis can be deceptively similar to childhood-onset genetic skeletal dysplasias and juvenile idiopathic arthritis. We aimed to report the syndrome’s clinical and radiologic features with emphasis on skeletal manifestations. And establish relevant phenotype-genotype correlations. We evaluated two boys, 4-and-7-years-old with MONA syndrome. Both patients had consanguineous parents. We verified the diagnosis by correlating the outcomes of clinical, radiologic and molecular analysis. We specifically evaluated the craniofacial morphology and clinical and radiographic skeletal abnormalities. We contextualized the resultant phenotype-genotype correlations to publications on MONA and its differential diagnosis. Skeletal manifestations were the presenting symptoms and mostly restricted to hands and feet in terms of fixed extension deformity of the metacarpophalangeal and flexion deformity of the interphalangeal joints with extension deformity of big toes. There were arthritic symptoms in the older patient especially of the wrists and minute pathologic fractures. The skeletal radiographs showed osteopenia/dysplastic changes of hands and feet. Both patients had variants in the matrix metalloproteinase2 gene which conformed to phenotype of previously reported literature in one patient while the other had a novel variant which conformed to MONA phenotype. Craniofacial abnormalities were present. However, minimal extra-skeletal manifestations. Overall, there is an emerging distinctive skeletal pattern of involvement in terms of both clinical and radiographic features. This includes age of onset and location of presenting skeletal manifestations, chronological order of joint affection, longitudinal disease progression, specifics of skeletal radiographic pathology and craniofacial features. Nevertheless, physicians are cautioned against differential diagnosis of similar genetic skeletal dysplasias and juvenile idiopathic arthritis.
Keywords: Torg-Winchester syndrome, MONA syndrome, MMP2 gene mutations, Frank-Ter Haar syndrome, Juvenile idiopathic arthritis, Childhood osteoporosis
Graphical abstract
MONA Syndrome, Orthopedic Manifestations.

Highlights
-
•
Presenting manifestations erupt simultaneously in the hands and feet.
-
•
Skeletal manifestations proceed fairly rapidly in a distal-to-proximal fashion.
-
•
Radiographic features are a mixture of osteopenia, joint destruction and fractures.
-
•
Major disability may ensue in late childhood/adolescence, arthrogenic dysplasia
-
•
Non-skeletal manifestations are variable in terms of age of appearance and frequency
1. Introduction
Multicentric Osteolysis, Nodulosis, and Arthropathy (MONA) syndrome or spectrum disorders is an umbrella term for ultra-rare subtypes of primary skeletal dysplasias that are predominantly osteolytic with characteristic involvement of the carpal and tarsal bones. According to the latest nosology and classification of genetic skeletal disorders, MONA syndrome includes diseases involving two genes: the Matrix Metalloproteinase 2 (MMP2) gene [MIM # 259600], type IV collagenase or gelatinase and Matrix Metalloproteinase 14 (MMP14) [MIM # 277950]. Both genes are assumed to cause phenotype variants of the same disease. Additionally the spectrum includes Winchester syndrome (WNCHRS [MIM # 277950]) (Winchester et al., 1969), which is also known as Torg osteolysis syndrome, nodulosis arthropathy osteolysis (NAO) syndrome and Torg-Winchester syndrome (de Vos et al., 2019; Bhavani et al., 2016; Zankl et al., 2007; Al-Mayouf et al., 2000; de Vos et al., 2018). MONA syndrome is inherited through an autosomal recessive manner. Only 46 patients with 24 different variants in MMP2 are reported (Bhavani et al., 2016; Kröger et al., 2019). Only four patients with two variants in MMP14 have been reported (de Vos et al., 2019). The pathological variants cause disruption of normal collagenous turnover and loss of gelatinolytic activity of the metalloproteinase enzyme (Bhavani et al., 2016). The characteristic skeletal manifestations of MONA syndrome includes progressive deformities of hands and feet associated with carpal and tarsal bone osteolysis and osteopenia. Subcutaneous nodules may accompany deformities of the hands and feet and patients suffer arthropathy-like manifestations as pain, stiffness and swelling (de Vos et al., 2019; Al Aqeel et al., 2000). In the hands, the flexion deformity of the proximal interphalangeal joints (camptodactyly) is characteristically noted. Contractures ultimately spread proximally to involve larger joints, causing greater disability. A similar pattern usually occurs in the feet (de Vos et al., 2019; Bhavani et al., 2016; Al-Mayouf et al., 2000; Al Aqeel et al., 2000; Al Kaissi et al., 2011; Vanatka et al., 2011; Tuysuz et al., 2009; Rouzier et al., 2006). Non-skeletal manifestations include facial dysmorphism and oral (de Vos et al., 2019; Bhavani et al., 2016; Tuysuz et al., 2009; Temtamy et al., 2012; Ekbote et al., 2014; Jeong et al., 2010), dermatologic (de Vos et al., 2019; Al Kaissi et al., 2011; Rouzier et al., 2006; Ekbote et al., 2014; Jeong et al., 2010; Bader-Meunier et al., 2016), ophthalmological and cardiac manifestations (de Vos et al., 2019; Bhavani et al., 2016; Kröger et al., 2019; Tuysuz et al., 2009; Temtamy et al., 2012; Jeong et al., 2010; Castberg et al., 2013; Gok et al., 2010).
However, the rarity of MONA syndrome does not allow for complete uncovering of the phenotype pattern. Besides, the reported phenotype-genotype correlations have shown inconsistent and inconclusive findings (de Vos et al., 2019; Bhavani et al., 2016; Kröger et al., 2019; Rouzier et al., 2006; Azzollini et al., 2014). Some authors include Ter Haar syndrome (FTHS [MIM # 249420]) which is caused by homozygous mutation in the TKS4 gene among the MONA syndrome spectrum disorders (Chang et al., 2017; Bendon et al., 2012; Iqbal et al., 2010; van Steensel et al., 2007; Borrone et al., 1993). The autosomal dominant rare multicentric carpotarsal osteolysis syndrome (MCTO [MIM # 166300]) with or without nephropathy is another important differential diagnosis of MONA syndrome (Li et al., 2020; Park et al., 2018; Upadia et al., 2018; Stajkovska et al., 2018; Mumm et al., 2014; Zankl et al., 2012). The overlapping skeletal and non-skeletal features of these two genetic skeletal dysplasias compounds the diagnostic challenges. Additionally, MONA and Winchester syndromes (Winchester et al., 1969; Al Aqeel et al., 2000) among others (Al Kaissi et al., 2011; Li et al., 2020) can have a misleading radioclinical resemblance to juvenile idiopathic arthritis and mucopolysaccharidosis (MPS) type-I as Scheie syndrome (Hampe et al., 2020; Gökay et al., 2018). The treatment of MONA syndrome is palliative (Pichler et al., 2016) and little is known about the long-term prognosis apart from tendency to progression of skeletal features. Contrastingly, unattended MPS type-I usually runs a fatal course. The fact that MPS type-I as Scheie syndrome has effective bone therapeutics as enzyme replacement and hematopoietic stem cell transplantation (Jameson et al., 2019), puts further emphasis on making clear clinical distinctions between MPS type-I and MONA syndrome.
The complicated diagnostic workup of MONA syndrome necessitates a re-review of the reported phenotype-genotype correlations. By expanding the phenotype of MONA syndrome including the skeletal manifestations, we hope that a consensus on its diagnostic criteria could be reached. This report aimed to systematically describe the clinical manifestations in two children with molecularly-confirmed MONA syndrome with emphasis on the skeletal manifestations, and to establish relevant phenotype-genotype correlations.
2. Material and methods
This was an observational retrospective cross sectional study. The records of two unrelated and clinically, radiographically and genetically verified patients with Multicentric Osteolysis, Nodulosis, and Arthropathy (MONA) syndrome were reviewed. We retrieved relevant documents pertaining to history of present illness, general pediatric and neuro-orthopedic examination, skeletal radiology surveys, laboratory reports among other visceral imaging investigations. The genetic analysis reports were assessed and correlated with the clinical – especially skeletal – and radiologic profile to confirm the diagnosis. We revised all the literature on MONA syndrome comprehensively. In that regard we focused specifically on the skeletal clinical and radiographic features, and craniofacial abnormalities. Publications on MMP2 gene variants similar to those identified in our study were assembled and compared to contextualize the genetic-skeletal relationships. The study was approved by the Research Ethics Committee of Faculty of Medicine, Ain Shams University, Egypt. All the procedures followed in this study were in accordance with the ethical standards of the responsible committee on human experimentation and with the Declaration of Helsinki. Assessment of craniofacial dysmorphism is integral to the diagnosis and management of genetic syndromes (Allanson et al., 2009). Consequently, a documented permission to publish identifiable (unmasked) patient images was obtained from the patient's parent and supplied as supplemental material and uploaded with the submission.
3. Results
3.1. Patient 1
A six-and-a-half-year-old boy presented to the outpatient clinic with progressive joint deformities and stiffness of both hands and feet that erupted during the early years of life. He was a product of a consanguineous marriage and has three healthy siblings. He was provisionally diagnosed as one form of juvenile idiopathic arthritis and was subjected to orthopedic physical and occupational therapy with unsatisfactory results. At age three years he received a genetic testing in the form of whole exome sequencing. It confirmed the presence of a homozygous pathogenic missense variant in the Matrix Metalloproteinase 2 MMP2 gene (reference sequence, NM_004530.5), namely c.302G>A p.(Arg101His) causingan amino acid change from Arg to His at position 101 (Table 1). Shortly after genetic diagnosis, he received intravenous bisphosphonate therapy in the form of zoledronic acid at regular three-monthly intervals which later transitioned to four-monthly. At age five years he underwent single-event multiple surgical soft tissue releases of the hands and feet to correct the existing deformities. However, deformities recurred rapidly after surgery and new deformities appeared on top of the existing ones especially in the right hand. The patient had no significant functional limitations despite the existing deformities. The child's parents report an awkward gait occasionally and tenderness on mild trauma to the affected extremities but no frank pain. The patient had no history of motor developmental delay nor neurological manifestations nor history suggestive of other system affection.
Table 1.
Clinical and skeletal phenotype of c.302G>A p.(Arg101His) recurrent MMP2 gene variant identified in patient 1 of this study and previously reported literature.
| Author (year) | Patients N (sex) | Age at presentation (AOO) years | Cranio-facial | Subcutaneous nodules | MPJ, PIPJ, DIPJ | Wrist | Foot/ankle | Proximal/large joints, spine | Radiography | Other systems |
|---|---|---|---|---|---|---|---|---|---|---|
| Martignetti et al. (2001) | 3 (1 family, 2 M, 1 F) | 12 (5) 19 (6) 10 (6) |
Frontal bossing, hypertelorism, but no coarse facial features | Mobile, non-tender, hard, smooth surfaced plantar nodules | MPJ, PIPJ in the form of hand clawing | Involved | Involved | Elbowsa, knees, shoulders | Diffuse osteopenia, subcortical erosions, medullary expansion, arthritis/deformity of carpus, wrist IPJ(s) MPJ(s) and feet. Pathologic fractures of right humerus and femur (one patient) | None |
| Zankl et al. (2007) | 1 (F) | 13.5 years (8 mos) | Coarse facial features and flat nasal bridge | Painless plantar nodules | All involving (arthritis/fusion) | Involved | Toes (hyperextension) and ankle stiffness, the right foot showed arthritis/joint erosions of IPJ of big toeb, hallux valgus and marked varus (inversion) deformity. | Knees, elbows, sacroiliac joints | Diffuse osteopeniac, subcortical erosions, medullary expansion, arthritis of carpus, wrist. IPJ(s) and some MPJ(s), fusion of thumb IPJ and patella-femoral joint, striated distal femoral and proximal femur metaphyses. Non-united pathologic fracture proximal phalanx of index of left hand and of undisplaced (greenstick) fractures of bases of metacarpals of right hand. The distal ends of the metacarpals were broad in the right and tapering in the left hand (at same point in time). | High arched palate and epicanthal folds |
| Bhavani et al. (2016) | 1 (F) | 9 years (by birth) | Coarse facial features and bullous nose | Plantar nodules | MPJ, and PIPJ of the right hand especially little finger | Involved | Bilateral hallux valgus especially the right foot and hyperextension of the greater toe of left foot. Mild clawing of lesser toes bilaterally | None | Diffuse osteoporosis, subcortical erosions, medullary expansion of metacarpals, metatarsals and to lesser extent the phalanges. Mild carpotarsal osteolysis. Generally, hands more affected than feet | Hirsutism |
| Patient 1 of this study | 1 (M) | 6.5 years (2 years) | Prominent forehead, hypertelorism, a depressed nasal bridge, long philtrum, large ears, thick protruding lower lip, but no coarse facial features | None | MPJ and PIPJ (deformity, no arthritis) | Involved | Greater toes, extension deformity and right hallux valgus | None clinically (radiography not done) | Hands show diffuse osteopenia, bilateral carpal osteolysis more pronounced on the right, minute pathologic fractures of metacarpals and feet (with osteopenia and midtarsal osteolysis), tapering of the proximal ends of metacarpals and transverse metaphyseal striations. | Dentition problems, asymptomatic, mild congenital cardiac defect |
Zankl et al. (2007), patient had a compound heterozygote for two mutations in the MMP2 gene; NR, not reported; AOO, age of onset; MPJ, metacarpophalangeal joint; PIPJ, proximal interphalangeal joint; DIPJ, distal interphalangeal joint; IPJ, interphalangeal joint.
The article reported proximal joint involvement collectively in a case series with various mutations. We were unable to associate this involvement with our target mutation/patients.
Radiographs of the right foot were well-described while those of the left were neither shown nor detailed in the text.
Striations of femoral and tibial metaphyses and phalanx non-union are most likely secondary to alendronate therapy (a subtype of bisphosphonate) and not necessary disease-specific. The same applies to the striations found in patient 1 of this study.
3.1.1. Examination
By inspection the patient had no coarse facial features, prominent forehead, hypertelorism, a depressed nasal bridge, long philtrum, large ears, thick protruding lower lip. He had dentition problems in the form of premature loss and delayed eruption of teeth. On examination he had normal height and weight for age. The metacarpophalangeal joints of the right hand showed an extension deformity whereas the left hand showed a similar deformity in the little finger only. The proximal interphalangeal joints of the right hand showed severe flexion deformity of 90° whereas those of the left hand showed a similar deformity in the little finger only (Figs. 1 2a–c). Both wrists were markedly stiff. The big toes showed a semi rigid extension deformity of 30° at the metacarpophalangeal and interphalangeal joints of the right and left foot respectively. The right foot also showed a hallux valgus deformity. Apart from the described abnormalities in the hands and feet we did not detect any other skeletal affection. On palpation there were no subcutaneous nodules. However, a previous ultrasound examination of the hands and feet demonstrated two small and fairly defined subcutaneous nodules in relation to the dorsal aspect of fourth metacarpophalangeal joint and plantar aspect of the first metacarpophalangeal of left hand and foot respectively. And diffuse thickening and edema of the subcutaneous fat. Radiographs of the hand showed marked osteopenia and carpal osteolysis more pronounced in right hand. Metaphyseal striations of distal forearm bones were seen. Radiographs of the foot showed marked osteopenia and midtarsal osteolysis more pronounced in the right foot. Radiographs of the pelvis, long bones and larger joints and spine were unremarkable. Echocardiography revealed a small septal defect that was closing, and pelvi-abdominal ultrasound and complete blood count were unremarkable.
Fig. 1.

(a, b): Patient 1. Right hand. Note the claw hand attitude created by fixed extension deformity of metacarpophalangeal joints and flexion of the interphalangeal joints of lesser fingers. Note the scars of previous soft tissue releases. Realize the thumb is spared.
Fig. 2.

(a, b, c): Patient 1. Radioclinical presentation of hands and feet. (a) Right hand shows extension and flexion deformity of the metacarpophalangeal and proximal interphalangeal joints respectively. Left hand shows a similar deformity but restricted to the little finger. (b) Radiographs show diffuse osteopenia, bilateral carpal osteolysis more pronounced on the right (hollow arrows) with minute pathologic fractures of metacarpals and feet (not shown). Note tapering of the proximal ends of some metacarpals. Note the transverse metaphyseal striations (arrows) which indicate previous bisphosphonate therapy. (c) Note the diffuse osteopenia of both feet and the osteolysis of the midtarsal joints especially of the right foot which also shows a hallux valgus deformity.
3.2. Patient 2
A four-year-old boy presented to the outpatient clinic with multiple joint swellings and an occasional painful limping gait over the past two years. The boy was a product of a first degree consanguineous marriage and has three healthy siblings. The skeletal symptomatology was restricted to both hands and the left foot. The child complained of left foot pain especially on exertion with seasonal fluctuations. The patient was originally diagnosed as juvenile idiopathic arthritis and received regular medical treatment in the form of methotrexate and steroids with an inconclusive response. And was considered for biological therapy at another institution just before referral to our clinic. His RF and ANA were negative. He had no history of motor developmental delay nor neurological manifestations nor history suggestive of other system affection. All enzymatic activities for MPS types I, II, IIIB, IVA, VI and VII including alpha-L-iduronidase were within normal. However, results of whole exome sequencing confirmed the presence of a homozygous frameshift variant of uncertain significance in the MMP2 gene (reference sequence, NM_004530.5), c.40del p.(Leu14fs*) creating a shift in the reading frame starting at codon 14. The new reading frame ends in a stop codon 0 positions downstream. The patient is scheduled to receive intravenous zoledronic acid injections for osteopenia.
3.2.1. Examination
By inspection the patient had dysmorphism in the form of triangular face with frontal bossing, heavy thick eye brows with partial synophrys, depressed nasal bridge, bilateral asymmetric ptosis with down-slanting palpebral fissures and strabismus, hypertelorism, thick protruding lower lip with no gingival hypertrophy, low set posteriorly rotated bat ears and a relatively thin chin (Fig. 3). On examination his weight was 14 kg (−1.8 SD), height was 98 cm (−1.6 SD) and occipito-frontal circumference of 50 cm (−0.9 SD). There was mild diffuse swelling of both knees but no palpable synovial thickening. The metacarpophalangeal joints of both hands -especially the fourth and fifth fingers- were stiff in extension. The proximal interphalangeal joints of the fourth and fifth fingers of both hands showed a severe fixed flexion deformity (Fig. 4). The left foot was undersized with respect to girth and length and showed a convex sole which was confirmed radiographically with clawing of the big toe. Apart from the described abnormalities in the hands, feet and knees we did not detect any other deformities. On palpation there were no subcutaneous nodules. Radiographs of the hand showed thin cortex, osteopenia and dysplastic changes especially the 5th metacarpal and the 4th and 5th proximal phalanges (Fig. 5). Radiographs of the feet showed osteopenia and osteolysis especially of the left hind and midfoot namely calcaneus and talus (Fig. 6a–c). Radiographs of the pelvis, long bones and larger joints and spine anterior-posterior views were unremarkable except for the clavicles (supplementary file). However, the lateral view of the dorsolumbar spine showed mild anterior notching of lumbar 1 and lower dorsal vertebrae (Fig. 7). MRI of the left foot showed thickening of plantar fascia, signs suggestive of Achilles tendonitis and mild effusion of the ankle and tarsal joints. Bone density measurement using dual energy X-ray absorptiometry revealed osteopenia. Apart from the ptosis and strabismus no other ophthalmologic abnormalities were detected. Echocardiography revealed two tiny inter-atrial septum fenestrations shunting from left to right. The pelvi-abdominal ultrasound was unremarkable.
Fig. 3.

Patient 2. Craniofacial features. Dysmorphism in the form of triangular face with frontal bossing, heavy thick eye brows with partial synophyrus, depressed nasal bridge, bilateral asymmetric ptosis with down-slanting palpebral fissures and strabismus, hypertelorism, thick protruding lower lip, low set posteriorly rotated bat ears and a relatively thin chin.
Fig. 4.

(a, b, c, d): Patient 2. Clinical presentations of both hands. (a, b) Volar view. Note the claw hand attitude created by fixed extension deformity of metacarpophalangeal joints and flexion of the interphalangeal joints of the 4th and 5th fingers. (c, d) Dorsal view. Note the same clawing features seen in the previous figures.
Fig. 5.

Patient 2. Radiographic presentation of hands. Radiographs of both hands show widespread osteopenia (cortical thinning and hypodensity), coarse trabecular pattern, tapering of the proximal ends of the metacarpals (arrows) and confirm the clinical deformity.
Fig. 6.

(a, b, c): Patient 2. Radioclinical presentation of feet. (a) Note the undersized left foot, convexity of the sole i.e. loss of arch concavity (rocker bottom deformity) and clawing of big toe i.e. extension of metacarpophalangeal joint and flexion of the interphalangeal. (b) Note the diffuse osteopenia, cortical irregularities, disappearance of head of talus and dysplastic changes of left hindfoot with a vertical talus deformity (demarcation lines). The right foot is unremarkable. (c) Left foot. Note the loss of normal concavity of the foot arch and even emergence of a sole convexity. Mark the clawing of big toe in the form of extension deformity at the first metacarpophalangeal joint and flexion deformity at the interphalangeal the joint.
Fig. 7.

Patient 2. Close-up lateral view of Lumber 1 and lower dorsal vertebrae. Note the mild anterior notching or scalloping of the anterior bodies (arrows).
4. Discussion
This study's findings confirmed or at least harmonized with some of the previously reported skeletal manifestations of MONA syndrome (de Vos et al., 2019; Bhavani et al., 2016) in terms of age of onset, predominant and early radioclinical affection of hands and feet, fairly symmetrical lesions especially in the hands and feet of the second and first patients respectively, presence of craniofacial abnormalities and the potential for proximal spread to large joints as the knees. The characteristic extension deformity of the metacarpophalangeal joints of the hands has not been focused upon explicitly. Together with the characteristic flexion deformity of the proximal interphalangeal joints they make up the hand clawing attitude described in literature (de Vos et al., 2019). The importance of delineating a clear and detailed radioclinical skeletal phenotype pattern in genetic skeletal dysplasia in general and MONA spectrum disorders in specific cannot be overemphasized (EL-Sobky et al., 2017a; El-Sobky et al., 2017b). The literature on MONA syndrome has characterized the skeletal manifestations in the hands and feet broadly. However, further detailing is needed with respect to ray involvement, type and number of joints affected, laterality, type and severity of deformity and characteristic sparing of individual joints if any. Longitudinal studies can track the chronological order of joint involvement especially of the larger/proximal joints (de Vos et al., 2019). The early recurrence following orthopedic surgery in our first patient reaffirms the progressive nature of the syndrome and argues against early surgical intervention. We reported expansion of the middle third of clavicles which was reported sporadically in literature (de Vos et al., 2019; Tuysuz et al., 2009).
4.1. Differential diagnosis
The history of present illness of our patients underscores once again the importance of making a clear distinction between some genetic skeletal dysplasias including MONA syndrome and multicentric carpotarsal osteolysis syndrome, and the spectrum of juvenile idiopathic arthritis (Barut et al., 2017). And conforms to history of present illness described repeatedly in the literature on the above-named genetic skeletal syndromes respecting initial misdiagnosis as juvenile idiopathic arthritis (Zankl et al., 2007; Al Aqeel et al., 2000; Al Kaissi et al., 2011; Castberg et al., 2013; Gok et al., 2010; Li et al., 2020; Park et al., 2018; Upadia et al., 2018; Gökay et al., 2018; Zhuang et al., 2017; Sun et al., 2016). This applies also to a wider array of genetic skeletal dysplasias which can mimic juvenile idiopathic arthritis clinically as progressive pseudorheumatoid dysplasia (EL-Sobky et al., 2017a; Gamal et al., 2017; Gaboon et al., 2019; Torreggiani et al., 2019; Madhusudan et al., 2016; Taspinar et al., 2016; Kaya Akca et al., 2021; Al Kaissi et al., 2020). This has two considerable implications. Firstly, the misdiagnosis of a genetic skeletal dysplasia as a juvenile idiopathic arthritis may deprive patients of potentially curable/life-saving bone therapeutics (Otaify, 2019) as enzyme replacement (Khalifa et al., 2011), hematopoietic stem cell transplantation (El-Sobky et al., 2017b) and gene therapy (Otaify, 2019), genetic counselling of family and expose patients to potential toxicity of -non-indicated- rheumatologic pharmacotherapy. Although an effective treatment is still not available for MONA syndrome, the concept still holds. Secondly, the misdiagnosis of juvenile idiopathic arthritis as a genetic skeletal dysplasia may lead to rheumatologic disease exacerbation and irreversible joint destruction that may have otherwise been preventable with timely institution of therapy. On the other hand, MONA syndrome and multicentric carpotarsal osteolysis syndrome seem to have overlapping clinical and radiologic features. However, the autosomal dominant inheritance pattern and the frequent presence of renal affection in the latter are important differentiating tools. Besides, the presence of MAFB gene mutations on molecular testing confirms the diagnosis of multicentric carpotarsal osteolysis syndrome (Park et al., 2018; Upadia et al., 2018; Stajkovska et al., 2018). The characteristics of non-skeletal manifestations as oral and dental abnormalities may provide an additional differential diagnosis tool of MONA syndrome (Mehrez et al., 2019). Genetic analysis remains important to the differential diagnosis of skeletal dysplasia with childhood-onset osteoporosis as pathogenic variants in SGMS2 gene (Al Kaissi et al., 2011; Pekkinen et al., 2019; Robinson et al., 2020) and pathogenic variants of PLS3 gene (Kämpe et al., 2017), to skeletal dysplasias simulating juvenile idiopathic arthritis as pathogenic variants in WISP3 gene (Gaboon et al., 2019; Torreggiani et al., 2019) and to the differential diagnosis of MPS type-I (Hampe et al., 2020).
4.2. Genetic skeletal correlations
The pathogenic variant of MMP2 gene c.302G>A p.(Arg101His) of patient 1 has been previously reported in three families in three different countries namely Saudi Arabia (3 children) (Martignetti et al., 2001), South America (one child) (Zankl et al., 2007), and India (10 year old female) (Bhavani et al., 2016). The phenotype of the above-mentioned recurrent MMP2 gene variant is summarized in (Table 1). The clinical and radiographic profile of patient 1 in our study and similar variant reported in literature overlap considerably. This implies a fairly consistent phenotype-genotype pattern. Unlike previous reports, patient 1 did not have subcutaneous nodules. A less consistent pattern is drawn in regard to dissimilar MMP2 gene mutations reported in the literature (de Vos et al., 2019; Bhavani et al., 2016).
The homozygous frameshift variant of MMP2 gene c.40del p.(Leu14fs*) of patient 2. This variant was not reported before and is considered a variant of unknown significance. It is novel variant with characteristic features of bilateral ptosis, downward slanting of palpebral fissures, no gingival hypertrophy or subcutaneous nodule but bilateral hand deformities and early unilateral osteolysis of hind and midfoot bones etc. The previous description conforms to the phenotype of MONA syndrome (Fig. 8) (Table 2). Parents were unavailable for molecular testing. We consider this a study Limitation. Generally, the clinical –including non-skeletal manifestations- and radiographic profile of children with MONA syndrome may vary with respect to prevalence and severity and distribution of each manifestation. This is particularly explicable by the occasional discrepancy between the age of onset - chronology - of a particular manifestation and age of the patient at time of presentation, and the presence of intrafamilial variations (Kröger et al., 2019; Rouzier et al., 2006). This may explain the absence of pain in our two patients. It may also explain the relatively more aggressive osteolysis and wrist joint stiffness in our older patient. Together with absence of proximal –large- joint and spine involvement in our young patients. That is, absence of pain in our patients may be attributed to their relatively young age at the time of presentation. However, joint destruction, progressive osteolysis and subsequent emergence of pain are highly likely manifestations, but late in the natural course of disease, namely in late childhood or adolescence. Nevertheless, there seems to be an all-embracing clinical and radiologic pattern in patients with MONA syndrome. This is based upon our findings and a thorough literature review (Graphical abstract);
-
a)
Presenting symptoms are fundamentally skeletal in nature and occur in early childhood.
-
b)
Presenting manifestations occur simultaneously in the hands and feet especially fingers and toes e.g. clawing.
-
c)
Skeletal manifestations proceed fairly rapidly in a distal-to-proximal fashion to involve proximal joints and spine eventually.
-
d)
Radiographic features are a mixture of bone resorption, joint destruction and pathologic fractures i.e. an “arthrogenic” skeletal dysplasia.
-
e)
Considerable disability may ensue in late childhood or early adolescence.
-
f)
Non-skeletal manifestations are variable in terms of age of appearance and frequency.
Fig. 8.
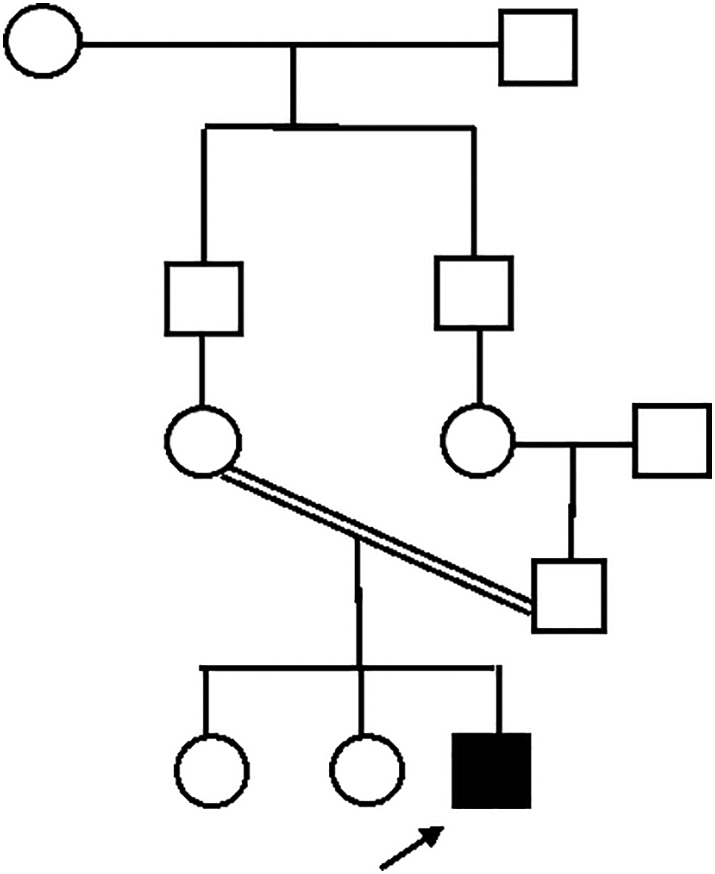
Family pedigree of patient 2.
Table 2.
Clinical and skeletal phenotype of novel variant of MMP2 gene identified in patient 2 of this study.
| Author (year) | Patients N (sex) | Age at presentation (AOO) years | Cranio-facial | Subcutaneous nodules | MPJ, PIPJ, DIPJ | Wrist | Foot/ankle | Proximal/large joints, spine | Radiography | Other systems |
|---|---|---|---|---|---|---|---|---|---|---|
| Patient 2 of this study | 1 (M) | 4 (2) | Triangular face with frontal bossing, heavy thick eye brows with partial synophyrus, depressed nasal bridge, bilateral asymmetric ptosis with down-slanting palpebral fissures and strabismus, hypertelorism, thick protruding lower lip, low set posteriorly rotated bat ears and a relatively thin chin | None | MPJ extension def., PIPJ flexion def. especially of the 4th and 5th fingers. No arthritis | None | Left foot was undersized with convex sole and clawing of big toe. No arthritis | Mildly affected knees, otherwise None | Hands and feet showed osteopenia and dysplastic changes, especially of the left hindfoot. Spine lateral views showed mild anterior scalloping of dorsolumbar junction vertebrae. Expansion of middle third of clavicles | Asymptomatic, mild congenital cardiac defect |
AOO, age of onset; MPJ, metacarpophalangeal joint; PIPJ, proximal interphalangeal joint; DIPJ, distal interphalangeal joint; IPJ, interphalangeal joint;
Although defective MMP2 enzyme activity is closely linked to osteolysis and abnormal extracellular matrix and bone remodeling found in MONA syndrome, the exact mechanism by which these pathological changes are induced remains controversial. Correlating MMP2 enzyme activity with genotype-phenotype variability noticed in MONA syndrome may provide insights into the various suggested roles of MMP2 in health and disease, and may inspire future experimental animal research (Li et al., 2021; Lieu et al., 2011).
5. Conclusion
The two reported children with multicentric osteolysis nodulosis arthropathy (MONA) syndrome caused by variants in MMP2 gene align with and expand the existing literature on skeletal phenotype in terms of the particulars of clinical and radiologic pattern and the novel variant detected in patient 2. Further meticulous reporting of patient phenotype in general and radioclinical skeletal phenotype in specific can produce a remarkable disease signature and resolve the unsettled current phenotype-genotype correlations. In that regard longitudinal studies have the potential to reveal the chronology and pattern of joint involvement, and assess the efficacy and complications of potential bone therapies like bisphosphonates. The importance of a multidisciplinary approach to diagnosis and management cannot be overemphasized.
6. Recommendations
The description of skeletal manifestations in the literature tended to be arbitrary. It occasionally lacked details as to the exact number of involved small joints of the hands or feet and the severity of involvement and incomplete information on the involvement of proximal joints or spine. We attempted to extrapolate the specifics of skeletal involvement from the posted clinical images. However, this may limit the accuracy of the reported findings. We therefore recommend accurate and systematic reporting of all skeletal manifestations preferably by a pediatric rheumatologist or orthopedic surgeon.
CRediT authorship contribution statement
Hanan Elsebaie; Conceptualization and design, revision of the manuscript critically for important intellectual content.
Mohamed Abdelhafiz Mansour: Conceptualization and design, revision of the manuscript critically for important intellectual content.
Solaf M. Elsayed: Interpretation of data for the work, revision of the manuscript critically for important intellectual content.
Shady Mahmoud: Acquisition and analysis of data, revision of the manuscript critically for important intellectual content.
Tamer A. El-Sobky: Design, interpretation of data for the work, drafting, writing and editing the manuscript.
All authors approved the final approval of the version to be published and are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Transparency document
Transparency document.
Declaration of competing interest
Nothing to declare.
Acknowledgments
Acknowledgement
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit domain.
Footnotes
The Transparency document associated with this article can be found, in online version.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bonr.2021.101106.
Contributor Information
Hanan Elsebaie, Email: hanan_salh@med.asu.edu.eg.
Tamer A. El-Sobky, Email: tamer.ahmed@med.asu.edu.eg.
Appendix A. Supplementary data
Supplementary material 1
References
- Al Aqeel A., Al Sewairi W., Edress B., Gorlin R.J., Desnick R.J., Martignetti J.A. Inherited multicentric osteolysis with arthritis: a variant resembling Torg syndrome in a Saudi family. Am. J. Med. Genet. 2000;93:11–18. doi: 10.1002/1096-8628(20000703)93:1<11::AID-AJMG3>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Al Kaissi A., Scholl-Buergi S., Biedermann R., Maurer K., Hofstaetter J.G., Klaushofer K. The diagnosis and management of patients with idiopathic osteolysis. Pediatr. Rheumatol. Online J. 2011;9:31. doi: 10.1186/1546-0096-9-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Kaissi A., Kenis V., Jemaa L.B., Sassi H., Shboul M., Grill F. Skeletal phenotype/genotype in progressive pseudorheumatoid chondrodysplasia. Clin. Rheumatol. 2020;39:553–560. doi: 10.1007/s10067-019-04783-z. [DOI] [PubMed] [Google Scholar]
- Allanson J.E., Cunniff C., Hoyme H.E., McGaughran J., Muenke M., Neri G. Elements morphology: standard of terminology for the head and face. Am. J. Med. Genet. A. 2009;149A:6–28. doi: 10.1002/ajmg.a.32612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Mayouf S.M., Majeed M., Hugosson C., Bahabri S. New form of idiopathic osteolysis: nodulosis, arthropathy and osteolysis (NAO) syndrome. Am. J. Med. Genet. 2000;93:5–10. doi: 10.1002/1096-8628(20000703)93:1<5::AID-AJMG2>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Azzollini J., Rovina D., Gervasini C., Parenti I., Fratoni A., Cubellis M.V. Functional characterisation of a novel mutation affecting the catalytic domain of MMP2 in siblings with multicentric osteolysis, nodulosis and arthropathy. J. Hum. Genet. 2014;59:631–637. doi: 10.1038/jhg.2014.84. [DOI] [PubMed] [Google Scholar]
- Bader-Meunier B., Bonafé L., Fraitag S., Breton S., Bodemer C., Baujat G. Mutation in MMP2 gene may result in scleroderma-like skin thickening. Ann. Rheum. Dis. 2016;75 doi: 10.1136/annrheumdis-2015-208182. [DOI] [PubMed] [Google Scholar]
- Barut K., Adrovic A., Şahin S., Kasapçopur Ö. Juvenile Idiopathic Arthritis. Balkan Med. J. 2017;34:90–101. doi: 10.4274/balkanmedj.2017.0111. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5394305/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendon C.L., Fenwick A.L., Hurst J.A., Nürnberg G., Nürnberg P., Wall S.A. Frank-ter Haar syndrome associated with sagittal craniosynostosis and raised intracranial pressure. BMC Med. Genet. 2012;13:104. doi: 10.1186/1471-2350-13-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhavani G.S., Shah H., Shukla A., Gupta N., Gowrishankar K., Rao A.P. Clinical and mutation profile of multicentric osteolysis nodulosis and arthropathy. Am. J. Med. Genet. A. 2016;170A:410–417. doi: 10.1002/ajmg.a.37447. [DOI] [PubMed] [Google Scholar]
- Borrone C., Di Rocco M., Crovato F., Camera G., Gambini C. New multisystemic disorder involving heart valves, skin, bones, and joints in two brothers. Am. J. Med. Genet. 1993;46:228–234. doi: 10.1002/ajmg.1320460225. [DOI] [PubMed] [Google Scholar]
- Castberg F.C., Kjaergaard S., Mosig R.A., Lobl M., Martignetti C., Martignetti J.A. Multicentric osteolysis with nodulosis and arthropathy (MONA) with cardiac malformation, mimicking polyarticular juvenile idiopathic arthritis: case report and literature review. Eur. J. Pediatr. 2013;172:1657–1663. doi: 10.1007/s00431-013-2102-8. [DOI] [PubMed] [Google Scholar]
- Chang T.C., Bauer M., Puerta H.S., Greenberg M.B., Cavuoto K.M. Ophthalmic findings in Frank-ter Haar syndrome: report of a sibling pair. J AAPOS. 2017;21:514–516. doi: 10.1016/j.jaapos.2017.07.216. [DOI] [PubMed] [Google Scholar]
- Ekbote A.V., Danda S., Zankl A., Mandal K., Maguire T., Ungerer K. Patient with mutation in the matrix metalloproteinase 2 (MMP2) gene - a case report and review of the literature. J. Clin. Res. Pediatr. Endocrinol. 2014;6:40–46. doi: 10.4274/Jcrpe.1166. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3986738/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- EL-Sobky T.A., El-Haddad A., Elsobky E., Elsayed S.M., Sakr H.M. Reversal of skeletal radiographic pathology in a case of malignant infantile osteopetrosis following hematopoietic stem cell transplantation. Egypt. J. Radiol. Nucl. Med. 2017;48:237–243. doi: 10.1016/j.ejrnm.2016.12.013. [DOI] [Google Scholar]
- El-Sobky T.A., Shawky R.M., Sakr H.M., Elsayed S.M., Elsayed N.S., Ragheb S.G. A systematized approach to radiographic assessment of commonly seen genetic bone diseases in children: a pictorial review. J. Musculoskelet. Surg. Res. 2017;1:25–32. doi: 10.4103/jmsr.jmsr_28_17. [DOI] [Google Scholar]
- Gaboon N.E.A., Parveen A., El Beheiry A., Al-Aama J.Y., Alsaedi M.S., Wasif N. A Novel Homozygous frameshift mutation in CCN6 causing progressive pseudorheumatoid dysplasia (pprd) in a consanguineous Yemeni family. Front. Pediatr. 2019;7:245. doi: 10.3389/fped.2019.00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamal R., Elsayed S.M., TA EL-Sobky, Elabd H.S. Pseudoachondroplasia in a child: the role of anthropometric measurements and skeletal imaging in differential diagnosis. Egypt. J. Radiol. Nucl. Med. 2017;48:245–250. doi: 10.1016/j.ejrnm.2016.10.007. [DOI] [Google Scholar]
- Gok F., Crettol L.M., Alanay Y., Hacihamdioglu B., Kocaoglu M., Bonafe L. Clinical and radiographic findings in two brothers affected with a novel mutation in matrix metalloproteinase 2 gene. Eur. J. Pediatr. 2010;169:363–367. doi: 10.1007/s00431-009-1028-7. [DOI] [PubMed] [Google Scholar]
- Gökay S., Kardaş F., Kendirci M., Sözeri B. Arthropathy-like findings and a carpal tunnel syndrome as the presenting features of Scheie syndrome: three cases from the same family. Turk. J. Pediatr. 2018;60:344–347. doi: 10.24953/turkjped.2018.03.020. [DOI] [PubMed] [Google Scholar]
- Hampe C.S., Eisengart J.B., Lund T.C., Orchard P.J., Swietlicka M., Wesley J. Mucopolysaccharidosis type I: a review of the natural history and molecular pathology. Cells. 2020;9:1838. doi: 10.3390/cells9081838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal Z., Cejudo-Martin P., de Brouwer A., van der Zwaag B. Disruption of the podosome adaptor protein TKS4 (SH3PXD2B) causes the skeletal dysplasia, eye, and cardiac abnormalities of Frank-Ter Haar Syndrome. Am. J. Hum. Genet. 2010;86:254–261. doi: 10.1016/j.ajhg.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jameson E., Jones S., Remmington T. Enzyme replacement therapy with laronidase (Aldurazyme®) for treating mucopolysaccharidosis type I. Cochrane Database Syst. Rev. 2019;6 doi: 10.1002/14651858.CD009354.pub5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong S.Y., Kim B.Y., Kim H.J., Yang J.A., Kim O.H. A novel homozygous MMP2 mutation in a patient with Torg-Winchester syndrome. J. Hum. Genet. 2010;55:764–766. doi: 10.1038/jhg.2010.102. [DOI] [PubMed] [Google Scholar]
- Kämpe A.J., Costantini A., Mäkitie R.E. PLS3 sequencing in childhood-onset primary osteoporosis identifies two novel disease-causing variants. Osteoporos. Int. 2017;28:3023–3032. doi: 10.1007/s00198-017-4150-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya Akca U., Simsek Kiper P.O., Urel Demir G., Sag E., Atalay E., Utine G.E., Alikasifoglu M. Genetic disorders with symptoms mimicking rheumatologic diseases: a single-center retrospective study. Eur. J. Med. Genet. 2021;64:104185. doi: 10.1016/j.ejmg.2021.104185. [DOI] [PubMed] [Google Scholar]
- Khalifa A.S., Tantawy A.A., Shawky R.M., Monir E., Elsayed S.M., Fateen E. Outcome of enzyme replacement therapy in children with Gaucher disease: the Egyptian experience. Egypt. J. Med. Hum. Genet. 2011;12:9–14. doi: 10.1016/j.ejmhg.2011.02.008. [DOI] [Google Scholar]
- Kröger L., Löppönen T., Ala-Kokko L., Kröger H., Jauhonen H.M., Lehti K. A novel mutation in the matrix metallopeptidase 2 coding gene associated with intrafamilial variability of multicentric osteolysis, nodulosis, and arthropathy. Mol. Genet. Genom. Med. 2019;7 doi: 10.1002/mgg3.802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Shi L., Lau K., Ma Y., Jia S., Gao X. Identification of a novel mutation in the MAFB gene in a pediatric patient with multicentric carpotarsal osteolysis syndrome using next-generation sequencing. Eur. J. Med. Genet. 2020;63:103902. doi: 10.1016/j.ejmg.2020.103902. [DOI] [PubMed] [Google Scholar]
- Li X., Jin L., Tan Y. Different roles of matrix metalloproteinase 2 in osteolysis of skeletal dysplasia and bone metastasis (Review) Mol. Med. Rep. 2021;23(1):70. doi: 10.3892/mmr.2020.11708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieu S., Hansen E., Dedini R. Impaired remodeling phase of fracture repair in the absence of matrix metalloproteinase-2. Dis. Model. Mech. 2011;4(2):203–211. doi: 10.1242/dmm.006304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhusudan S., Gupta A., Prakash M., Matta D., Suri D., Singh S. Camptodactyly-arthropathy-coxa vara-pericarditis (CACP) syndrome: a mimicker of juvenile idiopathic arthritis. Scand. J. Rheumatol. 2016;45:77–78. doi: 10.3109/03009742.2015.1085085. [DOI] [PubMed] [Google Scholar]
- Martignetti J.A., Aqeel A.A., Sewairi W.A., Boumah C.E., Kambouris M., Mayouf S.A. Mutation of the matrix metalloproteinase 2 gene (MMP2) causes a multicentric osteolysis and arthritis syndrome. Nat. Genet. 2001;28:261–265. doi: 10.1038/90100. [DOI] [PubMed] [Google Scholar]
- Mehrez M.I., Hassib N., Sayed I., Aboul-Ezz E., Ramzy M.I., El-Hadidi S.M. Genetic syndromes with premature loss of teeth: a retrospective study and a suggested classification. Middle East J. Med. Genet. 2019;8:100–106. doi: 10.4103/MXE.MXE_23_19. [DOI] [Google Scholar]
- Mumm S., Huskey M., Duan S., Wenkert D., Madson K.L., Gottesman G.S. Multicentric carpotarsal osteolysis syndrome is caused by only a few domain-specific mutations in MAFB, a negative regulator of RANKL-induced osteoclastogenesis. Am. J. Med. Genet. A. 2014;164A:2287–2293. doi: 10.1002/ajmg.a.36641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otaify G.A. Bone-specific therapeutic modalities for genetic skeletal diseases. Middle East J. Med. Genet. 2019;8:69–82. doi: 10.4103/MXE.MXE_3_20. [DOI] [Google Scholar]
- Park P.G., Kim K.H., Hyun H.S., Lee C.H., Park J.S., Kie J.H. Three cases of multicentric carpotarsal osteolysis syndrome: a case series. BMC Med. Genet. 2018;19:164. doi: 10.1186/s12881-018-0682-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekkinen M., Terhal P.A., Botto L.D., Henning P., Mäkitie R.E., Roschger P. Osteoporosis and skeletal dysplasia caused by pathogenic variants in SGMS2. JCI Insight. 2019;4 doi: 10.1172/jci.insight.126180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichler K., Karall D., Kotzot D., Steichen-Gersdorf E., Rümmele-Waibel A., Mittaz-Crettol L. Bisphosphonates in multicentric osteolysis, nodulosis and arthropathy (MONA) spectrum disorder - an alternative therapeutic approach. Sci. Rep. 2016;6:34017. doi: 10.1038/srep34017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.E., Bardai G., Veilleux L.N., Glorieux F.H., Rauch F. Musculoskeletal phenotype in two unrelated individuals with a recurrent nonsense variant in SGMS2. Bone. 2020;134:115261. doi: 10.1016/j.bone.2020.115261. [DOI] [PubMed] [Google Scholar]
- Rouzier C., Vanatka R., Bannwarth S., Philip N., Coussement A., Paquis-Flucklinger V. A novel homozygous MMP2 mutation in a family with Winchester syndrome. Clin. Genet. 2006;69:271–276. doi: 10.1111/j.1399-0004.2006.00584.x. [DOI] [PubMed] [Google Scholar]
- Stajkovska A., Mehandziska S., Stavrevska M., Jakovleva K., Nikchevska N., Mitrev Z. Trio clinical exome sequencing in a patient with multicentric carpotarsal osteolysis syndrome: first case report in the Balkans. Front. Genet. 2018;9:113. doi: 10.3389/fgene.2018.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel M.A., Ceulen R.P., Delhaas T., de Die-Smulders C. Two Dutch brothers with Borrone dermato-cardio-skeletal syndrome. Am. J. Med. Genet. A. 2007;143A:1223–1226. doi: 10.1002/ajmg.a.31719. [DOI] [PubMed] [Google Scholar]
- Sun K., Barlow B., Malik F., Inglis A., Figgie M., Goodman S. Total hip arthroplasty in a patient with multicentric carpotarsal osteolysis: a case report. HSS J. 2016;12:177–181. doi: 10.1007/s11420-015-9478-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taspinar O., Kelesoglu F., Keskin Y., Uludag M. Progressive pseudorheumatoid dysplasia misdiagnosed as seronegative juvenile idiopathic arthritis. Ethiop. J. Health Sci. 2016;26:397–400. doi: 10.4314/ejhs.v26i4.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temtamy S.A., Ismail S., Aglan M.S., Ashour A.M., Hosny L.A., El-Badry T.H. A report of three patients with MMP2 associated hereditary osteolysis. Genet. Couns. 2012;23:175–184. https://pubmed.ncbi.nlm.nih.gov/22876575/ [PubMed] [Google Scholar]
- Torreggiani S., Torcoletti M., Campos-Xavier B., Baldo F., Agostoni C., Superti-Furga A. Progressive pseudorheumatoid dysplasia: a rare childhood disease. Rheumatol. Int. 2019;39:441–452. doi: 10.1007/s00296-018-4170-6. [DOI] [PubMed] [Google Scholar]
- Tuysuz B., Mosig R., Altun G., Sancak S., Glucksman M.J., Martignetti J.A. A novel matrix metalloproteinase 2 (MMP2) terminal hemopexin domain mutation in a family with multicentric osteolysis with nodulosis and arthritis with cardiac defects. Eur. J. Hum. Genet. 2009;17:565–572. doi: 10.1038/ejhg.2008.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadia J., Gomes A., Weiser P., Descartes M. A familial case of multicentric carpotarsal osteolysis syndrome and treatment outcome. J. Pediatr. Genet. 2018;7:174–179. doi: 10.1055/s-0038-1657760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanatka R., Rouzier C., Lambert J.C., Leroux C., Coussement A. Winchester syndrome: the progression of radiological findings over a 23-year period. Skelet. Radiol. 2011;40:347–351. doi: 10.1007/s00256-010-1033-y. [DOI] [PubMed] [Google Scholar]
- de Vos I.J.H.M., Tao E.Y., Ong S.L.M., Goggi J.L., Scerri T., Wilson G.R. Functional analysis of a hypomorphic allele shows that MMP14 catalytic activity is the prime determinant of the Winchester syndrome phenotype. Hum. Mol. Genet. 2018;27:2775–2788. doi: 10.1093/hmg/ddy168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vos I.J.H.M., Wong A.S.W., Welting T.J.M., Coull B.J., van Steensel M.A.M. Multicentric osteolytic syndromes represent a phenotypic spectrum defined by defective collagen remodeling. Am. J. Med. Genet. A. 2019;179:1652–1664. doi: 10.1002/ajmg.a.61264. [DOI] [PubMed] [Google Scholar]
- Winchester P., Grossman H., Lim W.N., Danes B.S. A new acid mucopolysaccharidosis with skeletal deformities simulating rheumatoid arthritis. Am. J. Roentgenol. Radium Therapy, Nucl. Med. 1969;106:121–128. doi: 10.2214/ajr.106.1.121. [DOI] [PubMed] [Google Scholar]
- Zankl A., Pachman L., Poznanski A., Bonafé L., Wang F., Shusterman Y. Torg syndrome is caused by inactivating mutations in MMP2 and is allelic to NAO and Winchester syndrome. J. Bone Miner. Res. 2007;22:329–333. doi: 10.1359/jbmr.061013. [DOI] [PubMed] [Google Scholar]
- Zankl A., Duncan E.L., Leo P.J., Clark G.R., Glazov E.A., Addor M.C. Multicentric carpotarsal osteolysis is caused by mutations clustering in the amino-terminal transcriptional activation domain of MAFB. Am J Hum Genet 2012;90:494–501. Erratum. Am. J. Hum. Genet. 2012;94:643. doi: 10.1016/j.ajhg.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang L., Adler S., Aeberli D., Villiger P.M., Trueb B. Identification of a MAFB mutation in a patient with multicentric carpotarsal osteolysis. Swiss Med. Wkly. 2017;147:w14529. doi: 10.4414/smw.2017.14529. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Transparency document.
Supplementary material 1