

Abstract
Many neurodegenerative diseases such as Alzheimer’s disease are characterized by pathological β-sheet filaments of the tau protein, which spread in a prion-like manner in patient brains. To date, high-resolution structures of tau filaments obtained from patient brains show that the β-sheet core only includes portions of the microtubule-binding repeat domains and excludes the C-terminal residues, indicating that the C-terminus is dynamically disordered. Here we use solid-state NMR spectroscopy to identify the β-sheet core of full-length 0N3R tau fibrillized using heparin. Assignment of 13C and 15N chemical shifts of the rigid core of the protein revealed a single β-sheet conformation, which spans not only the R3, R4, R’ repeats, but also the entire C-terminal domain (CT) of the protein. This massive β-sheet core qualitatively differs from all other tau fibril structures known to date. Using long-range correlation NMR experiments, we found that the R3 and R4 repeats form a β-arch, similar to that seen in some of the brain-derived tau fibrils, but the R1 and R3 domains additionally stack against the CT, reminiscent of previously reported transient interactions of the CT with the microtubule-binding repeats. This expanded β-sheet core structure suggests that the CT may have a protective effect against the formation of pathological tau fibrils by shielding the amyloidogenic R3 and R4 domains, preventing side-on nucleation. Truncation and post-translational modification of the CT in vivo may thus play an important role in the progression of tauopathies.
Keywords: Solid-state NMR, steric zipper, water-edited NMR, β-arch
Graphical Abstract
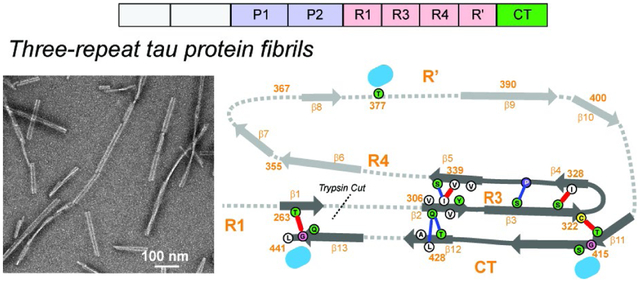
Introduction
Tau is an intrinsically disordered microtubule-binding protein that aggregates into intracellular inclusions in a number of neurodegenerative diseases1. These aggregates are cross-β amyloid fibrils, whose structured cores are comprised of parts of the microtubule-binding repeat domains (R) R1–R4 and R’ (Fig. 1a). The remainder of the protein is dynamically disordered and appears as a “fuzzy coat” around the fibril core in electron micrographs2–4. Adult human brains express six tau isoforms, distinguished by whether they contain zero, one or two 29-residue acidic inserts (0N, 1N, or 2N tau) in the N-terminal region and the presence or absence of the 31-residue R2 repeat (3R or 4R tau). Different tauopathies are characterized by tau proteins with different isoform compositions. For example, tau fibrils in Alzheimer’s disease (AD) and chronic traumatic encephalopathy (CTE) are comprised of both three-repeat (3R) and four-repeat (4R) tau, in Pick’s disease (PiD) are solely 3R tau, while in corticobasal degeneration (CBD) are solely 4R tau5. The β-sheet structures of tau fibrils in AD6, CTE7, PiD8 and CBD9 brains have been determined using cryo-electron microscopy (cryo-EM). These results show that each tauopathy is characterized by a distinct β-sheet core conformation10, which is the same for patients with the same disease. Moreover, these conformationally specific tau aggregates can spread between neurons in connected brain regions by recruiting soluble tau to adopt the same pathological conformation11–14. Therefore, mapping the structures of tau aggregates is important for understanding the structure-disease relationship and for designing imaging and therapeutic agents to diagnose and treat tauopathies.
Figure 1.
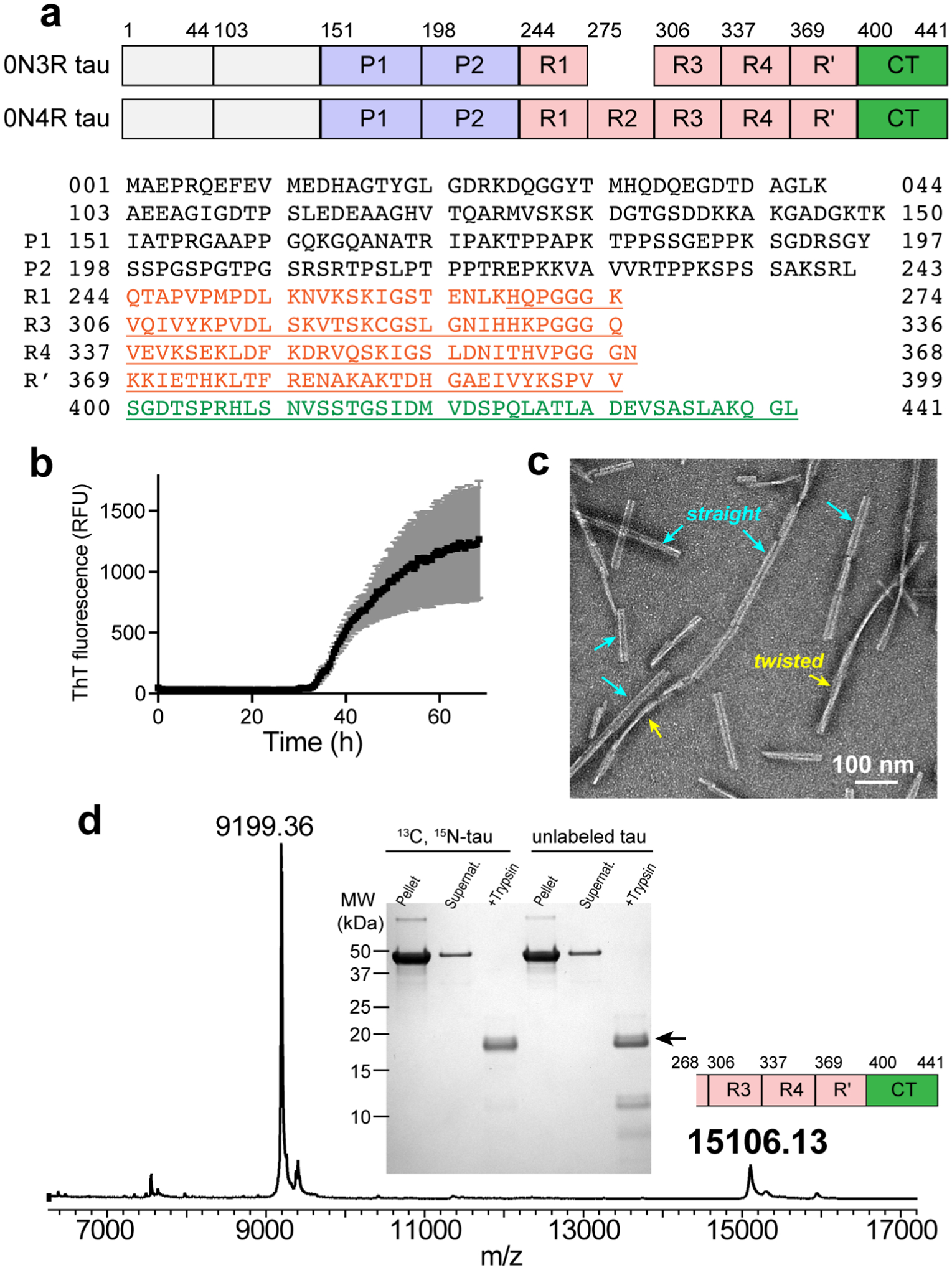
Heparin-fibrillized 0N3R tau forms well-ordered amyloid fibrils with a single ultrastructural morphology. (a) Comparison of the domains of 0N3R and 0N4R tau and the sequence of 0N3R tau. Due to the absence of N-terminal inserts, residue 44 precedes 103 in both 0N4R and 0N3R tau. 0N3R tau does not contain R2, thus residue 274 is followed by residue 306. Underlined residues correspond to the trypsin-resistant core of 0N3R tau. (b) ThT fluorescence of heparin-induced fibrillization of 0N3R tau, showing a lag time of ~33 hours. (c) Representative negative-stain TEM image of 0N3R tau, showing that the fibrils are predominantly straight with a width of ~23 nm, together with a small fraction of twisted fibrils. (d) SDS-PAGE gel and mass spectrum of 0N3R tau fibrils after trypsin digestion. In the gel, both 13C, 15N-labeled tau and unlabeled tau show a dominant band, whose molecular weight is 15.1 kDa, corresponding to residues 268–441. A much weaker band with a molecular weight of 9199 (residues 354–441) is sometimes observed. Pellet and supernatant lanes before trypsin digestion show a single band at the intact protein molecular weight, confirming the purity of the protein used for solid-state NMR measurements. The mass spectrum corresponds to unlabeled tau.
To date, the known rigid cores of tau filaments from patient brains comprise 73–107 residues (Table 1), all spanning R3, R4 and part of R’. Additional β-sheet residues are found in the R2 region in CBD tau and the R1 region in PiD tau. No residues before K254 or after E380 have been observed in any of the brain tau fibril cores. The β-strands in the core hydrogen-bond in a parallel-in-register fashion across hundreds of molecules along the fibril axis, giving rise to the spine of the long filaments seen in transmission electron microscopy (TEM) images. In addition to high-resolution cryo-EM structures, trypsin digestion followed by mass spectrometry has provided useful information about the size of the tau fibril cores in diseased brains15. These trypsin-resistant regions are overall consistent with the cryo-EM results, but can be larger than the structurally resolved β-sheet core: the most C-terminal residue reported so far is R406 at the beginning of the C-terminal domain (CT)15.
Table 1.
Locations of the β-sheet core of full-length tau fibrils reported so far.
| Tau fibril source | PDB code | Structurally assigned β-sheet core | No. residues | Microtubule-binding repeats | Longest trypsin resistant domain |
|---|---|---|---|---|---|
| AD tau (3R & 4R) | 6HRE6 | V306–F378 | 73 | R3, R4, R’ | H268-R40615 |
| PiD tau (3R) | 6GX521 | K254–F378 | 94 | R1, R3, R4, R’ | L243-K38515 |
| CTE tau (3R & 4R) | 6NWP7 | K274/S305–R379 | 75 | R3, R4, R’ | – |
| CBD tau (4R) | 6VHA9 | K274–E380 | 107 | R2, R3, R4, R’ | H268-K39515 |
| In vitro 0N4R tau24 | - | K274–K340 | 67 | R1, R2, R3, R4 | H268–K34024 |
| In vitro 2N4R tau, snake | 6QJH23 | G272–H330 | 28 | R1, R2, R3 | – |
| In vitro 0N3R tau | - | G262–L441 | 149 | R1, R3, R4, R’, CT | H268–L441 |
In addition to aggregating in patient brains, tau can also form fibrils in vitro with the help of anionic cofactors such as polyglutamate, RNA, and the sulfonated glycosaminoglycan heparin16–17. Extensive biochemical and biophysical studies of these in vitro fibrillized tau revealed structural similarities to brain tau fibrils, but the full three-dimensional fold usually differs from the brain tau fibrils, and the structure and morphology of these in vitro tau fibrils are also sensitive to the protein construct length and fibrillization conditions. Solid-state NMR studies of K19, a three-repeat tau construct that spans residues Q244-E372, found a β-arch formed by R3 and R418, with a rigid β-strand at V306–S320 and a disordered segment at K321–S32419–20. This motif was later observed in PiD tau21 and CBD tau9. Solution NMR studies of heparin-fibrillized 2N4R tau found residues 212–399 to be undetectable, indicating that this domain is either rigid or undergoes intermediate-timescale motion22. Cryo-EM studies of heparin-fibrillized 2N4R tau detected a β-sheet core consisting of residues G272–H330 in the dominant polymorph23, but several other polymorphs with moderately different structures were also found. In comparison, a solid-state NMR study of heparin-fibrillized 0N4R tau found a single conformation for the β-sheet core24, manifested by a single set of peaks in 2D and 3D correlation NMR spectra. Spectral assignment revealed that this β-sheet core spans residues H268–K340, and adopts a β-arch that places the R3 hexapeptide motif in close proximity to the 291CGS293 triplet in R2, similar to the CBD tau fold23. Dynamics NMR experiments showed that the protein becomes increasingly mobile from this rigid core towards the two termini: the R1 to R4 domains are semi-rigid, the proline-rich region and the R’ domain are semi-mobile with order parameters of ~0.4, whereas the N-and C-termini are isotropically mobile24.
Although 4R tau fibrils of both in vivo and in vitro origins have now been investigated in detail, in vitro 3R tau is much less understood. A single study of heparin-fibrillized 2N3R tau23 showed a small β-sheet core spanning residues G272–H330, which packs in parallel with another protofibril to form an asymmetric dimer. The unique cysteine of the protein, C322, is packed closely between the two monomers, suggesting the formation of an intermolecular disulfide bond23. This cryo-EM structure differs dramatically from the in vivo PiD 3R tau structure, which exhibits a larger, 93-residue, β-sheet core folded into a strand-turn-strand topology as a single protofibril. Given the unusual features of the in vitro 2N3R tau structure, and the fact that proteolysis during fibril formation has been shown to cause polymorphism to in vitro tau fibrils24, we set out to investigate the heparin-fibrillized 3R tau structure in more detail. Such in vitro studies of both 4R and 3R tau filaments are also necessary for understanding how 3R and 4R tau proteins mix in dual-isoform tauopathies such as AD.
Here we present a study of the molecular conformation and hydration of heparin-fibrillized 0N3R tau using magic-angle-spinning (MAS) solid-state NMR and complementary biophysical measurements. Trypsin fingerprinting and J-coupling-based 2D correlation experiments indicate that the entire CT of the protein is immobilized, in stark contrast to 0N4R tau and all in vivo tau fibrils known to date. 2D and 3D dipolar correlation NMR spectra confirm that the CT is part of the β-sheet core, together with R3, R4, and R’. Long-mixing time spectra indicate that the CT folds over the R3 domain. The incorporation of the entire CT into the fibril core suggests important biological roles of the CT in regulating the formation of pathological tau filaments.
Materials and Methods
Expression of unlabeled 0N3R tau
The 0N3R gene was cloned into a pET-28a vector, and the plasmid was transfected into E. coli. BL21 (DE3) competent cells (Novagen). A starter culture was grown in 20 mL LB medium containing 50 μg/mL kanamycin. After overnight growth at 37°C under shaking at 215 rpm, a 10 mL culture was inoculated into 1 L LB medium containing 50 μg/mL kanamycin. Cells were grown at 37°C and 215 rpm until the OD600 reached 0.8–0.9 and were induced with 1 mM IPTG for 3 h.
Expression of isotopically labeled 0N3R tau
Uniformly 13C, 15N-labeled 0N3R tau with reverse-labeled lysine was expressed in E. coli and purified by heat denaturation, cation exchange chromatography and reversed-phase HPLC. Specifically, a starter culture was grown in 20 mL LB medium containing 50 μg/mL kanamycin. After overnight growth at 37°C under shaking at 215 rpm, a 10 mL overnight culture was inoculated into 1 L LB medium containing 50 μg/mL kanamycin. Cells were grown at 37°C and 215 rpm until the OD600 reached 0.5. Cells were spun down at 3,000 g and 4°C for 5 minutes, and the cell pellet was resuspended in 1 L minimal media containing M9 salts (1x), 0.5 g/L 15NH4Cl, 3 g/L of 13C6 labeled D-glucose, 2 mM MgSO4, 0.1 mM CaCl2, 50 mg/L kanamycin, and vitamin/mineral supplements. We also added 100 mg/L unlabeled lysine to reduce spectral congestion. This concentration suppressed the lysine intensities to ~20% of those of fully labeled residues, thus weak intensities are detected for some of the Lys residues. Cells were then re-grown in minimal medium at 37°C and 215 rpm for 1.5 h before induction with 0.5 mM IPTG for 4 h.
Purification of 0N3R tau
Cells from 1 L expression were pelleted and resuspended in 50 mL lysis buffer containing 20 mM MES (pH 6.8), 1 mM EGTA, 0.2 mM MgCl2, 5 mM DTT, and 1x cOmplete™ protease inhibitor cocktail (Roche). Cells were lysed using a probe sonicator. The cell lysate was then boiled in a hot water bath for 20 min and spun at 40,000 g for 40 min to remove cell debris and protein aggregates. The supernatant containing 0N3R tau was first purified with a cation exchange column (self-packed with SP Sepharose Fast Flow resin, GE Healthcare). Fractions containing 0N3R tau were collected and further purified by reversed-phase HPLC (Agilent Zorbax 300SB-C3 column, 21.2 × 250 mm, 7 μm particle size) using an acetonitrile gradient of 5–50% in 45 min. HPLC fractions (>95% pure, determined by SDS-PAGE with Coomassie blue staining) were pooled and lyophilized to yield 0N3R tau as a powder. The yields of purified proteins are ~10 mg and ~3.5 mg per liter culture for unlabeled and labeled 0N3R tau, respectively.
0N3R tau fibrillization and kinetics
0N3R tau was fibrilized in sealed 1.5 mL eppendorf tubes containing 0N3R tau (1 mg/mL), TCEP (1 mM, pH adjusted to pH 7), and heparin (0.16 mg/mL, Santa Cruz Biotech, sc203075, 8,000–25,000 Da) in 1 × PBS solution (pH 7.4). The solution was shaken at 37°C and 1200 rpm for 3 days. To monitor the aggregation kinetics, 10 μM ThT was added to a small aliquot (300 μL) of the mixture, and the fluorescence signal (λex = 444 nm, λem = 485 nm) was monitored on a Spectramax M5 plate reader (Molecular Devices).
Electron microscopy
Fibril samples were adsorbed onto freshly glow-discharged, 200-mesh formvar/carbon-coated copper grids (Ted Pella), washed with 0.1 M ammonium acetate followed by 0.01 M ammonium acetate, and stained with 2% (wt/vol) uranyl acetate. TEM images were taken on an FEI Tecnai T12 electron microscope (UCSF EM core).
Trypsin fingerprinting
100 μL of unlabeled 0N3R tau fibrils (1 mg/mL) were treated with freshly prepared trypsin (Sigma T8003, 1 mg/mL) in 1x PBS by incubation at 37°C and 1400 rpm for 30 min. The trypsin-resistant material was pelleted at 100,000 g and 4°C for 30 min, washed once with 1 × PBS, and resuspended in 24 μL 1 × PBS for SDS-PAGE and mass spectrometry analysis. For mass analysis, 12 μL resuspended solution was mixed with 100 μL denaturing buffer (7.2 M GdnHCl, 100 mM Tris pH 7.5, 10 mM TCEP) and incubated at 95°C for 1 h. The sample was desalted using the StageTip (Pierce C18 Tips, Thermo Scientific) and dried by speedVac. The resuspended solution was mixed with sinapinic acid and analyzed on a Shimazu AXIMA Performance MALDI TOF/TOF mass spectrometer. To identify the sequence of the trypsin-resistant material, the average masses were searched against a sequence database generated using ProteinProspector (http://prospector.ucsf.edu/prospector/mshome.htm) with the following settings: protein 0N3R tau, enzyme trypsin, max. missed cleavages 99, mass type average, mass tolerance 200 ppm.
Solid-State NMR samples and experiments
The fibrillized tau aggregates were collected by ultracentrifugation at 100,000 × g for 30 min at 4°C. Pellets were combined and washed twice with water to reduce salt concentration to an estimated ~10 mM. The final pellet contained ~20 mg of dry tau mass with 3-fold excess water. This was reduced to 1-fold excess through slow desiccation, then the hydrated fibrils were transferred into a 3.2 mm thin-wall Revolution NMR pencil rotor using tabletop centrifugation with a series of funnels made of plastic pipette tips. The final sample contained ~18 mg 0N3R tau and ~17 mg solution.
All solid-state NMR spectra were measured on a Bruker Avance 800 MHz (18.8 T) spectrometer using a BlackFox 3.2 mm HCN MAS probe. 13C chemical shifts were referenced externally to the adamantane CH2 chemical shift at 38.48 ppm on the tetramethylsilane (TMS) scale, and 15N chemical shifts were referenced to the 15N peak of 15N-acetylvaline at 122.0 ppm on the liquid ammonia scale. Typical rf field strengths were 54–83 kHz for 1H, 16–50 kHz for 13C, and 25–36 kHz for 15N. 1H chemical shifts of a hydrated DSS-containing POPC membrane sample was used as an external standard to calibrate the 1H chemical shifts and measure the sample temperature. Reported temperatures are estimated sample temperatures based on the probe thermocouple and the measured water 1H chemical shift. Spectra that preferentially detect mobile residues were measured using scalar couplings. These include 1H-15N and 1H-13C INEPT spectra and 2D 13C-13C TOCSY spectra. Spectra that preferentially detect rigid residues were measured using cross polarization from 1H to 13C, 1H to 15N and 15N to 13C. In addition, the mixing sequences for these rigid-residue selective 2D and 3D experiments are dipolar in nature, including 13C-13C CORD25 and a modified CO-Cα BSH-CP sequence26 (Fig. S1). Most experiments were conducted under 10.5 kHz or 14 kHz MAS (Table S1). For many experiments, a short 1H-13C CP contact time of 70 μs was used to select only the most rigid β-sheet core signals while excluding the cumulative dynamic signals from mobile regions of the protein. Long NMR experiments were run in blocks of 1–3 days with field drift corrected between each block. Multiple blocks of these 2D and 3D spectra were added in the time domain before Fourier transformation.
All MAS NMR spectra were acquired and processed using TopSpin, and chemical shift assignment was conducted in SPARKY. Typical processing used either QSINE apodization with SSB = 3, or GM apodization with LB = −20 Hz and GB = 0.05, all in TopSpin. Multidimensional spectra are plotted with a 1.2 × scale between successive contour lines. Backbone (ϕ, ψ) dihedral angles were calculated from the assigned chemical shifts using the TALOS-N software27, after adding 2.0 ppm to all TMS-referenced experimental 13C chemical shifts to convert to the DSS scale. Long-range contacts were obtained from four long-mixing 2D and 3D spectra.
Water-edited 2D CC and 2D NCA spectra were measured using a water-selective 1H echo that consists of a 180° Gaussian pulse lasting 10 rotor periods in duration, surrounded by one rotor period before and after the pulse. This water-selective T2 period was followed by a 1H mixing period of 4 ms to transfer the water 1H polarization to protein protons. With vanishing mixing time, less than 0.1% of the protein signal remained in the spectrum, while a 100 ms 1H mixing period produced an equilibrated protein spectrum with 50% of the full intensity of the unedited protein spectra. This is roughly consistent with the similar mass of protein and water in the rotor. Therefore, for calculating the water transfer ratio S/S0, we used the unedited 2D CC and NCA spectra multiplied by a factor of 0.50 as the S0 intensity. The S/S0 intensity ratios were extracted from the individual peaks in the 2D spectra, after correcting for the number of scans, and after averaging over both sides of the diagonal for the 2D CC spectra. Error bars were propagated from the signal-to-noise ratios of 1D cross sections of resolved peaks in the 2D spectra, using empty regions of the 2D spectra as the noise.
Results
Heparin-fibrillized 0N3R tau exhibits a single predominant ultrastructural morphology
We expressed recombinant 0N3R tau in E. coli and purified it by heat denaturation, cation exchange chromatography, and reverse-phase HPLC. These purification conditions minimize trace contaminants, which can affect tau nucleation and hence fibril morphology. Fibrils were formed by adding 0.16 mg/ml heparin to 1.0 mg/ml tau monomers in phosphate buffer that contains 1 mM of the reducing agent TCEP. The solution was shaken at 1200 rpm at 37°C for three days. The tau and heparin concentrations were optimized to maximize the pelletable mass, as assessed by SDS-PAGE. These conditions gave a fibrillization yield of > 90%. Thioflavin-T fluorescence data (Fig. 1b) showed that fibrils formed with a lag time of ~33 hours, much longer than the ~1 hour lag time of 0N4R tau fibrils prepared under similar conditions. TEM images show ~23-nm wide filaments that are predominantly straight, together with a small fraction (< 15%) of twisted fibrils (Fig. 1c). Trypsin digestion followed by mass spectrometry revealed that the dominant trypsin-resistant fragment has a molecular weight of 15.1 kDa, which corresponds to residues K268 to L441 (Fig. 1d). This segment encompasses the end of R1, the full R3, R4 and R’ repeats, and the entire CT. This expanded core is unexpected, as no tau fibrils known to date have a β-sheet core that extends beyond E380.
2D NMR spectra confirm that the rigid domain of 0N3R tau extends to the C-terminus
To verify the size of the fibril core and obtain site-specific structural information, we measured one- and two-dimensional MAS NMR spectra of 13C, 15N-labeled protein. To simplify the spectra, we suppressed Lys labeling by adding 100 mg/L unlabeled Lys to 13C, 15N-labeled growth media. 1D 15N spectra (not shown) indicate a residual Lys labeling level of ~20%. We preferentially detect dynamic residues using J-coupling based experiments and immobilized residues using cross-polarization (CP) based experiments. 1D 13C and 15N INEPT spectra (Fig. 2a) show narrow linewidths of 0.4 ppm for 13C (resolution limited by 13C-13C J-coupling) and 0.4 ppm for 15N, and the intensity envelopes are similar to that of the 0N4R tau spectra. A 2D 1H-15N INEPT correlation spectrum (Fig. 2b) of 0N3R tau further resolved the amide 1H signals, and its overlay with the previously reported solution-state 1H and 15N chemical shifts of 2N4R protofibrils22 shows excellent agreement after removing the N1 and N2 residues, which are absent in 0N3R tau. This agreement indicates that the conformation of the dynamic portion of 0N3R tau is similar to that of 2N4R tau. The 1H-15N 2D INEPT spectrum of 0N4R tau fibrils24 also agrees well with the 2N4R tau chemical shifts. These consistencies allow us to transfer the solution-state chemical shifts of 2N4R tau to the solid-state spectra of 0N3R and 0N4R tau fibrils’ mobile domains.
Figure 2.
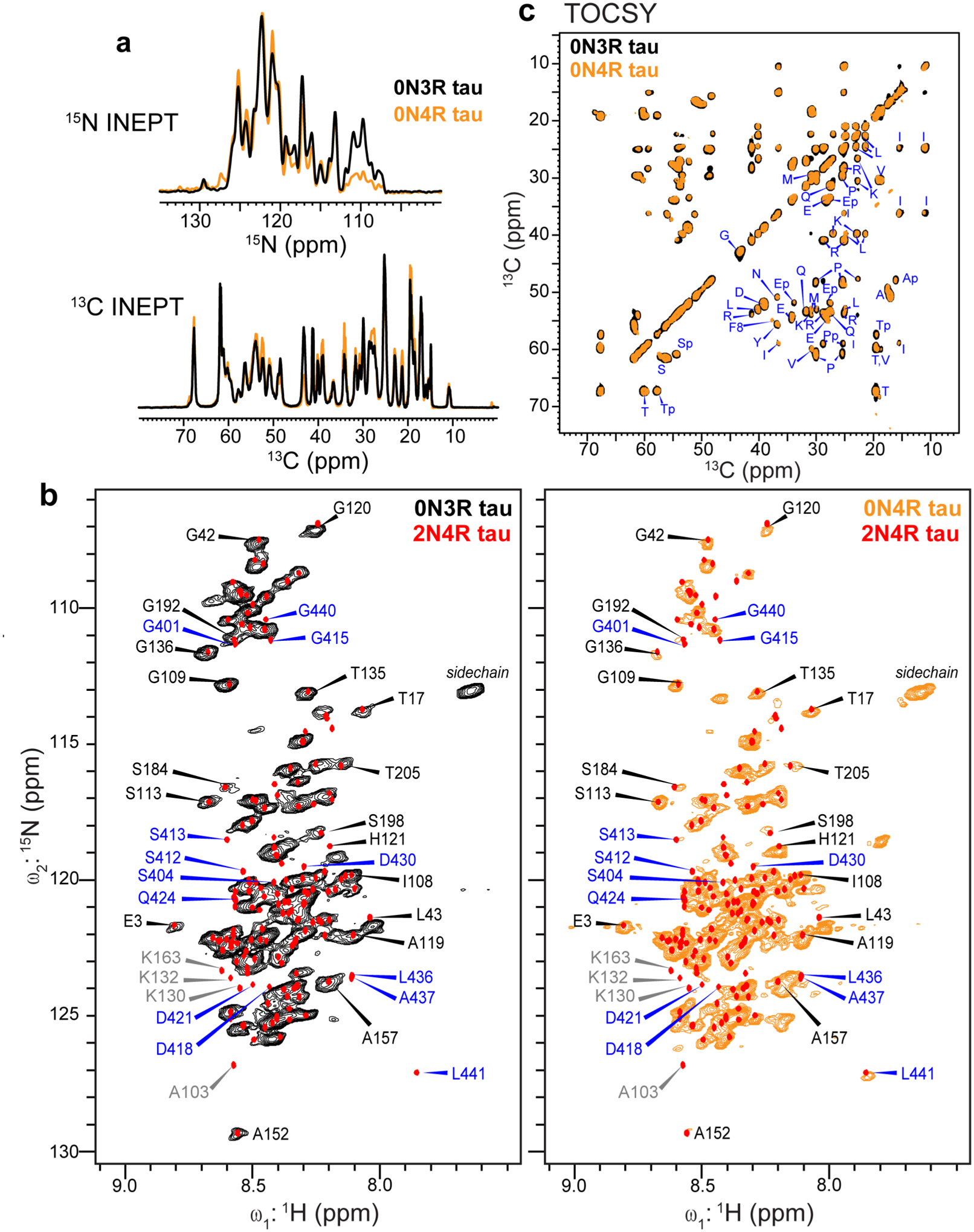
The C-terminal domain of 0N3R tau fibrils is immobilized. (a) 1D refocused INEPT 15N and 13C spectra of 0N3R tau (black), overlaid with the 0N4R tau spectra (orange). These INEPT spectra selectively detect highly mobile residues. (b) 2D 1H-15N INEPT correlation spectra of 0N3R tau fibrils (left) and of 0N4R tau fibrils (right)24. Superimposed onto each MAS spectrum are the solution NMR chemical shifts of 2N4R tau protofibrils (red points)22. Assignments for the N1 and N2 residues are removed since these domains are absent in 0N3R and 0N4R tau. Resolved N-terminal residues are assigned in black. Resolved 2N4R CT signals (assigned in blue) are missing in the 0N3R tau spectrum, but many of them are present in the 0N4R tau spectrum, indicating that the rigid core of 0N3R tau extends to the C-terminus. Several Lys peaks (assigned in grey) are too weak to be detected in the spectra due to reduced labeling of lysines. The Gly peaks are weaker in the 0N4R tau spectrum than the 0N3R tau spectrum due to reverse labeling of Gly in the former. (c) 2D 13C-13C TOCSY spectrum of 0N3R tau (black), showing residue-type specific random coil 13C chemical shifts. The spectrum agrees well with the 0N4R tau spectrum (orange). Residues that precede Pro, for example the serine residues marked as Sp, manifest different chemical shifts from those that do not precede Pro.
It can be seen that many N-terminal residues’ peaks in 2N4R tau are present in both 0N3R and 0N4R tau spectra, and their chemical shifts agree well among all three tau samples. These residues include, for example, T17, G42, G109, S113, and S184 (Fig. 2b). In contrast, the resolved C-terminal residues’ peaks (assigned in blue) are missing in the 0N3R tau spectrum, while many are present in the 0N4R tau spectrum, indicating that the C-terminal domain is not isotropically mobile in 0N3R tau. These missing signals include, for example, those of L441, A437, and L436. Several other peaks such as D418, Q424 and D430 are weak in the 0N4R tau spectrum but are completely missing in the 0N3R tau spectrum. As expected, residue A103, which is preceded by T102 in 2N4R tau but by K44 in 0N3R tau, resonates at different chemical shifts between 2N4R tau and 0N3R tau. Finally, several Lys peaks such as K130 and K163 are absent in the 0N3R spectrum due to the reduced Lys labeling. A 2D 13C-13C TOCSY spectrum (Fig. 2c) mostly exhibits residue-type random coil chemical shifts, confirming the presence of isotropically dynamic residues in 0N3R tau. The only exceptions are residues preceding Pro, whose chemical shifts differ from non-Pro-preceding residues28. The TOCSY spectrum of 0N3R tau is superimposable with the TOCSY spectrum of 0N4R tau within the spectral linewidth, confirming that the lack of R2 does not change the dynamically averaged conformation of the fuzzy coat.
To directly observe rigid β-sheet residues, we turned to 13C and 15N CP-MAS experiments (Fig. 3a). The fingerprint 2D 15N-13Cα (NCA) correlation spectrum shows linewidths of 0.5–0.7 ppm for 13C and 1.2–1.5 ppm for 15N for resolved peaks (Fig. 3b and Fig. S2), indicating that the β-sheet core has a relatively homogeneous molecular conformation. Compared to 0N4R tau, 0N3R tau exhibits more Ala peaks, which resonate at 47–50 ppm for Cα and 117–130 ppm for 15N. Tau has no Ala between residues 250–380 (spanning R1, R3, R4, and R’), whereas residues 380–441 (spanning R’ and the CT) contain seven Ala (Fig. 1a, Table S2). Thus, the large number of Ala peaks in the NCA spectrum is consistent with the inclusion of the C-terminus into the β-sheet core. Analysis of 3D correlation spectra (vide infra) allowed us to assign all seven Ala, confirming this conclusion.
Figure 3:
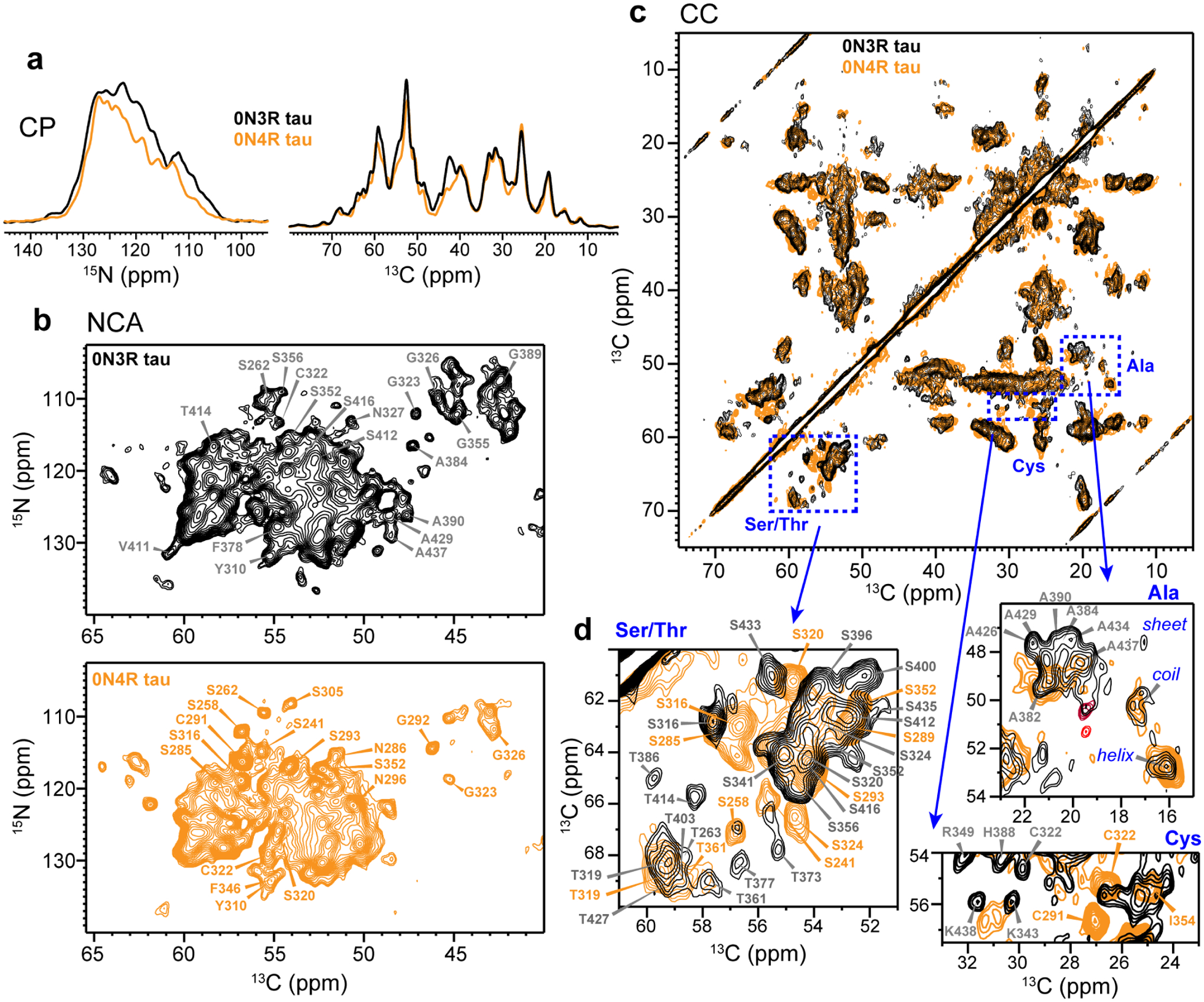
Fingerprint CP-MAS NMR spectra of 0N3R tau, showing the signals of immobilized residues. (a) 1D 13C and 15N CP spectra of 0N3R (black) and 0N4R (orange) tau fibrils. The 13C spectra were measured using a 70 μs 1H-13C CP contact time to suppress the signals of mobile residues. (b) 2D NCA spectra of 0N3R (black) and 0N4R (orange) tau. The 0N3R tau spectrum displays many more Ala peaks than the 0N4R tau spectrum, consistent with the inclusion of the R’ and CT in the rigid core. (c) 2D 13C-13C correlation spectrum of 0N3R tau (black), overlaid with the 0N4R tau spectrum (orange). (d) Insets of the Ser/Thr, Ala and Cys regions of the 2D CC spectrum. Assignments obtained from 3D correlation data are indicated for some of the signals.
Complementing the 2D NCA spectrum, we measured a 2D 13C-13C (CC) correlation spectrum using 23 ms 13C spin diffusion and a short 1H-13C CP contact time of 70 μs to suppress the signals of dynamic residues (Fig. 3c and Fig. S3). We focus on the Cys, Ser/Thr, and Ala regions of the spectrum to assess the size and conformational homogeneity of the β-sheet core (Fig. 3d). Assignment of some of the well resolved peaks based on 3D correlation spectra (vide infra) is indicated. The longest isoform of tau contains only two cysteines, C291 and C322, at equivalent positions in the R2 and R3 repeats. C322 is present in both 3R and 4R tau whereas C291 only exists in 4R tau. Based on the characteristic chemical shifts of reduced Cys and connectivities in the 3D correlation spectra, we assigned the C322 Cα-Cβ peak at (54.8, 30.0) ppm. This peak is weaker and has different chemical shifts from the C322 peak in 0N4R tau24, indicating that the C322 structure and dynamics differ between the two proteins. In the Ser/Thr region of the spectrum, 0N3R tau exhibits a similar number of Ser peaks as in 0N4R tau but more Thr peaks than 0N4R tau24. Full-length 3R and 4R tau isoforms both contain 4 Thr’s in R1–R4 repeats and 6 Thr’s in the R’ and CT (Table S2). Thus, the detection of a larger number of Thr peaks in the CC spectrum supports an expanded β-sheet core in 0N3R tau. In the well resolved Ala region of the spectrum, both 0N3R and 0N4R tau exhibit random coil, α-helical and β-sheet Cα-Cβ cross peaks. However, the coil and helical Ala signals are absent in the 3D spectra of 0N3R tau, indicating that the non-β-sheet Ala residues are semi-mobile. In the β-sheet Ala chemical shift region, more cross peaks are observed for 0N3R tau than for 0N4R tau, further supporting the larger β-sheet core in 0N3R tau.
Chemical shift assignment of 0N3R tau from 3D correlation spectra
To determine the site-specific backbone conformation of the 0N3R tau fibril core from 13C and 15N chemical shifts, we measured and assigned four 3D 15N-13C-13C correlation spectra. The NCACX spectrum correlates intra-residue 15N, 13Cα and sidechain 13C chemical shifts, whereas the NCOCX, CONCA, and CAN(CO)CA spectra correlate the chemical shifts of two sequential residues26, 29–32. An 80 ms 13C CORD mixing period was used in the NCACX and NCOCX experiments. Using standard reverse backbone walks from residue i to i-1 and forward walks from residue i to i+1, we readily assigned 20 residues (SI Methods). This includes, for example, 400SG401, which exhibits a well resolved CONCA peak at (172, 114.3, 41.9) ppm for the S400 13CO, G401 15N and Cα resonances (Fig. 4, Fig. S4), which are confirmed by peaks in the other three 3D spectra. Additional assignments were obtained by augmenting the backbone walk with several additional strategies, including exploiting the sequence uniqueness of amino acid triplets for residues of the same type, and using non-sequential short and medium-range inter-residue correlations to validate the assignment (SI Methods). Using this integrated approach, we assigned 104 out of 149 residues between S262 and L441 (Table S3, Table S4). The percentage of assigned residues is similar to that of 0N4R tau24, even though the core of 0N3R tau is twice as large. Importantly, all intense and resolved peaks in the 3D spectra are assigned. As a result, any minor conformation would represent at most 10–15% of the fibrils based on the spectral sensitivity, in agreement with the TEM data (Fig. 1c). The 45 unassigned residues out of 149 either give weak signals due to low labeling levels and dynamics or are unresolved due to sequence repetition such as PGGG (Fig. S5).
Figure 4.
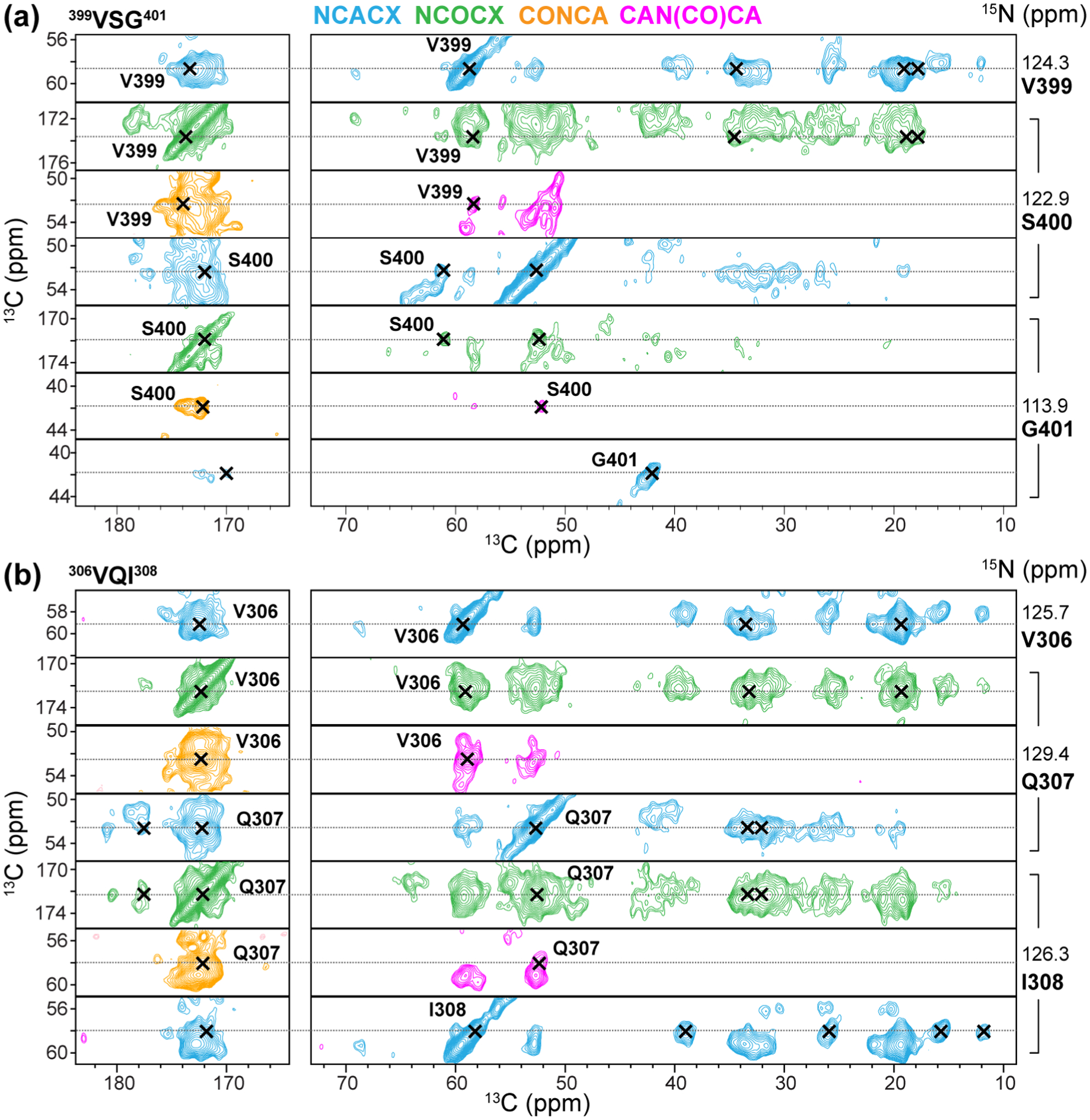
Representative strips of four 3D correlation spectra of 0N3R tau fibrils for chemical shift assignment. (a) Assignment of the 399VSG401 triplet. (b) Assignment of the 306VQI308 segment in the hexapeptide motif at the beginning of R3. The NCACX (blue) spectrum shows the intra-residue chemical shifts of residue i, while the NCOCX (green), CAN(CO)CA (lavender), and CONCA (orange) spectra correlate the chemical shifts of residue i-1 with the chemical shifts of residue i. Additional strips can be found in Fig. S4.
The resolved and assigned peaks in the aliphatic region of the 2D CC spectrum show a single set of chemical shifts per residue, indicating a single conformation for the β-sheet core (Fig. S3). The sole cysteine, C322, has a Cβ chemical shift (29.9 ppm) indicating a reduced thiol. We observed 30 out of 42 residues in the CT, including the terminal 437AKQGL441, confirming the rigidity of the C-terminal residues. The intensities of R3, R4, R’, and CT residues are similarly high in these dipolar correlation spectra, while the R1 signals are less intense (Fig. 5a), consistent with the start of the core from the middle of R1. Relative peak intensities of assigned residues are categorized qualitatively as strong, intermediate and weak based on comparison between resolved peaks of the same residue type. Most Cα chemical shifts are smaller than the random coil chemical shifts whereas most Cβ chemical shifts are larger, indicating that the core adopts a predominantly β-sheet conformation (Fig. 5b). The (ϕ, ψ) torsion angles obtained from these chemical shifts27 indicate 13 β-strands in the trypsin-resistant core (Fig. 5c). The 0N3R tau shares common β-strands with 0N4R tau at V306-Y310 and V313-H330 in R3, V337-S341, V350-K353, and N359-T361 in R4, and G261-S262 in R1 (Fig. 5d). However, we did not detect β-sheet residues past T361 in 0N4R tau, while 0N3R tau exhibits additional β-strands at K385-S400, V411-S422, A426-A429, and A434-G440 (Fig. 5c).
Figure 5.
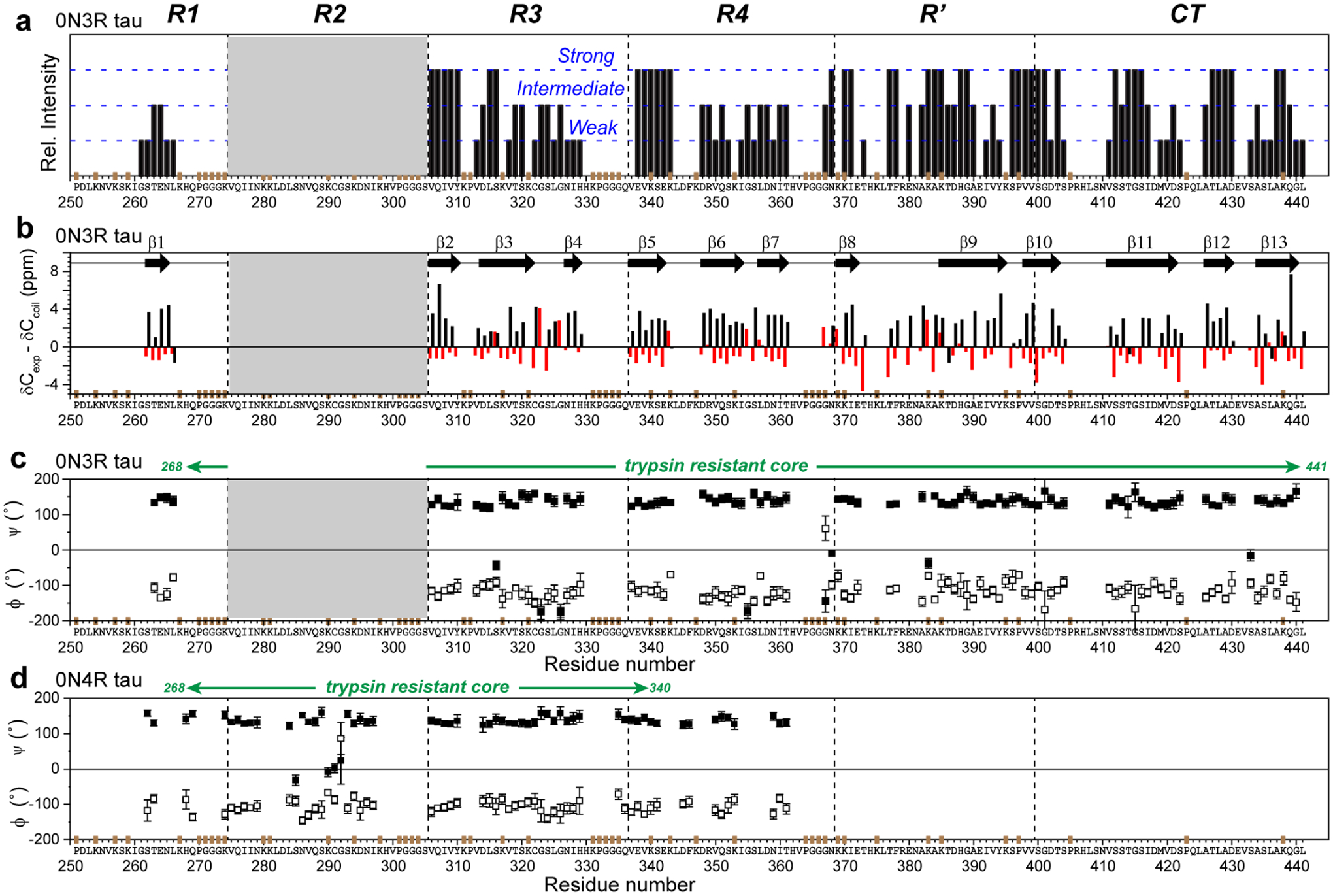
Backbone conformation of the 0N3R tau fibril core obtained from 13C and 15N chemical shifts. (a) Relative peak intensities of assigned residues, categorized qualitatively as strong, intermediate and weak based on comparing resolved peaks of the same type. Strong peaks indicate rigid and ordered residues. R1 residues have weaker intensities than residues in other domains. The positions of Pro (P), GGG triplets, and unlabeled Lys (K) residues are indicated in brown on the x-axis. (b) Cα and Cβ secondary chemical shifts. Negative Cα and positive Cβ secondary shifts are indicative of a β-strand conformation. Thirteen β-strands can be identified (arrows). Since not all residues are assigned, the exact number of distinct β-strands in the protein may differ. (c) Backbone torsion angles predicted from the measured chemical shifts27. (d) (ϕ, ψ) torsion angles of heparin-fibrillized 0N4R tau for comparison24.
Water accessibility of 0N3R tau fibrils
We investigated site-specific water accessibility of 0N3R tau to gain further insights into the fibril core structure. Water polarization transfer to amyloid proteins reveals which sidechains are located in dry steric zippers and which ones are well hydrated and likely surface-exposed, thus constraining the three-dimensional fold of the fibril core. We measured water-edited 2D CC and NCA spectra of the protein (Fig. S6a, c)33–35 by selecting the water 1H magnetization using a 1H T2 filter, then transferring it to the protein during a 1H mixing period. For 2D CC-detected water polarization transfer, hydrogen exchange with labile hydroxy protons is expected to favor the intensities of hydroxy-containing residues such as Ser and Thr. To minimize this effect, we conducted the water-edited 2D experiments at a sample temperature of −5°C to slow down the chemical exchange rates to less than 10 s−1 for Ser and Thr35–37. For the water-edited 2D NCA spectrum, water polarization transfer to amide protons is probed, which is relatively insensitive to the presence or absence of sidechain protons. We evaluate the water-transferred intensity ratios (S/S0) between a 4-ms water-edited spectrum (S) and a 100-ms control spectrum (S0), shown as hydration maps (Fig. S6b, d). Using the well-resolved 2D CC cross peaks of Ser and Thr and the well-resolved 2D NCA peaks of Gly and Ala, we compared the relative hydration of these residues. No systematic intensity differences are detected between Ser/Thr and Ala/Gly, confirming that chemical exchange is slow under our experimental conditions and water-edited CC and NC spectra similarly probe the exposure of the protein backbone to water. Interestingly, the water accessibilities show significant variations among residues of the same type. Among Ser residues, S416 has the highest water accessibility, with an S/S0 value of 0.38 ± 0.013, while other Ser residues have significantly lower S/S0 values of 0.23–0.27 (Fig. S6e). The water accessibility of S416 can be readily seen in the water-edited 2D CC spectrum as an intense Cα-Cβ cross peak (Fig. S6b). Among Thr residues, T377 exhibits the highest water accessibility. The G415 and G440 N-Cα cross peaks also show much higher water-transferred intensities than the other Gly. Taken together, these results indicate that the 415GS416 segment and the C-terminus are two well-hydrated regions of the protein (Fig. S6f).
Long-range contacts reveal the three-dimensional fold of the rigid core of 0N3R tau
To constrain the tertiary structure of the fibril core, we measured four long-mixing spectra: two 2D CC spectra with 200 ms and 450 ms 13C spin diffusion; a 3D NCACX spectrum with 450 ms spin diffusion, and a 3D 13C-13C-13C (CCC) correlation spectrum with 400 ms spin diffusion (Fig. 6 and Fig. S7). Inter-residue correlations in these spectra indicate that the respective carbons are within ~8 Å in space. We count multiple correlations between the same two residues as one contact. Analysis of these four spectra yielded 90 medium-range contacts; four unambiguous long-range contacts; and four ambiguous long-range contacts, which derive from chemical shifts with partial overlap but cannot be explained by any short-range contacts (Table S5). Originally, we found 93 putative long-range peaks in the 3D correlation spectra; however, most of them could be potentially explained by a short-range contact between residues with resonances within 0.5 ppm of the peak. We thus present only the small number of long-range peaks that cannot be explained by any short-range contacts.
Figure 6.
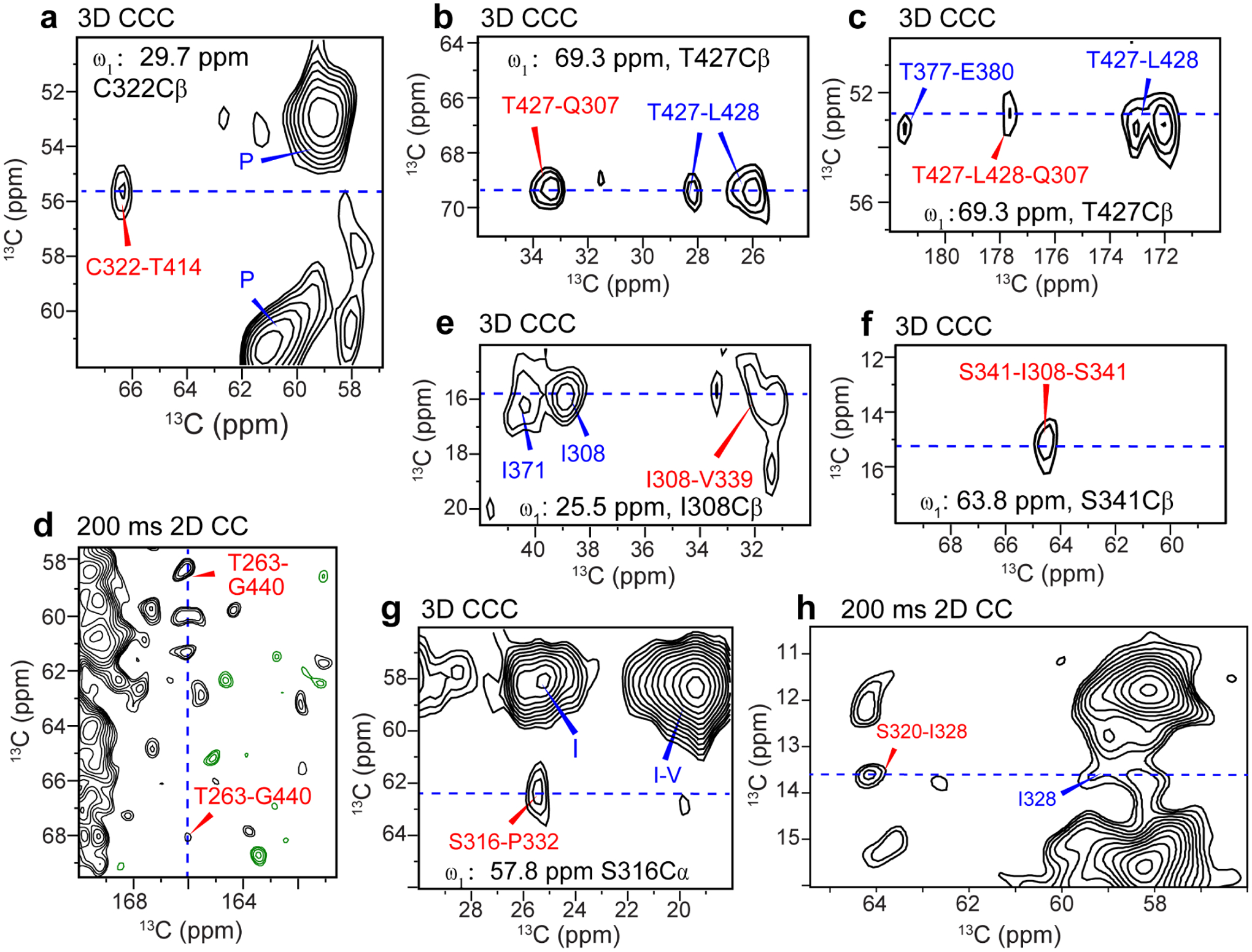
Long-range correlation peaks (assigned in red) that define the tertiary fold of the 0N3R tau fibril. Some of the short-range cross peaks and amino-acid type assignment of strong peaks are also indicated (blue). (a) C322-T414 correlation. (b) T427-Q307 correlation. (c) L428-Q307 correlations. (d) T263-G440 correlations. (e) I308-V339 correlation. (f) S341-I308 correlation. (g) S316-P332 correlation. (h) S320-I328 correlation. Cross sections extracted from the dashed blue lines are given in Fig. S7.
These eight long-range contacts provided clear constraints on the tertiary fold of the 0N3R tau core. One set of long-range contacts indicates that R3 is packed against the CT. The 3D CCC spectrum shows a C322-T414 contact at (ω1, ω2, ω3) chemical shifts of (29.5, 55.4, 66.4) ppm (Fig. 6a), which can be assigned to a C322Cβ-C322Cα-T414Cβ correlation. This C322-T414 contact is supported by cross peaks for Q307 polarization transfer to T427 and L428 in the 3D CCC spectrum (Fig. 6b, c). Since Q307 is 15 residues before C322 while T427 and L428 are 13–14 residues after T414 in the sequence, these two long-range contacts indicate antiparallel packing of R3 with the CT. In addition, we observed multiple correlations between G440 and T263 (Fig. 6d), which constrain the penultimate residue of the protein to be in close proximity with the N-terminus of the trypsin-resistant core. Together, these long-range contacts establish that the CT is stacked against the R1–R3 segment in an antiparallel fashion.
The second set of long-range contacts, between I328 and S320 in R3 and between I308 of R3 and V339 of R4, defines the packing of R3 and R4 repeats. Two cross peaks define these contacts: a (64.1, 13.6) ppm peak in the 200 ms 2D CC spectrum (Fig. 6h) and a (25.9, 16.1, 31.7) ppm peak in the 3D CCC spectrum (Fig. 6e). For each peak, the first residue’s assignment is unique, while the second residue’s assignment can be disambiguated by eliminating assignments that violate the β-strand geometry and contradict the unambiguously assigned peaks. In this way, we can assign the first peak to an S320Cβ-I328Cδ1 contact and the second peak to an I308Cδ1-V339Cβ contact (Table S5).
These long-range contacts, although sparse, define the major qualitative features of the three-dimensional fold of the 0N3R tau fibril core (Fig. 7a). In this provisional model, the fibril core has an elongated C-shape resembling an alligator head: the R3 hexapeptide motif 306VQIVYK311 forms a steric zipper with the R4 pseudo-hexapeptide motif 337VEVKSE342, whereas the other side of R3 stacks against CT. The R3-R4 β-arch is accomplished by a turn at G323-G326 and resembles the R3-R4 β-arch in PiD tau (Fig. 7b). However, the steric zipper registry in the heparin-fibrillized 0N3R tau may be shifted by two residues from the PiD tau, in which V306 is in close contact with V339 instead of S341. In 0N3R tau, the CT packs against the other side of R3, stretching from T414, which contacts C322, past T427, which contacts the R3 hexapeptide motif, to the C-terminal G440 and L441, which contact T263 in R1. The hydration data are consistent with this provisional model, showing that the last 30 residues of the CT domain have higher hydration than the rest of the fibril core (Fig. S6f). This tertiary fold does not account for the stretch of residues from E342 to N411, which form the top of the C-shape, even though the majority of these residues are assigned and are relatively rigid based on their intensities. The lack of tertiary structure information for these residues is due to a lack of information about long-range contacts involving these residues. The R’ region is expected to be packed against the second half of R3 to establish the R3-CT contacts. Peaks defining these long-range contacts involving R’ most likely exist in our current spectra, but are overlapped with resonances representing potential short-range contacts, and thus cannot be used as evidence of the tertiary fold. Further experiments are needed to test this hypothesis and define the structure of this region of the protein.
Figure 7.
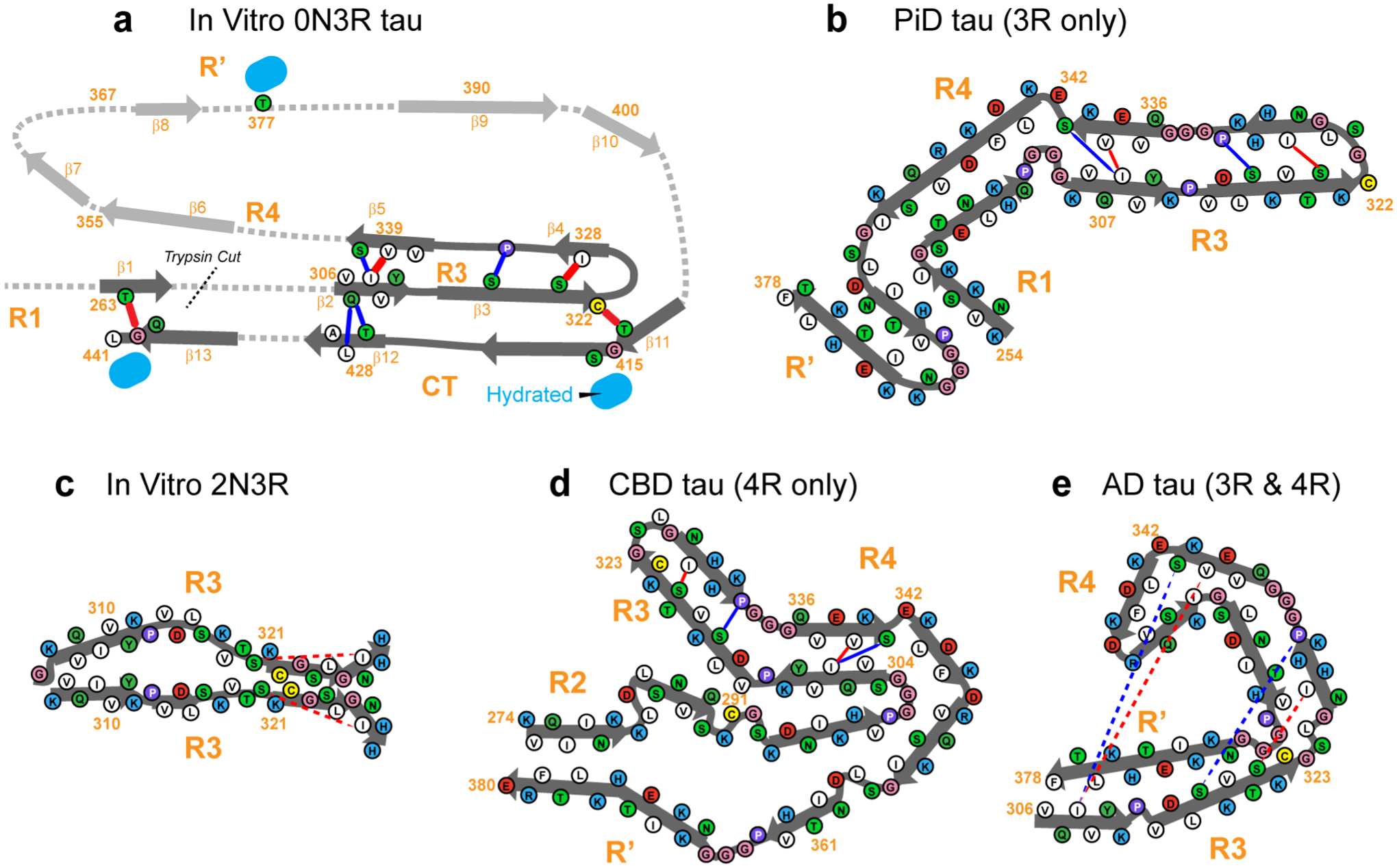
Provisional structural model of 0N3R tau, compared to other tau fibril structures known to date. (a) Tertiary fold of heparin-fibrillized 0N3R tau. Unambiguous long-range contacts are shown as red lines while ambiguous contacts are shown as blue lines. The tertiary fold of residues 342–411 is not known and is indicated by grey arrows and dotted lines, although most of these residues are rigid, ordered, and β-sheet. The R3-CT interactions could also be intermolecular, leading to a domain-swapped dimer, an example of which is shown in Fig. S8. (b) Tertiary fold of the Pick’s disease 3R tau fibril core (PDB code: 6GX5)21. (c) Tertiary fold of 2N3R tau fibril core (PDB code: 6QJQ)23. (d) Tertiary fold of the CBD tau fibril core (PDB code: 6TJO)7. (e) Tertiary fold of the AD tau fibril core (PDB code: 5O3L)6. For the structures in (b-e), the long-range contacts seen in 0N3R tau are annotated where possible, to indicate the different folds of the proteins.
Discussion
The NMR and biochemical data presented here unambiguously indicate that the β-sheet core of in vitro 0N3R tau fibrils adopt a single predominant molecular conformation that contains a reduced C322, and this β-sheet core encompasses the entire R3, R4, R’ and CT domains. The latter differs qualitatively from all in vivo and in vitro tau fibril core structures known to date. Compared to in vivo tau structures, the present 0N3R tau fold shows the highest resemblance to CBD 4R tau9 (Fig. 7d) and partial resemblance to PiD 3R tau21 (Fig. 7b). All three proteins share a common R3-R4 sidechain-stacked β-arch. CBD tau also possesses the same topology of an elongated C-shape as 0N3R tau, despite having four microtubule-binding repeats. Due to the additional R2 repeat, the outermost β-strand in CBD tau is in R’ instead of the CT. In both CBD and PiD tau structures, the R3 residues 322CGSLG326 participate in the turn of the strand-turn-strand fold, and the R3 and R4 hexapeptide motifs stack to form a steric zipper, with I308 contacting V337 and V339. The current 0N3R tau model has a similar steric zipper, but I308 shifts in registry slightly to contact V339 and S341 (Fig. 7a). In CBD tau, the R3 hexapeptide residues Q307 and V309 form another steric zipper with R2 residues around I297. In comparison, the current heparin-fibrillized 0N3R tau displays a second steric zipper between Q307/V309 and T427 in the CT. AD tau6 (Fig. 7e) also exhibits a C-shaped fold, but the core is much smaller. Future characterization of the most stable steric zippers in these tau fibrils will be of interest to obtain predictive insights into the folding landscape of the tau protein.
A recently reported heparin-fibrillized 2N3R tau structure23 (Fig. 7c) found a very small fibril core containing only the R3 domain; moreover the fibril core forms an asymmetric dimer, potentially stabilized by an intermolecular C322-C322 disulfide bond. However, this same study reported polymorphic 0N4R tau fibrils, which were subsequently reproduced under conditions of proteolysis during fibril formation24. Addition of protease inhibitors removed the polymorphism and yielded a single morphology for the 0N4R tau fibrils. Based on these observations, we suggest that the unusually small 2N3R tau β-sheet core from this study may have arisen from proteolysis during fibril formation, or may have resulted from differences in fibrillization conditions such as protein concentration, buffer composition, and shaking.
The most significant and striking finding of the current study is the incorporation of the entire C-terminal domain of tau into the fibril core, which has not been observed in any other tau fibril structures to date. However, transient interactions between CT and the rest of the protein have been reported in many studies. For example, fluorescence resonance energy transfer (FRET) data and electron paramagnetic resonance data of site-specifically labeled 2N4R tau monomers indicate interactions between the CT and R2-R3 residues, as well as between the CT and the N terminus of the protein38. For example, a FRET distance of ~20 Å was measured between V432 in the CT and Y310 in R3 in monomeric tau. This is qualitatively consistent with the Q307-T427/L428 contacts seen in the 0N3R tau fibrils. Solution NMR studies of 2N4R tau protofibrils showed that residues 409–426 in the CT transiently contact C322 in R322, 39, also consistent with the current results. These data suggest that the CT-R3 interaction observed in the 0N3R tau fibril is a solidification of the transient contacts of the protein sampled in solution.
Since a larger amyloid is harder to break up than smaller β-sheet cores, why has this large β-sheet core not been observed in tau filaments from patient brains? Would this expanded core of 3R tau be toxic or protective, if it is present in vivo? We hypothesize that the CT-incorporated β-sheet core is protective against pathogenic fibril formation for the following reasons. The CT is chemically heterogeneous in vivo due to posttranslational modification and proteolytic truncation. Heterogeneous phosphorylation of 13 residues in the CT40, along with caspase cleavage at D421 and other residues41, are well documented in diseased brains. Caspase cleavage that removes the last ~20 residues of the protein has been found to be an early event in aging and AD pathology41. Given this chemical heterogeneity, it is difficult to imagine that a C-terminal amyloid could be as cooperatively formed into infinite sheets in vivo as the less heterogeneous inner core that contains the more amyloidogenic R3-R4 regions. Instead, it would appear that the CT would be protective by folding back over the inner core, preventing side-on nucleation, a key step in amyloid formation42. Indeed, a large body of biochemical data has shown that truncations of the CT as well as the N-terminal disordered region accelerate fibril formation in vitro43–44, accelerate fibril propagation between cells45, enhance seeding by AD brain tau43, and increase tau phosphorylation1, 43, 46. These biochemical data thus support the notion that the CT is protective against pathological aggregation, by shielding the aggregation-prone microtubule-binding repeats from interacting with each other and from hyperphosphorylation. This protective effect implies that any CT-containing fibril core that might nucleate in vivo would be out-competed in growth by smaller and faster-aggregating species, hence making a CT-including expanded β-sheet core minimally present in diseased brains. This hypothesis is supported by the fact that 0N3R tau fibrillized with a 30-fold longer lag time than 0N4R tau under similar in vitro experimental conditions24, consistent with a high kinetic barrier for including the CT into the β-sheet core. We speculate that the lack of the C291-containing R2 domain in 3R tau might play a significant role in raising the energy barrier and hence slowing down the rate of fibril formation. C291 participates in crucial steric zipper interactions with the R3 hexapeptide motif in both heparin-fibrillized 0N4R tau24 and CBD 4R tau9. Despite the high kinetic barrier, the fact that we captured this CT-R3 interaction in our 0N3R tau sample suggests that the energetic stability of this structure, when the CT is free of posttranslational modifications, is not substantially weaker than the stability of the smaller cores seen in diseased brains.
Since the long-range R3-CT correlations we observed were obtained from uniformly 13C, 15N labeled protein without mixing, we cannot rule out that some of the cross peaks might be intermolecular instead of intramolecular in origin. However, even if this were true, for example caused by protofibril assembly into dimeric species, the same R3-CT interactions would still exist, but in a domain-swapped manner (Fig. S8). This would have no impact on the conclusion that the R3 domain is protected and structurally stabilized by the CT.
In conclusion, we have employed solid-state NMR spectroscopy and complementary biophysical measurements to characterize the structure of 0N3R tau fibrils formed using heparin. We find that the rigid core of 0N3R tau fibrils extends from the end of R1 all the way to the C-terminus of the protein in a single molecular conformation. An R3-R4 β-arch and an R3-CT antiparallel stacking give rise to an elongated C-shaped fold, which bears similarities to the CBD 4R tau fold. The incorporation of the entire CT into the β-sheet core suggests that the previously reported transient interactions between the CT and other regions of tau can be stabilized under suitable conditions. The lack of such C-terminus-including fibril cores in the brains of tauopathies suggests that cellular mechanisms exist to prevent the CT from aggregating in diseased brain. Elucidation of these mechanisms will be important to provide a fuller understanding of the misfolding pathways of tau in diseases, cellular determinants of distinct tau strains, as well as the folding of tau in healthy brains.
Supplementary Material
Acknowledgements
This work is supported by NIH grant AG059661 to M.H. and AG002132 to W.F.D. A.J.D. is supported by an NIH Ruth L. Kirschstein Individual National Research Service Award (F31AG069418). This study made use of NMR spectrometers at the MIT-Harvard Center for Magnetic Resonance, which is supported by NIH grant P41 GM132079.
Footnotes
Supporting Information Available
An NMR pulse sequence (Fig. S1), additional 2D and 3D correlation spectra of 0N3R tau (Fig. S2-S4, S7), assignment summary (Fig. S5), water-edited spectra (Fig. S6), and tables of NMR experimental conditions (Table S1), amino acid sequence distribution (Table S2), assigned chemical shifts (Tables S3, S4), and inter-residue correlations (Table S5), are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
Data availability
NMR chemical shifts have been deposited in the Biological Magnetic Resonance Bank (BMRB) with ID number 50785.
The authors declare no competing financial interest.
References
- 1.Wang Y; Mandelkow E, Tau in physiology and pathology. Nat. Rev. Neurosci 2016, 17, 5–21. [DOI] [PubMed] [Google Scholar]
- 2.Wischik CM; Novak M; Edwards PC; Klug A; Tichelaar W; Crowther RA, Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A 1988, 85, 4884–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crowther RA, Straight and Paired Helical Filaments in Alzheimer-Disease Have a Common Structural Unit. Proc. Natl. Acad. Sci. U.S.A 1991, 88, 2288–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wegmann S; Medalsy ID; Mandelkow E; Muller DJ, The fuzzy coat of pathological human Tau fibrils is a two-layered polyelectrolyte brush. Proc. Natl. Acad. Sci. U.S.A 2013, 110, E313–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goedert M; Eisenberg DS; Crowther RA, Propagation of Tau Aggregates and Neurodegeneration. Annu. Rev. Neurosci 2017, 40, 189–210. [DOI] [PubMed] [Google Scholar]
- 6.Fitzpatrick AWP; Falcon B; He S; Murzin AG; Murshudov G; Garringer HJ, … Scheres SHW, Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Falcon B; Zivanov J; Zhang W; Murzin AG; Garringer HJ; Vidal R, … Scheres SHW, Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 2019, 568, 420–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Falcon B; Zhang W; Schweighauser M; Murzin AG; Vidal R; Garringer HJ, … Goedert M, Tau filaments from multiple cases of sporadic and inherited Alzheimer’s disease adopt a common fold. Acta Neuropathol 2018, 136, 699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang W; Tarutani A; Newell KL; Murzin AG; Matsubara T; Falcon B, … Scheres SHW, Novel tau filament fold in corticobasal degeneration. Nature 2020, 580, 283–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arakhamia T; Lee CE; Carlomagno Y; Duong DM; Kundinger SR; Wang K, … Fitzpatrick AWP, Posttranslational Modifications Mediate the Structural Diversity of Tauopathy Strains. Cell 2020, 180, 633–644.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goedert M; Spillantini MG, Propagation of Tau aggregates. Mol. Brain 2017, 10, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gibbons GS; Lee VMY; Trojanowski JQ, Mechanisms of Cell-to-Cell Transmission of Pathological Tau A Review. Jama Neurology 2019, 76, 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morozova OA; March ZM; Robinson AS; Colby DW, Conformational features of tau fibrils from Alzheimer’s disease brain are faithfully propagated by unmodified recombinant protein. Biochemistry 2013, 52, 6960–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanders DW; Kaufman SK; DeVos SL; Sharma AM; Mirbaha H; Li A, … Diamond MI, Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 2014, 82, 1271–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taniguchi-Watanabe S; Arai T; Kametani F; Nonaka T; Masuda-Suzukake M; Tarutani A, … Hasegawa M, Biochemical classification of tauopathies by immunoblot, protein sequence and mass spectrometric analyses of sarkosyl-insoluble and trypsin-resistant tau. Acta Neuropathol 2016, 131, 267–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goedert M; Jakes R; Spillantini MG; Hasegawa M; Smith MJ; Crowther RA, Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 1996, 383, 550–553. [DOI] [PubMed] [Google Scholar]
- 17.Kampers T; Friedhoff P; Biernat J; Mandelkow EM; Mandelkow E, RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett 1996, 399, 344–9. [DOI] [PubMed] [Google Scholar]
- 18.Andronesi OC; von Bergen M; Biernat J; Seidel K; Griesinger C; Mandelkow E; Baldus M, Characterization of Alzheimer’s-like paired helical filaments from the core domain of tau protein using solid-state NMR spectroscopy. J. Am. Chem. Soc 2008, 130, 5922–5928. [DOI] [PubMed] [Google Scholar]
- 19.Daebel V; Chinnathambi S; Biernat J; Schwalbe M; Habenstein B; Loquet A, … Lange A, beta-Sheet core of tau paired helical filaments revealed by solid-state NMR. J. Am. Chem. Soc 2012, 134, 13982–9. [DOI] [PubMed] [Google Scholar]
- 20.Xiang S; Kulminskaya N; Habenstein B; Biernat J; Tepper K; Paulat M, … Linser R, A Two-Component Adhesive: Tau Fibrils Arise from a Combination of a Well-Defined Motif and Conformationally Flexible Interactions. J. Am. Chem. Soc 2017, 139, 2639–2646. [DOI] [PubMed] [Google Scholar]
- 21.Falcon B; Zhang W; Murzin AG; Murshudov G; Garringer HJ; Vidal R, … Goedert M, Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 2018, 561, 137–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bibow S; Mukrasch MD; Chinnathambi S; Biernat J; Griesinger C; Mandelkow E; Zweckstetter M, The dynamic structure of filamentous tau. Angew. Chem. Int. Edit 2011, 50, 11520–4. [DOI] [PubMed] [Google Scholar]
- 23.Zhang W; Falcon B; Murzin AG; Fan J; Crowther RA; Goedert M; Scheres SH, Heparin-induced tau filaments are polymorphic and differ from those in Alzheimer’s and Pick’s diseases. Elife 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dregni AJ; Mandala VS; Wu H; Elkins MR; Wang HK; Hung I, … Hong M, In vitro 0N4R tau fibrils contain a monomorphic β-sheet core enclosed by dynamically heterogeneous fuzzy coat segments. Proc. Natl. Acad. Sci. U.S.A 2019, 116, 16357–16366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hou GJ; Yan S; Trebosc J; Amoureux JP; Polenova T, Broadband homonuclear correlation spectroscopy driven by combined R2(n)(v) sequences under fast magic angle spinning for NMR structural analysis of organic and biological solids. J. Magn. Reson 2013, 232, 18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi CW; Fasshuber HK; Chevelkov V; Xiang SQ; Habenstein B; Vasa SK, … Lange A, BSH-CP based 3D solid-state NMR experiments for protein resonance assignment. J. Biomol. NMR 2014, 59, 15–22. [DOI] [PubMed] [Google Scholar]
- 27.Shen Y; Bax A, Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 2013, 56, 227–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wishart DS; Bigam CG; Holm A; Hodges RS; Sykes BD, 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J. Biomol. NMR 1995, 5, 67–81. [DOI] [PubMed] [Google Scholar]
- 29.Baldus M; Petkova AT; Herzfeld J; Griffin RG, Cross polarization in the tilted frame: assignment and spectral simplification in heteronuclear spin systems. Mol. Phys 1998, 95, 1197–1207. [Google Scholar]
- 30.Hong M, Resonance Assignment of 13C/15N Labeled Proteins by Two- and Three-Dimensional Magic-Angle-Spinning NMR. J. Biomol. NMR 1999, 15, 1–14. [DOI] [PubMed] [Google Scholar]
- 31.Rienstra CM; Hohwy M; Hong M; Griffin RG, 2D and 3D 15N-13C-13C NMR chemical shift correlation spectroscopy of solids: assignment of MAS spectra of peptides. J. Am. Chem. Soc 2000, 122, 10979–10990. [Google Scholar]
- 32.Hung I; Gan ZH, Spin-locking and cross-polarization under magic-angle spinning of uniformly labeled solids. J. Magn. Reson 2015, 256, 23–29. [DOI] [PubMed] [Google Scholar]
- 33.Wang T; Jo H; DeGrado WF; Hong M, Water Distribution, Dynamics, and Interactions with Alzheimer’s β-Amyloid Fibrils Investigated by Solid-State NMR. J. Am. Chem. Soc 2017, 139, 6242–6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gelenter MD; Smith KJ; Liao S-Y; Mandala VS; Dregni AJ; Lamm MS, … Hong M, The peptide hormone glucagon forms amyloid fibrils with two coexisting β-strand conformations. Nat. Struc. Mol. Biol 2019, 26, 592–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dregni AJ; Duan P; Hong M, Hydration and Dynamics of Full-Length Tau Amyloid Fibrils Investigated by Solid-State Nuclear Magnetic Resonance. Biochemistry 2020, 59, 2237–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liepinsh E; Otting G, Proton exchange rates from amino acid side chains - Implications for image contrast. Magnetic Resonance in Medicine 1996, 35, 30–42. [DOI] [PubMed] [Google Scholar]
- 37.Williams JK; Hong M, Probing membrane protein structure using water polarization transfer solid-state NMR. J. Magn. Reson 2014, 247, 118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jeganathan S; von Bergen M; Brutlach H; Steinhoff HJ; Mandelkow E, Global hairpin folding of tau in solution. Biochemistry 2006, 45, 2283–2293. [DOI] [PubMed] [Google Scholar]
- 39.Mukrasch MD; Bibow S; Korukottu J; Jeganathan S; Biernat J; Griesinger C, … Zweckstetter M, Structural Polymorphism of 441-Residue Tau at Single Residue Resolution. Plos Biol 2009, 7, 399–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Noble W; Hanger DP; Miller CC; Lovestone S, The importance of tau phosphorylation for neurodegenerative diseases. Front. Neurol 2013, 4, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wesseling H; Mair W; Kumar M; Schlaffner CN; Tang S; Beerepoot P, … Steen JA, Tau PTM Profiles Identify Patient Heterogeneity and Stages of Alzheimer’s Disease. Cell 2020, 183, 1699–1713.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Törnquist M; Michaels TCT; Sanagavarapu K; Yang X; Meisl G; Cohen SIA, … Linse S, Secondary nucleation in amyloid formation. Chem. Commun 2018, 54, 8667–8684. [DOI] [PubMed] [Google Scholar]
- 43.Gu JL; Xu W; Jin NN; Li LF; Zhou Y; Chu DD, … Liu F, Truncation of Tau selectively facilitates its pathological activities. J. Biol. Chem 2020, 295, 13812–13828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gamblin TC; Chen F; Zambrano A; Abraha A; Lagalwar S; Guillozet AL, … Cryns VL, Caspase cleavage of tau: Linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 10032–10037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stöhr J; Wu H; Nick M; Wu Y; Bhate MP; Condello C, … DeGrado WF, A 31-residue peptide induces aggregation of tau’s microtubule-binding region in cells. Nat. Chem 2017, 9, 874–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berry RW; Abraha A; Lagalwar S; LaPointe N; Gamblin TC; Cryns VL; Binder LI, Inhibition of tau polymerization by its carboxy-terminal caspase cleavage fragment. Biochemistry 2003, 42, 8325–8331. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.