

Abstract
SUMMARY
The fidelity of inhibitory neurotransmission is dependent on the accumulation of γ-aminobutyric acid type A receptors (GABAARs) at the appropriate synaptic sites. Synaptic GABAARs are constructed from α(1–3), β(1–3), and γ2 subunits, and neurons can target these subtypes to specific synapses. Here, we identify a 15-amino acid inhibitory synapse targeting motif (ISTM) within the α2 subunit that promotes the association between GABAARs and the inhibitory scaffold proteins collybistin and gephyrin. Using mice in which the ISTM has been introduced into the α1 subunit (Gabra1–2 mice), we show that the ISTM is critical for axo-axonic synapse formation, the efficacy of GABAergic neurotransmission, and seizure sensitivity. The Gabra1–2 mutation rescues seizure-induced lethality in Gabra2–1 mice, which lack axo-axonic synapses due to the deletion of the ISTM from the α2 subunit. Taken together, our data demonstrate that the ISTM plays a critical role in promoting inhibitory synapse formation, both in the axonic and somatodendritic compartments.
In Brief
Molecular mechanisms regulating specific synaptic GABAAR accumulation are critical for the fidelity of inhibitory neurotransmission. Nathanson et al. show that strengthening the interaction between α1-GABAARs and collybistin via genetic manipulation results in augmented synaptic targeting of these receptors, enhanced inhibitory neurotransmission, and seizure resilience.
Graphical abstract
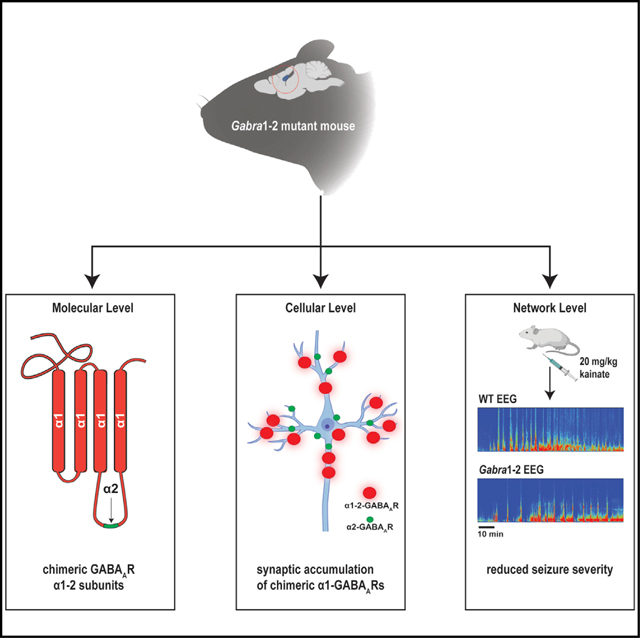
INTRODUCTION
Brain function in the mammalian nervous system depends on a dynamic relationship between excitatory and inhibitory neurotransmission (Fritschy, 2008; Selten et al., 2018). Inhibitory interneurons precisely control the firing of excitatory pyramidal neurons and thus regulate network activity patterns (Roux and Buzsáki, 2015). Interneurons inhibit the activity of pyramidal cells by releasing the neurotransmitter γ-aminobutyric acid (GABA) onto synapses containing GABA type A receptors (GABAARs). GABAARs are heteropentameric, ligand-gated ion channels permeable to chloride. GABAARs can be composed from 19 different subunits, but synaptic GABAARs are understood to be primarily assembled from 2 α(1–3), 2 β(1–3), and 1 γ2 subunits (Olsen and Sieghart, 2008; Sigel and Steinmann, 2012). Although the structure of these subunits is highly conserved, the specific combination of subunits in a given GABAAR does confer distinct physiological and pharmacological properties on the receptor (Goldstein et al., 2002; Rudolph et al., 1999). Subunit types are also differentially localized within the neuron. For example, GABAARs containing the α1 subunit tend to be found in dendritic synapses, while those containing α2 are highly enriched in axo-axonic synapses at the axon initial segment (AIS) (Klausberger et al., 2002; Nusser et al. 1996; Nyıŕi et al. 2001), the site of action potential (AP) initiation (Kole and Stuart, 2012). Recently, it has been shown that disturbing this subunit distribution in a mouse model (Gabra2–1) can disrupt inhibitory control of excitation and lead to seizures and early mortality (Hines et al., 2018).
Clearly, then, pyramidal neurons must precisely control the allocation of GABAAR subtypes to specific subcellular sites. The exact mechanisms by which this distribution is attained, however, remain unclear. Previous work suggests that interactions between the a subunit and intracellular proteins may be a determinant of GABAAR localization (Mukherjee et al., 2011; Tretter et al., 2008; Tretter and Moss, 2008). Although the sequences of α subunits are largely homologous, there is a significant area of divergence in the intracellular domain (ICD) that lies between transmembrane domains 3 and 4 (Olsen and Tobin, 1990). A 15–amino acid sequence within the ICD of α2 has been shown in vitro to mediate preferential binding to the inhibitory synaptic protein collybistin (CB). Replacing this α2-specific motif with the analogous sequence of α1 causes loss of α2-containing synapses at the AIS, suggesting that CB may play a role in the formation of subtype-specific inhibitory synapses via its interaction with α2 (Hines et al., 2018).
CB is a guanine nucleotide exchange factor that activates the small GTPase Cdc42, a regulator of the actin cytoskeleton. There are a number of CB isoforms, but the protein is generally composed of 3 domains: the pleckstrin homology (PH) phosphoinositide-binding domain, the catalytic RhoGEF (DH) domain, and the N-terminal Src-homology 3 (SH3) domain (Harvey et al., 2004; Xiang et al., 2006). CB was first identified as a gephyrin-interacting protein (Kins et al., 2000). Gephyrin (GPN) is an integral component of the inhibitory synapse that is known to bind directly to α subunits of GABAARs, forming a submembrane scaffold that anchors the receptors at the synapse (Mukherjee et al., 2011; Tretter et al., 2008, 2012). CB binds GPN via the DH domain and facilitates the aggregation and stabilization of GPN in the submembrane space (Grosskreutz et al., 2001; Kins et al., 2000; Tyagarajan et al., 2011). CB-knockout mice display a loss of both GPN and GABAARs at postsynaptic structures in the hippocampus (Papadopoulos et al., 2008, 2007), and mutations in the human CB gene ARHGEF9 are associated with epileptic diseases (Kalscheuer et al., 2009; Papadopoulos et al., 2015; Shimojima et al., 2011).
CB, therefore, is an attractive candidate for the regulation of subtype-specific synapse formation, via preferential interactions with the ICD of α2. However, no study has examined the association between CB and the α2 ICD motif in brain tissue or fully assessed the effect that this association has on the trafficking and stabilization of α1- and α2-containing GABAARs at synapses. To this end, we developed a knockin mouse in which the CB binding motif in the α2 loop is substituted into α1 (the Gabra1–2 mouse). We observed that this mutation does indeed enhance the association of the α1 subunit with CB in brain lysate. The mutation also alters the expression of α1, causes the enrichment of α1-GABAARs at the AIS, and increases the size and density of α1-containing synapses in cultured hippocampal cells. In addition, the Gabra1–2 mutation alters both phasic inhibition in the CΑ1 and baseline cortical electroencephalographic (EEG) activity, resulting in resistance to kainate-induced seizures. To demonstrate the strength of the seizure-resistant phenotype, we crossed Gabra1–2 mice with Gabra2–1 seizure-prone mice and found that the double heterozygous animals are spared from the lethality observed in Gabra2–1 mutants.
RESULTS
The Creation of the Gabra1–2 Mutant Mouse
To probe the interaction between CB and the α2 ICD-binding motif and determine its importance in synapse formation, we generated a knockin mouse in which amino acids 360–375 of the α1 ICD were replaced by those of the α2 subunit (Figure 1A; Figure S1A). Mice were generated using homologous recombination in ES cells (Genoway), as outlined previously (Moore et al., 2018; Terunuma et al., 2014; Vien et al., 2015). To confirm the Gabra1–2 mutation, we amplified the relevant portion of genomic DNA from wild-type (WT) and homozygous Gabra1–2 mice and sequenced the resulting PCR product, confirming the insertion of the α2 amino acid sequence into α1 (Figure S1B). Mice were genotyped by PCR using primers that detected the α2 insertion (Figure S1C). Gabra1–2 homozygotes were viable and did not display any overt phenotypes. Motor behavior, as measured by latency to fall off a rotarod beam and the total distance traveled in the open field test, was unaffected (Figure S2). In addition, cresyl violet staining showed no differences in gross hippocampal anatomy between WT and mutant animals (Figure S1D).
Figure 1. The Gabra1–2 Mutation Alters the Expression and Surface Stability of Specific GABAAR Subtypes.

(A) Cartoon showing the Gabra1–2 mutation, in which amino acids 360–375 of the α1 subunit of the GABAAR are replaced with that of the α2 subunit.
(B) Representative western blots examining expression of key components of the inhibitory synapse in total hippocampal lysates. GAPDH served as a loading control.
(C) Pooled quantification (n = 5 biological and technical replicates) reveals that the Gabra1–2 mutation leads to a decrease in the total expression of the α1 subunit, a trending decline in total α2 subunit expression (p = 0.077), and an increase in total CB expression.
(D) Representative western blots from biotinylated hippocampal slices examining the surface expression of the α1 and α2 subunits. Total α1/2 subunit was normalized to GAPDH. Surface α1/2 subunit was normalized to pan-cadherin.
(E) Pooled quantification (n = 5 biological and technical replicates) shows no change in the surface:total ratio of the α1 subunit in Gabra1–2 mutants but reveals a significant decrease in the surface expression of the α2 subunit.
Unpaired t tests with Welch’s correction used for statistical analysis. (*p < 0.05; **p < 0.01). All data are expressed as the percentage of WT and error bars represent mean ± SEM.
Changes in GABAAR Trafficking and Surface Stability in the Gabra1–2 Mouse
Next, we investigated the effect the Gabra1–2 mutation has on the expression and trafficking of inhibitory synapse components. To this end, we immunoblotted total hippocampal lysates for the α1 and α2 subunits, GPN, CB, and synapsin (Figure 1B), as well as the β3 and γ2 subunits (Figure S1E). There were no changes in the total expression of any of these proteins in heterozygous Gabra1–2 mice compared with WT (data not shown). In homozygous animals, total levels of synapsin were unchanged, indicating no widespread changes in synapse formation (Figure 1C). We found that the total CB was increased in homozygous mutants compared with controls, while GPN was unchanged (Figure 1C). We found no changes in either β3 or γ2 expression in homozygous animals compared to WT (Figure S1E). Immunoblotting showed that the total α1 expression decreased in homozygous Gabra1–2 mutants compared to controls, and total expression of α2 also trended toward a decrease (Figure 1C). To examine these findings in more depth, we performed surface biotinylation experiments in WT and Gabra1–2 homozygous hippocampal slices (Figure 1D). To better detect the low levels of surface α2 subunit, these experiments were performed using the previously described pHlourin-α2 mice (Nakamura et al., 2016)—in which the α2 subunit is GFP-tagged—crossed with Gabra1–2 animals. We found that the ratio of surface to total protein for α1 was unchanged, while that for the α2 subunit was reduced in mutants compared with controls (Figure 1E), consistent with the decline in total α2 expression (Figure 1C). Collectively, these results indicate a change in the trafficking of α subunits in Gabra1–2 mutants: α2-containing GABAARs are displaced by α1-containing receptors accumulating in the plasma membrane. The data indicating a depression in total α1 and α2 expression (Figure 1C) was obtained from crude hippocampal lysates, which contain the large intracellular pool of α subunits. Thus, the reduction in the total expression of α subunits may reflect a compensatory reduction in the intracellular pool in response to additional plasma membrane accumulation of α1-GABAARs.
The Gabra1–2 Mutation Enhances the Interaction between α1 and CB
It has been established that the α2 ICD motif mediates a preferential interaction with CB over GPN in vitro (Hines et al., 2018). To confirm this finding in vivo, we performed co-immunoprecipitation (coIP) experiments on forebrain plasma membrane lysates from WT and Gabra1–2 mice. Lysates were incubated with magnetic beads cross-linked to an N-terminal α1 antibody. Bound material was eluted from the beads and subjected to SDS-PAGE, followed by immunoblotting for either α1, CB, or GPN. α1 immunoblots showed excellent purification of the subunit from the total lysate and revealed reduced α1 in homozygotes, in both the input and the immunoprecipitate (IP), consistent with our earlier results (Figure 1C). Total and immunoprecipitated CB levels, particularly the high molecular weight isoforms, were increased in the Gabra1–2 IP compared with WT, confirming that the α2 ICD motif does indeed mediate a preferential association with CB in vivo (Figure 2). Interestingly, GPN levels were also increased in the mutant IP compared with WT, indicating an enhanced interaction between GPN and the mutant α1 subunit (Figure 2). As it has been previously shown that GPN can bind both a subunit ICDs and CB (Grosskreutz et al., 2001; Mukherjee et al., 2011; Tyagarajan et al., 2011), these data suggest that, via binding at the α2 ICD motif, GABAARs, CB, and GPN form a synergistic tripartite complex in which strengthening the interaction between two partners enhances the stability of the entire complex.
Figure 2. The Gabra1–2 Mutation Strengthens the Interactions between the Chimeric α1–2 Subunit and CB.

(A) Representative western blots showing co-immunoprecipitation levels of CB and GPN with the WT and mutant α1 subunit.
(B) Pooled quantification (n = 4 biological and technical replicates) of protein immunoprecipitation shows a significant reduction in the pull-down of the α1 subunit and significant increases in the pull-down of CB and GPN with the chimeric α1–2 subunit. Total protein expression data were normalized to GAPDH, and IP expression data were normalized to total α1 due to the decrease in total α1 expression in Gabra1–2 mutants.
Unpaired t tests with Welch’s correction were used to analyze data (*p < 0.01; **p < 0.05; #p < 0.005). Data are expressed as the percentage of WT and error bars represent mean ± SEM.
The Gabra1–2 Mutation Is Sufficient to Target α1-Containing GABAARs to the AIS
Our previous work has demonstrated that the α2 ICD motif is necessary for the accumulation of GABAARs at the AIS (Hines et al., 2018). To determine if this motif is sufficient for the formation of these precisely localized synapses, we performed immunocytochemistry (ICC) on WT and Gabra1–2 homozygous DIV21 hippocampal neurons. ICC was selected to allow for superior resolution of subcellular compartments, including the AIS. Neurons were co-labeled with antibodies against AIS markers (pan-Na+ and ankyrin G, together) and either the GABAAR α1 or α2 subunit. Imaging revealed a striking phenotype: chimeric α1-containing GABAARs formed significantly denser and larger puncta on Gabra1–2 AIS segments, compared with WT AIS segments (Figures 3A–3B). In addition, the number and size of synapses positive for the α1 subunit was far larger in the somatodendritic compartments of mutant neurons (Figures 3A–3B). Experiments examining the localization of α2-containing GABAARs showed neither changes in the levels of α2 at the mutant AIS nor alterations in the density or size of α2 positive synaptic puncta (Figures 3C–3D). These results indicate that the α2 360–375 motif does not simply allow GABAARs to access and remain at the AIS, but it also stabilizes receptors at synapses versus the extrasynaptic space in the soma and dendrites, providing a mechanism for the previously observed greater synaptic clustering of the α2 subunit (Tretter et al., 2008). This interpretation is strengthened by data from a co-localization analysis performed on whole neurons showing that the co-localization of α1 puncta and the vesicular GABA transporter (VGAT, presynaptic marker), as measured by the Pearson’s correlation coefficient, was significantly increased in Gabra1–2 neurons (Figures S3A–S3B). In addition, the co-localization of α1 puncta and GPN puncta was significantly increased in mutant neurons (Figures S3C–S3D). In contrast, the Gabra1–2 mutation does not affect the co-localization of α2 and VGAT (Figures S4A–S4B).
Figure 3. The Gabra1–2 Mutation Induces the Accumulation of Chimeric α1–2 Subunits at Synapses in the Soma, Dendrites, and AIS.

(A and C) Immunocytochemical (ICC) analysis of the subcellular distribution of the α1 and α2 subunits of the GABAAR in WT and homozygous DIV21 hippocampal neurons. Neurons were stained with antibodies against AIS markers (green) and either α1 (A, red) or α2 (C, red). Insets show magnification of representative AIS segments. White arrows indicate synaptic clusters of mutant α1–2 on the AIS.
(B) Pooled quantification (n = 3 biological and technical replicates, N = 30 cells/genotype) of puncta density, defined as the number of puncta per 10 μm of neurite or soma, reveals that in Gabra1–2 mutants, α1 puncta are significantly denser and larger compared with WT in all neuronal compartments examined.
(D) Pooled quantification (n = 3 biological and technical replicates, N = 30 cells/genotype) demonstrates no change in the distribution of the α2 subunit in Gabra1–2 mutants compared to WT. Scale bar, 10 μm (low mag); scale bar, 5 μm (AIS inset).
Unpaired t tests were used to analyze data (*p < 0.0001). Error bars represent mean ± SEM.
To ensure that there were no gross changes in inhibitory synapse formation, neurons were co-labeled for AIS markers, GPN, and VGAT. Co-localization between GPN and VGAT puncta was unchanged by the Gabra1–2 mutation (Figures S4C–S4D). Further analysis revealed a significant increase in the density and size of VGAT puncta specifically in the soma and dendrites of Gabra1–2 neurons, with VGAT distribution at the AIS unchanged (Figure S5). Analysis of GPN puncta showed the opposing effect: GPN puncta at the AIS were significantly smaller and more numerous in Gabra1–2 neurons when compared with WT controls, with no changes in the soma and dendrites (Figure S6). These results indicate that, in response to the increase in α1 positive synapses on pyramidal neurons, only inhibitory interneurons contacting the soma and dendrites create more presynaptic terminals. The change in AIS GPN from the one or two large cartridges seen in WT (Figure S6A) neurons to small, numerous puncta in mutants suggests that increased stabilization of the chimeric α1-GABAARs at the AIS requires a reconfiguration of the postsynaptic structure. To investigate this phenomenon further, neurons were labeled with antibodies against AIS markers and CB. Unfortunately, CB staining produced a diffuse stain throughout the neuron, impeding analysis of any changes in the subcellular distribution of CB (Figure S7).
Faster Mini Inhibitory Postsynaptic Current (mIPSC) Decay Times and a Shift in mIPSC Amplitude Distribution in Gabra1–2 Neurons
Our biochemical and immunostaining experiments revealed changes in the expression and clustering of mutant α1-containing GABAARs, creating larger and denser inhibitory synapses. To assess any effects these alterations could have on inhibitory synaptic signaling, we examined the kinetics of mIPSCs in CΑ1 pyramidal neurons (Figure 4A). The frequency of mIPSCs was unaffected by the Gabra1–2 mutation (Figure 4B). mIPSC decay times were significantly decreased in mutant neurons compared with WTs (Figure 4C). Examination of tonic inhibition in the CΑ1 showed no changes between WT and homozygous mice (Figure 4D). While the average mIPSC amplitude was unchanged (data not shown), further analysis showed that the Gabra1–2 mutation increased the proportion of high amplitude events (Figures 4E–4F). Fitting the data to Gaussian curves showed that the mIPSC amplitude distribution in WT mice was best described by the sum of 3 Gaussian components (Figure 4G); in Gabra1–2 mice, however, the mIPSC amplitude distribution was best described as the sum of 4 Gaussian components, with the first three similar in amplitude to the WT components and the fourth consisting of a high amplitude component exclusive to Gabra1–2 mutants (Figure 4H). These data demonstrate that enhancing the size and density of inhibitory synapses promotes inhibitory neurotransmission.
Figure 4. Miniature Inhibitory Postsynaptic Currents (mIPSC) Kinetics Are Altered in the Gabra1–2 Hippocampus.

(A) Representative mIPSC recordings from pyramidal neurons in the CΑ1 of WT and Gabra1–2 hippocampal slices, with superimposed spikes (right) representing the average WT (black) and Gabra1–2 traces (red).
(B and C) Quantification of mIPSC kinetics (n = 6 mice/genotype in 3 cohorts) shows no effect of the mutation on mIPSC frequency (B), but (C) reveals a significant decrease in the mIPSC decay time.
(D) Analysis of tonic inhibition in the CΑ1 (n = 8 mice/genotype in 4 cohorts) showed no difference between WT and homozygous animals. In-depth analysis of CΑ1 pyramidal neuron mIPSC amplitudes from WT and Gabra1–2 hippocampal slices (n = 6 mice/genotype in 3 cohorts) revealed a shift toward more high amplitude events in Gabra1–2 mutants.
Alterations in Baseline EEG Activity in Gabra1–2 Mice
Gabra1–2 mice experience changes in the distribution of subtype-specific GABAARs and presynaptic inputs, as well as enhanced inhibitory neurotransmission. Given that inhibitory control of pyramidal neurons is known to play an important role in the generation of cortical oscillations measured by EEG (Buzsáki and Chrobak, 1995; Mann et al., 2005; Roux and Buzsáki, 2015), we assessed baseline cortical EEG activity in WT and Gabra1–2 mice. Mice were recorded for at least 1 hour of wakefulness one week following EEG/electromyogram (EMG) implantation. Representative spectrograms and fast-Fourier transformation of 10 minutes of this awake EEG activity showed changes in specific frequency bands between WT and Gabra1–2 littermate controls (Figures 5A–5B). To examine these changes in more depth, we parsed the EEG activity into the following frequency bands: δ (1–4 Hz), θ (4–8 Hz), α (8–13 Hz), β (13–30 Hz), low γ (30–50 Hz), and high γ (50–100 Hz). Comparison of the relative power of each of these bands between WT and Gabra1–2 animals revealed a significant elevation in the α range in mutants compared with WT (Figure 5C).
Figure 5. The Gabra1–2 Mutation Produces Baseline Abnormalities in Cortical Electroencephalographic (EEG) Recordings.

(A) Spectrograms of representative WT and Gabra1–2 (n = 9 mice/genotype in 3 cohorts) EEG recordings without pharmacological manipulation.
(B) Power spectral density (PSD) plot of 10 minutes of awake baseline EEG activity.
(C) Pooled quantification of the relative power of binned frequency bands revealed a significant increase in the relative power of the α band (8–13 Hz) in Gabra1–2 mutants compared with WT littermate controls. Data were analyzed using unpaired t tests (*p < 0.05). Error bars represent mean ± SEM.
(E and F) The relative (E) and cumulative (F) frequency of mIPSC events of different amplitudes.
(G and H) Fitting the data to Gaussian curves and creating mIPSC histograms for WT (G) and Gabra1–2 (H) demonstrates the existence of a fourth, higher amplitude (Peak 4, blue, mean = —80.7 pA) curve in mutants that does not exist in WT. This result indicates that a population of high amplitude mIPSCs occurs exclusively in Gabra1–2 hippocampal slices.
Mann-Whitney tests were used to analyze mIPSC kinetics and tonic current data (#p < 0.05) and error bars represent mean ± SEM for these experiments. For (G) and (H), data are expressed as mean ± SEM amplitudes (pA) for each of the Gaussian components used to obtain optimal fits to the data.
Resistance to Kainate-Induced Seizures in Gabra1–2 Mice
Electrophysiological experiments show that inhibitory neurotransmission is changed in Gabra1–2 mice. To determine if these physiological changes affect seizure susceptibility, known to be at least partially dependent on inhibitory control of excitation (Fritschy, 2008), we employed the kainate model. Kainate (KA, 20 mg/kg, i.p.) was administered to WT and homozygous adult male littermates that were previously implanted with cortical EEG and EMG monitors. Using the EEG and EMG data to determine the onset of epileptic activity and status epilepticus (SE) (Figures 6A–C), we found that homozygous animals experienced their first tonic-clonic seizure later than WT littermate controls (Figure 6D). In addition, the latency to entering SE was longer in homozygous mice (Figure 6E). Mutant animals also exhibited resilience to SE-induced mortality: no homozygous animals died in the two hours following KA injection, while about 40% of WTs died during SE (Figure 6F). To explore the nature of epileptic activity in Gabra1–2 animals further, we performed fast-Fourier transformation on EEG recordings of SE and parsed the recordings into the specific frequency bands outlined above. This analysis revealed that Gabra1–2 mice experienced a specific reduction in the relative power of the high γ band (50–100 Hz) during SE (Figures 6G–6H). Collectively, these data show that the molecular and physiological changes induced by the Gabra1–2 mutation provide resilience to KA-induced seizures by blunting high γ power.
Figure 6. The Gabra1–2 Mutation Confers Resilience to Kainate-Induced Seizures.

(A) Representative EEG recordings and spectrograms from WT and Gabra1–2 mice injected with 20 mg/kg of kainate (n = 9 mice/genotype in 3 cohorts). The beginning of the trace represents 30 s after the time of injection.
(B and C) Arrows indicate traces expanded in (B) for WT (black) and (C) for homozygote (red). For both (B) and (C), trace 1 (top) represents baseline activity shortly after kainate injection, and trace 2 (bottom) represents the first tonic-clonic seizure.
(D) The onset of the first tonic-clonic seizure event is delayed in Gabra1–2 mutants compared with WT littermate controls. The time of onset is displayed as a percentage of WT.
(E) The onset of status epilepticus (SE) was delayed in Gabra1–2 mutants. Data are displayed as a percentage of WT littermate control.
(F) Kainate-induced mortality was reduced in Gabra1–2 animals compared with WT littermates. The survival plot shows the percentage of death of WT and mutant mice at the corresponding time after kainate injection.
(G) PSD plot of the first hour of EEG activity after kainate injection.
(H) Pooled quantification of the relative power of binned frequency bands during 5 minutes of SE revealed a specific decrease in the contribution of high γ band (50–100 Hz) to total EEΓ power during SE in Gabra1–2 mutants.
Data from (D), (E), and (H) were analyzed with unpaired t tests (*p < 0.05). Data from (F) was analyzed with a log-rank (Mantel-Cox) test (*p < 0.05).Error bars represent mean ± SEM.
The Gabra1–2 Mutation Rescues Early Mortality in Gabra2–1 Mice
Gabra2–1 mice possess a chimeric α2 subunit in which residues 360–375 from the α1 subunit have been knocked in to replace the normal α2 ICD motif. This mutation leads to a loss of inhibitory synapses at the AIS. Both heterozygous and homozygous Gabra2–1 pups display early mortality, with death peaking at postnatal day 20 (PND20) due to spontaneous seizure activity (Hines et al., 2018). To determine if the Gabra1–2 mutation rescues the Gabra2–1 mortality phenotype, homozygous Gabra1–2 mice were crossed with homozygous Gabra2–1 mice. The resulting offspring were genotyped by PCR with primers that detected both the α1 and α2 insertions and were confirmed to be heterozygous for both mutations (Figure 7A). Gabra2–1 heterozygous pups and Gabra1–2/Gabra2–1 double heterozygous pups were monitored each day after birth until PND40. Of the double heterozygous pups, 100% survived to PND40 compared with only 61% of the Gabra2–1 heterozygous animals (Figure 7B), demonstrating that the Gabra1–2 mutation and subsequent phenotype can rescue early mortality in Gabra2–1 animals, likely by promoting the synaptic localization of chimeric α1-GABAARs.
Figure 7. The Gabra1–2 Mutation Rescues the Gabra2–1 Lethal Seizure Phenotype.

(A) Genotyping for a Gabra1–2 homozygous (+/+) positive control, a Gabra2–1 homozygous (+/+) positive control, and two Gabra1–2/Gabra2–1 double heterozygous (+/−) animals. Primers to detect the α1–2 and the α2–1 mutation were used on both double heterozygotes.
(B) The Gabra1–2 mutation rescues the lethal seizure phenotype that leads to early mortality in Gabra2–1 heterozygotes. Data are shown as a percentage of Gabra2–1 and double heterozygous animals alive at a given time after birth (n = 20 mice/genotype).
DISCUSSION
We have provided evidence that the α2 360–375 ICD motif mediates a preferential interaction with CB in vivo and that the presence of this motif in a given GABAAR is sufficient to stabilize that receptor at the AIS. In addition, the α2 motif may constitute an “inhibitory synaptic targeting” signal, generally enhancing the anchoring of GABAARs in the synaptic space, as the Gabra1–2 mutation caused the accumulation of chimeric α1-GABAARs at somatodendritic synapses, altering inhibitory neurotransmission and baseline network oscillations. These changes were sufficient to ameliorate the severity of kainate-induced seizure activity. The Gabra1–2 mutation was also sufficient to rescue the lethal seizure phenotype previously described in Gabra2–1 mice (Hines et al., 2018).
Unexpectedly, given that recent in vitro data showed a relatively low affinity between GPN and the α2 ICD motif (Hines et al., 2018), the Gabra1–2 mutation also led to an enhanced in vivo interaction between the chimeric α1 subunit and GPN. Previous work in Y2H systems has suggested that GPN, CB, and the α2 subunit form a trimeric complex; interactions between GPN/α2 and CB/α2 were potentiated in the presence of CB and GPN, respectively (Saiepour et al., 2010). In addition, α2-GABAARs are more clustered at synapses than those containing α1, a phenomenon at least partially dependent on the presence of GPN (Tretter et al., 2008). Our results support the notion of a “tripartite complex” composed of GPN, CB, and an α2 subunit, in which strengthening the interaction between two of the complex’s partners stabilizes the entire trimer. GABAARs are known to be initially inserted into the plasma membrane extrasynaptically (Bogdanov et al., 2006; Thomas et al., 2005); they then laterally diffuse via Brownian motion until they contact scaffolding proteins in the synaptic space. These scaffolding proteins, including GPN (Jacob et al., 2005), contact the receptors, slowing their diffusion rate and immobilizing receptors at the synapse (Choquet and Triller, 2013; Renner et al., 2008; Triller and Choquet, 2005, 2008). Previous work has shown that the γ2 subunit is necessary for the synaptic anchoring process (Alldred et al., 2005); however, the γ2 subunit is promiscuous and is an unlikely candidate for GABAAR subtype-specific synaptic targeting. We demonstrate that a GPN-CB complex could act as a subtype-selective structural anchor at the synapse, specifically interacting with only those α subunits containing the α2 ICD motif, creating a super-stable trimer that anchors the GABAAR component at the synapse. The increased synaptic clustering of mutant α1-GABAARs seen in the Gabra1–2 mouse is likely due to increased recruitment of these receptors from the extrasynaptic space to the synapse via the α2 motif-CB-GPN interaction. In the AIS, where α2 is particularly enriched, the presence of the trimeric complex would be especially important for the stabilization of α2-GABAARs. α1-GABAARs may be able to access the AIS but will not stabilize as effectively at axo-axonic synapses due to α1’s weaker interaction with CB. Other proteins that are essential in AIS structure—including Ankyrin G and βIV-spectrin (Susuki and Rasband, 2008)—may also interact with the trimeric complex and/or the α2 ICD motif. Future research should investigate potential interactions between the α2 subunit and these AIS components and test the necessity of such interactions to the presence of GABAARs at the AIS.
In the chimeric Gabra1–2 mouse, α1-GABAARs containing the α2 ICD motif acquire an “α2-like” pattern of distribution, with greater synaptic clustering resulting in the proliferation of inhibitory synapses. Affected neurons undergo some compensation in response to these changes: reducing total levels of α1 subunit expression, increasing CB expression, and displacing surface α2-GABAARs. Still, our data demonstrate that the Gabra1–2 mutation enhances inhibitory neurotransmission. Larger proportions of high-amplitude mIPSCs are a result of the significant increase in large, stable inhibitory synapses in Gabra1–2 neurons, while changes in decay kinetics likely reflect an alteration in the type of GABAARs on the surface. Different a subunit compositions have been previously shown to affect mIPSC decay time (Goldstein et al., 2002); an increase in the proportion of α1-GABAARs at the synapse could shift the type of decay seen in mIPSC recordings.
Interestingly, our data demonstrate that the alterations in inhibitory neurotransmission caused by the Gabra1–2 mutation perturb network activity and result in abnormal EEG activity at baseline. Specifically, Gabra1–2 mice show an increase in the power of the α band. Inhibitory interneurons are thought to shape neuronal oscillations (Klausberger and Somogyi, 2008), including the α band (Lozano-Soldevilla, et al., 2014), and it is likely that the constitutive Gabra1–2 mutation also affects interneurons themselves, as shown by the observed increase in VGAT positive presynaptic contacts onto mutant pyramidal neurons. The Gabra1–2 mutation may also affect the interplay between interneurons and pyramidal neurons within neuronal networks. Either of these two putative effects could cause changes in network synchrony. Future research into the specific effect of the Gabra1–2 mutation on interneurons, particularly experiments examining changes in the types of interneurons contacting pyramidal cells, would elucidate the mechanisms at play. Furthermore, in both humans and mice the α frequency band is associated with behavioral inhibition, a process that underlies a variety of cognitive tasks, including attention and working memory (Knyazev, 2007). In addition, benzodiazepine sedation induces a decrease in α band power (Hotz et al., 2000). Future research should examine the possibility that Gabra1–2 mice may be resistant to the sedative effects of these GABAergic pharmacological agents and may also perform better at tasks measuring behavioral inhibition, such as the Go/No-Go task (Gubner et al., 2010).
While the mechanisms of ictogenesis remain unclear, it is generally accepted that inhibitory control of excitation is a contributing factor (Fritschy, 2008). In addition, AIS dysfunction and mutations in the human CB gene ARHGEF9 are both linked to epileptic disorders in humans (Buffington and Rasband, 2011; de Groot et al., 2017; Kalscheuer et al., 2009; Shimojima et al., 2011; Wimmer et al., 2010). The Gabra1–2 mutation enhances inhibitory neurotransmission, resulting in a reduction in the susceptibility of Gabra1–2 mice to kainate-induced seizures. Intriguingly, neuronal activity during SE seems to be altered: the Gabra1–2 mutation blunts the increase in γ band power commonly seen during SE (Sharma et al., 2018). Inhibitory interneurons are thought to shape neuronal oscillations, including the γ band (Mann et al., 2005; Wang et al., 2016), via synchronization of large pyramidal cell networks (Klausberger and Somogyi, 2008) and have been found to undergo morphological and functional changes in animal models of epilepsy (Smith, 2014). The Gabra1–2 mutation may prevent or compensate for KA-induced aberrant interneuron activity, thus mitigating pathological neuronal synchronization, ameliorating SE severity, and promoting survival. Interestingly, other genetic manipulations that affect GABAergic transmission have been shown to blunt γ power and SE severity in the same way as the Gabra1–2 mutation (Moore et al., 2018), suggesting a possible common pathway to reducing SE severity and mortality.
In the previously described Gabra2–1 mouse model, the ICD motif of the α2 subunit is replaced with that of α1. Consistent with the proposed importance of the α2 ICD motif in the stabilization of GABAARs at the AIS and elsewhere, Gabra2–1 animals lose both the majority of inhibitory synapses at the AIS and inhibitory synapses that oppose parvalbumin positive inputs in the somatodendritic compartments. Gabra2–1 mice, both heterozygous and homozygous, experience spontaneous seizures in the postnatal period, often leading to early mortality around PND20 (Hines et al., 2018). Given that the Gabra1–2 mutation seems to lead to the opposite effects of the Gabra2–1 mutation, it is perhaps unsurprising that crossing these two strains results in a rescue of the early mortality phenotype of Gabra2–1 animals. The restoration of inhibitory synapses at the AIS and the increase in size and density of α1-containing synapses due to the Gabra1–2 mutation likely compensates for the loss of α2-mediated inhibitory neurotransmission in Gabra2–1 mice. Importantly, the Gabra2–1 mutation reproduces the effects of many epileptogenic ARHGEF9 mutations in humans (Hines et al., 2018); the rescue of Gabra2–1 by Gabra1–2 suggests that manipulating the distribution of α1-GABAARs or targeting the CB-GPN-α2 complex could represent new avenues for research into therapies for ARHGEF9-associated disorders.
Taken together, we report evidence of a preferential interaction in vivo between CB and the α2 ICD motif and show that this interaction is sufficient to stabilize GABAARs at the AIS. Our data support the theory of a trimeric CB-GPN-α2 subunit complex at inhibitory synapse and present a possible explanation for the higher synaptic clustering of α2-GABAARs over α1-GABAARs. These insights improve our understanding of the subtype-specific inhibitory synaptogenesis that is so important for normal brain function. Finally, Gabra1–2 mice are resilient to kainate-induced seizures and rescue the ictogenic effects of the Gabra2–1 mutation, revealing a new avenue of investigation into treatments of epileptic disorders.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Stephen J. Moss (Stephen.moss@tufts.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal Studies
Animals were cared for in accordance with the NIH Guide for the Care and Use of Laboratory Animals, and all protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of Tufts University School of Medicine. Gabra1–2 animals were generated by Genoway (Lyon, France) and maintained in the vivarium at Tufts University’s Boston campus on a 12-hour light/dark cycle with constant temperature and humidity. All animals except those that underwent EEG/EMG surgery were group-housed and had access to food and water ad libitum, with once weekly cage changes. Germline transmission of the transgene was detected using PCR with primers spanning the intronic region containing the remaining LoxP site. After founder Gabra1–2 mice were received from Genoway, mice were backcrossed onto the C57Bl6J line for at least 10 generations prior to experiments. For all experiments, nontransgenic (wild-type, WT) littermates were used as controls. α2 surface expression experiments involved the generation of a Gabra1–2/Myc-pHlourin-α2 (pHα2) double homozygote line. Gabra1–2 homozygotes and preexisting pHα2 homozygotes were bred for several generations until double homozygotes were born. In pHα2 animals, a pHlourin and Myc tag is incorporated into the N-terminal of the mature α2 subunit protein (Nakamura et al., 2016), allowing for better detection of low levels of α2 expression. In experiments involving the double homozygotes, WT/pHα2 littermates were used as controls. Rescue and survival experiments involved the generation of Gabra1–2/Gabra2–1 heterozygotes, which was achieved by breeding Gabra1–2 homozygotes and preexisting Gabra2–1 homozygotes (Hines et al., 2018) together. All experiments save cell culture immunocytochemistry (ICC) and co-immunoprecipitation employed only male mice, aged 8–12 weeks. Co-immunoprecipitation experiments used age- and sex-matched 8–12-week old male and female mice.
Cell Culture
Primary cultures of hippocampal neurons for ICC were prepared from monogenotype WT and homozygous Gabra1–2 litters. Hippocampi from anaesthetized WT and Gabra1–2 P0 male and female neonates were dissected in ice-cold Hank’s buffered salt solution (HBSS, ThermoFisher, #14185052) with 10 mM HEPES and subsequently pooled and incubated with 0.1% (v/v) trypsin (ThermoFisher, #15090046) in HBSS for 20 minutes at 37◦C. Cells were then washed one time with HBSS and triturated in fresh media to dissociate neurons. After filtering with a 40 μm nylon mesh strainer (ThermoFisher, #22363547) to remove non-dissociated tissue, resulting hippocampal neurons were counted on a hemocytometer and plated on poly-L-lysine (1 mg/mL, Sigma, #P1274)- and laminin (1mg/mL, Sigma, #L2020)-coated glass coverslips in 35 mm dishes with 3 mL of Neurobasal-A culture medium (ThermoFisher, #10888022) containing 2% B27 (v/v; ThermoFisher, #17504044), 1% penicillin/streptomycin (v/v; ThermoFisher, #15140122), 0.6% D-glucose (w/v; Sigma, #G8270), and 1% GlutaMAX (v/v; ThermoFisher, #35050061) at a density of 5 × 105 cells/dish. Cells were grown at 37◦C, 5% CO2 and 95% humidity, and allowed to mature for 21 days (Day in vitro 21, DIV21).
METHOD DETAILS
Cresyl violet staining
8–12-week-old male WT and Gabra1–2 mice were transcardially perfused with 30 mL of phosphate buffered saline (PBS, pH 7.4), followed by 60 mL of 4% (v/v) paraformaldehyde in 0.15 M sodium phosphate buffer (PFA). Brains were post-fixed in PFA for 3 hours, then washed in PBS and transferred to 30% sucrose solution for cryoprotection. Brains were embedded in optimal cutting temperature compound (ThermoFisher, #23730571) and frozen at −80◦C. Brains were subsequently sliced on a cryostat (ThermoFisher, #HM 525) into 40 μm sections and mounted onto gelatin-coated glass slides (Southern Biotech, #SLD01-CS). Mounted sections were sequentially washed for 1 minute in (all v/v) 75% ethanol, 95% ethanol, 100% ethanol, distilled water, and then incubated in filtered cresyl violet solution for 10 minutes (0.3% glacial acetic acid v/v, 0.5% cresyl violet acetate w/v). Staining was followed by 1-minute sequential washes in distilled water, 75% ethanol, 95% ethanol, 100% ethanol, and xylene. Images were acquired using a Nikon E800 microscope (Nikon) with a 4x objective. 3 animals per genotype were imaged.
Western blotting
8–12-week-old male WT and Gabra1–2 mice were anaesthetized with isofluorane and their brains were rapidly removed. Whole hippocampi were dissected out and mechanically homogenized with a 26G needle in ice-cold lysis buffer containing 5 mM EDTA, 5 mM EGTA, 1 mM sodium orthovanadate, 100 mM NaCl, 10 mM NaH2PO4, 25 mM NaF, 2% Triton-X (v/v), and protease (mini cOmplete, Roche, #11836153001) and phosphatase inhibitor cocktails (PhosStop, Roche, #4906845001). Tissue was allowed to lyse on a rotator for 30 minutes at 4◦C. Samples were then centrifuged at 13,000rmp for 15 minutes at 4◦C to pellet insoluble material. Following lysis, protein concentration of the supernatant was assessed with a bicinchoninic acid (BCA) assay (Pierce, #23225) and samples (50 μg) were boiled for 10 minutes at 70◦C in NuPAGE LDS Sample Buffer (Invitrogen, #NP0007) containing 1% (v/v) β-mercaptoethanol. Samples were loaded onto NuPAGE 4%–12% Bis-Tris gels (Invitrogen, #NP0335) and run with MES SDS running buffer (Invitrogen, #NP0002) at 100 V for 2 hours. After separation by SDS-PAGE, proteins were transferred to nitrocellulose membranes using a wet transfer system (transfer buffer: 25 mM Tris, 192 mM glycine, 20% methanol) for 1 hour at 100 V. After transfer, membranes were blocked in 5% milk in tris-buffered saline with 1% (v/v) Tween-20 (TBS-T) for 1 hour at room temperature. Membranes were then incubated with their respective primary antibodies diluted into blocking solution overnight at 4◦C (see Table S1 for antibody sources and concentrations). After 3 washes in TBS-T, membranes were probed with HRP-conjugated secondary antibodies diluted into blocking solution (1:5000, Jackson ImmunoResearch Laboratories, see Table S1 for complete list) for 1 hour at room temperature. Blots were washed 3 times in TBS-T and developed with a chemiluminescence system as per manufacturer’s instructions (Pierce ECL, ThermoFisher, #32106). Blots were imaged (ChemiDoc XRS, Bio-Rad; paired with Image Lab 5.0, Bio-rad) and band densitometry was measured with ImageJ v.2.0.0 (NIH). For quantification, specific protein levels were determined by normalizing to GAPDH loading control densitometry results. Gabra1–2 protein levels were normalized to WT control (100%). 5 independent experiments were performed for all western blotting experiments. Unpaired t tests with Welch’s correction were performed on GraphPad Prism v.7.01 to compare mean values between WT and Gabra1–2, with values below *p < 0.05 considered significant. Graphs were plotted as mean ± standard error of the mean (SEM).
Biotinylation of cell surface proteins
8–12-week-old male mice of all genotypes (WT and Gabra1–2 homozygous for α1; pHα2 and pHα2/Gabra1–2 homozygous for α2) were anaesthetized with isofluorane and their brains were rapidly removed. 350 mm-thick coronal hippocampal slices were prepared and sectioned on a vibratome (Leica VT1000S) in ice-cold cutting solution containing 87 mM NaCl, 2.5 mM KCl, 0.5 mM CaCl2, 25 mM, NaHCO3, 1.25 mM NaH2PO4, 7 mM MgCl2, 50 mM sucrose, and 25 mM glucose. After sectioning, slices were allowed a 1-hour recovery period in 32◦C artificial cerebrospinal fluid (aCSF) containing 126 mM NaCl, 26 mM NaHCO3, 2.5 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 1mM glutamine, 1.25 mM NaH2PO4, 1.5 mM sodium pyruvate, and 10 mM glucose. Following recovery, slices were incubated for 45 minutes in ice-cold, 1 mg/mL EZ-Link Sulfo-NHS-SS-biotin (ThermoFisher, #21328T) in aCSF. All solutions described above were oxygenated with 95% (v/v) O2/5% (v/v) CO2. After 3 rinses in ice-cold 100 mM glycine in aSCF to halt the biotin reaction and 2 washes in ice-cold plain aCSF, samples were mechanically homogenized with a 26G needle and lysed in the previously described lysis buffer. Lysis was carried out as described above. After adjusting for protein concentration measured by BCA assay, hippocampal lysates (500 μg) were incubated with 50 μL of Strepavidin Agarose beads (ThermoFisher, #20347) on a rotator at 4◦C overnight, with a portion (50 μg) reserved for total protein analysis. The supernatant was removed, and beads were washed in lysis buffer 3 times. All samples were mixed with NuPAGE LDS Sample Buffer (Invitrogen, #NP0007)/1% (v/v) β-mercaptoethanol. Samples were then boiled for 10 minutes at 70◦C. Bound proteins and total protein samples were subjected to SDS and immunoblotting (see Table S1 for antibody sources and concentrations), developed, and imaged as described above. Band densitometry was measured with ImageJ v.2.0.0 (NIH) and protein levels were determined by normalizing to GAPDH (total protein) and/or pan-cadherin (surface protein) loading controls. The ratio of surface protein:total protein was calculated, and Gabra1–2 values were normalized to WT or pHα2 control (100%). 5 independent experiments were performed for all biotinylation experiments. Unpaired t tests with Welch’s correction were performed on GraphPad Prism v.7.10 to compare mean values between WT and Gabra1–2 or pHα2 and Gabra1–2/pHα2, with data below p < 0.05 considered significant. Graphs were plotted as mean ± SEM.
Co-Immunoprecipiation (coIP)
Forebrains (cortex and hippocampus) were rapidly removed from isofluorane-anaesthetized 8–12-week-old male and female WT and Gabra1–2 mice and collected in an ice-cold cryoprotectant buffer (225 mM mannitol, 75 mM sucrose, 30 mM Tris-HCl, pH 7.4). Tissue was mechanically homogenized using 14 strokes of a dounce homogenizer in homogenization buffer (225 mM mannitol, 75 mM sucrose, 0.5% (w/v) BSA, 0.5 mM EGTA, 30 mM Tris-HCl, pH 7.4) and subjected to the following sequential centrifugation, all at 4◦C: two 5-minute spins at 800 x g to remove unbroken cells; two 10-minute spins at 10,000 x g to remove mitochondria; and two 20-minute spins at 25,000 x g to isolate plasma membrane (PM) fractions. PM fractions were re-suspended and lysed in a lysis buffer containing 150 mM NaCl, 10 mM Tris, 0.5% Triton-X, and protease and phosphatase inhibitor cocktails, as above. These PM lysates (5000 mg protein) were incubated with 150 mL Protein A Dynabeads (ThermoFisher, #10001D) cross-linked to 15 μg of N-terminal α1 antibody (Abcam, #aβ33299) overnight at 4◦C. Supernatant was removed, and beads were washed 3 times in PBS with 0.05% (v/v) Tween. All samples were eluted in SDS-PAGE sample buffer. Immunoprecipitation samples were first boiled at 55◦C for 10 minutes to remove protein complexes from the beads, and then both total lysate and immunoprecipitation samples were boiled at 95◦C for 5 minutes. Samples were run through SDS-PAGE and western blotting for the appropriate proteins and developed and imaged as detailed above (see Table S1 for antibody sources and concentrations). For quantification using ImageJ v.2.0.0 (NIH), relative immunoprecipitation levels were determined by first normalizing to α1 densitometry results to account for differences in the amount of α1 present in WT versus Gabra1–2 samples. Gabra1–2 protein levels were then normalized to WT control (100%). 4 independent experiments were performed for the coIP experiments. Unpaired t tests with Welch’s correction were performed on GraphPad Prism v.7.10 to compare mean values between WT and Gabra1–2, with data below p < 0.05 considered significant. Graphs were plotted as mean ± SEM.
Immunocytochemistry (ICC)
DIV21 primary hippocampal WT and Gabra1–2 cells were washed once in PBS and fixed for 20 minutes in a solution of 4% (w/v) sucrose and 4% (v/v) paraformaldehyde in PBS. After 3 PBS washes to remove fixative, cells were blocked in a solution containing 5% (w/v) bovine serum albumin (BSA) for 1 hour at room temperature. Cells were then incubated with the appropriate primary antibodies (see Table S1 for details) diluted into a modified blocking solution containing 2.5% (w/v) BSA, 2% (v/v) normal goat serum (NGS), and 0.1% (v/v) Triton-X for 2 hours at room temperature. After 10 washes in PBS to remove unbound antibody, cells were incubated with Alexa conjugated secondary antibodies (1:5000, ThermoFisher, see Table S1 for details) in modified blocking solution for 1 hour at room temperature. Cells were washed with PBS 10 times and mounted on glass slides (Electron Microscopy Sciences, #71864) using ProLong Gold Antifade mounting media (ThermoFisher, #P36930).
Images were acquired with a Nikon Α1 confocal scanning laser microscope coupled with NIS Elements software v.4.20 (Nikon). Images were taken at a 512 × 512 resolution (17 nm/pixel) with a 60x (oil, numerical aperture: 1.00) objective, with the imager blind to genotype. Settings were optimized to ensure low background and sufficient signal/noise ratio, and within experiments settings were unchanged between genotypes. Images were saved as single-channel and overlay 8-bit TIF-files. Immunostaining density (the number of puncta per 10 μm of neurite/soma) and area were quantified using MetaMorph software (Molecular Devices). For definition of image thresholds, the brightness of single-channel images was adjusted with the “inclusive thresholding” function. Within each experiment, threshold adjustments were unchanged between genotypes. Puncta number and size was assessed using the MetaMorph-based “Integrated Morphometry Analysis” tool, which calculated the number, area, and average intensity of single objects > 0.05 μm. Co-localization analysis (calculation of the Pearson’s correlation coefficient) between single-channel images was achieved with the Coloc2 macro on ImageJ v.2.0.0/Fiji (https://imagej.net/ImageJ). A region of interest (ROI) containing both the soma and neurites of each neuron was defined on 1 channel using the polygon drawing tool and transferred to the other channel. Background was subtracted using a Rolling-Ball Background Subtraction of 50. All analysis was performed blinded to genotype. 3 independent experiments were performed for each antibody combination and within each experiment 10 neurons were imaged and analyzed per genotype. Results were analyzed on GraphPad Prism v.7.10 with unpaired t tests to compare mean values between WT and Gabra1–2, with data below p < 0.05 considered significant. Graphs were plotted as the mean ± SEM.
Electrophysiology
Brain slices were prepared from 4–6-week-old WT and Gabra1–2 male, isofluorane-anaethetized mice. 300 μm sections were cut in ice-cold, oxygentated saline (92 mM NMDG, 2.5 mM KCl, 1.25 mM NaH2PO4, 30 mM NaHCO3, 20 mM HEPES, 25 mM glucose, 2 mM thiourea, 5 mM Na-ascorbate, 3 mM Na-pyruvate, 0.5 mM CaCl2, 10 mM MgSO4) using a vibratome (VT1000S, Leica), then incubated at 32◦C for 10 minutes in holding aSCF (92 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 30 mM NaHCO3, 20 mM HEPES, 25 mM glucose, 2 mM thiourea, 5 mM Na-ascorbate, 3 mM Na-pyruvate, 2 mM CaCl2, 2 mM MgSO4). Brain slices were then transferred to the recording chamber and were continually perfused with normal aCSF (126 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 26 mM NaHCO3, 1.25 mM NaH2PO4, 10 mM glucose, 1.5 mM Na-pyruvate, 1 mM glutamine, 3 mM kynurenic acid, 0.005 M GABA) at 32◦C. All solutions were bubbled with 95% (v/v) O2/5% (v/v) CO2. Patch pipettes (5–7 MΩ) were pulled from borosilicate glass (World Precision Instruments) and filled with intracellular solution (140 mM CsCl, 1 mM MgCl2, 0.1 mM EGTA, 10 mM HEPES, 2 mM Mg-ATP, 4 mM NaCl, 0.3 mM Na-GTP, pH to 7.2 with CsOH). To examine mini inhibitory postsynaptic currents (mIPSCs), tetrodotoxin (TTX; 0.5 μM) was added to the aCSF. A 5-minute stabilization period was allowed after obtaining the whole-cell recording conformation (holding potential of −60 mV) in the hippocampal CΑ1. Subsequently, currents were recorded using an Axopatch 200B amplifier (Molecular Devices), low-pass filtered at 2 kHz, digitized at 20 kHz (Digidata 1440A; Molecular Devices), and stored for offline analysis. Series resistance and whole-cell capacitance were continually monitored and compensated for throughout the course of the experiment. Recordings were eliminated from data analysis if series resistance increased by > 20%. mIPSCs were analyzed using mini-analysis software v.5.6.4 (Synaptosoft). Minimum threshold detection was set to 3x the value of baseline noise signal. To assess mIPSC kinetics, the recording trace was visually inspected and only events with a stable baseline, sharp rising phase, and single peak were used to negate artifacts from event summation. Only recordings with a minimum of 200 events fitting these criteria were analyzed. mIPSC amplitude and frequency from each experimental condition were pooled and expressed as mean ± SEM. To measure mIPSC decay, we averaged 100 consecutive events, fit the decay to a double exponential, and took the weight decay constant (σ). Statistical analysis for mean mIPSC kinetics was performed using a Mann-Whitney test, where p < 0.05 is considered significant. To display the distribution of mIPSC amplitudes, data was fitted with a Gaussian function:
Where A is the amplitude, μ is the Gaussian mean amplitude current, σ the Gaussian standard deviation, and C is the constant for each component i.
For tonic current measurements in the CΑ1, once the response reached a plateau level an all-points histogram was plotted for a 10 s period before and during 100 μM picrotoxin application. Recordings with unstable baselines were discarded. Fitting the histogram with a Gaussian distribution gave the mean baseline current amplitude. The difference between the amplitudes before and during picrotoxin application was considered to be the tonic current. The negative section of the all-points histogram which corresponds to the inward IPSCs was not fitted with a Gaussian distribution (Kretschmannova et al., 2013; Nusser and Mody, 2002). Tonic current was analyzed with a Student’s t test, where p < 0.05 is considered significant.
Motor behavior
Genotype-blinded behavioral testing was performed on group-housed 8–12-week-old male WT and Gabra1–2 mice. All behavioral testing occurred during the light phase between 9am and 4pm, following at least 1 hour of habituation to the temperature controlled (70–74◦C) behavioral testing facility. Littermates were used for all experiments. Protocols for all testing were based on standard protocols. Equipment was cleaned between each mouse using 70% ethanol followed by Clidox (chlorine dioxide based sterilant).
For assessment of motor behavior, the rotarod test and open field test (OFT) were employed. During the rotatrod test, mice were placed on the rotarod apparatus (Med Associates; rubber-coated cylinder 4 cm in diameter, fixed 30 cm above ground) and trained in the task in three 3-minute trials at a speed of 16 rotations per minute (rpm), with a 15-minute inter-trial interval. After completion of training, the animals were returned to their homecage and allowed to recover for an additional 15 minutes before testing began. Rotarod testing was conducted on an accelerated speed setting, with rotarod speeds increasing from 4 to 40 rpm over the course of 5 minutes. Animals were subject to three 5-minute-maximum tests, again with an inter-trial interval of 15 minutes. The latency to fall off the rotarod beam or to lose control (defined as 3 rotations around the cylinder without active movement) was measured for each test, and the average latency was calculated for each animal.
In the OFT, mice were individually placed in the center of a 60 cm x 60 cm white-walled box and allowed to freely explore for 10 minutes. The OFT box was positioned inside a photobeam frame with 16 × 16 equally spaced photo emitters and detectors (Kinder Scientific). The frames connected to MotorMonitor software (Kinder Scientific), which measured the total distance traveled for each mouse. All behavioral tests used 10 mice per genotype in at least 3 independent cohorts. All results were analyzed on GraphPad Prism v.7.10 with one-way analysis of variance (ANOVA), with posthoc testing comparing the means between WT and Gabra1–2 hetero- and homozygotes. Data below p < 0.05 was considered significant. Graphs were plotted as mean ± SEM.
Electroencephalography (EEG)
Surgery and recording
8–12-week-old male WT and Gabra1–2 littermates were used for EEG studies. Prefabricated EEG and electromyography (EMG) headmounts (2-channel, Pinnacle Technology, #8201) were implanted under isofluorane anesthesia (3%–5% for induction, 1%–2% for maintenance). Mice were given 0.1 mg/kg buprenorphine i.p. as an additional analgesic. All surgical instruments were heat-sterilized. The fur covering the scalp was shaved off and the scalp itself was sterilized with 3 applications of β-iodine, with a wash of 70% ethanol between each application. Scissors were used to make an incision down the vertical midline of the scalp to expose the skull. The skull was then washed with a sterile Q-tip soaked in 70% ethanol. The EEG/EMG headmount was aligned with lambda and secured to the skull with two 0.10 inch and two 0.12-inch ground screws (Pinnacle Technology, #8209 and #8212, respectively). Two of the screws served as EEG leads in the frontal cortex, 1 as a reference ground, and 1 as an animal ground. Dental cement was used to close the scalp and secure the headmount. Mice were singly-housed post-surgery. After a minimum of 7 days of recovery, EEG and EMG recordings were collected with Sirenia Acquisition software v.1.7.10 (Pinnacle Technology). Recordings were performed in awake, behaving WT and Gabra1–2 mice. Animals were given 1 hour of habituation to the recording chamber and the preamplifier. For baseline EEG activity, EEG/EMG recordings were then collected for 2 hours. For epileptic EEG activity, EEG/EMG recordings were collected for 1 hour pre- and 2 hours post-injection of the chemoconvulsant kainic acid (KA; 20 mg/kg i.p.; Sigma, #K0250). All recordings were processed and analyzed blind with pClamp v.10.3 (Molecular Devices) and Labchart v.7.3.8 (AD Instruments) software.
Baseline analysis
Power analysis was performed using LabChart software v.7.3.8. Fast-Fourier transform (FFT) was used to transform 10 minutes of awake baseline activity from the time domain to the frequency domain to generate a power spectral density plot (FFT size 4K, 93.7% overlap, Hanns window function). Activity was then binned into the following frequency bands: delta (1–4 Hz), theta (4–8 Hz), alpha (8–13 Hz), beta (13–30 Hz), low gamma (30–50 Hz), and high gamma (50–100 Hz). 9 animals per genotype were used in at least 3 separate cohorts. The power of each frequency band was expressed as a percentage of the total EEG power and plotted as the mean ± SEM. Unpaired t tests were performed with GraphPad Prism v.7.10 to compare genotype means, and data below p < 0.05 was considered significant.
Seizure analysis
The latency to the first seizure event and the latency to the onset of status epilepticus (SE) was measured with pClamp software v.10.3. Epileptiform activity was defined as having an amplitude of at least 2.5x the standard deviation of baseline activity and lasting for at least 30 s. Latency to the first seizure was defined as the time from KA injection to the start of the first detected electrographic seizure. SE was defined as continuous epileptiform activity lasting at least 5 minutes, with no more than 30 s between epileptiform events. Latency to SE was defined as the time from KA injection to the start of the first period of such activity. As above, FFT was used to transform the first hour after KA injection from the time domain to the frequency domain to generate a power spectral density plot (FFT size 4K, 93.7% overlap, Hanns window function). Activity was then binned into the same frequency bands as above. Mortality during the post-KA period was also assessed. 9 animals per genotype were used in at least 3 separate cohorts. Gabra1–2 data (latency to first seizure and latency to SE) was normalized to WT littermate controls to account for variability due to differences in KA potency between cohorts. Results were analyzed with GraphPad Prism v.7.10 using either unpaired t tests or unpaired t tests with Welch’s correction, where appropriate, to compare mean values between WT and Gabra1–2. Graphs were plotted as the mean ± SEM. Mortality data was plotted as a survival curve and analyzed with a log-rank (Mantel-Cox) test. Data below p < 0.05 was considered significant.
Survival assessments
Assessment of postnatal survival comparing Gabra2–1 heterozygotes and Gabra1–2/Gabra2–1 heterozygotes was performed. Litters were observed by the investigator once per day until weaning at postnatal day 21 (PND21) to check for pup death. After weaning, offspring were observed once per day to check for mortality until PND40. At least 20 pups per condition were assessed. Results were plotted with GraphPad Prism v.7.10 as the percent of animals alive from PND0-PND40.
QUANTIFICATION AND STATISTICAL ANALYSIS
All programs used for quantification and statistical analysis are outlined in the Method Details section. Statistical analyses are described in the corresponding figure legends and also in the Method Details section.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Ankyrin-G (staining) scaffold protein monoclonal mouse | UC Davis/NIH NeuroMab Facility | Cat# 73-146; RRID:AB_10697718 |
| Anti-GABAAR β3 mouse monoclonal | UC Davis/NIH NeuroMab Facility | Cat# 75-149; RRID: AB_2109585 |
| Anti-Cadherin, pan Rabbit polyclonal | Cell Signaling | Cat# 4068; RRID:AB_2158565 |
| Anti-Collybistin rabbit polyclonal | Synaptic Systems | Cat# 261 003; RRID:AB_2619977 |
| GABAAR α1 N-terminal rabbit polyclonal | Abcam | Cat# ab33299; RRID:AB_732498 |
| Anti-GABAAR α2 N-terminal rabbit polyclonal | Synaptic Systems | Cat# 224 103; RRID:AB_2108839 |
| GABAAR α2 C-terminal rabbit polyclonal | Phosphosolutions | No longer available |
| Anti-GABAAR γ2L mouse monoclonal | UC Davis/NIH NeuroMab Facility | Cat #75-442; RRID: AB_2617122 |
| GAPDH rabbit polyclonal | Santa Cruz | No longer available |
| Anti-Gephyrin 3B11 mouse monoclonal | Synaptic Systems | Cat# 147 111; RRID:AB_887719 |
| Anti-Gephyrin mAb7a mouse monoclonal | Synaptic Systems | Cat# 147 011; RRID:AB_887717 |
| Anti-Gephyrin RbmAb7a rabbit monoclonal | Synaptic Systems | Cat# 147 008; RRID:AB_2619834 |
| Anti-GFP Antibody, Unconjugated chicken polyclonal | Cell Signaling | Cat# 2555; RRID:AB_10692764 |
| Anti-Sodium Channel, Pan antibody mouse monoclonal | Sigma Aldrich | Cat# S8809; RRID:AB_477552 |
| Anti-Synapsin 1 antibody mouse monoclonal | Synaptic Systems | Cat# 106 011; RRID:AB_2619772 |
| Anti-VGAT antibody guinea pig polyclonal | Synaptic Systems | 131 004; RRID:AB_887873 |
| Donkey anti-mouse peroxidase conjugated | Jackson ImmunoResearch | Cat# 715-036-151; RRID:AB_2340774 |
| Donkey anti-rabbit peroxidase conjugated | Jackson ImmunoResearch | Cat# 711-036-152; RRID:AB_2340590 |
| Goat anti-guinea pig Alexa Fluor 647 | ThermoFisher Scientific | Cat# A-21450; RRID:AB_2735091 |
| Goat anti-mouse Alexa Fluor 488 | ThermoFisher Scientific | Cat# A28175; RRID:AB_2536161 |
| Goat anti-rabbit Alexa Fluor 568 | ThermoFisher Scientific | Cat# A-11011; RRID:AB_143157 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 1 M CaCl2 | Sigma Aldrich | Cat#: 21114 |
| 1 M KCl | Sigma Aldrich | Cat#: 60121 |
| 1 M MgCl2 | Sigma Aldrich | Cat#: 63020 |
| 10x Phosphate Buffered Saline (PBS) | ThermoFisher Scientific | Cat#: BP399-4 |
| 32% PFA stock solution | Electron Miscroscopy Sciences | Cat#: 15714S |
| Acetic acid, glacial | ThermoFisher Scientific | Cat#: A38-212 |
| Bovine Serum Albumin (BSA) | Sigma Aldrich | Cat#: A7906 |
| Cresyl violet acetate | Sigma Aldrich | Cat#: C-5042 |
| CsCl | Sigma Aldrich | Cat#: C4036 |
| CsOH | Sigma Aldrich | Cat#: C8518 |
| D-Glucose | Sigma Aldrich | Cat#: G8270 |
| Dehydrated milk | Amazon | https://amazon.com/Emergency-Essentials-Instant-Nonfat-Milk/dp/B003SQ98EC |
| Ethanol, 200 proof | VWR | Cat#: 89125-172 |
| Ethylene glycol-bis(2-aminoethylether)- N,N,N′,N′-tetraacetic acid (EGTA) | Sigma Aldrich | Cat#: E3889 |
| Ethylenediaminetetraacetic acid (EDTA) | Sigma Aldrich | Cat#: E9884 |
| EZ-Link Sulfo-NHS-SS-Biotin | ThermoFisher Scientific | Cat#: 21328T |
| Fisher Healthcare Tissue-Plus O.C.T. Compound | ThermoFisher Scientific | Cat#: 23-730-571 |
| GABA | Sigma Aldrich | Cat#: A2129 |
| GIBCO B27 (50x) | ThermoFisher Scientific | Cat#: 17504044 |
| GIBCO GlutaMAX Supplement | ThermoFisher Scientific | Cat#: 35050061 |
| GIBCO Penicillin/streptomycin (10,000 U/mL) | ThermoFisher Scientific | Cat#: 15140122 |
| GIBCO Trypsin (2.5%), no phenol red | ThermoFisher Scientific | Cat#: 15090046 |
| Glycine | VWR | Cat#: 0167 |
| Invitrogen 1 M HEPES buffer solution | ThermoFisher Scientific | Cat#: 15630080 |
| Invitrogen HBSS, 10X, no phenol red, no calcium, no magnesium | ThermoFisher Scientific | Cat#: 14185-052 |
| Invitrogen MES SDS Running Buffer 20x | Thermofisher Scientific | Cat#: NP0002 |
| Invitrogen NuPAGE LDS Sample Buffer 4x | ThermoFisher Scientific | Cat#: NP0007 |
| Invitrogen ProLong Gold Antifade Mounting Reagent | Thermo Fisher Scientific | Cat#: P36930 |
| Invitrogen Protein A Dynabeads | ThermoFisher Scientific | Cat#: 10001D |
| Isofluorane | Piramal Healthcare | Cat#: NDC 66794-013-25 |
| Kainic Acid (Kainate) | Sigma Aldrich | Cat#: K0250 |
| Kynurenic Acid | Sigma Aldrich | Cat#: K337 |
| L-glutamine | Sigma Aldrich | Cat#: G8540 |
| Laminin | Sigma Aldrich | Cat#: L2020 |
| Mannitol | Sigma Aldrich | Cat#: M4125 |
| Meglumine (NMDG) | Sigma Aldrich | Cat#: M9179 |
| Methanol | Sigma Aldrich | Cat#: 179337 |
| Mg-ATP | Sigma Aldrich | Cat#: A9187 |
| MgSO4 | Sigma Aldrich | Cat#: M3409 |
| Na-ascorbate | Sigma Aldrich | Cat#: A7631 |
| Na-GTP | Sigma Aldrich | Cat#: G8877 |
| NaCl | Sigma Aldrich | Cat#: S7653 |
| NaF | Sigma Aldrich | Cat#: S7920 |
| NaH2PO4 | Sigma Aldrich | Cat#: S9638 |
| NaHCO3 | Sigma Aldrich | Cat#: S6297 |
| Neurobasal-A Medium, serum-free | ThermoFisher Scientific | Cat#: 10888022 |
| Normal Goat Serum (NGS) | Abcam | Cat#: ab7481 |
| Pierce Streptavidin Agarose beads | ThermoFisher Scientific | Cat#: 20347 |
| Pierce BCA Protein Assay Kit | ThermoFisher Scientific | Cat#: 23225 |
| Pierce ECL Western Blotting Substrate | ThermoFisher Scientific | Cat#: 32106 |
| Poly-L-Lysine hydrobromide | Sigma Aldrich | Cat#: P1274 |
| Roche cOmplete, Mini Protease Inhibitor Cocktail | Sigma Aldrich | Cat#: 11836153001 |
| Roche PhosSTOP | Sigma Aldrich | Cat#: 4906845001 |
| Sodium orthovanadate | Sigma Aldrich | Cat#: 450243 |
| Sodium pyruvate | Sigma Aldrich | Cat#: P3662 |
| Sucrose | VWR | Cat#: 0335 |
| Tetrodotoxin (TTX) | Tocris | Cat#: 1069 |
| Thiourea | Sigma Aldrich | Cat#: 8658 |
| Tris | VWR | Cat#: 0497 |
| Triton X-100 | Sigma Aldrich | Cat#: T8787 |
| Tween-20 | Sigma Aldrich | Cat#: P2287 |
| β-mercaptoethanol | Sigma Aldrich | Cat#: 63689 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Gabra1-2(−/−) Mouse: Gabra1-2(+/−) Mouse: Gabra1-2(+/+) | This paper | N/A |
| Mouse: pHlourin-α2(+/+) Mouse: pHlourin-α2(+/+)/Gabra1-2(+/+) | Nakamura et al., 2016 | N/A |
| Mouse: Gabra2-1(+/−)/Gabra1-2(+/−) | Hines et al., 2018; This paper | N/A |
| Oligonucleotides | ||
| Gabra1-2 Genotyping Forward Primer: 5′-GGGTC GACAACTATTTCACCAAGAGAGG-3′ | Integrated DNA Technologies | N/A |
| Gabra1-2 Genotyping Reverse Primer: 5′-GGGC GGCCGCTTATCGGTCGATTTTGCTGACGC-3′ | Integrated DNA Technologies | N/A |
| Gabra1-2 Sequencing Primer 1: 5′-TGCCAGGGA GTCTAACCGT-3′ | Integrated DNA Technologies | N/A |
| Gabra1-2 Sequencing Primer 2: 5′-CAAATAGCA GCGGAAAGGCT-3′ | Integrated DNA Technologies | N/A |
| Gabra1-2 Sequencing Primer 3: 5′-CTGCCAGG GAGTCTAACCGT-3′ | Integrated DNA Technologies | N/A |
| Gabra1-2 Sequencing Primer 4: 5′-GACAGTCG GTCGATTTTGCTG-3′ | Integrated DNA Technologies | N/A |
| Software and Algorithms | ||
| GraphPad Prism, v.7.01 | GraphPad Software | RRID:SCR_002798 https://www.graphpad.com |
| Image Lab, v.5.0 | Bio-Rad | RRID:SCR_014210 http://www.bio-rad.com |
| ImageJ v.2.0.0 | Fiji | RRID:SCR_002285 https://imagej.net |
| LabChart v.7.3.8 | AD Instruments | RRID:SCR_001620 https://www.adinstruments.com/products/labchart |
| MetaMorph, v.7.8.0.0 | Molecular Devices | RRID:SCR_002368 https://www.moleculardevices.com |
| Mini-Analysis Software v.5.6.4 | Synaptosoft | http://www.synaptosoft.com/MiniAnalysis/ |
| MotorMonitor | Kinder Scientific | http://kinderscientific.com/products/open_field |
| NIS-Elements, v.4.20 | Nikon | RRID:SCR_014329 https://www.microscope.healthcare.nikon.com/products/software |
| pClamp v.10.3 | Moleclular Devices | RRID:SCR_011323 https://www.moleculardevices.com |
| Sirenia Acquisition, v.1.7.10 | Pinnacle Technology | RRID:SCR_016183 https://www.pinnaclet.com |
| Other | ||
| Axon Axopatch 200B Microelectrode Amplifier | Molecular Devices | Cat#: 200B https://www.moleculardevices.com/sites/default/files/en/assets/data-sheets/dd/cns/axon-axopatch-200b-microelectrode-amplifier.pdf |
| ChemiDoc XRS | Bio-Rad | Cat#: 1708265 http://www.bio-rad.com/en-uk/product/chemidoc-xrs-system?ID=NINJHRKG4 |
| Crytostat | ThermoFisher Scientific | Cat#: HM525 No longer available |
| Leica VT1000 S Vibrating blade microtome | Leica | Cat#: VT1000 S https://www.leicabiosystems.com/histology-equipment/sliding-and-vibrating-blade-microtomes/vibrating-blade-microtome/products/leica-vt1000-s/ |
| Nikon A1 confocal scanning laser microscope | Nikon | Cat#: A1HD25 https://www.microscope.healthcare.nikon.com/products/confocal-microscopes/a1hd25-a1rhd25 |
| Nikon Eclipse E800 Brightfield Microscope | Nikon | No longer available |
Highlights.
Strengthening specific α1-GABAAR/collybistin interactions increases synaptic size
Augmenting the synaptic targeting of α1-GABAARs increases inhibitory transmission
Enhancing the specific synaptic targeting of α1-GABAARs provides seizure resilience
ACKNOWLEDGMENTS
This work was supported by funding from the NIH, National Institute of Mental Health (NIMH) grant MH097446 (P.A.D. and S.J.M.); National Institute of Neurological Disorders and Stroke (NINDS) grants NS051195, NS081735, NS080064, and NS087662 (S.J.M.); the Department of Defense (DOD) grant AR140209 (P.A.D. and S.J.M.); and the Simons Foundation grant 206026 (S.J.M.).
Footnotes
DECLARATION OF INTERESTS
H.J.W. and N.J.B. are full-time employees and shareholders of AstraZeneca. S.J.M. serves as a consultant for AstraZeneca, Bain Capital and SAGE Therapeutics, relationships that are regulated by Tufts University. S.J.M. is also a shareholder of SAGE Therapeutics.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.06.014.
REFERENCES
- Alldred MJ, Mulder-Rosi J, Lingenfelter SE, Chen G, and Lüscher B. (2005). Distinct gammα2 subunit domains mediate clustering and synaptic function of postsynaptic GABAA receptors and gephyrin. J. Neurosci 25, 594–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov Y, Michels G, Armstrong-Gold C, Haydon PG, Lindstrom J, Pangalos M, and Moss SJ (2006). Synaptic GABAA receptors are directly recruited from their extrasynaptic counterparts. EMBO J 25, 4381–4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffington SA, and Rasband MN (2011). The axon initial segment in nervous system disease and injury. Eur. J. Neurosci 34, 1609–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G, and Chrobak JJ (1995). Temporal structure in spatially organized neuronal ensembles: a role for interneuronal networks. Curr. Opin. Neurobiol 5, 504–510. [DOI] [PubMed] [Google Scholar]
- Choquet D, and Triller A. (2013). The dynamic synapse. Neuron 80, 691–703. [DOI] [PubMed] [Google Scholar]
- de Groot C, Floriou-Servou A, Tsai Y-C, Früh S, Kohler M, Parkin G, Schwerdel C, Bosshard G, Kaila K, Fritschy JM, and Tyagarajan SK (2017). RhoGEF9 splice isoforms influence neuronal maturation and synapse formation downstream of α2 GABAA receptors. PLoS Genet. 13, e1007073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritschy J-M (2008). Epilepsy, E/I Balance and GABA(A) Receptor Plasticity. Front. Mol. Neurosci 1, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein PA, Elsen FP, Ying S-W, Ferguson C, Homanics GE, and Harrison NL (2002). Prolongation of hippocampal miniature inhibitory postsynaptic currents in mice lacking the GABA(A) receptor alpha1 subunit. J. Neurophysiol 88, 3208–3217. [DOI] [PubMed] [Google Scholar]
- Grosskreutz Y, Hermann A, Kins S, Fuhrmann JC, Betz H, and Kneussel M. (2001). Identification of a gephyrin-binding motif in the GDP/GTP exchange factor collybistin. Biol. Chem 382, 1455–1462. [DOI] [PubMed] [Google Scholar]
- Gubner NR, Wilhelm CJ, Phillips TJ, and Mitchell SH (2010). Strain differences in behavioral inhibition in a Go/No-go task demonstrated using 15 inbred mouse strains. Alcohol. Clin. Exp. Res 34, 1353–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey K, Duguid IC, Alldred MJ, Beatty SE, Ward H, Keep NH, Lingenfelter SE, Pearce BR, Lundgren J, Owen MJ, et al. (2004). The GDP-GTP exchange factor collybistin: an essential determinant of neuronal gephyrin clustering. J. Neurosci 24, 5816–5826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines RM, Maric HM, Hines DJ, Modgil A, Panzanelli P, Nakamura Y, Nathanson AJ, Cross A, Deeb T, Brandon NJ, et al. (2018). Developmental seizures and mortality result from reducing GABAA receptor α2-subunit interaction with collybistin. Nat. Commun 9, 3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotz MA, Ritz R, Linder L, Scollo-Lavizzari G, and Haefeli WE (2000). Auditory and electroencephalographic effects of midazolam and α-hydroxymidazolam in healthy subjects. Br. J. Clin. Pharmacol 49, 72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob TC, Bogdanov YD, Magnus C, Saliba RS, Kittler JT, Haydon PG, and Moss SJ (2005). Gephyrin Regulates the Cell Surface Dynamics of Synaptic GABAA Receptors. J. Neurosci 25, 10469–10478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalscheuer VM, Musante L, Fang C, Hoffmann K, Fuchs C, Carta E, Deas E, Venkateswarlu K, Menzel C, Ullmann R, et al. (2009). A balanced chromosomal translocation disrupting ARHGEF9 is associated with epilepsy, anxiety, aggression, and mental retardation. Hum. Mutat 30, 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kins S, Betz H, and Kirsch J. (2000). Collybistin, a newly identified brain-specific GEF, induces submembrane clustering of gephyrin. Nat. Neurosci 3, 22–29. [DOI] [PubMed] [Google Scholar]
- Klausberger T, and Somogyi P. (2008). Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science 321, 53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, Roberts JDB, and Somogyi P. (2002). Cell Type- and Input-Specific Differences in the Number and Subtypes of Synaptic GABAA Receptors in the Hippocampus. J. Neurosci 22, 2513–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knyazev GG (2007). Motivation, emotion, and their inhibitory control mirrored in brain oscillations. Neurosci. Biobehav. Rev 31, 377–395. [DOI] [PubMed] [Google Scholar]
- Kole MHP, and Stuart GJ (2012). Signal processing in the axon initial segment. Neuron 73, 235–247. [DOI] [PubMed] [Google Scholar]
- Kretschmannova K, Hines RM, Revilla-Sanchez R, Terunuma M, Tretter V, Jurd R, Kelz MB, Moss SJ, and Davies PA (2013). Enhanced tonic inhibition influences the hynoptic and amnestic actions of the intravenous anesthetics etomidate and propofol. J. Neurosci 33, 7264–7273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano-Soldevilla D, ter Huurne N, Cools R, and Jensen O. (2014). GABAergic modulation of visual gamma and alpha oscillations and its consequences for working memory performance. Curr. Biol 24, 2878–2887. [DOI] [PubMed] [Google Scholar]
- Mann EO, Radcliffe CA, and Paulsen O. (2005). Hippocampal gamma-frequency oscillations: from interneurones to pyramidal cells, and back. J. Physiol 562, 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore YE, Deeb TZ, Chadchankar H, Brandon NJ, and Moss SJ (2018). Potentiating KCC2 activity is sufficient to limit the onset and severity of seizures. Proc. Natl. Acad. Sci. USA 115, 10166–10171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee J, Kretschmannova K, Gouzer G, Maric H-M, Ramsden S, Tretter V, Harvey K, Davies PA, Triller A, Schindelin H, and Moss SJ (2011). The residence time of GABA(A)Rs at inhibitory synapses is determined by direct binding of the receptor α1 subunit to gephyrin. J. Neurosci 31, 14677–14687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Morrow DH, Modgil A, Huyghe D, Deeb TZ, Lumb MJ, Davies PA, and Moss SJ (2016). Proteomic Characterization of Inhibitory Synapses Using a Novel pHluorin-tagged γ-aminobutyric Acid Receptor, Type A (GABAA), α2 Subunit Knock-in Mouse. J. Biol. Chem 291, 12394–12407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusser Z, and Mody I. (2002). Selective modulation of tonic and phasic inhibitions in dentate gyrus granule cells. J. Neurophysiol 87, 2624–2628. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Sieghart W, Benke D, Fritschy JM, and Somogyi P. (1996). Differential synaptic localization of two major gamma-aminobutyric acid type A receptor alpha subunits on hippocampal pyramidal cells. Proc. Natl. Acad. Sci. USA 93, 11939–11944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyíri G, Freund TF, and Somogyi P. (2001). Input-dependent synaptic targeting of α(2)-subunit-containing GABA(A) receptors in synapses of hippocampal pyramidal cells of the rat. Eur. J. Neurosci 13, 428–442. [DOI] [PubMed] [Google Scholar]
- Olsen RW, and Sieghart W. (2008). International Union of Pharmacology. LXX. Subtypes of gamma-aminobutyric acid(A) receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol. Rev 60, 243–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen RW, and Tobin AJ (1990). Molecular biology of GABAA receptors. FASEB J. 4, 1469–1480. [DOI] [PubMed] [Google Scholar]
- Papadopoulos T, Korte M, Eulenburg V, Kubota H, Retiounskaia M, Harvey RJ, Harvey K, O’Sullivan GA, Laube B, Hülsmann S, et al. (2007). Impaired GABAergic transmission and altered hippocampal synaptic plasticity in collybistin-deficient mice. EMBO J. 26, 3888–3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos T, Eulenburg V, Reddy-Alla S, Mansuy IM, Li Y, and Betz H. (2008). Collybistin is required for both the formation and maintenance of GABAergic postsynapses in the hippocampus. Mol. Cell. Neurosci 39, 161–169. [DOI] [PubMed] [Google Scholar]
- Papadopoulos T, Schemm R, Grubmu€ller H, and Brose N. (2015). Lipid binding defects and perturbed synaptogenic activity of a Collybistin R290H mutant that causes epilepsy and intellectual disability. J. Biol. Chem 290, 8256–8270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner M, Specht CG, and Triller A. (2008). Molecular dynamics of postsynaptic receptors and scaffold proteins. Curr. Opin. Neurobiol 18, 532–540. [DOI] [PubMed] [Google Scholar]
- Roux L, and Buzsáki G. (2015). Tasks for inhibitory interneurons in intact brain circuits. Neuropharmacology 88, 10–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph U, Crestani F, Benke D, Brünig I, Benson JA, Fritschy JM, Martin JR, Bluethmann H, and Möhler H. (1999). Benzodiazepine actions mediated by specific gamma-aminobutyric acid(A) receptor subtypes. Nature 401, 796–800. [DOI] [PubMed] [Google Scholar]
- Saiepour L, Fuchs C, Patrizi A, Sassoè-Pognetto M, Harvey RJ, and Harvey K. (2010). Complex role of collybistin and gephyrin in GABAA receptor clustering. J. Biol. Chem 285, 29623–29631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selten M, van Bokhoven H, and Nadif Kasri N. (2018). Inhibitory control of the excitatory/inhibitory balance in psychiatric disorders. F1000Res. 7, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Puttachary S, Thippeswamy A, Kanthasamy AG, and Thippeswamy T. (2018). Status Epilepticus: Behavioral and Electroencephalography Seizure Correlates in Kainate Experimental Models. Front. Neurol 9, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimojima K, Sugawara M, Shichiji M, Mukaida S, Takayama R, Imai K, and Yamamoto T. (2011). Loss-of-function mutation of collybistin is responsible for X-linked mental retardation associated with epilepsy. J. Hum. Genet 56, 561–565. [DOI] [PubMed] [Google Scholar]
- Sigel E, and Steinmann ME (2012). Structure, function, and modulation of GABA(A) receptors. J. Biol. Chem 287, 40224–40231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BN (2014). Inhibitory neurons cut a new path in epilepsy development. Epilepsy Curr. 14, 213–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susuki K, and Rasband MN (2008). Spectrin and ankyrin-based cytoskeletons at polarized domains in myelinated axons. Exp. Biol. Med. (Maywood) 233, 394–400. [DOI] [PubMed] [Google Scholar]
- Terunuma M, Revilla-Sanchez R, Quadros IM, Deng Q, Deeb TZ, Lumb M, Sicinski P, Haydon PG, Pangalos MN, and Moss SJ (2014). Postsynaptic GABAB receptor activity regulates excitatory neuronal architecture and spatial memory. J. Neurosci 34, 804–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas P, Mortensen M, Hosie AM, and Smart TG (2005). Dynamic mobility of functional GABAA receptors at inhibitory synapses. Nat. Neurosci 8, 889–897. [DOI] [PubMed] [Google Scholar]
- Tretter V, and Moss SJ (2008). GABA(A) Receptor Dynamics and Constructing GABAergic Synapses. Front. Mol. Neurosci 1, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretter V, Jacob TC, Mukherjee J, Fritschy J-M, Pangalos MN, and Moss SJ (2008). The clustering of GABA(A) receptor subtypes at inhibitory synapses is facilitated via the direct binding of receptor alpha 2 subunits to gephyrin. J. Neurosci 28, 1356–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretter V, Mukherjee J, Maric H-M, Schindelin H, Sieghart W, and Moss SJ (2012). Gephyrin, the enigmatic organizer at GABAergic synapses. Front. Cell. Neurosci 6, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triller A, and Choquet D. (2005). Surface trafficking of receptors between synaptic and extrasynaptic membranes: and yet they do move!. Trends Neurosci. 28, 133–139. [DOI] [PubMed] [Google Scholar]
- Triller A, and Choquet D. (2008). New concepts in synaptic biology derived from single-molecule imaging. Neuron 59, 359–374. [DOI] [PubMed] [Google Scholar]
- Tyagarajan SK, Ghosh H, Harvey K, and Fritschy J-M (2011). Collybistin splice variants differentially interact with gephyrin and Cdc42 to regulate gephyrin clustering at GABAergic synapses. J. Cell Sci 124, 2786–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vien TN, Modgil A, Abramian AM, Jurd R, Walker J, Brandon NJ, Terunuma M, Rudolph U, Maguire J, Davies PA, and Moss SJ (2015). Compromising the phosphodependent regulation of the GABAAR beta3 subunit reproduces the core phenotypes of autism spectrum disorders. PNAS 112, 14805–14810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang P, and Wyskiel DR (2016). Chandelier Cells in Functional and Dysfunctional Neural Circuits. Front. Neural Circuits 10, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer VC, Reid CA, So EY-W, Berkovic SF, and Petrou S. (2010). Axon initial segment dysfunction in epilepsy. J. Physiol 588, 1829–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang S, Kim EY, Connelly JJ, Nassar N, Kirsch J, Winking J, Schwarz G, and Schindelin H. (2006). The Crystal Structure of Cdc42 in Complex with Collybistin II, a Gephyrin-interacting Guanine Nucleotide Exchange Factor. J. Mol. Biol 359, 35–46. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.