

Abstract
Oxygen availability is a key factor regulating microbiota composition and the homeostatic function of cells present in the intestinal mucosa of vertebrates. Microbiota-derived metabolites increase oxygen consumption by intestinal epithelial cells (IECs), reducing its availability in the gut and leading to hypoxia. This physiological hypoxia activates cellular hypoxic sensors that adapt the metabolism and function of IECs and mucosa-resident cells such as type-3 innate lymphoid cells (ILC3). In this review, we discuss recent evidence suggesting that the intricate and multidirectional interactions among the microbiota, hypoxia/ hypoxic sensors, and mammalian host cells (IECs and ILC3) determine how the intestinal barrier and host-microbiota-pathogens connections are molded. Understanding these interactions might provide new treatment possibilities for dysbiosis, as well as certain inflammatory and infectious diseases.
Keywords: microbiota, short-chain fatty acids, intestinal epithelial cells, innate lymphoid cells
Hypoxia inducible factors: key adaptors to hypoxia
Oxygen (O2) acts as a double edge sword in the human body. On the one hand, as the final acceptor of electrons in aerobic respiration and participant in several reactions for the formation of molecules, it is essential to cells. On the other hand, it can also be converted to reactive oxygen species (ROS), which may have detrimental cellular effects [1]. Considering this dual aspect, it is not surprising that organisms have evolved several mechanisms devoted to regulating the uptake, transport, and use of this molecule.
At the cellular level, there are systems that sense and respond to variations of oxygen concentrations -- an important contributing factor for the survival and success of multicellular organisms [2]. One of the best-known sensors acting in this context is the Hypoxia Inducible Factor (HIF, see Glossary). The activity of HIF is primarily regulated by the stability and transactivation potential of HIFα, which is modulated by O2-regulated proteins, including the prolyl-hydroxylases (PHDs) and the factor-inhibiting HIF (FIH) (Figure 1) [3].
Figure 1. Hypoxia in the mammalian intestine.
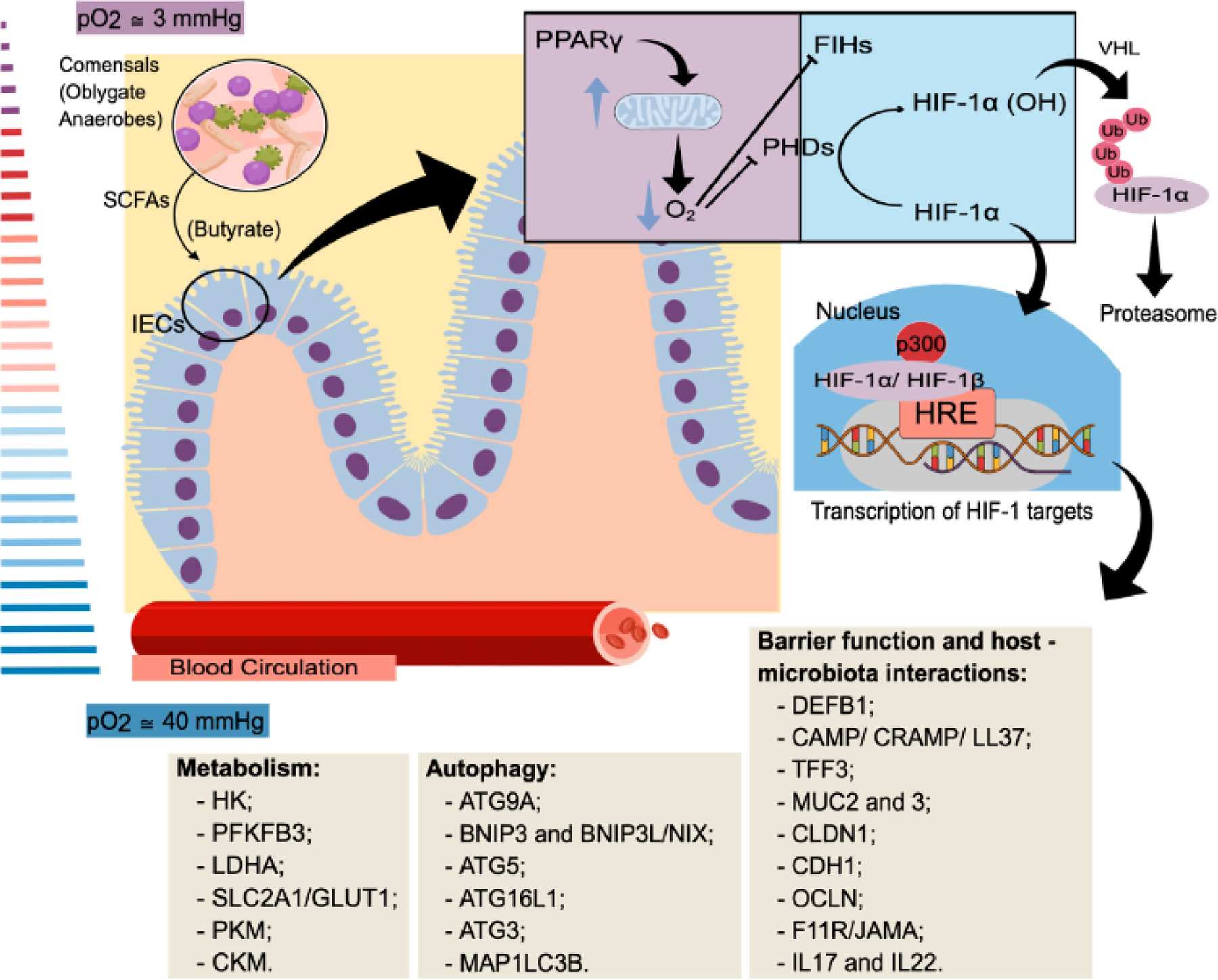
The hypoxic environment of the intestine can induce the expression of several HIF-1 target genes in intestinal epithelial cells, thus impacting their metabolism, barrier function, and survival. The intestinal lumen is characterized by low availability of oxygen. Physiological hypoxia is maintained in part by the intestinal microbiota production of short-chain fatty acids (SCFAs). These metabolites, particularly butyrate, regulate several signaling pathways on intestinal epithelial cells (IECs) including the activation of the peroxisome proliferator-activated receptor γ (PPARγ), whose signaling reinforces mitochondrial β-oxidation and consumption of oxygen. The decreased oxygen availability inhibits the prolyl-hydroxylases (PHDs) and factor-inhibiting HIF (FIH), reducing the degradation of HIF-1α and allowing its translocation to the nucleus. Once in the nucleus, HIF-1α forms a complex with HIF-1β and p300, and increases the transcription of several genes. Genes associated with adaptation of cell metabolism (i.e. inducing a shift from oxidative to glycolytic metabolism) and a variety of processes including barrier function and autophagy are among the list of HIF-1 target genes and play an important role in the adaptation of intestinal epithelial cells to the hypoxic environment found in the gut. Examples of HIF-1 target genes associated with changes in metabolism, induction of autophagy and increased barrier function and immunity are shown in the boxes. [5,17,30,31,34,42]. Abbreviations: pO2, Partial pressure of oxygen. DEFB1, Defensin beta 1. CAMP/CRAMP/LL3, Cathelicidin antimicrobial peptide. TFF3, Trefoil factor 3. MUC2, Mucin 2. MUC3, Mucin 3. CLDN1, Claudin 1. CDH1, E-cadherin. OCLN, Occludin. F11R/JAMA, F11 Receptor. IL17, Interleukin 17. IL22, Interleukin 22. ATG9A, Autophagy related 9A. BNIP3, BCL2 interacting protein 3. BNIP3L or NIX, BCL2 interacting protein 3 like. ATG5, Autophagy related 5. ATG16L1, Autophagy related 16 like 1. ATG3, Autophagy related 3. MAP1LC3B, Microtubule associated protein 1 light chain 3 beta. HK, Hexokinase 1. PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3. LDHA, Lactate dehydrogenase A. SLC2A1/Glut1, Solute carrier family 2 member 1. PKM, Pyruvate kinase M1/2. CKM, Creatine kinase, M-type. Human gene nomenclature was used because most of the genes presented in the boxes were described in human cell lines or tissues, but in several cases these genes were also modulated in mice, as described in the text.
Three isoforms of the α subunit have been reported in vertebrate species: HIF-1α; HIF-2α; and HIF-3α [3]. These translocate to the nucleus, form complexes together with the constitutively expressed β subunit and regulate the expression of genes impacting key cellular pathways, including metabolism and autophagy (Figure 1) [3].
Various factors contribute to the complexity of HIF-responses, including that: i) they can also be activated through O2-independent mechanisms and ii) they are cell-specific [4–9]. A recent study compared HIF-1α-target genes in four mouse cell types (i.e. oligodendrocytes, T-cells, melanocytes, and embryonic heart cells) and found that only 51 genes (canonical HIF-1 targets) were present in all of them [9]. In contrast, many more cell-specific HIF-1 targets were observed [9]. An enrichment of HIF-1α and the transcription factor Oligodendrocyte transcription factor (OLIG2), as well as an open chromatin status for oligodendrocyte-specific HIF-1 target genes (compared to other tissue-specific HIF-1 targets) suggested that chromatin accessibility and the interactions between chromatin and cell-specific transcription factors might contribute to explain certain cell-specific responses [9].
Although HIF activation is the most studied mechanism underpinning responses to hypoxia, it is not the only one involved. A recent study used a genome-wide pool of human gene knockout (KO) K562 cells that were exposed to 1%, 5%, or 21% O2 and were analyzed for the gradual depletion or accumulation of given gene KOs [10]. Using this approach, the authors found hundreds of genes relevant for hypoxia and normoxia [10]. Canonical hypoxic pathways were modulated in the short-term (3-day time point) but were not deemed to be essential for hypoxia fitness under long periods of hypoxia [10]. Therefore, the responses to hypoxia appear to be more complex than previously anticipated, given that they can also involve HIF-independent mechanisms, are cell-specific, and may play different roles under acute versus chronic periods of tissue hypoxia.
Herein, we discuss recent studies highlighting the interesting crosstalk between microbiota, hypoxia/HIF-1 activation, and two key cells involved in regulating host-microbiota-pathogens interactions in the gut, namely intestinal epithelial cells (IECs) and type-3 innate lymphoid cells (ILC3), which are relevant for the host immune system. We first describe the factors contributing to the low concentration of oxygen in the intestine and then address the relevance of hypoxia/HIF-1 to IECs and ILC3 functions and their presumed role in inflammatory and/or infectious diseases in mice and humans.
Intestinal hypoxia
Oxygen concentrations along the mammalian intestinal tract and particularly in the large intestine are lower than in other tissues (such as lung, liver, and heart) and in the atmosphere [11]. Studies performed in mice using either spectral–spatial electron paramagnetic resonance imaging or phosphorescence quenching have shown that, in addition to a drastic reduction in O2 along the gastrointestinal tract, there is also a transversal gradient along the tissue (e.g. from the vascularized submucosal layer to the anoxic lumen) (Figure 1) [12,13]. The low-oxygen availability is also not restricted to the intestinal lumen, thus affecting the epithelial layer and adjacent cells [12,13].
The gut oxygen environment results from two processes: i) oxygenation by the bloodstream; and ii) oxygen consumption by host cells and microbiota components [11]. In this regard, hyperoxia can increase intestinal luminal oxygen concentrations in mice [13], while the opposite response (decreased luminal O2) can be quickly induced in mice exposed to hypoxia relative to normoxic control animals [12,14]. In both cases, 16S rRNA sequencing has demonstrated changes in the gut microbiota composition, with a higher abundance of Proteobacteria and Actinobacteria in hyperoxic animals versus higher abundance of Firmicutes in hypoxic mice[12,14]. A recent study found a similar shift in the microbiota profile after hyperoxia exposure in mice, and in human patients receiving mechanical ventilation [15].
IECs provide a physical and chemical barrier to segregate the microbiota and to protect the organism against pathogens [16] (Box 1). They also contribute to the hypoxic environment of the intestine by consuming part of the oxygen provided from the circulation through mitochondrial oxidative phosphorylation -- a process that is essential for ATP production, and which is modulated by molecules from the microbiota, such as short-chain fatty acids (SCFAs) [17,18].
Box 1. Intestinal epithelial cells (IECs).
The intestinal epithelium is a dynamic monolayer composed of distinct cell types -- collectively named intestinal epithelial cells [84], that provide a first line of defense against pathogens in the gut [16]. IECs are originated from intestinal stem cells (ISCs) located at the bottom of the intestinal crypts (crypts of Lieberkϋhn), which are constantly proliferating and differentiating into mature cells. The daughter cells migrate up into de crypt/villi, being extruded once at the tip [85,86]. In this sense, IECs have an extremely short lifetime and the intestinal epithelium shows the fastest turnover rate in the human body, being completely renewed after every 3 to 5 days [87]. Goblet cells are a type of IEC responsible for the production and secretion of mucins, creating a mucus layer above the epithelium (single layer in the small intestine and double layer in the colon), carrying antibodies and antimicrobial peptides (AMPs) [16] (Figure 2). This architecture prevents a closer contact between luminal microorganisms and IECs. In addition, IECs comprise absorptive enterocytes/colonocytes, enteroendocrine cells that secrete hormones [88], Paneth cells that secrete AMPs and factors to nurture and maintain the ISCs [89], microfold (M) cells responsible for antigen sampling and presentation to immune cells [90], and tuft cells involved in anti-helminth defense [91] (Figure 2). Although IECs are mostly shared between the small intestine and colon, Paneth cells and M cells are exclusive to the small intestine [16]. The continuous physical barrier of the epithelium is then formed by IECs connected through tight junctions. These cells integrate luminal signals and respond by producing cytokines and chemokines that regulate immune cells, contributing to the constant surveillance and the maintenance of homeostasis [16] (Figure 2).
Host-microbiota interactions can maintain intestinal hypoxia
Dietary fibers are polymers that are resistant to digestion by host enzymes [19]. They reach the large intestine where they are partially converted from polymeric molecules (i.e. pectins and non-digestible oligosaccharides) to mono(di)meric one (i.e. glucose, fructose and galactose) by a diverse repertoire of microbial enzymes [19]. During this process, there is production and release of SCFAs. Acetate, propionate, and butyrate are the predominant SCFAs in the intestine, with colonic concentrations ranging from 20 to 140 mM [19]. They act on host cells through distinct mechanisms, including the activation of receptors such as FFAR2 and FFAR3, and/or the inhibition of histone deacetylases (HDACs); these mechanisms can in turn lead to several cellular responses including chemotaxis, proliferation, and apoptosis, thus modulating host-microbiota interactions [19]. In addition, SCFAs are also an important source of energy: butyrate is rapidly taken up by colonocytes and used as a main source of energy to produce ATP [19]. Butyrate also enhances mitochondrial oxygen consumption in Caco-2 cells in vitro, depleting O2, which consequently activates HIF-1 and its target genes such as LDHA and SLC2A1 (Figure 1); this in turn favors a barrier function, as evidenced from a 30% reduction in FITC-dextran permeability in butyrate-treated Caco-2 cells compared to controls [17]. Moreover, relative to control mice, germ-free and antibiotic-treated mice present lower SCFAs concentrations in the colon, as well as disrupted physiological hypoxia, and reduced HIF-1 activation; these effects are reverted by the oral administration of tributyrin – a butyrate pro-drug [17]. Indeed, lentiviral-mediated HIF-1β knockdown in T84 cells has been shown to abrogate the effects of butyrate and impair the epithelial barrier, with a 6 fold-greater FITC-dextran flux ratio compared to control cells [17]. This in turn, has revealed a close relationship between butyrate and HIF-1 activation in IECs [17].
In line with this, the exposure of mice to pre-inflammatory bowel disease (pre-IBD)-risk factors (i.e. a single oral dose of the antibiotic streptomycin combined with intake of a high-fat diet (HFD) (45% fat) impaired IEC mitochondrial function relative to mock-treated, low-fat diet (LFD) (10% fat) controls, thus leading to a shift from oxidative phosphorylation to a glycolytic metabolism in IECs [20]. This change in metabolism, which was assessed by lactate concentrations and the expression of genes related to mitochondrial respiration (Ndufs1, Uqcr and Atp5g1), resulted in increased epithelial oxygenation, triggering dysbiosis in mice relative to LFD controls [20]. The dietary use of a peroxisome proliferator-activated receptor-γ (PPARγ) agonist, 5-amino salicylic acid (5-ASA), restored intestinal hypoxia and furthermore, blunted or attenuated the dysbiosis and inflammation in HFD- and streptomycin-treated mice. This latter result implied that: i) changes in gut microbial communities observed in mice exposed to pre-IBD factors may be secondary to alterations in IECs metabolism and oxygen availability, and ii) that gut dysbiosis may be reversed by treating IEC mitochondrial dysfunction [20].
Of note, butyrate increases the activation of PPARγ in mice colonocytes [18]. This effect is associated with increased mitochondrial activity and fatty acid β-oxidation -- a process that consumes O2 (Figure 1), as demonstrated in mice lacking epithelial PPARγ signaling18]. Furthermore, depletion of mouse gut microbiota bacterial populations that produce butyrate (i.e. Clostridia species) via streptomycin treatment is associated with a shift in IEC metabolism from β-oxidation to anaerobic glycolysis, impacting both microorganisms [18,21,22] and host cells [23]. However, by restoring the butyrate-producer Clostridia sp. once the effect of antibiotics has diminished, supplementing butyrate directly in drinking water, or indirectly by feeding mice a high-fiber diet, or by activating butyrate’s downstream pathway (i.e. using PPARγ agonistssuch as 5-ASA and GW9662), it has been possible to reestablish intestinal hypoxia [17,18,20,23,24].
Together these studies highlight a cycle that maintains intestinal physiological hypoxia: commensal anaerobic bacteria release metabolites that are used by IECs to generate ATP via mitochondrial respiration -- a process that depletes oxygen from the environment. In this sense, the conditions (e.g. antibiotics or low-fiber diets) that interfere with either microbiota composition or epithelial metabolism can compromise this mutual beneficial interaction, disrupting hypoxia and favoring pathogen colonization.
Hypoxia-mediated regulation of IECs: the role of HIF-1 and SCFAs
The role of butyrate in enhancing gut epithelial barrier function has been extensively studied [25–29]. Indeed, several reports have linked the protective effect of butyrate to HIF-1 stabilization [5,17,24,30]. In mice, for example, the absence of HIF-1α in IECs (Hif1afl/flVillin-Cre+) worsened the pathological effects of chronic alcohol exposure, including dysfunction of the epithelial barrier, with increased permeability and higher concentrations of circulating LPS and hepatic Escherichia coli protein relative to WT mice [30]. In a separate murine model of Clostridioides difficile infection (CDI) -- a toxigenic Gram-positive anaerobic bacterium which can cause intense intestinal damage and inflammation, HIF-1 was shown to be necessary for butyrate’s protective effects on the intestinal epithelium [24]. Compared to WT mice, HIF-1 deficient mice (Hif1afl/flVillin-Cre+) showed no changes in bacterial or FITC-dextran translocation when treated with butyrate, aside from presenting a poorer clinical score after CDI [24]. Oral antibiotics are one of the main risk factors for CDI due to their negative impact on microbiota colonization resistance. In this sense, strategies that restored butyrate concentrations in the colon of antibiotic-treated mice, such as oral administration of butyrate or a high-fiber diet, protected the animals against CDI, significantly reducing disease intensity relative to mock-treated animals or to mice receiving a conventional diet [24]. Together, these results highlight the relevance of HIF-1 in the intestinal epithelium and provide evidence for its role as a potential therapeutic target in the preservation of intestinal barrier function.
From another angle, chromatin immunoprecipitation (ChIP) of HIF-1 in Caco-2 cells has revealed that HIF-1 can bind several autophagy-related genes, including human ATG9A [5]. ATG9A is an interesting factor as it has been shown to determine the localization of the adherens junctions and tight junctions in IECs: deletion of ATG9A in T84 human epithelial cells through short hairpin RNA (shRNA) nearly abolished barrier formation by inducing abnormalities in the actin cytoskeleton when compared to vector controls; indeed, this was an effect that could not be reversed by butyrate administration [5]. Moreover, a recent study in T84 cells showed that such intestinal barrier function could be improved when cultivated under hypoxic conditions; under this setting, higher transepithelial resistance and enhanced expression of tight-junction related genes were noted compared to cells cultivated under normoxic conditions [31]. The changes induced with hypoxia were abrogated when HIF-1α was knocked down via shRNAs in T84 cells [31]. Moreover, chemical stabilization of HIF-1α/HIF-2α with dimethyloxalylglycine (DMOG) in vitro was enough to mimic the phenotypes of the cells cultivated under normoxia [31]; this in turn suggested that the benefits of hypoxia to IECs might be attained with pharmacological approaches that target HIF-1 stabilization under normoxia. In addition, a whole-transcriptome miRNA profiling study identified several hypoxia-regulated miRNAs involved in this barrier effect. In particular, miRNA-320a, whose expression is controlled by HIF-1α, has been associated with barrier formation [31]. HIF-1 is also considered as being important during intestinal epithelial healing. Specifically, stabilization of HIF-1 by GB-004 (a PHD inhibitor) in T84 cells accelerated wound closure by increasing cell migration and proliferation in scratch assays relative to vehicle-treated controls [32]. A similar phenotype of wound closure using PHD inhibitors was seen in vivo in both pinch biopsy and trinitrobenzene sulfonic acid (TNBS)-induced colitis [32], highlighting the importance of epithelial HIF-1 on mucosal healing.
The mucus layer and production of antimicrobial peptides (AMP) are also regulated by hypoxia and HIF-1 [19,30,33–35]. For example, HIF-1α activation in HT-29 human colonocytes by the PHD inhibitor mimosine induces production of the cathelicidin antimicrobial peptide (CAMP) relative to mock-treated cells and drastically reduces intestinal colonization by Candida albicans compared with vehicle-treated mice -- an effect that was abrogated in Hif1afl/flVillin-Cre+ mice [33]. Also, the HDAC inhibitor entinostat induces AMP LL-37 expression in HT-29 cells through the activation of STAT3 and HIF-1α [34] -- an effect that can be linked to butyrate activity in the colon, since this metabolite is known as a potent HDAC inhibitor in colonocytes [19]. Moreover, Hif1afl/flVillin-Cre+ mice presented exacerbated dysbiosis after chronic treatment with a 5% alcohol liquid diet [30], a phenotype that could be partially explained by the deficient production of β-defensins and other AMPs after alcohol exposure [30], since those factors regulate bacteria composition and numbers. Lastly, stabilization of HIF-1 with DMOG was sufficient to restore the expression of β-defensins and LL-37 in alcohol-treated animals [30], confirming the relevance of HIF-1 in modulating the expression of these AMPs in mice.
Collectively, these studies demonstrate the relevance of SCFAs and hypoxia/HIF-1 activation for the intestinal epithelial barrier by enhancing tight junctions and decreasing gut permeability, as well as elevating mucus and the production of AMPs; these segregate the microbial community from the epithelium -- effects that together, serve to avoid bacterial overgrowth and translocation.
Hypoxia/HIF-1 and SCFAs regulating intestinal epithelial autophagy
Autophagy is required to maintain intestinal homeostasis since it plays a role in modulating epithelial barrier function and the mucosal immune response [36]. In one study, autophagy was identified as the major HIF-1 target pathway in hypoxia-treated Caco-2 cells following an unbiased approach of chromatin immunoprecipitation (ChIP) analysis [5]. Enrichment of several genes associated with autophagy (e.g. BNIP3, ATG5 and ATG16L1) was observed compared with input DNA [5]. Another report indicated that hypoxia could activate autophagy and reduce intestinal inflammation in IBD patients relative to non-IBD individuals [37]. In this study, healthy individuals and IBD patients were subjected to hypoxic conditions equivalent to an altitude of 4000 m above sea level. Colon biopsies obtained from IBD patients and incubated under hypoxic condition for 3 hours in vitro revealed downregulation of TNFA, IL6 and NLRP3, and induction of autophagy-associated SQSTM1 (also known as p62) relative to colon biopsies from healthy people [37]. Similar responses were observed in dextran sulfate sodium (DSS)-treated mice under hypoxic conditions, as well as in mice treated with DMOG to stabilize HIF-1. Moreover, in vitro, hypoxia increased HIF-1α-activated autophagy, and led to lysosome accumulation and NLRP3 degradation in human HT-29 cells cultivated with LPS, compared to cells mantainted under normoxic conditions [37]. A recent report suggested a relationship between SCFAs, HIF-1 activation, and induction of autophagy in IECs [38]. Butyrate treatment increased autophagy in HT-29 cells in vitro -- an effect that was significantly attenuated in the absence of HIF-1α [38]. Together, these results indicated that HIF-1 activation by SCFAs could induce autophagy in IECs, and that this process might counteract intestinal inflammation.
Altogether, the microbiota/SCFA axis appears to be essential for maintaining physiological hypoxia in the intestinal epithelium of both mice and humans, coordinating several aspects of its functionality that can impact directly in the host’s general health (Figure 2). However, there is a multitude of aspects that need to be investigated in more detail; namely, the relevance of other microbiota-derived metabolites on this axis, the role of HIF-1 on different cells comprising the intestinal epithelium, and the differences between short and long-term activation of HIF-1 in vivo.
Figure 2. Microbiota-derived metabolites can contribute to hypoxia and HIF-1 stabilization.
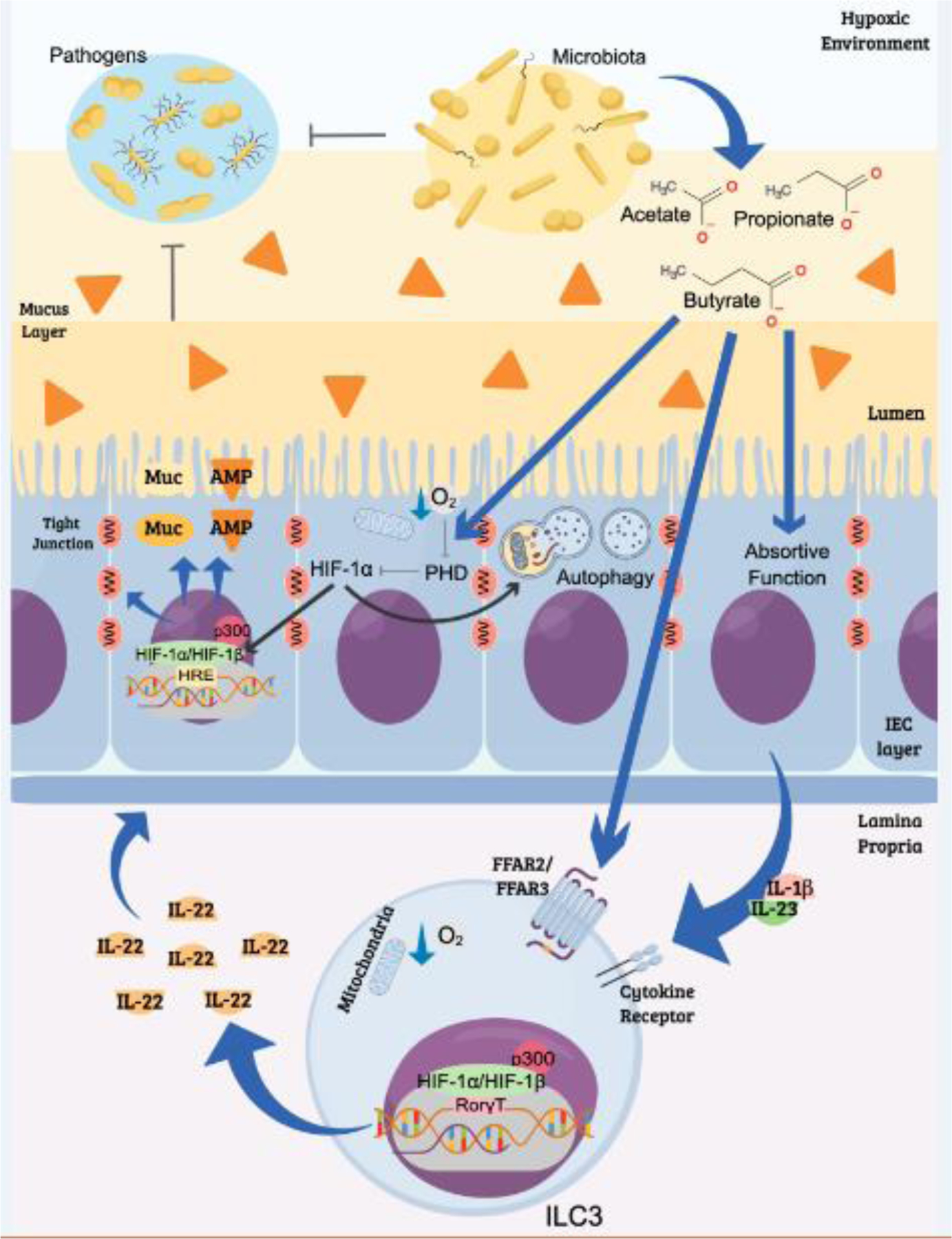
This process molds IEC and ILC3 functions that are relevant for host-microbiota interactions and the resistance/response to pathogens. The commensal microbiota competes and directly inhibits the growth of potential intestinal pathogens through different mechanisms. Among them, is the production of SCFAs, which can contribute to regulating host immune responses and the function of the intestinal epithelium. In humans and mice, these metabolites contribute to oxygen depletion and activation of HIF-1 responses in IECs, as well as in other cell types, such as the ILC3. HIF-1 regulates the expression of several genes including mucins (Muc), antimicrobial peptides (AMP) and tight junction proteins that are important for barrier function of the epithelium. Activation of HIF-1 induces autophagy in IECs -- a cellular process that is pivotal for innate immune defense. ILC3 also plays an important role in maintaining the epithelial barrier function through the production of IL-22. These cells express the SCFA receptors, FFAR2 and FFAR3, which contribute to their proliferation, responsiveness, and cytokine production. The activation of ILC3 is associated with increased mitochondrial activity and HIF-1α stabilization. In the nucleus, HIF-1 interacts with the transcription factor RORγt, increasing the production of IL-22 that regulates epithelial cell survival/proliferation, barrier integrity, and microbiota composition Thus, low oxygen concentration is crucial for cross-talk between cells in the lamina propria and maintains coordinated actions for the control of intestinal homeostasis.
Hypoxia-, HIF-1-, and SCFA-mediated regulation of ILC3s
Here, we focus on ILC3s (Box 2) because they are among the most predominant ILCs in the mammalian intestinal tract and present an intense and bidirectional crosstalk with IECs and the gut microbiota [39–41]. These interactions can contribute to the maintenance of intestinal hypoxia since they impact on microbiota composition and metabolite production and can be molded by changes in oxygen availability in the intestine [42,43,44].
Box 2. Innate lymphoid cells (ILC).
ILCs are considered the innate counterpart of adaptive helper and cytotoxic T-cells [45,92]. ILCs do not display rearranged receptors such as T cells, but share the expression of key transcription factors and effector cytokines with T cell subsets. These transcription factors are used to classify ILCs into three main subsets: 1) Group 1 ILCs includes natural killer (NK) cells and other cells that express the transcription factor T-bet and whose main effector cytokine is interferon (IFN)-γ (similar to T helper 1, Th1); ii) Group 2 ILCs express GATA3, secrete IL-4, IL-5, and IL-13, and resemble Th2 cells; and iii) Group 3 ILCs express retinoid-related orphan receptor γt (RORγt) and secrete IL-22 and IL-17, resembling Th17 cells in mammals [45]. These cells are important in both homeostatic and pathological conditions and, despite their similarities with T cells, they have unique and non-redundant functions, which are related to the timing and magnitude of their responses in humans and mice [93]. ILC3s constitute a heterogeneous population of cells with some distinct functions and high plasticity between them and other ILCs in humans and mice [92]. There are ILC3s expressing the NKp46 surface protein (also known as natural cytotoxicity receptor or NRC) and cells not-expressing this molecule (NRC negative) in mice [94]. One of the NRC-negative subsets, called LTi (lymphoid tissue inducer), expresses the chemokine receptor CCR6 and is present in lymphoid organs and the colon of mice [95]. These cells produce lymphotoxins and TNF-α and stimulate the mesenchymal cell production and expression of chemokines and adhesion molecules, participating in the formation of murine lymphoid organs [45,96]. The NCR− CCR6− ILC3s are distributed along the whole intestinal lamina propria and are a substantial source of IL-22 and IL-17, mainly acting on epithelial and stroma cells in mice [94]. However, the emerging murine RORγt+ CCR6− ILC3s acquire transcription factor T-bet for subsequent expression of NCR, showing an ILC1-like phenotype and producing TNF and IFN-γ, in addition to IL-22, yet produce less IL-17 [97]. This subtype plasticity has implications for the underlying mechanisms of ILC3 development and maintenance in murine and human tissues at steady state, and mainly during intestinal diseases [92].
ILC3s constitute a heterogeneous population of cells important for mucosal homeostasis and for maintaining the epithelial barrier (Box 2) [45]. These cells are abundant in the mouse intestinal lamina propria and are characterized by the production of cytokines in response to signals released by resident cells such as mononuclear phagocytes following tissue damage [46]. One of the main cytokines produced by ILC3s is IL-22, bearing a crucial role in the regulation of intestinal epithelium functions and host-microbiota interactions [47,48]. Specifically, IL-22 induces epithelial cell survival and proliferation in the thymus, skin, gut, and respiratory tract of mice and humans [49–52], and contributes to intestinal barrier function by promoting the expression of tight junction proteins, mucin, and enabling mucus glycosylation, as shown in murine colitis models and in human IECs [53,54]. Studies performed using antibodies to neutralize IL-22 and in IL-22 knockout mice have implicated this cytokine in shaping the gut microbiota, via stimulation of intestinal production of AMPs, such as with the regenerating protein RegIIIγ [55].
Conversely, microbiota derived signals, including SCFAs, can regulate ILC3 responses [42,56,57]. Oral administration of propionate or acetate to mice expanded colonic ILC3 population and their production of IL-22 at steady state and during DSS-induced colitis or intestinal Citrobacter rodentium infection [56]. Moreover, ILC3s express the receptor FFAR2 whose activation has selectively promoted increased colonic ILC3 numbers and activity in mice [56]. Indeed, the acetate-FFAR2 axis enhanced IL-1R expression in intestinal ILC3, thereby heightening ILC3 sensitivity to IL-1β and IL-22 production; together, this induction ameliorated the response to acute C. difficile infection in mice [57]. Similarly, SCFAs also regulated ILC3 numbers in peripheral tissues and attenuated intestinal C. rodentium infection in mice by boosting cytokine signaling and activating the mammalian target of rapamycin (mTOR) pathway [58].
In addition, butyrate upregulates IL-22 production by murine ILCs stimulated in vitro with IL-23 [42]. This study also showed that oral treatment of mice with 200 mM butyrate in drinking water for 3 weeks increased IL-22 production by ILC3 (and T helper populations) in the spleen, mesenteric lymph nodes, and intestinal lamina propria, compared to untreated mice [42]. Mechanistically, butyrate acted through the FFAR3 receptor on ILC3 and CD4+ T cells, and induced the expression of aryl hydrocarbon receptor (AhR) and HIF-1α -- differentially regulated by mTOR and Stat3 [42]. Butyrate also increased the accessibility of HIF-1α-binding sites in the Il22 promoter through histone modification, thus enhancing IL-22 expression [42]. Butyrate’s effect on IL-22 production was also demonstrated in human cells: this SCFA promoted HIF1A and AHR expression and increased IL-22 in vitro in human CD4+ T cells [42]. Together, these findings suggested that: i) SCFAs are a key link between the microbiota and the intestinal production of IL-22; ii) different mechanisms and target cells (T cells and ILC3) can account for an IL-22-promoting effect from SCFAs; and iii) SCFAs are relevant in the control intestinal infection and inflammation.
Although hypoxia and activated hypoxia sensors, such as HIF proteins, are key regulators of immune cells [11,43], their impact on ILC3s was only recently addressed. Using conditional knockout mice for a key component of the mTOR complex-1, the raptor gene (Rptorfl/flRorc-Cre+), and mTOR inhibition by rapamycin, a major role for this pathway in sustaining intestine ILC3 numbers, proliferation, and cytokine secretion was described in mice at steady-state and following C. rodentium infection [59]. mTOR activation in ILC3s increased the production of mitochondrial ROS -- an effect that was relevant for HIF-1α and RORγt expression, as well as for the proliferation and cytokine production of these cells, since these responses were abrogated when murine ILC3 were incubated with a reactive oxygen species scavenger (N-acetyl-cysteine), or with mitochondrial inhibitors (rotenone and antimycin A) [59]. Mechanistically, HIF-1 has been deemed to contribute to ILC3 activation by increasing glycolysis and RORγt expression [59]. These results reveal a key role of mTOR-HIF-1 signaling in the regulation of ILC3s responses.
Of note, HIF-1α directly promotes the transcription of the gene encoding RORγt and its target genes, such as IL22 and IL17 cytokines, in humans and mice [44,60]. Moreover, HIF-1 enhances Th17 differentiation by stimulating RORγt expression and collaborates with this transcription factor to induce Th17 signature genes, including Il17 in mice [60]. These mechanisms also seem to be relevant in ILC3. Hypoxia-induced HIF-1α activation enhanced the ex vivo proliferation and IL-22/IL-17 production of murine intestinal ILC3 through a RORγt-HIF-1α transcriptional program [44]. In addition, hypoxia/HIF-1α signaling improved murine resistance to C. difficile infection; this was shown from an improved clinical score and reduced intestinal bacterial translocation in infected wildtype (WT) mice compared to conditional RORyt-specific HIF-1α knockout mice (Hif1afl/flRorc-Cre+) [44].
Collectively, these mouse ILC3 studies demonstrated that hypoxia and mTOR-HIF-1 activation regulate the number and response of these cells, particularly their ability to produce IL-22 in the intestine (Figure 2) [44,59]. Thus, we hypothesize that SCFAs might also have a relevant role in modulating ILC3 responses in this tissue. We propose that SCFAs might indirectly contribute to the activation of HIF-1 in ILC3 through their effects on oxygen utilization by IECs, or via their impact on cells producing ILC3-activating mediators (e.g. increased neutrophil IL-1β production during C. difficile infection [57]). In addition, they might directly regulate the activation of HIF-1 and other pathways in ILC3s such as FFAR2 and FFAR3 receptor engagement, thus affecting ILC3 numbers and functions [42,57] (Figure 2). These responses would be expected to be beneficial for host-microbiota crosstalk, since ILC3s contribute to maintaining epithelial barrier function and innate immunity [47,48]. However, extensive, and robust studies are needed to test these hypotheses and to clearly define the pathways of hypoxia-induced HIF-1 in influencing the differentiation, maintenance, and migration of ILC3s in the intestine, as well as their effects on microbiota composition. Indeed, how the daily changes in oxygen supply provided by blood circulation might regulate this system is a relevant question for future inquiry (see Outstanding Questions).
OUTSTANDING QUESTIONS BOX.
The physiological concentration of O2 varies for distinct tissues within the mammalian body [11]. In this sense, HIF-1 is essential for transcriptional cellular adaptation under hypoxia, although there are other oxygen sensors present in the cells, such as HIF-2 and HIF-3 [3]. What are the roles of other hypoxia activated genes/sensors at steady state and during inflammatory conditions?
There is a gradient of O2 along the intestinal tissue as its concentrations decrease more than 10 times from the submucosa to the lumen (Figure 2) [12, 13]. Thus, IECs in different locations may be affected by hypoxia in different ways, demanding distinct adaptation. How can hypoxia/HIF-1 activation lead to distinct responses in different cells? What are the cell-specific genes/proteins involved? Are there HIF-1 cell-specific responses occurring via O2-independent mechanisms?
In addition to its effect on epithelial HIF-1 stabilization in mice [17,24,38], the SCFA butyrate inhibits histone deacetylase activity, resulting in general DNA hyperacetylation and increased chromatin accessibility [34,42]. How does increased chromatin acetylation affect HIF-1 interactions with DNA and other nuclear proteins? We anticipate that new technologies (e.g., ATAC-seq and ChIP-seq) might help address some of these questions.
Intestinal hypoxia plays an important role during acute intestinal inflammation in humans and mice, acting on several cells, including IECs and immune cells [44,98–101]. However, some studies have shown that chronic HIF-1 stabilization can be harmful and exacerbate certain inflammatory conditions [61–69]. What are the consequences of acute and chronic activation of HIF-1 in different cells/tissues and diseases?
The daily fasting/refeeding cycle alters the gut microbiota as well as genes related to SCFA production, in mice and humans [103,104]. How might daily variations in feeding rhythms and types of diet impact hypoxia and HIF-1expression/activity in intestinal cells as well as microbial components?
Relevance of hypoxia and HIF-1 in inflammatory and/or infectious conditions
Several studies have investigated the role of hypoxia and HIF-1 activation in different inflammatory and infectious experimental models. As can be appreciated in table 1, HIF-1α activation during inflammatory or immune responses can be beneficial or detrimental to the host depending on the type of model analyzed. In general, the activation of this pathway in immune or epithelial cells has so far had a positive impact on host immune response to pathogens and injury in mouse intestinal models of inflammation, such as DSS-, Clostridioides difficile- and Citrobacter rodentium-induced colitis (Figure 3) [24,42,44,60]. However, this has not been observed in other inflammatory or infectious models [61–69]. For example, HIF-1 activation worsened the immune response and outcomes observed for pulmonary conditions in mice, such as pneumonia caused by Klebsiella pneumoniae infection [65] and chronic obstructive pulmonary fibrosis [64], leading to increased lung infiltrates and tissue damage [64,65]. Similar findings have been reported in the context of Ischemia and Reperfusion Injury in mice [62,66].
Table 1. Reported outcomes of HIF-1α activation in diverse inflammation models.
Schematic review of studies (published in the last 5 years) investigating the impact of HIF-1α activation in several inflammatory/infectious models. The colored vertical bar indicates the overall effect of HIF-1α activation, from green (positive, beneficial effect for the organisms) to red (negative, detrimental effect to the organisms).Ref: reference.
| Inflammation model (species) | Mechanism | Reported Outcome | Ref | ||
|---|---|---|---|---|---|
| HIF-α Activation | DSS-induced colitis (mice). | Butyrate induced-HIF-1α stabilization in IECs promotes autophagy, thus increasing the renewal of the intestinal barrier and decreasing the production of pro-inflammatory mediators. | Alleviation of DSS-induced colitis by promoting autophagy via HIF-1α. | [38] | |
| DSS-induced colitis (mice). | Hif-1α deletion on epithelial cells impacted on their production of cytokines (reduced IL-7 and IL-15 and increased IL-10). Also affected the numbers and phenotype of intraepithelial lymphocytes (IELs). | Mice with Hif-α deletion on IECs presented an increased susceptibility to DSS-induced colitis. | [98 | ||
| DSS -induced colitis (mice). | HIF-1α, together with p-STAT3, regulates the transcription of CD11b on B cells. | Mice with HIF-1α deletion on B cells presented an exacerbated inflammatory response indicating that this transcription factor may contribute to B cell protective activity in this model. | [99] | ||
| Pinch biopsy and 2,4,6 - trinitrobenzenesulf onic acid (TNBS) colitis model (mice). | HIF-1 stabilization with PHDi induces α6 and α2 integrin expression, which may contribute to epithelial cell migration and proliferation. | HIF-1 regulates integrin-α6 expression and function, promoting intestinal epithelial healing. | [32] | ||
| Clostridioides difficile -induced colitis (mice) | Butyrate induced-HIF-1α stabilization in IECs increases tight junctions, reducing intestinal epithelial permeability, and bacterial translocation. | Butyrate induced-HIF-1 in IECs mitigates local inflammatory response and tissue damage. | [24] | ||
| Clostridioides difficile -induced colitis (mice). | HIF-1α stabilization induces ILC3 activation by modulating RORγt and target gene expression. | Hypoxia and HIF-1 activation in ILC3 attenuate the intestinal infection by C.difficile. | [44] | ||
| Citrobacter rodentium infection (mice). | mTORC1 induces the activation of mitochondrial metabolism and the generation of mROS, which stabilizes HIF-1α, reprogramming ILC3 metabolism toward glycolysis and sustaining the expression of RORγt. These responses are important for ILC3 proliferation and production of IL-17A and IL-22 after their activation. | Mice with deletion of mTORC1 subunit Raptor are more susceptible to C. rodentium infection. | [59] | ||
| Citrobacter rodentium infection (mice). | Short Chain Fatty Acids (SCFAs) upregulate IL-22 production by promoting AhR and HIF-1α expression, which are differentially regulated by mTOR and Stat3. HIF-1α binds directly to the II22 promoter, and SCFAs increase HIF1α binding to the II22 promoter through histone modification. | Oral supplementation of mice with SCFA enhances IL-22 production conferring protection from intestinal inflammation. | [42] | ||
| Alcoholic liver disease (ALD) model (mice). | HIF-1α activation regulates gut bacterial composition through the production of antimicrobial peptides and stabilizes barrier function via upregulation of P-glycoprotein and tight junction proteins. | The absence of HIF-1α in IECs worsened the pathological effects of chronic alcohol exposure such as gut dysbiosis and dysfunction of the epithelial barrier with increased permeability and bacterial product translocation. | [30] | ||
| Mycobacterium tuberculosis infection (mice). | HIF-1α stabilization by nitric oxide promotes macrophage activation through expression of pro-inflammatory and anti-microbial genes. | Stabilization of HIF-1α in macrophages is important for effective bacterial killing. | [70] | ||
| Leishmania donovani infection (mice). | Deficiency of HIF-1α in myeloid cells leads to increased lipid accumulation, by promoting de novo lipogenesis through the BNIP3/mTOR/SREBP-1c axis. | Deficiency of HIF-1α in the myeloid compartment impairs the host-protective anti-leishmanial functions of myeloid cells. | [100] | ||
| Calu-3 or primary bronchial epithelial cells (PBECs) SARS-CoV-2 pseudo-particle (pp) infection (In vitro). | Hypoxia and the HIF prolyl hydroxylase inhibitor Roxadustat reduce ACE2 expression and inhibit SARS-CoV-2 entry and replication in lung epithelial cells via an HIF-1α-dependent pathway. | Hypoxia inhibits SARS-CoV-2 entry, replication, and secretion of infectious particles in lung epithelial cells. | [101] | ||
| 2,4-dinitro-fluorobenzene (DNFB) -induced contact hypersensitivity (CHS) (mice). | CCR7 stimulation activates the HIF-1 pathway in dendritic cells (DCs), leading to metabolic reprogramming toward glycolysis, a response that is important for DC migration. The long non-coding RNA Inc-Dpf3 directly binds to HIF-1α and suppresses HIF-1α-dependent transcription of the glycolytic gene Ldha, thus inhibiting DC glycolytic metabolism and migratory capacity. | Inhibition of HIF-1α-dependent glycolysis impairs DC migratory capacity, attenuating the immune-activating function at the late stage of an inflammatory immune response. | [75] | ||
| Liver Ischemia/Reperfus ion Injury (IRI) (mice). | HIF-1α stabilization promotes Foxp3 downregulation and RORγt and IL-17A upregulation in IRI livers. | Aggravation of IR-induced liver inflammation. | [62] | ||
| DSS-induced colitis (mice). | HIF-1α stabilization on myeloid cells aggravates the mortality and weight loss of DSS treated mice, fastens the onset of rectal bleeding, and increases infiltration in the colon. | HIF-1α stabilization on myeloid cells worsened the progression of DSS - induced colitis. | [63] | ||
| Cigarette Smoke (CS) -induced Chronic obstructive pulmonary fibrosis (COPD) (mice) | H2S upregulates prolyl hydroxylase (PHD)2, suppressing HIF-1α expression and MAPK activation, downregulating pro-inflammatory cytokines, and upregulating the expression of tight junction proteins. | Inhibition of HIF-1α/MAPK signaling reduced inflammation, epithelial cell injury and apoptosis, abrogating CS-induced emphysema and improving pulmonary function in mice. | [64] | ||
| Klebsiella pneumoniae pneumonia (mice). | Epithelial HIF-1α stabilization by siderophores induces bacterial dissemination. | Enhanced systemic bacterial spread during infection. | [65] | ||
| Ischemia/reperfusi on (I/R) injury to the kidney and unilateral ureteral obstruction (UUO) (mice). | Increased expression of HIF-1α induces selective shedding of microRNA-23a (miRNA-23a)-enriched exosomes by tubular epithelial cells (TECs), triggering macrophages reprogramming into a pro-inflammatory state. | HIF-1α incites tubular inflammation in the kidney, via exosome - mediated intercellular communication between TECs and macrophages. | [66] | ||
| Chronic visceral leishmaniasis (VL) (mice) | Induction of HIF-1α in splenic DCs inhibits IL-12 and increases IL-10 expression. The imbalance between these cytokines may lead to inefficient priming of protective Th1 responses. | HIF-1α stabilization promotes L. donovani survival and limits the development of protective Th1 responses, resulting in the establishment of chronic infection. | [67] | ||
| Collagen-induced arthritis (CIA) (rat). | Intracellular succinate induces angiogenesis through HIF-1α dependent VEGF expression, while extracellular succinate acts on GPR91 activation. | Disturbance of energy metabolism, exacerbation of inflammation and angiogenesis in arthritis synovium. | [68] | ||
| Collagen-induced arthritis (CIA) (rat). | RNAi silencing of HIF-1α leads to a reduction in pro-inflammatory cytokine expression. | Inhibition of HIF-1 attenuated the systemic inflammatory condition and clinical changes observed in this model. | [69] | ||
| H1N1 virus infection (mice) | H1N1 activates the HIF-1 pathway by inhibiting proteasome activity and decreasing Factor Inhibiting HIF-1 (FIH) expression. | H1N1 activates the HIF-1 pathway under normoxic conditions mimicking a hypoxic response, supporting a more efficient viral replication. | [61] | ||
| SARS-CoV-2 infection (human monocytes) | SARS-CoV-2 promotes a switch to aerobic glycolysis in human monocyte metabolism by triggering mitochondrial ROS production and inducing HIF-1α stabilization. | HIF-1α stabilization in SARS-CoV-2 infected monocytes sustains the production of IL-1β and promotes T cell dysfunction and lung epithelial cell death. | [102] |
Figure 3. Comparison of the epithelial and immune responses to inflammatory/infectious insults in the presence or absence of hypoxia/HIF-1 signaling.
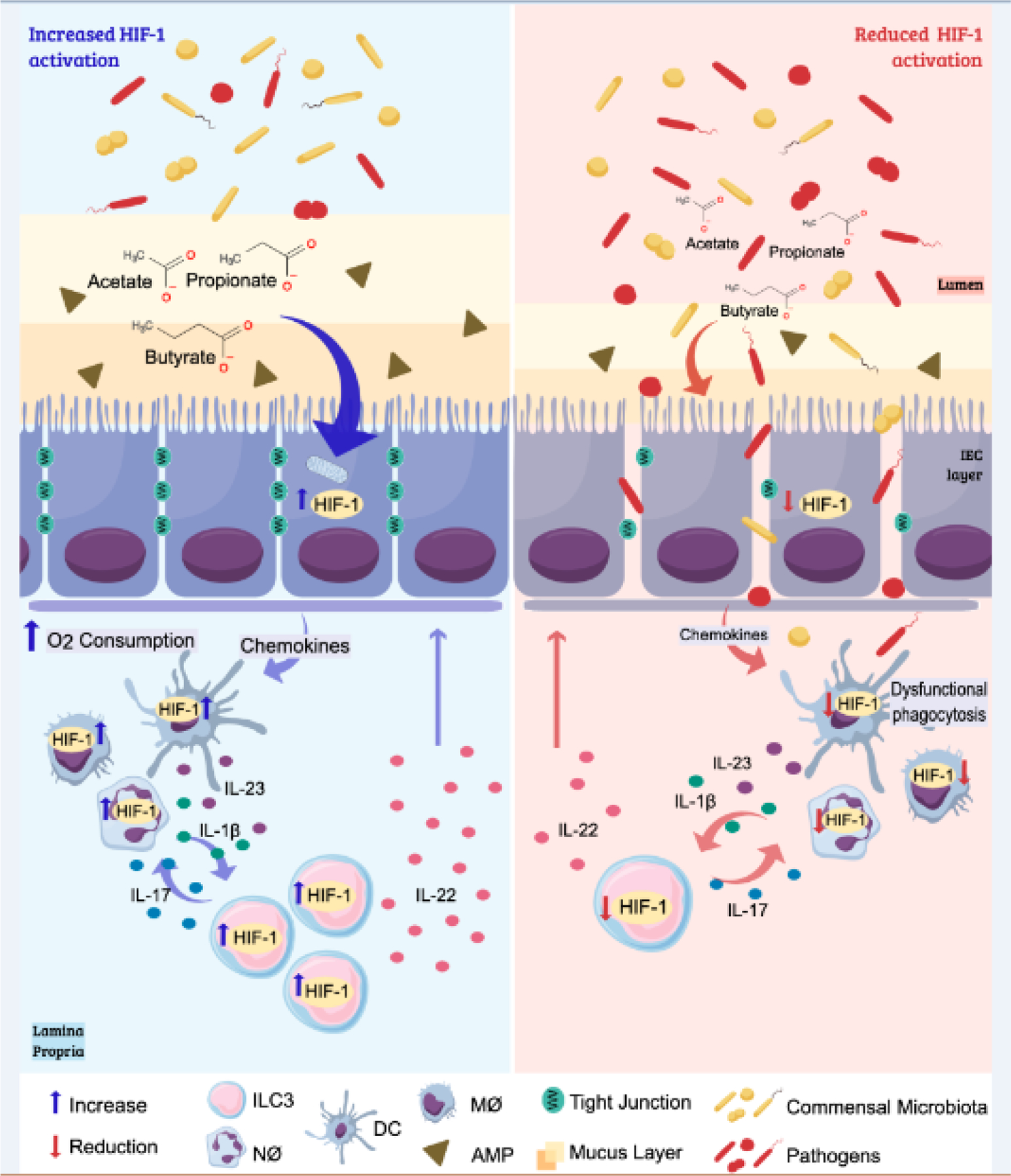
The intestinal microbiota contributes to the hypoxic environment of the intestinal lumen through the production of short-chain fatty acids (SCFAs). These metabolites increase the oxygen consumption by intestinal cells leading to stabilization of HIF-1 and induction of HIF-1 responses in this tissue. Activation of HIF-1 contributes to the barrier function through the production of mucus, antimicrobial peptides, and the expression of tight-junction proteins, in humans and mouse models. Together, these effects reduce bacterial translocation during an infection. The activation of HIF-1 also promotes positive effects on resident and recruited leukocytes that are relevant to an effective inflammatory/immune response in the intestine. Stabilization of HIF-1 and the HIF-1 associated metabolic reprogramming of neutrophils and macrophages contributes to their microbicidal effects and production of pro-inflammatory cytokines. In dendritic cells it is important for their capacity to migrate to the draining lymph nodes. ILC3 responds to IL-1β and IL-23 from resident cells. SCFAs and HIF-1 stabilization improves the production of IL-17 and IL-22 by these cells, thus enhancing the recruitment of neutrophils (which also contribute to oxygen consumption in the site) and strengthening the barrier function by their effects on IECs. In the absence of HIF-1, the production of mucus and AMPs by IECs is decreased, as well as the expression of tight junction proteins, compromising the epithelial barrier permeability. This dysfunctional epithelium is also associated with changes in composition of the microbiota and lower production of SCFAs. HIF-1 downregulation in leukocytes also diminishes their recruitment, production of inflammatory mediators, phagocytosis, and bacterial killing during an infection. Reduction of HIF-1 signaling impacts on ILC3 production of IL-22 and IL-17, contributing to an impaired immune response and epithelial healing in the intestine. See references in text.
During infections, HIF-1α stabilization can lead to the expression of pro-inflammatory genes, potentiating the activation of macrophages [70,71], the activation and survival of neutrophils [72–74], the maturation and migration of dendritic cells [75] and other effects that might contribute to immune defense. Together, these effects may be relevant in acute infections by increasing the efficiency of the immune system (Figure 3) [43,76]. However, we hypothesize that with the prolongation of the inflammatory process, the effects of HIF-1 on immune cells can contribute to tissue damage. Studies have reported the stabilization of HIF-1α as a complication predictor during inflammatory responses in the airways in mice [77–79] and humans [80], while its stabilization in the intestinal tissue is generally related to improved outcomes (Table 1) in mice [44,81,82] and humans [83]. These results may be due to the high partial pressure of oxygen found in the respiratory tract, which can lead to naturally low concentrations of HIF-1α in this tissue. Moreover, HIF-1α has also been implicated in the viral replication and pathogenesis of influenza H1N1 [61,77 and HIV [78] viruses in mice and in vitro [61,77], generally triggering cytokine production [61,77,78]. Together, these findings suggest that the activation of HIF-1 during infectious responses in the airways may lead to detrimental immune responses and tissue damage.
Concluding Remarks
Taken together, the studies discussed in this review provide relevant information about possible outcomes for HIF-1α stabilization under different inflammatory contexts, and is suggestive of the diverse and complex role these outcomes might play in humans. Accordingly, hypoxia and activation of the HIF-1 pathway might be considered putative therapeutic targets to treat certain inflammatory and/or infectious diseases, particularly those affecting the intestine, such as CDI, pre-IBD and IBD. However, such possibilities remain theoretical, and hypoxia/HIF-1 targeting approaches will need to be analyzed with extreme caution due to the pleiotropic and complex effects of this pathway on host physiology. We have yet to learn from these (see outstanding questions) and should be vigilant about the possibility of exerting unwanted detrimental effects when aiming to treat a variety of conditions.
HIGHLIGHTS BOX.
Oxygen consumption by mammalian intestinal epithelial cells (IECs) results in intestinal hypoxia, which is stimulated by metabolites (short-chain fatty acids, SCFAs) derived from the microbiota.
Hypoxia and activation of hypoxia inducible factor (HIF)-responses are relevant for IEC metabolism, regulation of epithelial barrier function, and gut microbiota segregation in humans and mice.
The activation of type-3 innate lymphoid cells (ILC3) and their key cytokine production are boosted in hypoxia through a HIF-1 dependent mechanism in mice -- an effect that may be partly due to SCFAs.
Strategies that contribute to the maintenance or re-establishment of epithelial hypoxia, luminal amounts of SCFAs and HIF-1 responses in humans and mice intestinal cells may be relevant for assessing inflammatory and infectious conditions.
Acknowledgments
This study was supported by National Institutes of Health (1R01DK126969-01) and Fundação de Amparo à Pesquisa do Estado de São Paulo (18/15313-8). The study was also financed by the National Council for Scientific and Technological Development (CNPq) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES)—Finance Code 001. L.P.P., J.L.F. and R.O.C are recipients of fellowships from FAPESP (2018/02208-1, 2017/06577-9 and 2016/23142-3, respectively). M.C. was supported by MIST (U01AI095542).
GLOSSARY
- Anoxic
state in which there is no oxygen availability to the organs and tissues (0% pO2).
- Antimicrobial peptides (AMPs)
small peptides (including defensins and cathelicidins) secreted in the intestine lumen mainly by Paneth cells to prevent bacterial colonization close to the intestinal epithelium.
- Aryl hydrocarbon receptor (AhR)
transcription factor activated by a wide range of endogenous (e.g. kynurenine) and exogenous ligands (e.g. natural plant flavonoids and polyphenols and xenobiotics); regulates the expression of genes associated with several biological processes including metabolism, immunity, cellular proliferation, and differentiation.
- Autophagy
conserved catabolic process in which cytoplasmic constituents and organelles are degraded in the lysosome.
- β-oxidation
catabolic fatty acid process that generates acetyl-CoA for the mitochondrial citric acid cycle and reduces coenzymes (NADH and FADH2), which provide energy for the electron transport chain production of ATP.
- Chromatin immunoprecipitation (ChIP)
immunoprecipitation method that combined with PCR or DNA sequencing is useful for identifying genome binding sites for transcription factors and other proteins.
- Colonocytes
colon absorptive epithelial cells.
- Dextran sulfate sodium (DSS)
administration of DSS, a highly sulfated polysaccharide, causes intestinal inflammation in rodents. DSS-induced colitis is a useful and widely used experimental model of intestinal inflammation that shares some similarities with human ulcerative colitis.
- Dysbiosis
pathological condition in which there are significant alterations in the profile of the normal microbiota (qualitatively and quantitatively).
- Germ-free mice
lack all microorganisms (microbiologically sterile) and are housed in a controlled sterile environment to avoid contamination.
- Glycolytic metabolism
one of the main cellular mechanisms for generating energy (ATP) in cells. This pathway involves several enzymatic reactions that convert glucose into pyruvate in the presence of oxygen, or lactate in the absence of oxygen (anaerobic glycolysis).
- Hyperoxia
oxygen availability is higher than the physiological condition (pO2 higher than 21%).
- Hypoxia Inducible Factor (HIF)
dimeric protein complex formed by an α and a β subunit that acts as a transcription factor for cellular adaptation in response to low oxygen concentration.
- HIF-1 stabilization
under hypoxic conditions, the activity of HIF prolyl-hydroxylases and ubiquitin ligases is inhibited, allowing the accumulation of the α subunit of HIF in the cytoplasm (stabilization). This subunit then translocates to the nucleus and binds to the β subunit, forming the HIF-1 complex that regulates the expression of several genes.
- Hypoxia
characterized by low oxygen tensions (pO2 lower than 10%). The oxygen availability is below the demands of organs and tissues requiring their adaptation. This condition can be considered physiological or pathological depending on context.
- Innate Lymphoid cells (ILCs)
immune cells known as the innate counterparts of adaptive T cells that secrete cytokines to modulate several immune responses in distinct tissues.
- Intestinal epithelial cells (IECs)
distinct cell types that form the intestinal epithelium, a dynamic monolayer that provides a physical/chemical barrier to the external environment within the intestinal lumen. It includes absorptive enterocytes/colonocytes, goblet cells, Paneth cells, enteroendocrine cells, microfold (M) cells, tuft cells, and intestinal stem cells.
- Microbiota
diverse group of commensal microorganisms (Archaea, bacteria, fungi, viruses and parasites) colonizing host tissues, e.g. the gastrointestinal tract.
- NLRP3 inflammasome
multimeric intracellular protein complex, whose activation leads to the maturation and secretion of pro-inflammatory cytokines IL-1β and IL-18.
- Normoxia
O2 level is similar to that found in the atmospheric air at sea level (20–21% pO2). A state of oxygen availability that exceeds the metabolic demands of organs and tissues.
- Peroxisome proliferator-activated receptor-γ (PPARγ)
member of the nuclear receptor family of transcription factors responsible for mediating ligand-dependent transcriptional activation and repression; activated by several endogenous and exogenous ligands including polyunsaturated fatty acids such as arachidonic acid and its metabolites, short-chain fatty acids, and thiazolidinediones.
- Pinch biopsy
model of mucosal wounding in which a 2 mm piece of the mucosal tissue is removed with the guidance of an endoscope.
- Partial pressure of oxygen (pO2)
the individual contribution of oxygen to the total pressure of a gas mixture.
- RAR-related orphan receptor gamma t (RORγt)
transcription factor expressed by immature CD4+CD8+ thymocytes, T helper 17 lymphocytes (Th17), and type 3 innate lymphoid cells (ILC3).
- Short-chain fatty acids (SCFAs)
metabolites produced and released by the microbiota during the process of bacterial fermentation of dietary fibers that play important roles in the host’s physiology.
- Tight junctions
cell-cell transmembrane adhesion complexes that play a role in the organization of epithelial tissue preventing molecules from passing in between the cells.
- Trinitrobenzene sulfonic acid (TNBS)-induced colitis
murine model of colonic inflammation with similar aspects to Crohn’s disease, Th1-mediated bowel inflammation. The disease develops after administration of 2,4,6-trinitrobenzene sulfonic acid (TNBS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- 1.Pizzino G et al. (2017) Oxidative stress: harms and benefits for human health. Oxid. Med. Cell. Longev DOI: 10.1155/2017/8416763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hammarlund EU et al. (2020) Oxygen-sensing mechanisms across eukaryotic kingdoms and their roles in complex multicellularity. Science 370, 6515. [DOI] [PubMed] [Google Scholar]
- 3.Ratcliffe PJ (2013) Oxygen sensing and hypoxia signalling pathways in animals: the implications of physiology for cancer. J. Physiol 591(8), 2027–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haeberle HA, et al. (2008) Oxygen-independent stabilization of hypoxia inducible factor (HIF)-1 during RSV infection. PLoS One, 3(10), e3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dowdell AS et al. (2020) The HIF target ATG9A is essential for epithelial barrier function and tight junction biogenesis. Mol. Biol. Cell 31, 2249–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parra-Izquierdo I et al. (2019) Lipopolysaccharide and interferon-γ team up to activate HIF-1α via STAT1 in normoxia and exhibit sex differences in human aortic valve interstitial cells. Biochim. Biophys. Acta. Mol. Basis Dis 1865(9), 2168–2179. [DOI] [PubMed] [Google Scholar]
- 7.Zhang S et al. (2020) Glial type specific regulation of CNS angiogenesis by HIFα-activated different signaling pathways. Nat. Commun 11(1), 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smythies JA et al. (2019) Inherent DNA-binding specificities of the HIF-1α and HIF-2α transcription factors in chromatin. EMBO Rep, 20(1), e46401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allan KC, et al. (2021) Non-canonical Targets of HIF1a Impair Oligodendrocyte Progenitor Cell Function. Cell stem cell 28, 257–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jain IH, et al. (2020) Genetic screen for cell fitness in high or low oxygen highlights mitochondrial and lipid metabolism. Cell 181, 716–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taylor CT, & Colgan SP (2017). Regulation of immunity and inflammation by hypoxia in immunological niches. Nat. Rev. Immunol 17(12), 774–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He G, et al. (1999) Noninvasive measurement of anatomic structure and intraluminal oxygenation in the gastrointestinal tract of living mice with spatial and spectral EPR imaging. Proc. Natl. Acad. Sci. U. S. A 96, 4586–4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Albenberg L, et al. (2014) Correlation between intraluminal oxygen gradient and radial partitioning of intestinal microbiota. Gastroenterology 147, 1055–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moreno-Indias I et al. (2015) Intermittent hypoxia alters gut microbiota diversity in a mouse model of sleep apnoea. Eur. Respir. J 45, 1055–1065 [DOI] [PubMed] [Google Scholar]
- 15.Ashley SL et al. (2020) Lung and gut microbiota are altered by hyperoxia and contribute to oxygen-induced lung injury in mice. Sci. Transl. Med 12. DOI: 10.1126/scitranslmed.aau9959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peterson LW and Artis D (2014) Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat. Rev. Immunol 14, 141–153 [DOI] [PubMed] [Google Scholar]
- 17.Kelly CJ et al. (2015) Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe 17, 662–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Byndloss MX et al. (2017) Microbiota-activated PPAR-γ signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 357, 570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Corrêa-Oliveira R et al. (2016) Regulation of immune cell function by short-chain fatty acids. Clin. Trans. Immunol 5, e73. DOI: 10.1038/cti.2016.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee JY et al. (2020) High-fat diet and antibiotics cooperatively impair mitochondrial bioenergetics to trigger dysbiosis that exacerbates pre-inflammatory bowel disease. Cell Host Microbe 28, 273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cevallos SA et al. (2021) 5-Aminosalicylic Acid Ameliorates Colitis and Checks Dysbiotic Escherichia coli Expansion by Activating PPAR-γ Signaling in the Intestinal Epithelium. Mbio 12(1). DOI: 10.1128/mBio.03227-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reese AT et al. (2018) Antibiotic-induced changes in the microbiota disrupt redox dynamics in the gut. Elife 7, e35987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rivera-Chávez F et al. (2016) Depletion of butyrate-producing Clostridia from the gut microbiota drives an aerobic luminal expansion of Salmonella. Cell Host Microbe 19, 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fachi JL et al. (2019) Butyrate protects mice from Clostridium difficile-induced colitis through an HIF-1-dependent mechanism. Cell Rep. 27, 750–761. [DOI] [PubMed] [Google Scholar]
- 25.Chen T et al. (2017). Dietary fibre-based SCFA mixtures promote both protection and repair of intestinal epithelial barrier function in a Caco-2 cell model. Food funct. 8, 1166–1173. [DOI] [PubMed] [Google Scholar]
- 26.Geirnaert A et al. (2017) Butyrate-producing bacteria supplemented in vitro to Crohn’s disease patient microbiota increased butyrate production and enhanced intestinal epithelial barrier integrity. Sci. Rep 7, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng L et al. (2017) Microbial-derived butyrate promotes epithelial barrier function through IL-10 receptor–dependent repression of claudin-2. J. Immunol 199, 2976–2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu ED et al. (2018) High fiber dietary and sodium butyrate attenuate experimental autoimmune hepatitis through regulation of immune regulatory cells and intestinal barrier. Cell. Immunol 328, 24–32. [DOI] [PubMed] [Google Scholar]
- 29.Wang RX et al. (2020) Microbiota-derived butyrate dynamically regulates intestinal homeostasis through regulation of actin-associated protein synaptopodin. Proc. Nati. Acad. Sci. U. S. A 117, 11648–11657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shao T et al. (2018) Intestinal HIF-1α deletion exacerbates alcoholic liver disease by inducing intestinal dysbiosis and barrier dysfunction. J. Hepatol 69, 886–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muenchau S et al. (2019) Hypoxic environment promotes barrier formation in human intestinal epithelial cells through regulation of microRNA 320a expression. Mol. Cell. Biol 39, e00553–18. DOI: 10.1128/MCB.00553-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goggins BJ et al. (2021) Pharmacological HIF-1 stabilization promotes intestinal epithelial healing through regulation of α-integrin expression and function. Am. J. Physiol. Gastrointest. Liver Physiol DOI: 10.1152/ajpgi.00192.2020. [DOI] [PubMed] [Google Scholar]
- 33.Fan D et al. (2015) Activation of HIF-1α and LL-37 by commensal bacteria inhibits Candida albicans colonization. Nat. Med 21, 808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miraglia E et al. (2016) Entinostat up-regulates the CAMP gene encoding LL-37 via activation of STAT3 and HIF-1α transcription factors. Sci. Rep 6, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dilly AK et al. (2016) Targeting hypoxia-mediated mucin 2 production as a therapeutic strategy for mucinous tumors. Transl. Res 169, 19–30. [DOI] [PubMed] [Google Scholar]
- 36.Haq S et al. (2019) Autophagy: roles in intestinal mucosal homeostasis and inflammation. J. Biomed. Sci 26, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cosin-Roger J et al. (2017) Hypoxia ameliorates intestinal inflammation through NLRP3/mTOR downregulation and autophagy activation. Nat. Commun 8, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou C et al. (2020) SCFAs induce autophagy in intestinal epithelial cells and relieve colitis by stabilizing HIF-1α. J. Mol. Med 98, 1189–1202. [DOI] [PubMed] [Google Scholar]
- 39.Talbot J et al. (2020) Feeding-dependent VIP neuron–ILC3 circuit regulates the intestinal barrier. Nature 579(7800), 575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mao K et al. (2018) Innate and adaptive lymphocytes sequentially shape the gut microbiota and lipid metabolism. Nature 554(7691), 255–259. [DOI] [PubMed] [Google Scholar]
- 41.Goto Y et al. (2014) Innate lymphoid cells regulate intestinal epithelial cell glycosylation. Science 345(6202). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang W et al. (2020) Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nat. Commun 11(1), 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Colgan SP, Furuta GT, & Taylor CT (2020) Hypoxia and innate immunity: keeping up with the HIFsters. Annu. Rev. Immunol 38, 341–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fachi JL et al. (2021) Hypoxia enhances ILC3 responses through HIF-1α-dependent mechanism. Mucosal Immunol 1–14. DOI: 10.1038/s41385-020-00371-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vivier E et al. (2018) Innate lymphoid cells: 10 years on. Cell, 174, 1054–1066. [DOI] [PubMed] [Google Scholar]
- 46.Longman RS et al. (2014) CX3CR1+ mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J. Exp. Med 211, 1571–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rankin LC et al. (2016) Complementarity and redundancy of IL-22-producing innate lymphoid cells. Nat. Immunol 17(2), 179–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guo X et al. (2014) Induction of innate lymphoid cell-derived interleukin-22 by the transcription factor STAT3 mediates protection against intestinal infection. Immunity 40(1), 25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dudakov JA et al. (2012) Interleukin-22 drives endogenous thymic regeneration in mice. Science 336, 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zheng Y et al. (2008) Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med 14(3), 282–289. [DOI] [PubMed] [Google Scholar]
- 51.Wolk K et al. (2006) IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur. J. Immunol 36, 1309–1323. [DOI] [PubMed] [Google Scholar]
- 52.Aujla SJ et al. (2008) IL-22 mediates mucosal host defense against Gram- negative bacterial pneumonia. Nat. Med 14, 275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sugimoto K et al. (2008) IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Invest 118, 534–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brand S et al. (2006) IL-22 is increased in active Crohn’s disease and promotes proinflammatory gene expression and intestinal epithelial cell migration. Am. J. Physiol. Gastrointest Liver Physiol 290, G827–G838. [DOI] [PubMed] [Google Scholar]
- 55.Lo BC et al. (2019) IL-22 preserves gut epithelial integrity and promotes disease remission during chronic Salmonella infection. J. Immunol 202(3), 956–965. [DOI] [PubMed] [Google Scholar]
- 56.Chun E et al. (2019) Metabolite-sensing receptor Ffar2 regulates colonic group 3 innate lymphoid cells and gut immunity. Immunity 51, 871–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fachi JL et al. (2020) Acetate coordinates neutrophil and ILC3 responses against C. difficile through FFAR2. J. Exp. Med 217, e20190489. DOI: 10.1084/jem.20190489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sepahi A et al. (2021) Dietary fiber metabolites regulate innate lymphoid cell responses. Mucosal Immunol. 14, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Di Luccia B et al. (2019) ILC3s integrate glycolysis and mitochondrial production of reactive oxygen species to fulfill activation demands. J. Exp. Med 216, 2231–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dang EV et al. (2011). Control of TH17/Treg balance by hypoxia-inducible factor 1. Cell, 146, 772–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ren L et al. (2019) Influenza A virus (H1N1) triggers a hypoxic response by stabilizing hypoxia-inducible factor-1α via inhibition of proteasome. Virology 530, 51–58. [DOI] [PubMed] [Google Scholar]
- 62.Zhu Q et al. (2018) Loss of ATF3 exacerbates liver damage through the activation of mTOR/p70S6K/HIF-1α signaling pathway in liver inflammatory injury. Cell Death Dis. 9, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim YE et al. (2018) HIF-1α activation in myeloid cells accelerates dextran sodium sulfate-induced colitis progression in mice. Dis. Model. Mech 11, dmm033241. DOI: 10.1242/dmm.033241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guan R et al. (2020) Hydrogen sulfide inhibits cigarette smoke-induced inflammation and injury in alveolar epithelial cells by suppressing PHD2/HIF-1α/MAPK signaling pathway. Int. Immunopharmacol 81, 105979. [DOI] [PubMed] [Google Scholar]
- 65.Holden VI et al. (2016) Klebsiella pneumoniae siderophores induce inflammation, bacterial dissemination, and HIF-1α stabilization during pneumonia. MBio 7. DOI: 10.1128/mBio.01397-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li ZL et al. (2019) HIF-1α inducing exosomal microRNA-23a expression mediates the cross-talk between tubular epithelial cells and macrophages in tubulointerstitial inflammation. Kidney Int. 95, 388–404. [DOI] [PubMed] [Google Scholar]
- 67.Hammami A et al. (2018) HIF-1α hampers dendritic cell function and Th1 generation during chronic visceral leishmaniasis. Sci. Rep 8, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li Y et al. (2018) Succinate induces synovial angiogenesis in rheumatoid arthritis through metabolic remodeling and HIF-1α/VEGF axis. Free Radic. Biol. Med 126, 1–14. [DOI] [PubMed] [Google Scholar]
- 69.Hu Y et al. (2020) Downregulation of Hypoxia-Inducible Factor-1a by RNA Interference Alleviates the Development of Collagen-Induced Arthritis in Rats. Mol. Ther 19 DOI: 10.1016/j.omtn.2020.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Braverman J, & Stanley SA (2017) Nitric oxide modulates macrophage responses to Mycobacterium tuberculosis infection through activation of HIF-1α and repression of NF-κB. J. Immunol 199, 1805–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang T et al. (2017) HIF1α-induced glycolysis metabolism is essential to the activation of inflammatory macrophages. Mediators Inflamm. DOI: 10.1155/2017/9029327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thompson AR (2017) Hypoxia determines survival outcomes of bacterial infection through HIF-1alpha dependent re-programming of leukocyte metabolism. Sci Immunol. 2, eaal2861. DOI: 10.1126/sciimmunol.aal2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Walmsley SR et al. (2005) Hypoxia-induced neutrophil survival is mediated by HIF-1α–dependent NF-κB activity. J. Exp. Med 201, 105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McInturff AM et al. (2012) Mammalian target of rapamycin regulates neutrophil extracellular trap formation via induction of hypoxia-inducible factor 1 α. Blood 120, 3118–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu J et al. (2019) CCR7 chemokine receptor-inducible lnc-Dpf3 restrains dendritic cell migration by inhibiting HIF-1α-mediated glycolysis. Immunity 50, 600–615. [DOI] [PubMed] [Google Scholar]
- 76.Cramer T, et al. (2003) HIF-1α is essential for myeloid cell-mediated inflammation. Cell 112(5), 645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guo X et al. (2017) Nuclear translocation of HIF-1α induced by influenza A (H1N1) infection is critical to the production of proinflammatory cytokines: HIF-1α nuclear translocation induced by H1N1. Emerg. Microbes Infect 6, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Duette G et al. (2018) Induction of HIF-1α by HIV-1 infection in CD4+ T cells promotes viral replication and drives extracellular vesicle-mediated inflammation. MBio9. DOI: 10.1128/mBio.00757-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sokulsky LA, et al. (2020) GSTO1–1 is an upstream suppressor of M2 macrophage skewing and HIF-1α-induced eosinophilic airway inflammation. Clin. Exp. Allergy 50(5), 609–624. [DOI] [PubMed] [Google Scholar]
- 80.Talreja J, et al. (2019) HIF-1α regulates IL-1β and IL-17 in sarcoidosis. Elife 8, e44519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cummins EP, et al. (2008) The Hydroxylase Inhibitor Dimethyloxalylglycine Is Protective in a Murine Model of Colitis. Gastroenterology 134, 156–165. [DOI] [PubMed] [Google Scholar]
- 82.Robinson A, et al. (2008) Mucosal Protection by Hypoxia-Inducible Factor (HIF) Prolyl Hydroxylase Inhibition. Gastroenterology 134(1), 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brown E, et al. (2020) Mucosal inflammation downregulates PHD1 expression promoting a barrier-protective HIF-1α response in ulcerative colitis patients. FASEB J. 34(3), 3732–3742. [DOI] [PubMed] [Google Scholar]
- 84.Haber AL et al. (2017) A single-cell survey of the small intestinal epithelium. Nature 551(7680), 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Barker N et al. (2007) Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449(7165), 1003–1007. [DOI] [PubMed] [Google Scholar]
- 86.Sato T et al. (2009) Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459(7244), 262–265. [DOI] [PubMed] [Google Scholar]
- 87.Darwich AS et al. (2014) Meta-analysis of the turnover of intestinal epithelia in preclinical animal species and humans. Drug Metab. Dispos 42(12), 2016–2022. [DOI] [PubMed] [Google Scholar]
- 88.Basak O et al. (2017) Induced quiescence of Lgr5+ stem cells in intestinal organoids enables differentiation of hormone-producing enteroendocrine cells. Cell stem cell 20(2), 177–190. [DOI] [PubMed] [Google Scholar]
- 89.Yilmaz ÖH et al. (2012) mTORC1 in the Paneth cell niche couples intestinal stem-cell function to calorie intake. Nature 486(7404), 490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nakamura Y et al. (2020) Microfold cell-dependent antigen transport alleviates infectious colitis by inducing antigen-specific cellular immunity. Mucosal Immunol. 13(4), 679–690. [DOI] [PubMed] [Google Scholar]
- 91.McGinty JW et al. (2020) Tuft-cell-derived leukotrienes drive rapid anti-helminth immunity in the small intestine but are dispensable for anti-protist immunity. Immunity, 52(3), 528–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gury-BenAri M et al. (2016) The Spectrum and regulatory landscape of intestinal innate lymphoid cells are shaped by the microbiome. Cell. 166, 1231–1246. [DOI] [PubMed] [Google Scholar]
- 93.Colonna M (2018) Innate lymphoid cells: diversity, plasticity, and unique functions in immunity. Immunity, 48, 1104–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Robinette ML et al. (2015) Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat. Immunol 16, 306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lügering A et al. (2010) CCR6 identifies lymphoid tissue inducer cells within cryptopatches. Clin Exp Immunol. 160(3): 440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bando JK et al. (2018). The tumor necrosis factor superfamily member RANKL suppresses effector cytokine production in group 3 innate lymphoid cells. Immunity 48(6), 1208–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Klose CSN et al. (2013) A T-bet gradient controls the fate and function of CCR6−RORγt+ innate lymphoid cells. Nature 494, 261–265. [DOI] [PubMed] [Google Scholar]
- 98.Sun L et al. (2019) Intestinal Epithelial Cells-Derived Hypoxia-Inducible Factor-1α Is Essential for the Homeostasis of Intestinal Intraepithelial Lymphocytes. Front. Immunol 10, 806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Qian T et al. (2019). Regulation of CD11b by HIF-1α and the STAT3 signaling pathway contributes to the immunosuppressive function of B cells in inflammatory bowel disease. Mol. Immunol 111, 162–171. [DOI] [PubMed] [Google Scholar]
- 100.Mesquita I et al. (2020) The absence of HIF-1α increases susceptibility to Leishmania donovani infection via activation of BNIP3/mTOR/SREBP-1c axis. Cell Rep. 30, 4052–4064. [DOI] [PubMed] [Google Scholar]
- 101.Wing Peter A. C., et al. (2021) Hypoxic and pharmacological activation of HIFs inhibits SARS-CoV-2 infection of lung epithelial cells. Cell Rep. 35, 109020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Codo AC et al. (2020) Elevated glucose levels favor SARS-CoV-2 infection and monocyte response through a HIF-1α/glycolysis-dependent axis. Cell Metab. 32, 437–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li L et al. (2020) The effects of daily fasting hours on shaping gut microbiota in mice. BMC Microbiol. 20, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Maifeld A et al. (2021) Fasting alters the gut microbiome reducing blood pressure and body weight in metabolic syndrome patients. Nat. Commun 12(1), 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]