

Abstract
The 5-year survival rate of pancreatic ductal carcinoma (PDAC) patients is <10% despite progress in clinical medicine. Strategies to prevent the development of PDAC are urgently required. The flavonoids Luteolin (Lut) and hesperetin (Hes) may be cancer-chemopreventive, but effects on pancreatic carcinogenesis in vivo have not been studied. Here, the chemopreventive effects of Lut and Hes on pancreatic carcinogenesis are assessed in the BOP-induced hamster PDAC model. Lut but not Hes suppressed proliferation of pancreatic intraepithelial neoplasia (PanIN) and reduced the incidence and multiplicity of PDAC in this model. Lut also inhibited the proliferation of hamster and human pancreatic cancer cells in vitro. Multi-blot and microarray assays revealed decreased phosphorylated STAT3 (pSTAT3) and dihydropyrimidine dehydrogenase (DPYD) on Lut exposure. To explore the relationship between DPYD and STAT3 activity, the former was silenced by RNAi or overexpressed using expression vectors, and the latter was inactivated by small molecule inhibitors or stimulated by IL6 in human PDAC cells. DPYD knock-down decreased, and overexpression increased, pSTAT3 and cell proliferation. DPYD expression was decreased by inactivation of STAT3 and increased by its activation. The frequency of pSTAT3-positive cells and DPYD expression was significantly correlated and was decreased in parallel by Lut in the hamster PDAC model. Finally, immunohistochemical analysis in 73 cases of human PDAC demonstrated that DPYD expression was positively correlated with the Ki-67 labeling index, and high expression was associated with poor prognosis. These results indicate that Lut is a promising chemopreventive agent for PDAC, targeting a novel STAT3-DPYD pathway.
This study describes the chemopreventive effects of luteolin on pancreatic carcinogenesis via STAT3 inactivation caused by DPYD down-regulation, resulting in suppression of cell proliferation. This is consistent with the association of high DPYD expression with the poor prognosis of pancreatic cancer.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) remains one of the most lethal therapeutically-nonresponsive cancers, despite developments and improvements of cancer therapy (1). In Japan, about 34 000 patients were newly diagnosed with pancreatic cancer in 2017, and almost the same numbers of deaths were recorded. The overall 5-year survival rate is only about 7.5% in Japan, imbuing PDAC with the most dismal prognosis of all solid tumors (2). This is attributed to its aggressive biological behavior, lack of symptoms enabling early clinical diagnosis, lack of early effective screening modalities and lack of effective treatments for advanced disease (3). Thus, there is an urgent need for new therapies, but effective chemoprevention is also imperative. Many potentially chemopreventive agents have been described, falling into the two main categories of phytochemicals or drugs. The latter include COX inhibitors, metformin and statins, which are at least partially effective in vitro and in vivo for PDAC chemoprevention but are of limited use in patients with drug adaptation (4). Phytochemicals such as curcumin, green tea, vitamins, retinoids and flavonoids in various foods and drinks were also reported to be effective in vitro and in vivo for PDAC chemoprevention (4,5). They have low toxicity, but also limited efficacy.
Luteolin (3’,4’,5,7-tetrahydroxy flavone; Lut) is an aglycone flavone, present in celery, green pepper, parsley and perilla leaf. It possesses many beneficial properties including antioxidant, anti-inflammatory, anti-microbial and anti-diabetic actions. In East Asian countries including Japan, leaves and seeds of perilla, known to contain high levels of Lut, are commonly consumed. Some anti-tumor effects of Lut on cancer in vivo including lung, colon, liver, ovary, breast, prostate, urinary bladder and non-alcoholic steatohepatitis (NASH)-related liver carcinoma have already been reported (6–10). Concerning pancreatic cancer, one study reported that combination therapy of Lut and gemcitabine inhibited tumor growth in an orthotopic mouse model, although differences between the effects of combination therapy-versus-gemcitabine alone did not achieve statistical significance (11). In vitro, some studies reported that Lut more effectively inhibited cell proliferation than other flavonoids such as apigenin, quercetin, kaempferol, or naringenin (12). One mechanism of inhibition of cell proliferation and the induction of apoptosis by Lut in human pancreatic cell lines is considered to be via the inhibition of GSK3β through NF-κB signaling and fatty acid synthase (12,13). Another study demonstrated that Lut inhibited angiogenesis by down-regulation of VEGF, and also inhibited the epithelial-mesenchymal transition (EMT) associated with down-regulation of STAT3 signaling in vitro (14,15).
Hesperetin (3’,5,7-trihydroxy-4’methoxy flavanone; Hes) is an aglycon flavonoid, present in citrus. One cohort study in Japan suggested that citrus consumption, especially every day, was associated with reduced pancreatic cancer occurrence (RR = 0.62, 95% CI = 0.38–1.00) (16). Some experimental in vivo studies reported that Hes inhibited oral, esophageal and colon carcinogenesis (17–19). In an in vitro study, combined treatment with naringenin and hesperetin, which mimic extracts of Citrus unshu peel, inhibited cell proliferation through attenuation of phosphorylated FAK and p38 expression (20). These data indicate that both Lut and Hes may be promising flavonoid dietary chemopreventive agents. However, there are no studies on the chemopreventive effects of these two flavonoids for pancreatic carcinogenesis.
Therefore, in the present study, we investigated the chemopreventive effects of Lut and Hes on pancreatic carcinogenesis, and sought to clarify the molecular mechanisms of their chemopreventive action against PDAC using in vitro experiments, in vivo models and human clinical cases.
Materials and methods
Details of the Materials and Methods are given in Supplementary Materials and Methods, available at Carcinogenesis Online.
Animal model
Five-week-old female Syrian golden hamsters were purchased from Japan SLC, Inc. (Shizuoka, Japan) and acclimated to the animal facility for one week. They were maintained in plastic cages on hardwood chips, in an air-conditioned, specific pathogen-free animal room at 22 ± 2°C and 50% humidity with a 12/12h light-dark cycle. The Quick Fat diet (crude fat, 13.6%; crude protein, 24.2%; total calories, 4.06 kcal/g) (CLEA Japan, Tokyo, Japan) was provided as high-fat chow. Lut and Hes were purchased from Tokyo Chemical Industry Co., Ltd (Tokyo, Japan) and N-nitroso-bis(2-oxopropyl)amine (BOP) was obtained from Santa Cruz Biotechnology (Dallas, Texas). All animal experiments were performed under protocols approved by the Institutional Animal Care and Use Committee of Nagoya City University School of Medical Sciences. Hamster models of BOP-induced pancreatic carcinogenesis on the Quick Fat diet were reported previously (21). Briefly, a total of 53 female hamsters at 6 weeks of age received four subcutaneous injections of BOP (on days 1, 3, 5 and 7) at a dose of 10 mg/kg body weight. All hamsters were kept on a high-fat diet. One week after the last BOP injection, they were randomly divided into four groups and given Lut or Hes in the diet for 13 weeks as follows: Lut 100 ppm (n = 13), Hes 100 ppm (n = 13), Hes 1000 ppm (n = 13) and Control (n = 14). Food and water were available ad libitum, and amounts consumed and body weights were measured weekly.
Histopathological analysis
At autopsy, major organs including liver, kidneys and lungs were excised and fixed in 10% buffered formalin, except for the pancreas, and routinely processed for hematoxylin and eosin (H&E)-stained sections. For the pancreas, the three anatomical parts (gastric, splenic and duodenal lobes) were spread out on filter paper and then fixed in 10% buffered formalin or frozen for RNA extraction. Pancreatic lesions were histopathologically diagnosed as PDAC or pancreatic intraepithelial neoplasias (PanINs) by two experienced pathologists (H.K, S.S). PanINs correspond to lesions in humans that are referred to as PanIN1, 2 and 3. The score for PanIN progression was calculated by weighting the lesion (normal = 0, PanIN1 = 1. PanIN2 = 2, PanIN3 = 3, PDAC = 4). Sections were also used for Azan staining or immunohistochemistry of Ki-67 (SP6, Abcam plc, Cambridge, UK), phosphorylated STAT3 (Tyr705) (pSTAT3) (Cell Signaling Technology, Danvers, MA, 1:200), dihydropyrimidine dehydrogenase (DPYD) (Abcam, 1:100), and αSMA (Agilent Technologies Inc., Santa Clara, CA, 1:4000) for hamster tissue. Labeling indices for Ki-67 and pSTAT3 were generated by counting over one thousand cells in hamster PDACs and PanINs and were expressed as percentages of positive cells. DPYD intensities were evaluated from five separate locations in PanIN lesions using a Biorevo BZ-9000 microscope and the associated software (KEYENCE, Osaka, Japan). αSMA-positive areas or blue-stained areas of Azan in PDAC were also measured by Biorevo BZ-9000 microscopy.
Cell culture
Human pancreatic cancer cell lines (MIAPaCa2, PANC1, BxPC3, KP4, HuPT3, PK1, PA-TU-8988T, TCCPAN2 and AsPC1) were obtained from the American Type Culture Collection (ATCC, Rockville, MD). They were maintained in RPMI-1640 (Wako Pure Chemical Industries Co. Ltd., Osaka, Japan) supplemented with 10% fetal bovine serum (FBS). The BOP-induced hamster pancreatic cancer cell line (HPD1NR) was a kind gift of Dr. Masahiro Tsutsumi. These cells were used in previous studies (5,22). They were maintained in DMEM (Wako) supplemented with 10% FBS. Cells were cultured at 37°C in 5% CO2 humidified air. Cell authentication (STR profile) of all human cancer cell lines was performed using the GenePrint System (10 loci) by BEX Co. Ltd within six months of use.
Cell proliferation assay
Cell proliferation was assessed by the 4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate tetrazolium salt (WST-1) assay (Roche Applied Science, Mannheim, Germany). Briefly, MIAPaCa2, PANC1, KP4 and HPD1NR cells were seeded into 96-well culture plates at a concentration of 1.0 × 104 cells/50 μL/well and incubated overnight. Cells were subsequently treated with Lut (final concentration: 0–100 μM) or siDPYD or Stattic (Abcam) as a selective STAT3 inhibitor (final concentration: 0–10 μM) (23). After 48 hours, cells were incubated for a further 2 hours with WST-1 reagent at 37°C, and the absorbance of each well was measured at 440 nm. Cell proliferation was expressed as a proportion of untreated control cells.
Cell cycle analysis
MIAPaCa2 and KP4 cells were treated with 25 or 50 μM Lut, and subsequently, suspensions were prepared and stained with propidium iodide (Guava Cell Cycle Reagent, Merck, Darmstadt, Germany) according to the Guava Cell Cycle Assay protocol. The absorbance of fluorescent dye was measured using the Guava easyCyte Single system (Merck).
Multi-blotting assay
The protocol for this assay was the same as for the usual Western blotting method illustrated in Supplementary Material and Methods, available at Carcinogenesis Online, up to the point where proteins from MIAPaCa2 or PANC1 cells were transferred to membranes, except that the applied amounts were 300 μg. Thereafter, the membrane was inserted into the Mini-PROTEAN II Multiscreen Apparatus (Bio-Rad Laboratories, Inc., Hercules, CA). Antibodies specific for phosphorylated molecules purchased from Cell Signaling Technology and Abcam listed are in Figure 2C. The protocol for detection was the same as for Western blotting.
Figure 2.

Luteolin suppresses pSTAT3 and DPYD expression. Cell proliferation quantified by WST-1 assay (n = 6 per dose) (A), and cell cycle analysis (n = 3 per dose) of MIAPaCa2 cells after Luteolin (Lut) treatment for 48 h (B). Multi-Western blotting of phosphorylated proteins associated with cell proliferation. On the left, no treatment and on the right 75 μM Lut treatment for 48 h. Circles and labels in blue or red depict Lut down- or up-regulated proteins in MIAPaCa2 cells, respectively (C). Western blotting for pSTAT3 (Tyr705), STAT3 after Lut (25, 50μM) treatment for 48 h in MIAPaCa2, PANC1, HPD1NR (D). Relative mRNA expression plotted from the result of microarrays comparing no treatment with 25 μM Lut treatment for 48 h in MIAPaCa2 cells. Blue lines are ratios of mRNA expression in controls: 25 μM Lut = 1 : 2 or 2 : 1 (E). The 20 genes most down-regulated by 25 μM Lut treatment (relative expression >100 in controls) were detected by microarray analysis (F). Overall survival (OS) (Log-rank test **P < 0.01) (G) and recurrence-free survival (RFS) (H) of PDAC patients extracted from the TCGA pancreatic cancer dataset (n = 159), which includes DPYD high expression (n = 79) and DPYD low expression (n = 80). Expression of DPYD mRNA after Lut (25, 50 μM) and Stattic (2 μM) treatment in MIAPaCa2 cells (I). Data are mean ± SD. *, **, *** P < 0.05, 0.01, 0.001 compared with no treatment. #, ##P < 0.05, 0.01 between 25 μM and 50 μM Lut treatment.
Microarray analysis
Gene expression analysis was performed using a human Oligo chip 20k (Toray Industries, Tokyo, Japan) according to the manufacturer’s instructions. RNA expression from MIAPaCa2 cells was compared between cells treated with or without 25 μM Lut for 48 hours.
siRNA transfection
Two human DPYD-siRNAs (Hs_DPYD_0538, Hs_DPYD_0539, MISSION siRNA; siDPYD) and the MISSION siRNA Universal Negative Control (siCont) were purchased from Sigma-Aldrich (St Louis, MO). KP4 cells were seeded into 6-well plates (2 × 105) for mRNA and protein, and in 96-well plates (2 × 105) for cell proliferation assays. These KP4 cells were transfected with siDPYD or siCont at a final concentration of 12 nM using Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturer’s instructions. After 48 hours, the silencing efficiencies of each of the siRNAs were evaluated by qRT-PCR and western blotting. The primer sequence is listed in Supplementary Table S1, available at Carcinogenesis Online.
Construction of the DPYD expression vector
DPYD-transfected and control LacZ-transfected cell lines were established using the lentivirus vector CSII-CMV-MCS-IRES2-Bds vector, which was provided by Dr. H. Miyoshi (RIKEN BioResource Center, Tsukuba, Japan). The coding sequences of DPYD were synthesized by PCR used PrimeSTAR HS DNA Polymerase (Takara Bio, Japan) and the primers containing the restriction enzyme sites (Supplementary Table S1, available at Carcinogenesis Online). CSII-CMV-MCS-IRES2-Bds with DPYD or the LacZ sequence was transfected into 293T cells with pCMV-VSV-G-RSV-Rev and pCAG-HIVgp by lipofectamine 3000 (Thermo Fisher Scientific). Supernatants concentrated by Lenti-XTM Concentrator (Takara Bio, Japan) and used to infect human PDAC cell lines (AsPC1, PATU-8988T), followed by selection using blasticidin (8 μg/ml for AsPC1, 20 μg/ml for PATU-8988T).
Tissue microarray analysis of human PDACs
We used the computerized database of our institution to identify 73 patients with pancreatic cancer who underwent surgery between April 2004 to December 2015 at the Department of Gastroenterological Surgery, Nagoya City University Hospital. The surgical specimens were fixed in 10% buffered formalin and embedded in paraffin. Two pathologists diagnosed all cases and selected two appropriate lesions for cutting out of the cylinders from the paraffin block, the diameter of which was 2 mm so that we could detect 48 cases on one slide. Immunohistochemical evaluation of Ki-67 (MIB-1, Agilent) was the same as for the hamster study. For DPYD (Abcam) evaluation, the degree of positivity of PDAC cells was divided into four groups as none (0), weak (1+), intermediate (2+), strong staining (3+). This study was approved by the Institutional Review Board at Nagoya City University Graduate School of Medical Sciences and conformed to the guidelines of Declaration of Helsinki.
Statistical analysis
Differences in the quantitative data, expressed as mean ± SD, between groups were compared by one-way ANOVA and Dunnett’s post-hoc test. Relativity score and P values were calculated by the Spearman’s test. Comparison of survival curves was compared by Grehan-Breslow-Wilcoxon test. All tests were analyzed by Graph Pad Prism 5 (GraphPad Software, Inc., La Jolla, CA). Multivariate analysis was calculated by EZR.
Results
Luteolin inhibits pancreatic carcinogenesis in a hamster model
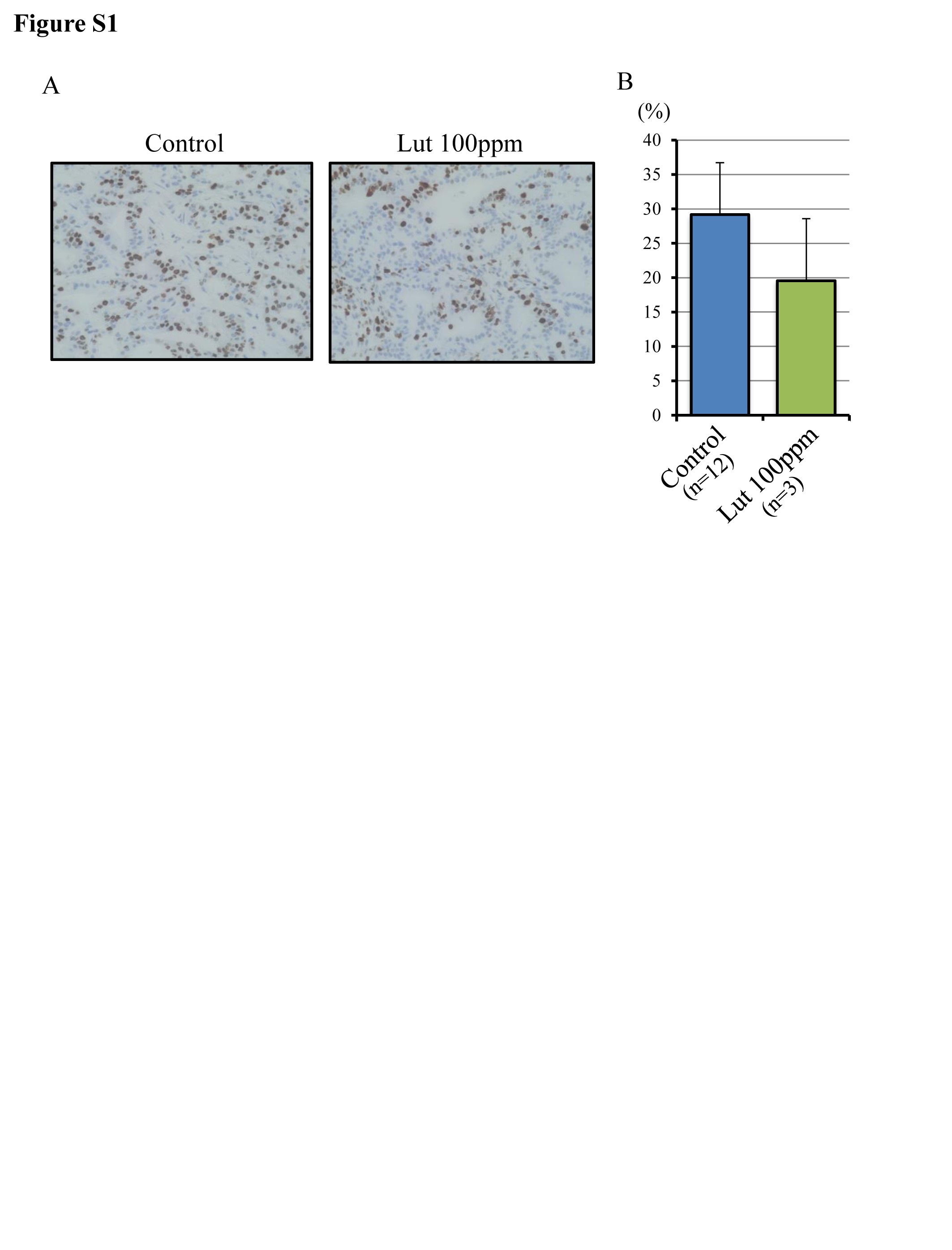
Treatment with Lut or Hes in the diet did not affect animal weight increases, and there was no significant difference in the final body weights, or liver and kidney weights between groups (Figure 1A and Supplementary Table S2A, available at Carcinogenesis Online). Serum data suggested that Lut down-regulated the level of total cholesterol and up-regulated the level of amylase but did not affect LDL-cholesterol, HDL-cholesterol, triglyceride. In contrast, Hes had no significant effects (Supplementary Table S2B, available at Carcinogenesis Online). The incidence and multiplicity of PDACs with similar histology to human PDAC (Figure 1B) was significantly decreased in the Lut group relative to controls (incidence, 23%-versus-71%, P < 0.05; and multiplicity, 0.23 ± 0.44-versus-0.93 ± 0.73, P < 0.05 respectively) (Figure 1C, D). On the other hand, the incidence and multiplicity of other tumors such as lung adenoma, cholangiocellular carcinoma and gallbladder adenoma due to BOP treatment were not different among the groups (Supplementary Table S3, available at Carcinogenesis Online). Lesions of all pancreatic ducts (diameter > 200 μm) in the duodenal lobe were classified into normal, PanIN1, PanIN2, PanIN3 and PDAC, resulting in the proportions of normal ducts being significantly decreased by Lut treatment compared with controls (Figure 1E). Progression scores were also significantly decreased by Lut treatment (Figure 1F). These data suggest that Lut retarded the progression of neoplastic lesions including PanINs and PDACs. The Ki-67 labeling index in PanIN lesions was also significantly suppressed by Lut but not changed by Hes treatment (Figure 1G, H). The Ki-67 labeling index in PDACs also tended to be decreased in the Lut group relative to controls, but this did not achieve statistical significance because the total number of PDACs was very small in the former (Supplementart Figure S1, available at Carcinogenesis Online). These results demonstrate that Lut inhibits pancreatic carcinogenesis in the BOP-induced hamster PDAC model.
Figure 1.

Luteolin inhibits pancreatic carcinogenesis and cell proliferation in a hamster model. Body-weights (A) and representative histology of hamster PDAC in controls; on the left, a low power field (×20), on the right, a high power field (×100) (B). The incidence in all lobes (C) and multiplicity in all, duodenal, splenic, and gastric lobes (D). The proportion of normal, PanIN1, PanIN2, PanIN3 and PDAC in all pancreatic ducts (diameter > 200 mm) of duodenal lobes (E) and the progression score calculated by weighting respective lesions (normal = 0, PanIN1 = 1, PanIN2 = 2, PanIN3 = 3, PDAC = 4) (F), Immunohistochemical findings of Ki-67 (G) and Ki-67 labeling index in PanINs (H). Data represented as mean ± SD, n = 14 (Control) and 13 (Lut 100 ppm, Hes 100 ppm, 1000 ppm). *P < 0.05 statistically significant compared with controls.
Luteolin suppresses PDAC cell proliferation and STAT3 activity in vitro
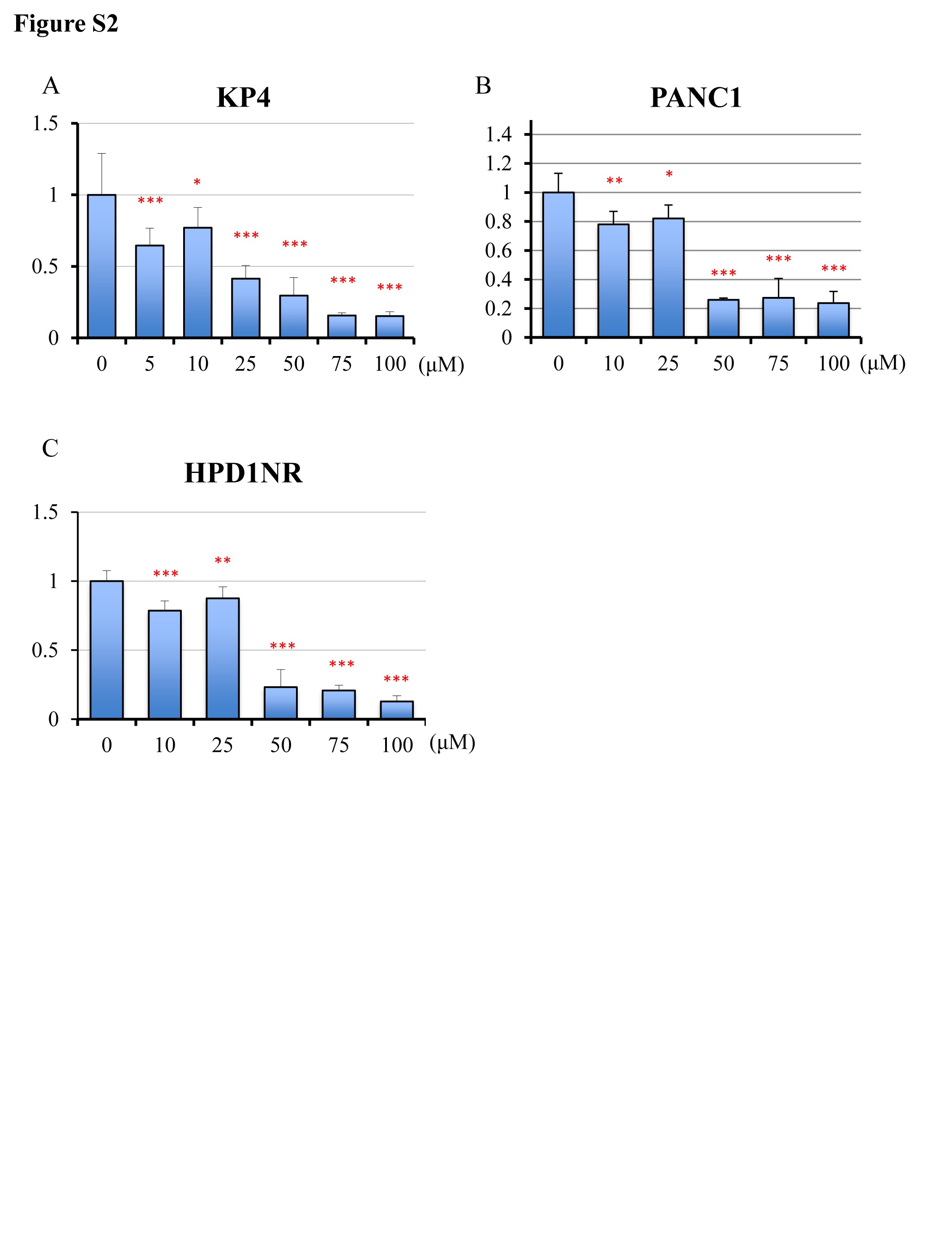
Because we found that Lut had an anti-proliferative effect in the hamster PDAC model, we further analyzed its mechanism of action using the human PDAC cell lines MIAPaCa2, KP4, PANC1, and a hamster PDAC cell line (HPD1NR). Lut significantly inhibited the growth of all these cell lines in a dose-dependent manner (Figure 2A and Supplementary Figure S2A–C, available at Carcinogenesis Online). Cell cycle analyses showed that MIAPaCa2 cells treated with Lut accumulated in the G1 phase, suggesting that Lut caused G1 arrest (Figure 2B).
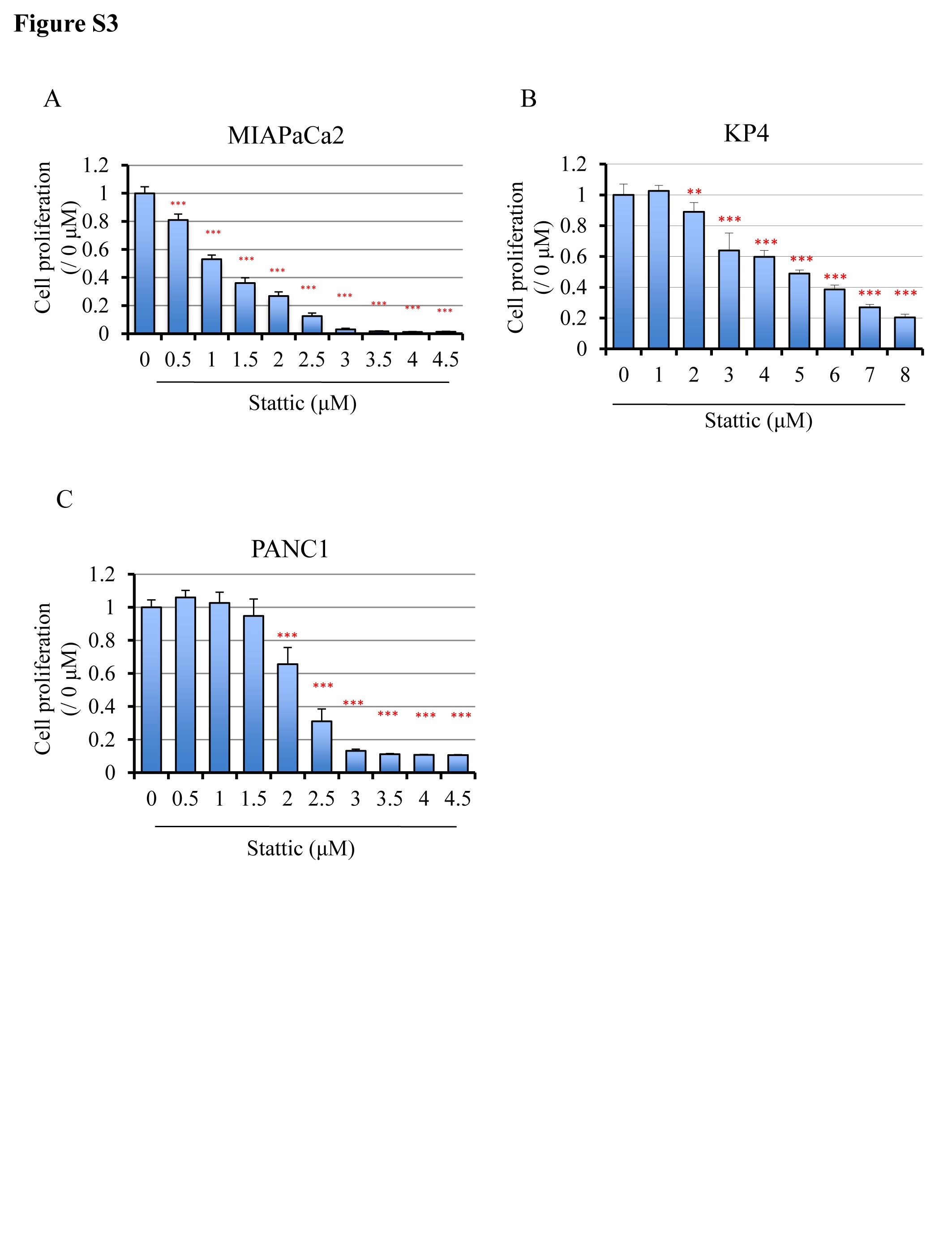
To clarify the mechanisms underlying the inhibition of cell proliferation by Lut treatment, we utilized multi-blot analyses to investigate the phosphorylation status of molecular in pathways associated with cell proliferation. As shown in Figure 2C, Lut suppressed the expression of phospho-AMPK (Thr172), phospho-p38 MAPK (Thr180/Tyr182) and phospho-STAT3 (Tyr 705), and increased the expression of phospho-GSK3β (Ser 9) in MIAPaCa2 cells. Dose-dependent suppression of pSTAT3 by Lut was confirmed in MIAPaCa2, PANC1 and HPD1NR cells (Figure 2D). To elucidate the relationship between pSTAT3 expression and cell proliferation in pancreatic cancer cells, PDAC cell lines were treated with Stattic, a small-molecule selectively inhibiting STAT3 activation and dimerization (23). We found that Stattic inhibited cell proliferation in a dose-dependent manner in the MIAPaCa2, PANC1 and KP4 cell lines (Supplementary Figure S3A–C, available at Carcinogenesis Online). These results suggest that Lut suppresses the proliferation of PDAC cells by decreasing pSTAT3.
Luteolin inhibits DPYD expression
To investigate chemopreventive mechanisms exerted by Lut on pancreatic cancer via suppression of PDAC cells in more detail, cDNA microarray analysis was performed using MIAPaCa2 cells treated with 25 mM Lut for 48h (Figure 2E). We found that 182 genes were down-regulated to less than a half of control following Lut treatment, with the 20 most decreased genes of which relative signal intensity is 100 at least are shown in Figure 2F. To explore genes targeted by Lut, we determined which were associated with poor prognosis in the human pancreatic cancer data (n = 159) of The Cancer Genome Atlas (TCGA) after excluding the 37 cases not diagnosed as PDAC. Of the 20 genes we identified, DPYD and CAV1 were significantly related to poor prognosis in PDAC in the TCGA (Figure 2G, H and Supplementary Figure S4A, B, available at Carcinogenesis Online). Multivariate analysis indicated that DPYD was an independent poor prognostic factor (HR = 1.738, P = 0.016) (Supplementary Table S4, available at Carcinogenesis Online). We confirmed that Lut inhibited the mRNA expression of these genes in a dose-dependent manner. Moreover, DPYD expression was significantly decreased by Stattic treatment, while CAV1 expression did not change (Figure 2I and Supplementary Figure S4C, available at Carcinogenesis Online). These results indicate that Lut inhibits DPYD expression, and DPYD expression may be regulated by STAT3 activation.
DPYD and pSTAT3 are mutual regulators
To clarify the relationship between DPYD and pSTAT3 expression, these factors were analyzed at the protein level in nine human pancreatic cell lines (KP4, MIAPaCa2, HupT3, PK1, PANC1, BxPC3, PA-TU-8988T, TCCPAN2, AsPC1). The results suggest that cells with high DPYD expression also had high pSTAT3 expression (rho = 0.883, P < 0.01) (Figure 3A, B). Lut and Stattic treatments reduced DPYD expression in parallel with decreased pSTAT3 in KP4 cells (Figure 3C). The cell line ASPC1 has low DPYD expression, but both pSTAT3 and DPYD expression were promoted by IL-6 stimulation (a known activator of STAT3 pathway in pancreatic cancer) and decreased subsequently by Lut treatments (Figure 3D). These results indicate that DPYD protein expression is regulated by pSTAT3 expression. To investigate whether DPYD expression affects pSTAT3 expression, RNA knock-down in KP4 cells by two siRNA (siDPYD1, siDPYD2) was tested. Knock-down efficiency was about 40 – 60% for either siDPYD (Figure 3E). When the expression of DPYD was decreased by siDPYDs, pSTAT3 and Cyclin D1 were also decreased, consistent with the notion that DPYD regulates STAT3 (Figure 3F). Knock-down of DPYD inhibited cell proliferation as seen in the WST-1 assay in Figure 3G, consistent with decreased Cyclin D1 expression. Further, the CSII-CMV-MCS-IRES2-Bds vector was used to transfect either LacZ or DPYD into PDAC cell lines with low-DPYD expression (AsPC1, PATU-8988T). We found that pSTAT3 expression was up-regulated, in parallel with DPYD over-expression (Figure 3H, I). Furthermore, the induced DPYD expression was reduced by pSTAT3 inhibitor (Stattic) treatment (Figure 3H, I). Cell proliferation was also enhanced in DPYD over-expressing cells compared with LacZ-transfected cells (Figure 3J, K). These results suggest that DPYD also regulates pSTAT3 expression, and taken together with the former results, suggest that DPYD and pSTAT3 expression regulate each other.
Figure 3.

DPYD and pSTAT3 expression affected one other. Western blotting for pSTAT3, STAT3, DPYD and β-Actin protein in nine human pancreatic cell lines (A). DPYD and pSTAT3 expression corrected by β-Actin and total STAT3 (Spearman rho = 0.883, P < 0.01) (B). Western blotting for pSTAT3, STAT3, DPYD and β-Actin after Lut (25, 50 μM) or Stattic (5 μM) treatment in the DPYD high-expressing line (KP4) (C) and after stimulation of the STAT3 pathway by IL 6 (50 ng/ml) for 24 h followed by addition of Lut (10, 25, 50 μM) in a DPYD low-expressing line (AsPC1) (D). Expression of DPYD mRNA (n = 3 per cell) (E) and DPYD protein for pSTAT3, STAT3, Cyclin D1 and β-Actin (F), and cell proliferation (n = 6 per condition) (G) after siDPYD (siDPYD1, siDPYD2) transfection into KP4 cells. Expression of DPYD, pSTAT3, STAT3 and β-Actin protein (H, I) and proliferation (J, K) when DPYD is overexpressed with or without Stattic treatment in AsPC1 and PATU-8988T cells (MIAPaCa cell protein as a positive control for DPYD). Data are mean ± SD. *, **, *** P < 0.001 compared with control or siControl or CSII-LacZ.
Luteolin inhibits DPYD and pSTAT3 signaling in vivo
We confirmed that DPYD expression correlated with pSTAT3 in vitro. To determine whether this is also the case in vivo, we analyzed pSTAT3 and DPYD expression in hamster PanINs and PDACs. The frequency of pSTAT3 nuclear positivity in PDACs and PanINs was significantly decreased on Lut treatment relative to controls (Figure 4A, B). STAT3 is strongly activated by some cytokines including IL 6 secreted by activated pancreatic stellate cells (PSCs) (24–26). PDAC is characterized by dense cancer-associated fibroblasts (CAFs), which contribute to blocking drug delivery and facilitate cancer cell growth. A major source of CAFs in PDACs in the PSCs, which secrete extracellular matrix-binding factors including pro-inflammatory cytokines (IL 6, TGFb, TNFa and PDGF) and mediate cross-talk to PDAC cells (27,28). In the hamster pancreas, levels of pro-inflammatory cytokine mRNAs were decreased on Lut treatment (Figure 4C). The area of collagen fiber detected by Azan staining and activated PSCs detected by αSMA staining tended to be decreased in PDACs of hamsters given Lut relative to no treatment. However, the difference did not achieve significance due to the small number of PDAC tissues in the Lut group (n = 3) (Supplementary Figure S5A–D, available at Carcinogenesis Online). DPYD intensity scores were significantly decreased by Lut intake in hamster PanINs (Figure 4D, E). Furthermore, there was a significant positive relationship between pSTAT3 positivity and DPYD intensity in PanINs (rho = 0.41, P < 0.05) (Figure 4F), indicating that Lut inhibited DPYD and STAT3 signaling in pancreatic cancer not only in vitro but also in vivo.
Figure 4.

Luteolin inhibits pSTAT3 and DPYD expression in the hamster pancreatic cancer model. Immunohistochemical findings of pSTAT3 in PanIN and PDAC (A) and nuclear pSTAT3 positivity of PanIN (Controls: n = 14, Lut 100 ppm: n = 13) and PDAC (Controls: n = 9, Lut 100 ppm: n = 3) in control and 100 ppm Lut treated animals (B). Expression of cytokine mRNA (Il6, Tgfβ, Tnfα, Il10) associated with the STAT3 pathway in hamster pancreas gastric lobe (n = 6 respectively, standardized by β2-microglobulin, Bmg) (C). DPYD expression detected in cytoplasm immunohistochemically (D) and its relative intensity in PanIN of 100 ppm Lut (n = 13) and controls (n = 14) (E). Graph of DPYD relative intensity and pSTAT3 nuclear positivity (Spearman rho = 0.41, P < 0.05) (F). Data are mean ± SD. *, ** P < 0.05, 0.01 as compared with controls.
DPYD expression in human PDAC tissue
To investigate the role of DPYD in human PDAC, we accessed 73 PDACs cases resected from 2009 to 2015 in Nagoya City University Hospital and constructed tissue microarrays. We coded DPYD staining into four categories dichotomized into low (0, 1 and 2+, n = 50) and high DPYD expression (3+, n = 23) (Figure 5A). Clinicopathological features are summarized in Table 1, suggesting that high DPYD expression is associated with poor differentiation (P < 0.001) but not TNM classification, stage, and metastasis (Table 1). Patients with high DPYD expression had a significantly poorer prognosis (median OS; 14.5 months) than those with low DPYD expression (median OS; 25 months) as seen for 3-year overall survival (OS) in Figure 5B. However, 3-year recurrence-free survival was no different (Figure 5C). Multivariate analysis indicated that strong DPYD expression was a risk factor for OS but this was not significant (HR = 1.538, P = 0.215) (Supplementary Table S5, available at Carcinogenesis Online). The DPYD score did significantly correlate with the Ki-67 labeling index (Spearman rho = 0.45. P < 0.01) (Figure 5D). Because DPYD is the first enzyme involved in 5-fluorouracil (5-FU) degradation, we separately analyzed patients who had received chemotherapy containing this drug (n = 45). Now the difference in OS between high and low DPYD was more marked (P = 0.018) in PDAC patients receiving 5-FU treatment than in the entire cohort of PDAC patients (n = 73, P = 0.048) (Figure 5E). However, in an in vitro study, DPYD-low cells (PATU-8988T, AsPC1) did not show higher susceptibility to 5-FU than DPYD-high cells (MIAPaCa2). Treatment of MIAPaCa2 with a combination of 5-FU (2, 4 μM) and Lut (10, 20 μM) inhibited cell proliferation additively but not synergistically (CI: 0. 953–1.179) (Supplementary Figure S6A–C, available at Carcinogenesis Online). Taken together, these results show that high DPYD expression is associated with poor prognosis and acceleration of cell proliferation in human PDACs.
Figure 5.

DPYD expression is associated with poor prognosis in human PDAC. Immunohistochemical staining of DPYD in a PDAC tissue microarray (n = 73) stratified into four categories (0; none, 1+; weak, 2+; moderate, 3+; strong) and coded as DPYD low (0, 1+, 2+, n =50) and high expression (3+, n = 23) (A). Three-year overall survival (OS) (B) and 3-year recurrence-free survival (RFS) (C) of PDAC patients with low DPYD (n = 50) or high DPYD expression (n = 23) *P = 0.045 between low and high DPYD expression groups. DPYD expression score and Ki-67 labeling index (Spearman rho = 0.445, P < 0.05) (D). Three-year OS of PDAC patients with S-1 adjuvant therapy (n = 45) including high DPYD (n = 15) and low DPYD expression groups (n = 30) *P = 0.018 between low and high DPYD expressors (E).
Table 1.
Clinicopathological characteristics of pancreatic ductal carcinoma according to DPYD expression
| DPYD | P value | ||
|---|---|---|---|
| low | high | ||
| n | 50 | 23 | |
| male/female | 39/11 | 13/10 | 0.094 |
| Age | 68 ± 9 | 70 ± 9 | 0.433 |
| Differentiation | |||
| well | 25 (50) | 3 (13) | |
| moderately | 10 (20) | 4 (17) | |
| poorly | 15 (30) | 16 (70) | <0.001 |
| T | |||
| 1 | 4 (8) | 2 (9) | |
| 2 | 6 (12) | 1 (4) | |
| 3 | 37 (74) | 20 (87) | |
| 4 | 3 (6) | 0 (0) | 0.270 |
| N (+) | 28 (56) | 15 (65) | 0.610 |
| M (+) | 1 (2) | 1 (4) | >0.999 |
| Stage | |||
| I A | 4 (8) | 1 (4) | |
| I B | 2 (4) | 2 (9) | |
| II A | 14 (28) | 5 (22) | |
| II B | 26 (52) | 14 (61) | |
| III | 3 (6) | 0 (0) | |
| IV | 1 (2) | 1 (4) | 0.862 |
Discussion
The concept of cancer prevention relies on a strategy for delaying or preventing carcinogenesis by suppressing progression from premalignant lesions to invasive cancer using natural or synthetic agents (29). PDAC is an attractive target for cancer prevention because it is one of the most lethal tumors which is difficult to detect at an early stage and is highly resistant to standard chemotherapeutic regimens. In the present study, we found that the aglycone flavone Lut impedes the progression of pancreatic carcinogenesis by affecting the STAT3-DPYD axis. These data suggest that Lut may serve as a novel chemopreventive and chemotherapeutic agent for PDAC. Several investigations regarding suppressive mechanisms for PDAC cell growth have been limited to testing pathways in vitro studies, including PI3K-AKT-NF-kB and JAK-STAT pathways (4). Here, the novel STAT3-DPYD pathway we describe both in vivo and in vitro is implicated in pancreatic cancer prevention strategies.
Properly designed in vivo animal experiments play a key role in bridging the gap between in vitro studies and clinical trials, especially for determining optimal doses and toxicity of new compounds before recommending their use in humans. In chemoprevention studies, there must be no systematic change in toxicity at the individual level. It has been reported that dietary luteolin intakes are 0.85 ± 0.02 mg/day in Korean adults (30), and 1.56 ± 1.36 mg/day in Mexican women (31). A diet of 100 ppm Lut (real intake 4.58 mg/kg/day) in the present study was selected as very similar to human exposure and was equivalent to 275 mg/day by a 60 kg person. It is considered that this amount of Lut can be taken as a nutritional supplement, and is not in any way an unrealistic amount. However, we need to take into consideration factors concerning the metabolism of Lut to translate our results to clinical treatments. A previous study demonstrated that luteolin-3’-glucuronide was the most abundant metabolic product in human and rat plasma, and not Lut aglycon (10). Thus, we need to further investigate the chemopreventive effects of these metabolites on pancreatic cancer. To resolve these issues, some recent studies demonstrated that delivery mediated by nanoparticles enables Lut to locate tissues as an aglycon, resulting in higher bioavailability, longer retention in the blood and higher efficiency (32). Thus, encapsulating Lut in nanoparticles may be the most effective mode of use for pancreatic cancer.
Considering potential chemopreventive effects of Lut, a previous study had shown that Lut facilitated apoptosis through inhibiting K-ras, Gsk3b and NF-κB pathways (11). In agreement with this previous study, we also observed up-regulation of inactivated Gsk3b in the multi-blotting assay, but in the hamster model, induction of apoptosis was not observed by TUNEL staining (data not shown). We also failed to detect the expression of pNF-κB or pIκB in the multi-blotting assay. A different published study had demonstrated that Lut suppressed acinar-ductal metaplasia in a mouse model of acute pancreatitis induced by caerulein, accompanied by reduced pSTAT3, SOX9, CK19 and Ki-67 expression in acinar cells (33). Lut reduced pSTAT3 expression in PanINs or PDACs and inhibited cell proliferation in the hamster model. Regarding the role of STAT3 activation in PDAC carcinogenesis, a previous study using conditional inactivation of STAT3 in a Kras-driven PDAC mouse model reported that it had a critical role in the multi-step carcinogenesis from acinar-ductal metaplasia and PanIN to PDAC (34). These results suggested that STAT3 is a key molecule for pancreatic cancer initiation and progression in vivo.
DPYD is the initial phase enzyme in the degradation pathway for pyrimidine bases such as thymine, uracil or the anti-cancer drug 5-FU. Inhibition of DPYD leads to maintenance of a higher concentration of 5-FU in blood and tumor tissue because it catabolizes 85% of the administered 5-FU to the non-toxic intermediate 5-dihydro fluorouracil (35). In human PDAC, some studies reported that high DPYD expression is a biomarker of poor PDAC prognosis (36–39), and the present study is consistent with this. In addition to the role of DPYD as an enzyme that affects the efficacy of drugs such as 5-FU, it may also facilitate the epithelial-mesenchymal transition (EMT) related to cancer progression (40). STAT3 signaling is known to be involved in EMT as well as in promoting cell proliferation, migration and invasion in several cancers (41). We found that knock-down of DPYD inhibited STAT3 activation, suggesting that Lut inhibited pancreatic carcinogenesis via EMT suppression through the STAT3-DPYD pathway.
Knowledge of mechanisms modulating DPYD expression is sparse, especially in the context of PDAC. Interferon-α (IFNα) has been reported to reduce the expression and activity of DPYD in some tumor cell lines including pancreatic cancer (40). In hepatocellular carcinoma, DPYD was down-regulated by IFNα and knock-down of DPYD inhibited tumor growth in a xenograft model through down-regulation of NF-κB signaling (42). Another study showed that knock-down of TWIST1 decreased DPYD expression in colon cancer cell lines (43). In addition to changes at the protein level, changes at the level of microRNAs (miR-302b in hepatocellular carcinoma, miR-494 in colon cancer, and miR-27a and -27b in hepatocytes), epigenetics (histone H3K27 trimethylation in HEK293T/c17 cell), and long non-coding RNA (LINC00261 in esophageal cancer) are known to be involved in the regulation of DPYD expression (15,44–47). Although it is not known whether these complex mechanisms are important in PDAC, our present data indicate interactions between STAT3 and DPYD, directly or indirectly, and document that STAT3 is a key molecule involved in multiple signaling pathways. DPYD is an enzyme, suggesting that it acts downstream of STAT3. However, it is unclear how DPYD is involved in the regulation of STAT3 expression, and further investigation is required.
In summary, the present study showed that Lut suppressed pancreatic carcinogenesis in a BOP-induced hamster model through inhibition of the STAT3-DPYD pathway that plays an important role in pancreatic cancer development. Thus, Lut may be an attractive candidate for pancreatic cancer chemoprevention as well as representing a new chemotherapeutic compound for combination treatments containing 5-FU as a DPYD inhibitor for pancreatic cancer.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Glossary
Abbreviations
- BOP
N-nitroso-bis(2-oxopropyl)-amine
- CAFs
cancer associated fibroblasts;
- DPYD
dihydropyrimidine dehydrogenase
- 5-FU
5-fluorouracil
- Hes
Hesperetin (3′,5,7-trihydroxy-4′ methoxy flavanone)
- Lut
Luteolin (3′,4′,5,7-tetrahydroxy flavone)
- PanINs
pancreatic intraepithelial neoplasias
- PDAC
pancreatic ductal carcinoma
- PSCs
pancreatic stellate cells
- pSTAT3
phosphorylated STAT3 (Tyr705)
- WST-1
4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate tetrazolium salt
Funding
This work was supported by a grant from Ono Pharmaceutical Co., Ltd to S.T., and by JSPS KAKENHI (Grant Number 17K15666 and 20K16225).
Conflict of Interest Statement: There are no potential conflicts of interest to declare.
Author contributions
H.K., A.N.-I. and S.T. conceived and designed the experiments and supervised the project. H.K., A.N.-I., S.S., T.N., S.I., K.N. and M.K. carried out the experiments. H.K., K.K., A.K. and Y.M. collected data and constructed the database for the human PDAC tissue array. H.K., A.N.-I. and S.T. wrote the paper. All authors discussed the results and contributed the manuscript.
References
- 1. Roth G and GBD 2017 Causes of Death Collaborators. (2018) Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet, 392, 1736–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ministry of Health, Labour and Welfare. (2017) Vital Statistics Japan. [Google Scholar]
- 3. Moffat, G.T., et al. (2019) Pancreatic cancer-a disease in need: optimizing and integrating supportive care. Cancer, 125, 3927–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Benzel, J., et al. (2018) Chemoprevention and treatment of pancreatic cancer: update and review of the literature. Digestion, 97, 275–287. [DOI] [PubMed] [Google Scholar]
- 5. Kato, A., et al. (2015) Chemopreventive effect of resveratrol and apocynin on pancreatic carcinogenesis via modulation of nuclear phosphorylated GSK3β and ERK1/2. Oncotarget, 6, 42963–42975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jiang, Z.Q., et al. (2018) Luteolin inhibits tumorigenesis and induces apoptosis of non-small cell lung cancer cells via regulation of MicroRNA-34a-5p. Int. J. Mol. Sci., 19, 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pandurangan, A.K., et al. (2014) Luteolin, a bioflavonoid inhibits colorectal cancer through modulation of multiple signaling pathways: a review. Asian Pac. J. Cancer Prev., 15, 5501–5508. [DOI] [PubMed] [Google Scholar]
- 8. Sagawa, H., et al. (2015) Connexin 32 and luteolin play protective roles in non-alcoholic steatohepatitis development and its related hepatocarcinogenesis in rats. Carcinogenesis, 36, 1539–1549. [DOI] [PubMed] [Google Scholar]
- 9. Naiki-Ito, A., et al. (2020) Recruitment of miR-8080 by luteolin inhibits androgen receptor splice variant 7 expression in castration-resistant prostate cancer. Carcinogenesis, 41, 1145–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Iida, K., et al. (2020) Luteolin suppresses bladder cancer growth via regulation of mechanistic target of rapamycin pathway. Cancer Sci., 111, 1165–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Johnson, J.L., et al. (2015) Luteolin and gemcitabine protect against pancreatic cancer in an orthotopic mouse model. Pancreas, 44, 144–151. [DOI] [PubMed] [Google Scholar]
- 12. Johnson, J.L., et al. (2011) Citrus flavonoids luteolin, apigenin, and quercetin inhibit glycogen synthase kinase-3β enzymatic activity by lowering the interaction energy within the binding cavity. J. Med. Food, 14, 325–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Harris, D.M., et al. (2012) Diverse mechanisms of growth inhibition by luteolin, resveratrol, and quercetin in MIA PaCa-2 cells: a comparative glucose tracer study with the fatty acid synthase inhibitor C75. Metabolomics, 8, 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang, X., et al. (2015) Luteolin decreases invasiveness, deactivates STAT3 signaling, and reverses interleukin-6 induced epithelial-mesenchymal transition and matrix metalloproteinase secretion of pancreatic cancer cells. Onco. Targets. Ther., 8, 2989–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cai, X., et al. (2012) The molecular mechanism of luteolin-induced apoptosis is potentially related to inhibition of angiogenesis in human pancreatic carcinoma cells. Oncol. Rep., 28, 1353–1361. [DOI] [PubMed] [Google Scholar]
- 16. Li, W.Q., et al. (2010) Citrus consumption and cancer incidence: the Ohsaki cohort study. Int. J. Cancer, 127, 1913–1922. [DOI] [PubMed] [Google Scholar]
- 17. Tanaka, T., et al. (1997) Chemoprevention of 4-nitroquinoline 1-oxide-induced oral carcinogenesis in rats by flavonoids diosmin and hesperidin, each alone and in combination. Cancer Res., 57, 246–252. [PubMed] [Google Scholar]
- 18. Tanaka, T., et al. (1997) Modulation of N-methyl-N-amylnitrosamine-induced rat oesophageal tumourigenesis by dietary feeding of diosmin and hesperidin, both alone and in combination. Carcinogenesis, 18, 761–769. [DOI] [PubMed] [Google Scholar]
- 19. Tanaka, T., et al. (1997) Chemoprevention of azoxymethane-induced rat colon carcinogenesis by the naturally occurring flavonoids, diosmin and hesperidin. Carcinogenesis, 18, 957–965. [DOI] [PubMed] [Google Scholar]
- 20. Lee, J., et al. (2019) Combined administration of naringenin and hesperetin with optimal ratio maximizes the anti-cancer effect in human pancreatic cancer via down regulation of FAK and p38 signaling pathway. Phytomedicine, 58, 152762. [DOI] [PubMed] [Google Scholar]
- 21. Hori, M., et al. (2011) Enhancement of carcinogenesis and fatty infiltration in the pancreas in N-nitrosobis(2-oxopropyl)amine-treated hamsters by high-fat diet. Pancreas, 40, 1234–1240. [DOI] [PubMed] [Google Scholar]
- 22. Inaguma, S., et al. (2013) GLI1 interferes with the DNA mismatch repair system in pancreatic cancer through BHLHE41-mediated suppression of MLH1. Cancer Res., 73, 7313–7323. [DOI] [PubMed] [Google Scholar]
- 23. Schust, J., et al. (2006) Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol., 13, 1235–1242. [DOI] [PubMed] [Google Scholar]
- 24. Shi, Y., et al. (2019) Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature, 569, 131–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Komar, H.M., et al. (2017) Inhibition of Jak/STAT signaling reduces the activation of pancreatic stellate cells in vitro and limits caerulein-induced chronic pancreatitis in vivo. Sci. Rep., 7, 1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Marzoq, A.J., et al. (2019) Impact of the secretome of activated pancreatic stellate cells on growth and differentiation of pancreatic tumour cells. Sci. Rep., 9, 5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Öhlund, D., et al. (2017) Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med., 214, 579–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Elahi-Gedwillo, K.Y., et al. (2019) Antifibrotic therapy disrupts stromal barriers and modulates the immune landscape in pancreatic ductal adenocarcinoma. Cancer Res., 79, 372–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sporn, M.B. (1976) Approaches to prevention of epithelial cancer during the preneoplastic period. Cancer Res., 36 (7 PT 2), 2699–2702. [PubMed] [Google Scholar]
- 30. Kim, Y.J., et al. (2015) Intake and major sources of dietary flavonoid in Korean adults: Korean National Health and Nutrition Examination Survey 2010-2012. Asia Pac. J. Clin. Nutr., 24, 456–463. [DOI] [PubMed] [Google Scholar]
- 31. Chavez-Suarez, K.M., et al. (2017) Phytoestrogen concentrations in human urine as biomarkers for dietary phytoestrogen intake in Mexican women. Nutrients, 9, 1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Majumdar, D., et al. (2014) Luteolin nanoparticle in chemoprevention: in vitro and in vivo anticancer activity. Cancer Prev. Res. (Phila)., 7, 65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang, X., et al. (2018) Luteolin inhibits pancreatitis-induced acinar-ductal metaplasia, proliferation and epithelial-mesenchymal transition of acinar cells. Mol. Med. Rep., 17, 3681–3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Corcoran, R.B., et al. (2011) STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res., 71, 5020–5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tatsumi, K., et al. (1987) Inhibitory effects of pyrimidine, barbituric acid and pyridine derivatives on 5-fluorouracil degradation in rat liver extracts. Jpn. J. Cancer Res., 78, 748–755. [PubMed] [Google Scholar]
- 36. Elander, N.O., et al. ; European Study Group for Pancreatic Cancer. (2018) Expression of dihydropyrimidine dehydrogenase (DPD) and hENT1 predicts survival in pancreatic cancer. Br. J. Cancer, 118, 947–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Murakawa, M., et al. (2017) Clinical implications of dihydropyrimidine dehydrogenase expression in patients with pancreatic cancer who undergo curative resection with S-1 adjuvant chemotherapy. Oncol. Lett., 14, 1505–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Miyake, K., et al. (2007) Role of thymidine phosphorylase and orotate phosphoribosyltransferase mRNA expression and its ratio to dihydropyrimidine dehydrogenase in the prognosis and clinicopathological features of patients with pancreatic cancer. Int. J. Clin. Oncol., 12, 111–119. [DOI] [PubMed] [Google Scholar]
- 39. Nakahara, O., et al. (2010) Clinical significance of dihydropyrimidine dehydrogenase and thymidylate synthase expression in patients with pancreatic cancer. Int. J. Clin. Oncol., 15, 39–45. [DOI] [PubMed] [Google Scholar]
- 40. Milano, G., et al. (1994) Inhibition of dihydropyrimidine dehydrogenase by alpha-interferon: experimental data on human tumor cell lines. Cancer Chemother. Pharmacol., 34, 147–152. [DOI] [PubMed] [Google Scholar]
- 41. Yuan, J., et al. (2015) Multiple regulation pathways and pivotal biological functions of STAT3 in cancer. Sci. Rep., 5, 17663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhu, W.P., et al. (2018) Dihydropyrimidine dehydrogenase predicts survival and response to interferon-α in hepatocellular carcinoma. Cell Death Dis., 9, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sakowicz-Burkiewicz, M., et al. (2016) Suppression of TWIST1 enhances the sensitivity of colon cancer cells to 5-fluorouracil. Int. J. Biochem. Cell Biol., 78, 268–278. [DOI] [PubMed] [Google Scholar]
- 44. Chai, J., et al. (2015) MicroRNA-494 sensitizes colon cancer cells to fluorouracil through regulation of DPYD. IUBMB Life, 67, 191–201. [DOI] [PubMed] [Google Scholar]
- 45. Offer, S.M., et al. (2014) microRNAs miR-27a and miR-27b directly regulate liver dihydropyrimidine dehydrogenase expression through two conserved binding sites. Mol. Cancer Ther., 13, 742–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wu, R., et al. (2016) Histone H3K27 trimethylation modulates 5-fluorouracil resistance by inhibiting PU.1 binding to the DPYD promoter. Cancer Res., 76, 6362–6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lin, K., et al. (2019) Long noncoding RNA LINC00261 induces chemosensitization to 5-fluorouracil by mediating methylation-dependent repression of DPYD in human esophageal cancer. FASEB J., 33, 1972–1988. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.