

Abstract
Fc glycosylation profoundly impacts the effector functions of antibodies and often dictates an antibody’s pro- or anti-inflammatory activities. It is well established that core fucosylation of the Fc domain N-glycans of an antibody significantly reduces its affinity for FcγRIIIa receptors and antibody-dependent cellular cytotoxicity (ADCC). Previous structural studies have suggested that the presence of a core fucose remarkably decreases the unique and favorable carbohydrate–carbohydrate interactions between the Fc and the receptor N-glycans, leading to reduced affinity. We report here that in contrast to natural core fucose, special site-specific modification on the core fucose could dramatically enhance the affinity of an antibody for FcγRIIIa. The site-selective modification was achieved through an enzymatic transfucosylation with a novel fucosidase mutant, which was shown to be able to use modified α-fucosyl fluoride as the donor substrate. We found that replacement of the core l-fucose with 6-azide- or 6-hydroxy-l-fucose (l-galactose) significantly enhanced the antibody’s affinity for FcγRIIIa receptors and substantially increased the ADCC activity. To understand the mechanism of the modified fucose-mediated affinity enhancement, we performed molecular dynamics simulations. Our data revealed that the number of glycan contacts between the Fc and the Fc receptor was increased by the selective core-fucose modifications, showing the importance of unique carbohydrate–carbohydrate interactions in achieving high FcγRIIIa affinity and ADCC activity of antibodies. Thus, the direct site-selective modification turns the adverse effect of the core fucose into a favorable force to promote the carbohydrate–carbohydrate interactions.
Graphical Abstract
INTRODUCTION
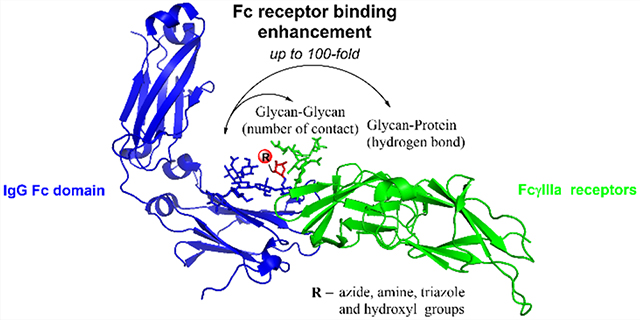
Glycosylation is one of the most important post-translational modifications to produce fully functional proteins in nature.1,2 Among different types of modifications on glycans, the attachment of a 1,6-linked fucose to the innermost N-acetylglucosamine (GlcNAc) moiety of N-glycans, named core fucosylation, plays a critical role in modulating the conformations and biological functions of N-glycans and N-glycoproteins, including therapeutic monoclonal antibodies.3–5 Compelling data have demonstrated that the presence of the core fucose on the Fc glycan significantly decreases the affinity of antibodies for FcγIIIa receptor (FcγRIIIa or CD16a), leading to decreased ADCC activity and decreased anticancer efficacy in vitro and in vivo.6–11 Human FcγRIIIa itself is a glycoprotein carrying five N-glycans at the Asn38, Asn45, Asn74, Asn162, and Asn169 glycosylation sites. Two previous crystallographic studies on the complexes of recombinant human FcγRIIIa with afucosylated IgG-Fc or core-fucosylated IgG-Fc have indicated that in the structure of the FcγRIIIa complexed with afucosylated IgG-Fc there are extensive intermolecular carbohydrate–carbohydrate interactions between the Asn162 glycan of FcγRIIIa and the afucosylated Fc glycan, but these favorable interactions are significantly decreased or not existing in the structures of glycosylated FcγRIIIa complexed with the core-fucosylated IgG-Fc.11,12 Further analysis suggests that the core-fucose residue of the Fc glycan is oriented toward the second core GlcNAc moiety in the Asn162-glycan of FcγRIIIa, and as a result, the Asn162 glycan must move away, significantly reducing its interaction with the Fc glycan. Recent solution NMR and crystallographic analyses suggest that antibody fucosylation lowers the FcγRIIIa affinity mainly through limiting the conformations sampled by the Asn162 glycan instead of disrupting the glycan–glycan interactions.13 However, it is not clear whether and how modification of the core-fucose structure could positively impact the affinity of antibodies to Fcγ receptors. We describe in the present work a chemoenzymatic method that allows introduction of selectively modified fucose analogs at the core position in intact antibodies using Herceptin as a model antibody (Figure 1). Using a novel fucoligase mutant that we have previously discovered,9 we found that the core fucose could be readily replaced with l-fucose analogs, including d-arabinose (demethyl-l-fucose), l-galactose (6-hydroxyl-l-fucose), and 6-azido-l-fucose, which could be further derivatized using a click reaction. Binding studies revealed that replacing with a d-arabinose, i.e., removal of the methyl group in the l-fucose, did not change the affinity for FcγRIIIa, suggesting the methyl group in the core fucose did not impact the antibody–receptor interactions. However, introduction of an azide group at the 6 position of the l-fucose resulted in 18-fold and over 100-fold enhancement in the affinity of the antibody to the high-affinity and low-affinity FcγIIIa receptor alleles, FcγRIIIa-V158 and FcγRIIIa-F158, respectively. Several other modifications at the 6 position of l-fucose also led to enhancement of the antibody’s affinity for FcγIIIa receptors but to a lesser extent. Cell-based assays confirmed that the 6-azide derivative had remarkably enhanced ADCC activity compared with the parent Herceptin. Molecular dynamics (MD) simulations showed that in contrast to the detrimental effect of the parent core fucose, the site-specific modification at the 6 position with an azide or hydroxyl moiety changed the orientation of the core-fucose moiety and increased the numbers of favorable carbohydrate–carbohydrate interactions between the Fc and the receptor glycans, thereby leading to the enhanced affinity for the FcγRIIIa receptors.
Figure 1.
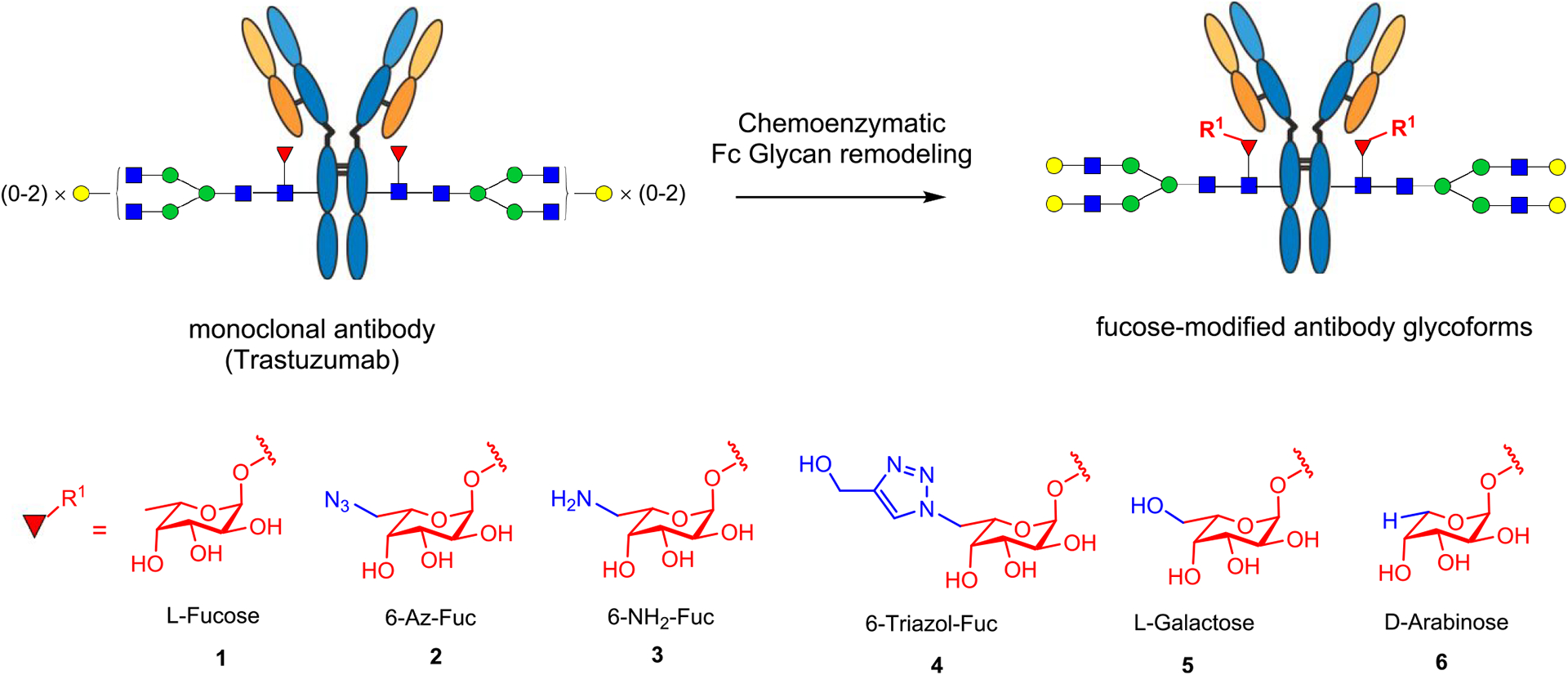
Chemoenzymatic approach to site-specific modification at the core fucose of a monoclonal antibody.
RESULTS AND DISCUSSION
Chemical Synthesis of Selectively Modified α-l-Fucosyl Fluoride Derivatives.
We previously reported that mutants such as E274A derived from the Lactobacillus casei α-fucosidase are able to use α-l-fucosyl fluoride as a simple substrate for core fucosylation.14 To examine if the mutant enzyme can take modified α-fucosyl fluoride derivatives as donor substrate for fucosylation, we synthesized several α-fucosyl fluoride derivatives in which the 6-methyl group was modified or removed. These included the α-glycosyl fluorides of 6-azido-l-fucose, l-galactose (i.e., 6-hydroxyl-l-fucose), and d-arabinose (i.e., the l-fucose derivative in which the methyl group was removed). Thus, treatment of the per-O-acetylated sugar with HF in pyridine, followed by de-O-acetylation gave the corresponding α-glycosyl fluorides (Scheme 1). The synthetic and selectively modified α-fucosyl fluoride derivatives (8, 12, and 13) were confirmed by 1H and 13C NMR analysis (Supporting Information).
Scheme 1.
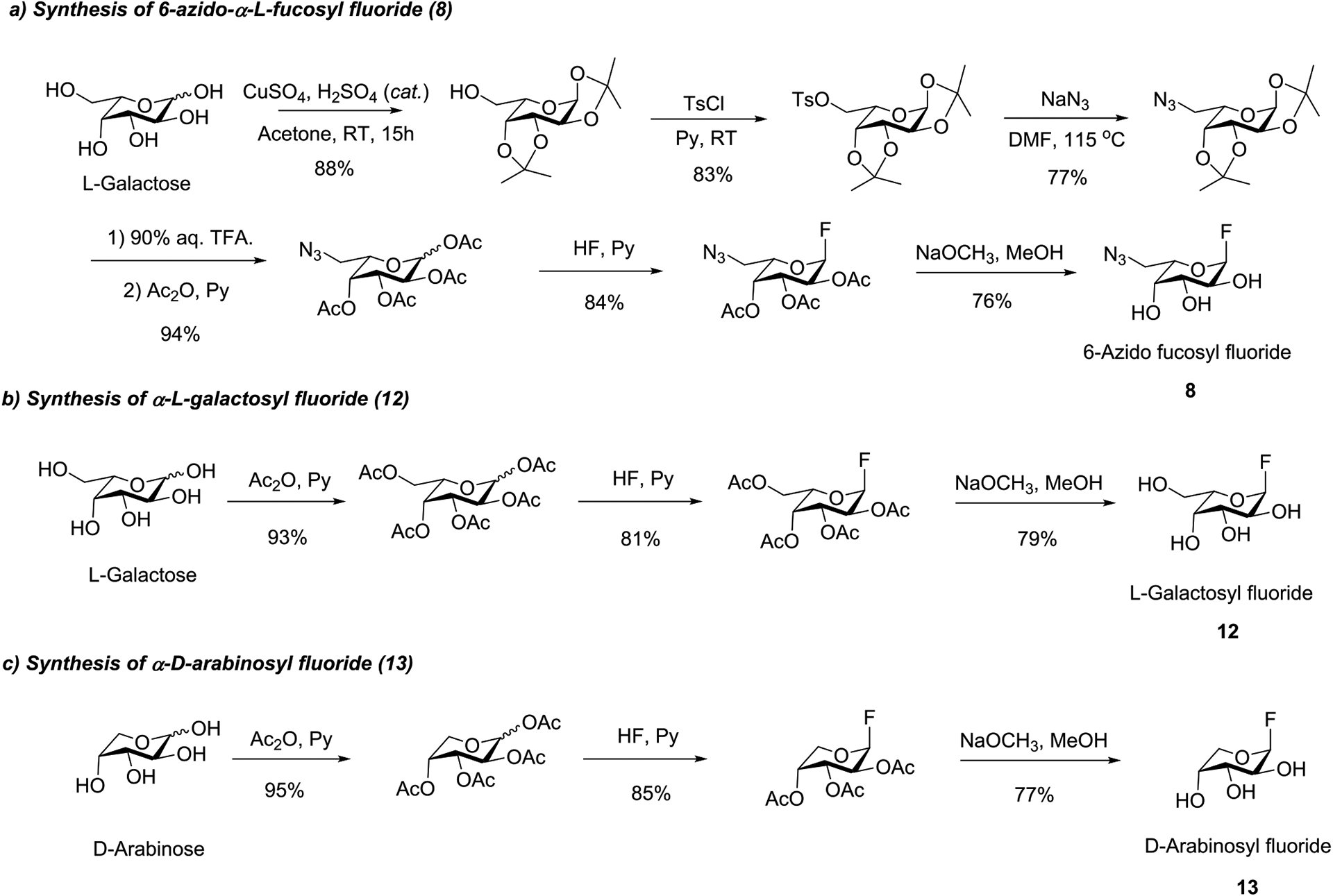
Synthesis of the Selectively Modified α-l-Fucosyl Fluoride Derivatives
Chemoenzymatic Synthesis of Homogeneous Glycoforms of Herceptin with Site-Specific Modifications on the Core Fucose.
Using Herceptin (Trastuzumab), a therapeutic monoclonal antibody used for the treatment of breast cancer as a model antibody, we developed a two-step approach for the site-specific modification on the core fucose. Herceptin was deglycosylated and defucosylated with endoglycosidase S2 (EndoS2) and the Lactobacillus casei α1,6-fucosidase (AlfC),15 respectively, to afford the GlcNAc–Herceptin (7) (Scheme 2). We previously reported a glycoligase mutant derived from AlfC, AlfC E274A, that can use α-l-fucosyl fluoride as the donor substrate to transfer a fucose moiety to a deglycosylated antibody (GlcNAc–Ab) to restore the core fucose.14,16 However, it was not clear if the fucosidase mutant could use modified α-fucosyl fluorides as substrates for transfucosylation. To test this, we incubated the deglycosylated antibody (7) and the 6-azido-l-fucosyl fluoride (8) with the AlfC E274A mutant at 30 °C and monitored the reaction by LC-ESI-MS analysis of the Fc domains released from IdeS protease digestion of the antibody. We found that AlfC E274A was able to transfer the 6-azido-fucose from the glycosyl fluoride (8) to give the 6-AzFucα1,6GlcNAc–Herceptin (9), which was isolated as a single product in an 86% yield after protein A affinity column chromatography. Subsequent transfer of a complex type N-glycan using the complex type N-glycan oxazoline as the donor substrate was achieved using Endo-S2 D184 M as the glycosynthase,17 affording the fully glycosylated AzFuc–Herceptin (2) in an excellent yield. As an alternative route, the afucosylated complex-type glycoform (10) was synthesized by EndoS2-D184 M-catalyzed glycosylation of the GlcNAc–Herceptin intermediate (7) with glycan oxazoline 10. Then direct fucosylation with 6-azido-α-fucosyl fluoride 8 was attempted with AlfC D274A mutant following our previously reported procedure.14 However, the direct enzymatic fucose transfer onto the intact Fc glycan was found to be very slow, and only a low yield of the desired azido-glycoform (2) was obtained after using a relatively large amount of enzyme and an excess amount of the donor substrate (Scheme 2a). The identity of the azide-tagged antibody (2) was confirmed by LC-ESI-MS analysis. The deconvolution data of the ESI-MS (found, M = 148 785 Da) were consistent with the expected molecular mass of the product (calculated for 2, M = 148 778 Da) (Figure S1). Reduction of the azide group in 2 was achieved by treatment with tris(2-carboxyethyl)phosphine (TCEP) to give the core 6-amino-fucosylated Herceptin (3). The 6-triazole-fucosylated Herceptin (4) was prepared by a copper(I)-catalyzed azide–alkyne cycloaddition between 2 and propargyl alcohol (Scheme 2a). We also tested two additional α-fucosyl fluoride derivatives, the α-l-galactosyl fluoride (12), where a hydroxyl group was introduced at the 6 position of l-fucose, and the α-d-arabinosyl fluoride (13), in which the methyl group in l-fucose was removed with the AlfC-E274A mutant. Again, we found that the E274A mutant could efficiently use the two α-glycosyl fluorides as donor substrates for transfucosylation to provide the core-fucose-modified antibody intermediates (14 and 15), respectively. Extension of the sugar chain using the EndoS2-D184 M mutant gave the full-length glycoforms (5 and 6) in which the core fucose was specifically replaced with an l-galactose and d-arabinose, respectively (Scheme 2b). These results indicated that the AlfC E274A mutant had a relaxed donor substrate specificity toward various α-fucosyl fluoride analogs. For the subsequent comparative analysis, core-fucosylated (G2F) and nonfucosylated (G2) Herceptin glycoforms carrying the respective biantennary complex-type N-glycan were prepared following our previously described procedures.9,17
Scheme 2.
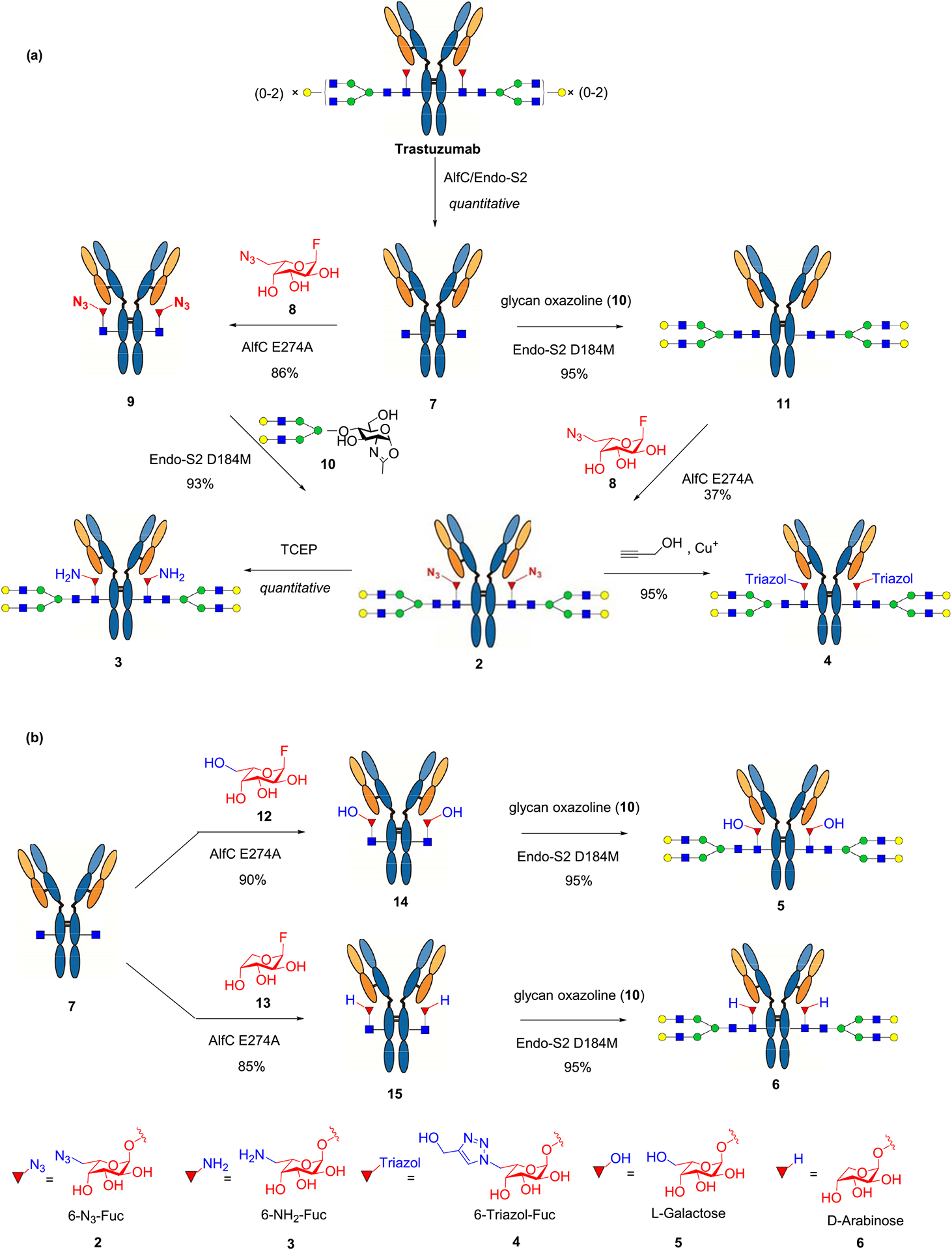
Chemoenzymatic Synthesis of Structurally Well-Defined Core-Fucose-Modified Glycoforms of Herceptin
All of the synthetic antibody glycoforms (1–6) were characterized by mass spectrometric analysis. The antibodies were treated with dithiothreitol (DTT) to disconnect the heavy chain and light chain to give monomeric heavy chains. MALDI-TOF MS analysis of the monomeric heavy chain indicated that the glycoengineered antibody heavy chains were single species with a molecular mass consistent with the calculated data (Figure S2). In addition, the Fc N-glycans in the glycoforms (1–6) were released by PNGase F treatment, and their identities were verified by MALDI-TOF MS analysis (Figure S3).
Binding Affinity of the Core-Fucose-Modified Glycoforms of Herceptin for FcγIIIa Receptors.
Fcγ receptor polymorphism is an important factor for the efficacy of antibody-based treatment.18 Two polymorphic FcγRIIIa variants are present in humans in which the amino acid residue at the 158 position is either a valine (V158) or a phenylalanine (F158) due to point mutation. The two FcγRIIIa alleles show significantly different affinity for antibodies with the V158 allele being a high-affinity FcγRIIIa with the F158 allele being a low-affinity receptor. Natural killer (NK) cells expressing the FcγRIIIa-V158 exhibit more potent NK cell-mediated killing than the F158 variant.19,20 Clinical studies of Rituximab (an anti-CD20 mAb) and trastuzumab (Herceptin) for the treatment of non-Hodgkin’s lymphoma and breast cancer, respectively, have shown that patients with the high-affinity FcγRIIIa-V158 polymorphism responded to the treatment with the respective rituximab and trastuzumab much better than those with the lower affinity FcγRIIIa-F158 polymorphism.21,22 Thus, identifying high-affinity antibody glycoforms particularly for the low-affinity FcgRIIIa-F158 allele is clinically relevant.
The affinity of the core-modified Herceptin glycoforms for two FcγRIIIa alleles, the high-affinity receptor FcγRIIIa-V158 and the low-affinity receptor FcγRIIIa-F158, was assessed by ELISA assays. We found that different core-fucose modifications showed significantly different affinities for the FcγIIIa receptors (Table 1; Figure S4). While all of the glycoforms carrying the natural core fucose demonstrated relatively low affinity for the FcγIIIa receptors, the 6-azido fucose glycoform (2) demonstrated 18-fold enhancement in affinity for the high-affinity receptor FcγRIIIa-V158 and over 100-fold enhancement in affinity for the low-affinity receptor FcγRIIIa-F158. This remarkable affinity enhancement was unexpected as the difference was only an attachment of a small azide group at the 6 position of the parent l-fucose. Modification of the 6 position of fucose with an amino, a hydroxyl, and a triazole group also increased the affinity for the FcγIIIa receptors but to a lesser extent. Thus, the above 3 modifications resulted in 3–7-fold increased affinity for the high-affinity receptor (FcγRIIIa-V158) and about 15-fold-enhanced affinity for the low-affinity receptor (FcγRIIIa-F158) in comparison with the parent core-fucosylated glycoform (G2F). Interestingly, the arabinose glycoform demonstrated essentially the same affinity as the parent G2F glycoform for both Fcγ receptors (Table 1). These results suggested that the methyl group itself in the l-fucose did not impact the antibody’s affinity for the Fc receptors, as its removal (as shown by its replacement with the d-arabinose) did not apparently change the affinity. However, an appropriate modification at the 6 position, such as introduction of an azide group, could rescue the adverse effect of core fucose and significantly enhance the antibody’s affinity for the FcγRIIIa receptors.
Table 1.
Binding Affinity of Core-Fucose-Modified Fc Glycans for the High-Affinity FcγIIIa V158 and Low-Affinity F158 Alleles by ELISAa
| IgG glycoform | Glycan structure | V158 | F158 | ||
|---|---|---|---|---|---|
| EC50 (M) | Fold change | EC50(M) | Fold change | ||
| Herceptin | 5.32 × 10−8 | 1.0 | 1.31 × 10−6 | 1.0 | |
| G2F (1) | 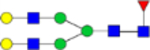 |
4.26 × 10−8 | 1.2 | 1.57 × 10−6 | 0.82 |
| G2(Az)F (2) | 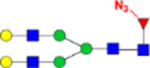 |
2.86 × 10−9 | 18.6 | 1.28 × 10−8 | 102 |
| G2(NH2)F (3) | 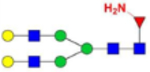 |
9.25 × 10−9 | 5.8 | 8.93 × 10−8 | 14.7 |
| G2(OH- triazol)F (4) | 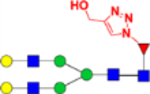 |
1.82 × 10−8 | 2.9 | 8.14 × 10−8 | 16.2 |
| G2Gal (5) | 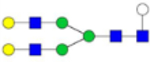 |
7.19 × 10−9 | 7.4 | 8.46 × 10−8 | 15.6 |
| G2Ara (6) | 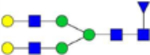 |
3.97 × 10−8 | 1.3 | 1.43 × 10−6 | 0.9 |
| G2 (11) | 1.21 × 10−9 | 44.0 | 1.15 × 10−8 | 114 | |
FcγRIIIa receptors were coated on 96-well plates. For the V158 allele, binding was tested with Herceptin glycoforms in concentrations ranging from 250 to 0.016 nM (5-fold serial dilutions). For the F158 allele, binding was tested with Herceptin glycoforms in concentrations ranging from 2000 to 0.128 nM (5-fold serial dilutions).
ADCC Activity of the Core-Fucose-Modified Antibody Glycoforms.
The antibody-dependent cellular cytotoxicity (ADCC) of different Herceptin glycoforms was assessed by a robust ADCC assay against the human breast carcinoma cell line SKBR3 (HER2+) using an engineered stable Jurkat cell line expressing the F allele of FcγRIIIa as the effector cells. This activation assay appears to be more sensitive and quantitative than the conventional in vitro cell killing assays.23 The 6-azido-fucose glycoform (2) exhibited dramatically enhanced ADCC activity as compared with the commercial Herceptin and the glycoform carrying unmodified l-fucose (Figure 2). Its ADCC activity was comparable to the best optimized Herceptin glycoform (G2) that is afucosylated and carrying a full-size galactosylated biantennary complex-type N-glycan.9,24,25 The glycoforms (5 and 3) in which the l-fucose is modified by attachment of hydroxyl and amino groups, respectively, also showed significant ADCC activity but to a lesser extent than that of the 6-azide-modified glycoform (2). These results are consistent with the binding affinity of the respective glycoforms for the Fcγ receptors. An exception was observed for the 6-triazole-modified glycoform (4), which showed enhanced affinity for the Fcγ receptors but did not show an apparent increase in ADCC in in this assay. The opposite was observed for the d-arabinose glycoform (6), which had similar affinity for Fcγ receptors as the Herceptin but showed much better ADCC activity than the parent antibody (Figure 2). An enhanced ADCC activity of the d-arabinose glycoform (6), which was produced via a metabolic glycoengineering method, was also observed in a previous report.26 The reason for the inconsistency between the in vitro binding affinity for Fcγ receptors and the cellular or in vivo ADCC activity for some antibody glycoforms was not clear, which was also observed in previous studies.9
Figure 2.
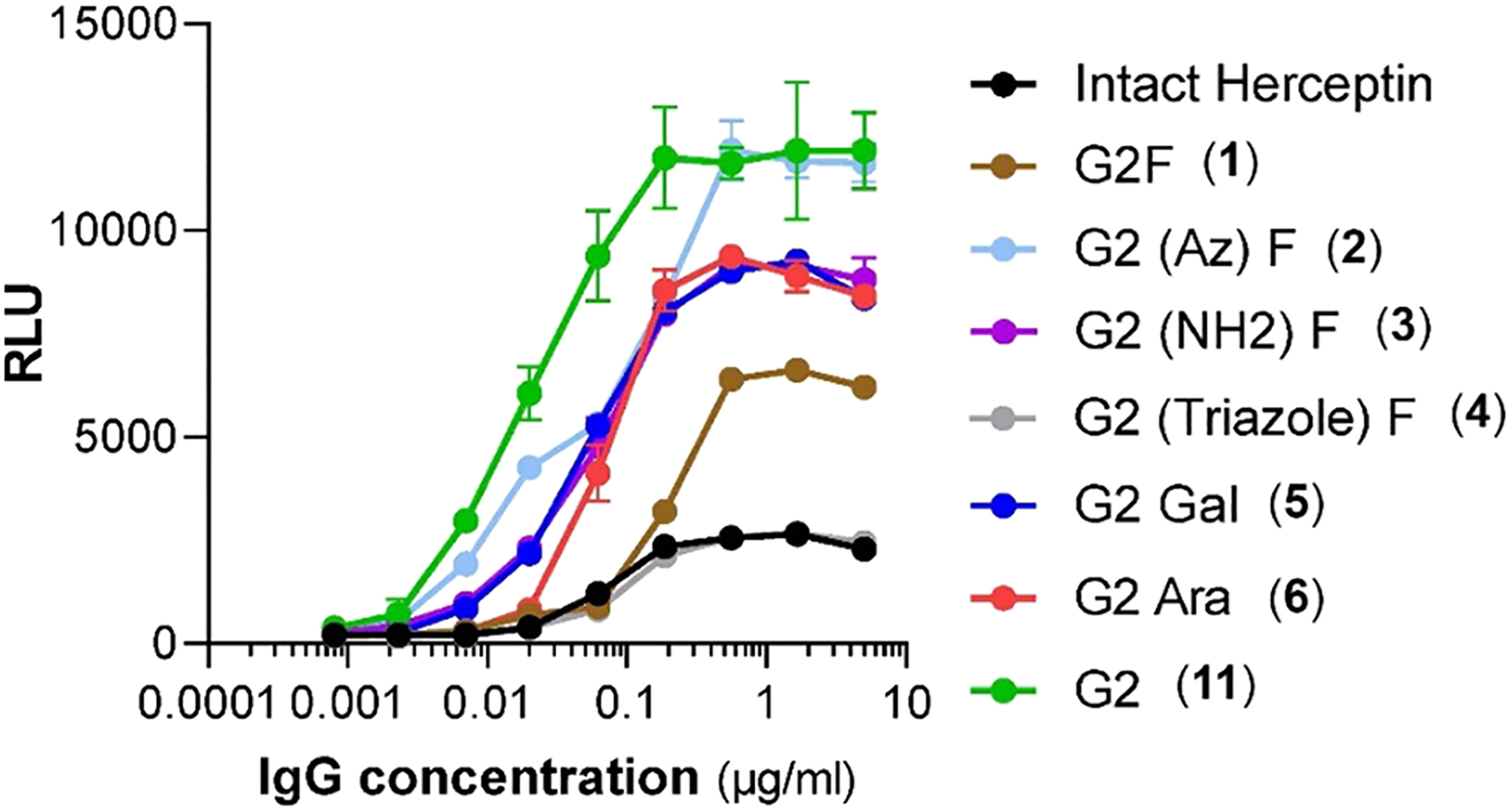
Antibody-dependent cellular cytotoxicity (ADCC) of different Herceptin glycoforms. ADCC activity was assessed by a robust ADCC assay against the human breast carcinoma cell line SKBR3 (HER2+, ATCC) using an engineered stable Jurkat cell line expressing the F allele of FcγRIIIa as the effector cells. Activation of the effector cells by the antibodies was probed by measurement of the luminescence with the Bio-Glo Luciferase assay reagent. Background luminescence from negative control samples was subtracted, and signal was expressed as relative luciferase units (RLU).
Molecular Dynamics (MD) Simulation Analysis of the Effect of Core-Fucose Modification on the Antibody–FcγIIIa Receptor Interactions.
To understand the mechanism by which the selected core-fucose modifications alter the affinity between the antibody and the FcγRIIIa receptor, we performed MD simulations based on Hamiltonian replica exchange with solute tempering and biasing potentials (HREST-BP)27,28 of the Fc–FcγRIIIa receptor complexes for the glycoforms (1, 2, and 5) and the afucosylated Fc (the G2 Fc glycoform). To obtain information on the extent of interactions between the Fc chain A and the FcγRIIIa glycans, the number of contacts between the glycans was determined based on the distance between the glycan centers of mass, excluding the core glycan, being <7 Å. Approximately 3–11-fold increases in the average number of contacts observed in the simulations occurred in the system with either core galactose or core azide-fucose and the afucosylated G2 form, respectively (Table S1, Figure S5). In addition, the interaction energy showed that the lack of a core fucose led to the most favorable interaction energies for Fc glycan conformations, while the presence of the unmodified core fucose gave the least favorable energies (Figure S6a). The addition of polar groups at C6 of the parent fucose, as in the case of core galactose and core azide-fucose glycoforms, expanded the hydrogen-bonding possibilities, leading to more favorable interaction energies with the FcR glycan (Figure S6b–d). These results are consistent with the experimental EC50 values, directly showing the importance of carbohydrate–carbohydrate interactions in the binding affinity of the antibody Fc to FcγRIIIa (Table 1).
To understand the molecular contributions to the changes in the extent of Fc and FcγIIIa receptor interactions, we first determined the conformations of the core glycans as measured by the O5′–C1′–O6–C6 dihedral angle ϕ in the core(′)–GlcNAc1 glycosidic linkage. The conformation of this dihedral orientation impacted interactions of the Fc glycan with its environment, offering insights into the molecular interaction contributing to the relative binding affinities of the antibody Fc to FcR (Figure 3a). Addition of a polar functional group at C6 in core fucose appears to enable hydrogen bonding between this group and other glycans, which could facilitate high-energy transitions about the dihedral angle ϕ. Dihedral angles of ϕ = 180–225° and 225–270° led to conformations in which the core sugar and its C6 functional group projected out of the Fc–FcR binding pocket (Figure 3b and 3c). At ϕ = 180–225°, the hydroxyl groups of the Fc chain A core sugar were oriented such that they could hydrogen bond with the FcR glycan. In addition, the azide group in the core azide-fucose was long enough, compared to the C6 hydroxyl in l-galactose, to hydrogen bond with FcR Arg155, stabilizing the Fc–FcR interface through both glycan–glycan and glycan–protein interactions (Figure 3b) and contributing to its greater affinity. The orientation of the hydroxyl groups in core sugars with ϕ = 225–270° limited hydrogen-bond contacts with the FcR glycan, and the C6 functional group was displaced away from FcR Arg155 (Figure 3c). Finally, the sampling of the ϕ = 0–45° region with the core azide-fucose led to a number of hydrogen bonds with the FcR glycan involving the hydroxyls of GlcNAc1, fucose, as well as the azide moiety (Figure 3d). These additional interactions led to the increased contacts and more favorable interaction energies of core azide-fucose over core galactose that contribute to the more favorable binding and ADCC activity.
Figure 3.
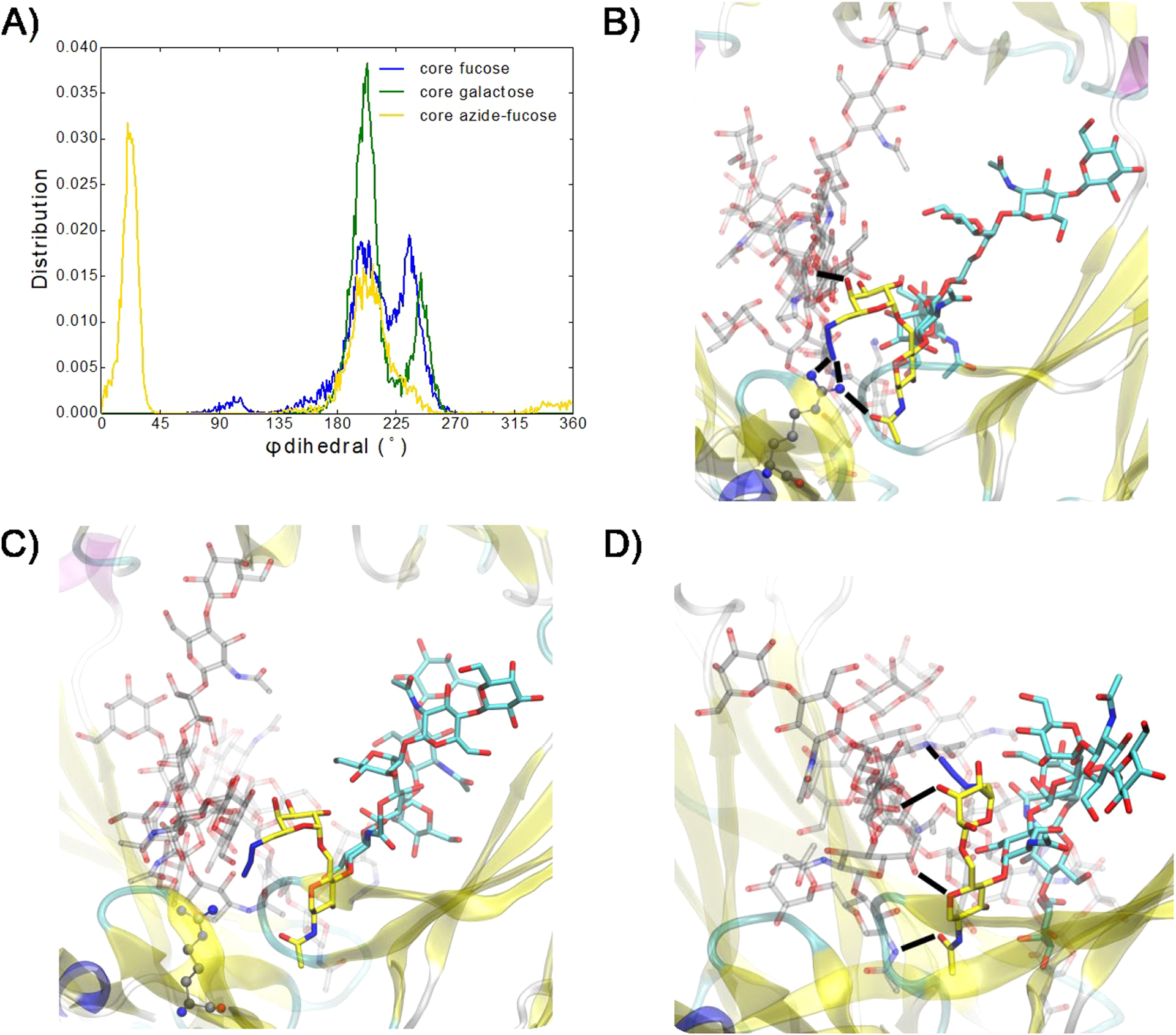
(A) Distribution of ϕ dihedral angles (O5′–C1′–O6–C6) in the core sugar(′)–GlcNAc1 glycosidic linkage. (B–D) Core azide-fucose of the Fc chain A glycan linked to GlcNAc1 (carbons in yellow, oxygens in red, and nitrogens in blue) with FcR Arg155 in gray CPK representation with (B) ϕ = 180–225°, (C) ϕ = 225–270°, and (D) ϕ = 0–45°. Remaining Fc chain A glycan is with carbons in cyan. Fc and FcR proteins are in cartoon representation, and Fc chain B and FcR glycans carbons are in transparent gray. Hydrogen bonds are indicated by black dashes.
It has been well established that the attachment of a core fucose leads to a significant decrease in the affinity of an antibody for its FcγIIIa receptors and, as a result, remarkably reduces the ADCC activity of the antibody in vivo. One explanation of this outcome is that the core fucose interferes with the favorable carbohydrate–carbohydrate interactions between the Fc glycan of the antibody and the Asn162 glycan from the FcγIIIa receptor.11,12 Thus, removal of the core fucose has become a practical way to enhance the ADCC activity of therapeutic antibodies such as Herceptin to enhance their therapeutic efficacy. However, it has not been clear if and how the core fucose can be modified to modulate the effector functions of antibodies. One challenge in this pursuit is the lack of technology in site-specific modification of the core fucose in intact antibodies. Although attempts have been made to incorporate modified fucose through metabolic glycoengineering, the sort of modification that can be achieved is limited.26,29–32 In this study, we established a chemoenzymatic method that allowed the introduction of selectively modified l-fucose in an intact antibody and enabled the structure–activity relationship study of the effects of the modified core fucose on FcγIIIa receptor binding and ADCC activity. Among the five selectively modified Herceptin glycoforms, we found that introduction of an azide group at the 6 position of the l-fucose resulted in 18-fold and over 100-fold enhancement in the antibody’s affinity for the high-affinity and low-affinity FcγIIIa receptor alleles, FcγRIIIa-V158 and FcγRIIIa-F158, respectively. Several other modifications at the 6 position of l-fucose also led to enhancement of the antibody’s affinity for FcγIIIa receptors but to a lesser extent. Cell-based assays confirmed that the 6-azide derivative had remarkably enhanced ADCC activity compared with the parent Herceptin. These unexpected experimental results reveal that site-selective modification of the core fucose, instead of its removal, constitutes a new molecular approach to modulating the effector functions of antibodies.
Previous crystallographic studies have shown that removal of the core fucose enables direct contact and hydrogen bonding between the FcR glycan GlcNAc1/GlcNAc2 monosaccharides with GlcNAc1 of the Fc chain A glycan.11,33 In addition, a lack of the core fucose gives Gln295 in Fc chain A access to hydrogen bonding with the FcR α1,3-branched glycan.11 In our present MD simulations, interactions between Fc glycan GlcNAc1 and FcR glycan GlcNAc1 and GlcNAc2 have a ~2.5-fold greater likelihood of having interaction energies more favorable than −10 kcal/mol when the Fc glycan has the core azide-fucose with ϕ = 0–45° vs core galactose in its preferred population at ϕ = 180–225° (Figure S6b). Core azide-fucose with ϕ = 0–45° is the only species in which the core sugar facilitates interaction of the FcR α1,3 branch with Fc Gln295, yielding interaction energies more favorable than −5 kcal/mol, which are more favorable than the same interaction with no core fucose (Figure S6c and S6d). FcR Lys128 and Fc chain A Tyr296 were observed to interact in the crystal structures of Fc–FcR with core afucosylated Fc glycans but not in Fc glycans with core fucose.11,12 The conformations obtained from the MD simulations for these species show that systems with Fc glycans lacking core fucose or carrying the core galactose have the largest populations of FcR Lys128–Fc Tyr296 distances of ~5–8 Å (Figure S5b). Comparison of the core azide-fucose and l-galactose species show the population of the shorter interactions to be greater with the latter species. As the experiments show that the binding affinity of Fc to FcR is greater when the Fc glycan is engineered with core azide-fucose compared to galactose, these results suggest that the FcR Lys128–Fc Tyr296 interaction contributes less to the binding affinity.
CONCLUSION
A chemoenzymatic method is described that permits site-selective modification on the core fucose of intact antibodies and enables the investigation of how the modified fucose affects the antibody’s functions. We have shown that introduction of a small functional group such as an azide at the 6 position can have a huge impact on the antibody’s binding to the FcγRIIIa receptors, leading to 18-fold and 100-fold enhancement in affinity for the high- and low-affinity FcγRIIIa (V-158 and F158) alleles, respectively. Cell-based assays have shown that the 6-azide- and 6-hydroxyl l-fucose-containing glycoforms demonstrate significantly enhanced ADCC activity over the antibody carrying the natural core fucose. These results are unexpected as previously structural studies indicated that the presence of the natural core fucose poses steric hindrance to block favorable carbohydrate– carbohydrate interactions between the Fc and the FcγR N-glycans, resulting in significantly decreased affinity of the antibody for the FcγR. Our MD simulations show that the introduction of an azide or hydroxyl group at the C-6 changes the orientation of the core fucose to promote favorable carbohydrate–carbohydrate and carbohydrate–protein interactions, leading to enhanced affinity between antibody Fc and FcγR receptor. Taken together, the present experimental and MD simulation studies reveal a new mechanism and a novel strategy to enhance the antibody’s Fcγ receptor binding and cellular cytotoxicity.
EXPERIMENTAL SECTION
Materials and Methods.
All chemicals, reagents, and solvents were purchased from Sigma-Aldrich and TCI and unless specially noted applied in the reaction without further purification. Monoclonal Herceptin was purchased from Premium Health Services Inc. (Columbia, MD). Deglycosylated GlcNAc–Herceptin was prepared using our previously reported method.25 Silica gel (200–425 mesh) for flash chromatography was purchased from Sigma-Aldrich. Liquid chromatography electrospray mass spectrometry (LC-ESI-MS) was performed on an Exactive Plus Orbitrap Mass Spectrometer (Thermo Scientific) equipped with a C-4 column (XBridge BEH300 C4, 2.1 × 50 mm, 3.5 μm, Waters) for antibody analysis. Matrix-assisted laser desorption ionization with a time-of-flight analyzer (MALDI-TOF) was performed using a Bruker UltrafleXtreme MALDI TOF/TOF Mass Spectrometer with a dihydroxybenzoic acid/dimethylamide (DHB/DMA) matrix and used to analyze transfer products including various core-modified N-glycoforms of Herceptin. 1H and 13C, NMR spectra were recorded on a 600 MHz spectrometer (Bruker, Tokyo, Japan) with D2O as the solvent. Endo-S2 D184 M glycosynthase derived from Streptococcus pyogenes and AlfC E274A fucoligase derived from Lactobacillus casei were expressed following our previously described method.14 Fast protein liquid chromatography (FPLC) was performed using an AKTA Explorer (GE Healthcare) equipped with a HisTrapTM HP column (1 mL) for protein purification or a HiTrapTM Protein A HP column (1 mL) for antibody purification. Protein concentration was determined with NanoDrop 2000c (Thermo Scientific).
Synthesis of 6-Azido-α-l-fucopyranosyl Fluoride (8).
A solution of l-galactose (500 mg, 2.78 mmol), CuSO4 (1.11 g, 6.93 mmol), and H2SO4 (50 μL) was stirred in acetone (11 mL) at room temperature for 15 h. Upon completion, the reaction mixture was neutralized with saturated NaHCO3 and the reaction mixture was concentrated. The residue was partitioned in CHCl3 and water, and the organic layer was separated, washed with brine, dried with anhydrous Na2SO4, and filtered. The filtrate was concentrated to give the 1,2:3,4-di-O-isopropylidene l-galactopyranose as a white solid (630 mg, 88%). To a solution of the intermediate (500 mg, 1.92 mmol) in anhydrous pyridine (4 mL) was added TsCl (1 g, 5.26 mmol), and the solution was stirred at room temperature for 12 h. Then the reaction mixture was diluted with CH2Cl2 and washed with HCl (10 mM), saturated NaHSO4 solution, and brine, sequentially. The organic layer was dried and filtered, and the filtrate was concentrated to give the tosylate derivative as a syrup. The resulting tosylate ester (760 mg, 1.91 mmol) was dissolved in DMF (13 mL), and to the solution was added NaN3 (480 mg, 7.38 mol), and the mixture was stirred under reflux at 115 °C for 15 h. The reaction mixture was concentrated under reduced pressure. The residue was suspended in CHCl3 and washed with brine and water. The organic layer was dried and filtered. The filtrate was concentrated, and the product was purified by silica gel flash chromatography (heptane/EtOAc = 2:1, v/v) to obtain the azide product as white powder (420 mg, 77%). The purified intermediate was deprotected with 90% TFA and then acetylated with acetic anhydride in pyridine. The peracetylated 6-azido-fucose (296 mg, 0.79 mmol) was fluorinated using HF pyridine (2 mL) and then de-O-acetylated with sodium methoxide (5 mM) in anhydrous MeOH (10 mL). The reaction mixture was concentrated, and the residue was dissolved in water and lyophilized to give the 6-azido-α-l-fucosyl fluoride (8) (157 mg, 64% in two steps) as a white powder. 1H NMR (D2O, 400 MHz): δ = 5.71 (dd, J1,2 = 2.0 Hz, J1,F = 53.2 Hz, 1H, H-1), 4.20 (dd, J = 4.0, 8.4 Hz, 1H, H-2), 4.02 (d, J = 2.4 Hz, 1H, H-3), 3.89 (d, J = 2.4 Hz, 1H, H-4), 3.81 (dd, J = 3.6, 11.2 Hz, 1H, H-5), 3.55 (m, 2H, H-6). ESI-MS: calcd for 6-Az-FucF, M = 207.07 Da; found (m/z), 208.11 [M + H]+.
Synthesis of α-l-Galactosyl Fluoride (12).
The synthesis was performed following the same procedure as the preparation of α-l-fucosyl fluoride14 using peracetylated l-galactopyranose as the starting material. 1H NMR (D2O, 600 MHz): 5.61 (dd, J1,2 = 3.0 Hz, J1,F = 65.4 Hz, 1 H, H-1), 4.00 (m, 1H, H-2), 3.98 (m, 1H, H-3), 3.79 (m, 1H, H-4), 3.76 (m, 1H, H-5), 3.69–3.63 (m, 2H, H-6a and H-6b). 13C NMR (D2O, 125 MHz): 107.75 (C-1), 72.93 (C-5), 68.43 (C-2), 67.51 (C-3), 67.35 (C-4), 60.51 (C-6). ESI-MS: calcd for α-l-GalF, M = 182.06 Da; found (m/z), 183.09 [M + H]+.
Synthesis of α-d-Arabinosyl Fluoride (13).
The synthesis of 13 was performed following the same procedure as the preparation of α-l-fucosyl fluoride14 using peracetylated d-arabinopyranose as the starting material. 1H NMR (D2O, 600 MHz): 5.59 (dd, J1,2 = 3.0 Hz, J1,F = 57.6 Hz, 1 H, H-1), 3.96 (m, 1H, H-2), 3.94 (m, 1H, H-3), 3.82–3.71 (m, 3H, H-4. H-5a, and H-5b). 13C NMR (D2O, 125 MHz): 107.95 (C-1), 68.42 (C-3), 67.85 (C-2), 67.69 (C-4), 65.01 (C-5). ESI-MS: calcd for α-AraF, M = 152.05 Da; found (m/z), 153.12 [M + H]+.
Synthesis of Core 6-Azido-Fucosylated Herceptin Carrying a Complex-Type N-Glycan (2).
To a mixture of the synthetic α-monosaccharyl fluoride (55.9 μg, 0.27 μmol) and the GlcNAc– Herceptin (1 mg, 6.6 nmol) in a buffer (PBS, 150 mM, pH 7.4, 100 μL) was added mutant AlfC E274A (200 μg, 2.0 μL). The solution was incubated at 30 °C, and the reaction was monitored by LC-ESI-MS analysis. Additional AlfC E274A and monosaccharide fluoride were added to drive the reaction to completion, as monitored by LC-ESI-MS. The mixture was then loaded on a protein A affinity column (HiTrap Protein A HP, GE Healthcare). After washing, the desired product was eluted with citrate buffer (30 mM, pH 3.5) and promptly dialyzed against a buffer (PBS, 150 mM, pH 7.4) at 4 °C. The solution was concentrated. The quantity of the core-modified Herceptin was determined with NanoDrop analysis (862 μg, 86%). To make the fully glycosylated core-modified Herceptin, a solution of antibody (1 mg, 6.6 nmol) and G2-ox (0.14 mg, 0.26 μmol) was incubated with Endo-S2 D184 M (20 μg, 0.2 mg/mL) at 30 °C in a buffer (PBS, 150 mM, pH 7.4, 100 μL) for 2 h. The reaction was monitored with LC-ESI-MS. The product was purified with a protein A column. MALDI-TOF-MS analysis: calcd for the heavy chain of 2, M = 50979 Da; found (m/z), 50971. The engineered glycoforms of Herceptin were further identified by PNGase F cleavage from the heavy chain. MALDI-TOF-MS: calcd for the N-glycan of 2, M = 1828.4 Da; found (m/z), 1851.5 [M + Na]+. The core modification of Herceptin using α-l-GalF and α-AraF was performed in a similar manner, and the reaction took 6–10 h to completion as monitored by LC-ESI-MS. The products were purified with protein A chromatography.
Core l-Galactosylated Herceptin (5, 82%).
MALDI-TOF-MS: calcd for the heavy chain of 5, M = 50953 Da; found (m/z) 50944. MALDI-TOF-MS: calcd for the N-glycan of 5, M = 1803.4 Da; found (m/z), 1826.4 [M + Na]+.
Core d-Arabinosylated Herceptin (6, 81%).
MALDI-TOF-MS: calcd for the heavy chain of 6, M = 50921 Da; found (m/z) 50914. MALDI-TOF-MS: calcd for the N-glycan of 6, M = 1771.8 Da; found (m/z), 1794.8 [M + Na]+.
Synthesis of Core 6-Amino-fucosylated Herceptin (3).
To the solution of 2 (1 mg, 6.6 nmol) in a buffer (Tris, 100 mM, pH 7.4, 100 μL) was added tris(2-carboxyethyl)phosphine hydrochloride (TCEP, 50 mM). The reaction was incubated at room temperature for 2 h and monitored with MALDI-TOF-MS. After completion, the reaction mixture was buffer exchanged with a PBS buffer (150 mM, pH 7.4) with a centrifugal filter unit (Amicon Ultra-4, Millipore).34 The product was purified with protein A chromatography as described above and dialyzed against a PBS buffer (150 mM, pH 7.4) to afford core 6-amino-fucosylated Herceptin (3) (435 μg, 87%). MALDI-TOF-MS: calcd for the heavy chain of 3, M = 50952 Da; found (m/z) 50941. MALDI-TOF-MS: calcd for the N-glycan of 3, M = 1802.1 Da; found (m/z), 1825.1 [M + Na]+.
Synthesis of Core 6-Triazole-fucosylated Herceptin (4).
Propargyl alcohol (0.1 μL, 1.8 μmol), CuSO4 (1 μL, 0.5 mM), trishydroxypropyltriazolylmethylamine (THPTA) (2 μL, 0.2 mM), l-ascorbic acid (5 μL, 0.5 mM), and aminoguanidine hydrochloride (1 μL, 50 μM) were added to a solution of 6-azido-fucosylated Herceptin (2) (500 μg, 3.3 nmol) in a buffer (PBS, 150 mM, pH 7.4, 100 μL). The reaction was incubated at room temperature overnight and monitored with LC-ESI-MS. The product was purified with protein A chromatography as described above and dialyzed against a PBS buffer (150 mM, pH 7.4) to afford core 6-triazole-fucosylated Herceptin (4) (423 μg, 84%). MALDI-TOF-MS: calcd for the heavy chain of 4, M = 51034 Da; found (m/z) 51026. MALDI-TOF-MS: calcd for the N-glycan of 4, M = 1884.3 Da; found (m/z), 1907.4 [M + Na]+.
ELISA Binding Tests.
FcγRIIIA V158 or F158 (0.5 μg/mL, 100 μL/well, Sino Biological) in a buffer (PBS, 150 mM, pH 7.4) was coated onto a high-binding 96-well plate (Santa Cruz Biotechnology) at 4 °C overnight. After washing twice with PBS buffer containing 0.5% Tween 20 (PBST, 200 μL/well), 1% Bovine serum albumin (BSA) in PBS (200 μL/well) was added to block the plate for 2 h. Subsequently, after being washed twice again, a serial dilution of core-modified Herceptin in PBST buffer (100 μL/well) was added, and the plate was incubated for 1 h. For the V158 plate, the concentration of each Herceptin glycoform ranged from 250 to 0.016 nM (5-fold serial dilutions). For the F158 plate, the concentration of each Herceptin glycoform ranged from 2000 to 0.128 nM (5-fold serial dilutions). After washing five times with PBST buffer, the plate was incubated with antihuman IgG F(ab′)2 HRP (0.16 μg/mL, 100 μL/well, Invitrogen) for 1 h. After washing again, 3,3′,5,5′-tetramethylbenzidine (100 μL/well, Thermo Scientific) was added for signal development. The reaction was stopped by adding sulfuric acid (2 N, 100 μL/well). Absorbance at 450 nm was measured with a SpectraMax M5 microplate reader (Molecular Devices). EC50 values were determined by fitting the ELISA binding curves (Figure S4) to the nonlinear regression dose–response equation in OriginLab 7.5.
ADCC Activity Assay.
Anti-HER2 antibody glycoforms were tested in a robust ADCC assay (PMID 25086226)23 against the human breast carcinoma cell line SKBR3 (HER2+, ATCC). In brief, SKBR3 cells were plated 24 h prior to the assay in opaque 96-well flat bottom plates (Costar) at 104 cells/well. On the day of the assay, each glycoform was prepared at 5 μg/mL and 3-fold serial dilutions were made in a separate 96-well plate. Each was then added, in duplicate, to the SKBR3 cells and incubated for 30 min. The effector cells (Jurkat cell line expressing the F allele of FcγRIIIA, Promega) were thawed per the manufacturer’s recommendations and added to the plate at a concentration of 7.5 × 104 cells/well. The plates were then incubated for 6 h at 37 °C (5% CO2). Following incubation, the plates were equilibrated to room temperature for 15 min prior to the addition of Bio-Glo Luciferase Assay Reagent (Promega). Cells were allowed to develop at room temperature for 15 min prior to reading on a SpectraMax Plus spectrophotometer (Molecular Devices). Background luminescence from negative control samples was subtracted, and signal was expressed as relative luciferase units.
Molecular Dynamics Simulations.
The Fc–FcR system with core-fucosylated FcR glycans and with either core-fucosylated or afucosylated Fc glycans were set up using CHARMM-GUI35,36 with PDB ID 3SGK.11 The Fc glycan core l-fucose was structurally modified by replacing the C6 methyl group with either a hydroxyl or an azide group to generate core l-galactose or core azide-l-fucose, respectively. Force-field parameters for the azide moiety in azide-l-fucose were generated using CGenFF37,38 to be compatible with the CHARMM36 additive force field.39,40 Using OpenMM,41 the systems were energy minimized, followed by molecular dynamics (MD) simulations at in the NVT ensemble for 0.25 ns at 298 K and then in the NPT ensemble for 1 ns at 298 K and 1 atm using the MonteCarloBarostat, to prepare them for Hamiltonian replica exchange with solute tempering-biasing potential (HREST-BP) simulations. MD and HREST-BP simulations all employed rigid constraints for covalent bonds with hydrogen, a 12 Å cutoff for Lennard–Jones interactions with a switching function to zero from 10 to 12 Å, and the particle mesh Ewald method for calculation of long-range electrostatic interactions beyond 12 Å.
To enhance the sampling of the glycans, we implemented the HREST-BP method27,28 in OpenMM. For our application of the HREST-BP method to the Fc–FcR systems, the potential energy functions (i.e., the Hamiltonian) for intramolecular dihedral and nonbonded interactions within the designated solute (the glycans) in the nth replica were scaled by βn/β0, where β0 is the Boltzmann constant, kB, × the ground-state temperature T0 (298 K) and βn is kB × the effective temperature Tn of the nth replica, and the potential energy functions for intermolecular nonbonded interactions between solute (glycans) and the designated solvent (proteins and water) were scaled by following the scaling parameters of the REST2 Hamiltonian replica exchange method.42 For the flexible glycosidic linkages between monosaccharides and the glycopeptide linkage between GlcNAc and Asn162 and Asn297 of the FcR and Fc proteins, respectively, an additional biasing potential was applied in the form of a CMAP term.43 The CMAP biasing potentials (bpCMAPs) were previously computed for the unique pairs of disaccharide and glycopeptide linkages in the Fc–FcR system.27 The bpCMAPs were scaled by βn/β0 × λn, with λn as an additional replica-dependent parameter. Scaling parameters for each replica were obtained from previously published results27 on the complex biantennary glycan sequences and were used in this work (Table S2).
Six replicas were run per system in the effective temperature range of 298–400 K for 40 ns each for a total of 240 ns of sampling time per system. HREST-BP simulations were run in the NVT ensemble with a 2 fs/time step with a replica exchange attempt made every 2000 time steps using the Metropolis criterion and data collected from the ground-state replica every 2000 time steps. The replica walks of the original ground-state replicas and highest replicas vs time for each system show a high exchange rate with frequent round trips for the Fc glycan with core-fucose system (Figure S7). The first 8 ns were not used in the analysis of the HREST-BP simulations, as the distribution of contacts between Fc and FcR glycans vs time did not converge until after 8 ns.
Supplementary Material
ACKNOWLEDGMENTS
We thank the members of the Wang lab for technical assistance and helpful discussions. This work was supported in part by the National Institutes of Health (NIH grants R01GM096973 and R01AI155716 to L.X.W., R35GM131710 to A.D.M., Jr., and R01AI129795 to J.V.R.) and the University of Maryland Computer-Aided Drug Design Center. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
The authors declare the following competing financial interest(s): Lai-Xi Wang is the Founder of GlycoT Therapeutics, LLC; Alexander D. MacKerell, Jr. is a cofounder and CSO of SilcsBio LLC. All other authors have no conflict of interest.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c03174.
Supplemental tables and figures; 1H and 13C NMR spectra of selectively modified α-glycosyl fluorides (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.1c03174
Contributor Information
Chao Li, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742, United States.
Gene Chong, Computer Aided Drug Design Center, Department of Pharmaceutical Sciences, School of Pharmacy, University of Maryland, Baltimore, Maryland 21201, United States.
Guanghui Zong, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742, United States.
Asaminew Haile Aytenfisu, Computer Aided Drug Design Center, Department of Pharmaceutical Sciences, School of Pharmacy, University of Maryland, Baltimore, Maryland 21201, United States.
Grace K. Henry, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742, United States
Jeffrey V. Ravetch, Laboratory of Molecular Genetics and Immunology, The Rockefeller University, New York 10065, United States
Alexander D. MacKerell, Jr., Computer Aided Drug Design Center, Department of Pharmaceutical Sciences, School of Pharmacy, University of Maryland, Baltimore, Maryland 21201, United States.
Lai-Xi Wang, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742, United States.
REFERENCES
- (1).Helenius A; Aebi M Intracellular functions of N-linked glycans. Science 2001, 291 (5512), 2364–9. [DOI] [PubMed] [Google Scholar]
- (2).Pinho SS; Reis CA Glycosylation in cancer: mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15 (9), 540–55. [DOI] [PubMed] [Google Scholar]
- (3).Andre S; Kozar T; Kojima S; Unverzagt C; Gabius HJ From structural to functional glycomics: core substitutions as molecular switches for shape and lectin affinity of N-glycans. Biol. Chem 2009, 390 (7), 557–565. [DOI] [PubMed] [Google Scholar]
- (4).Miyoshi E; Moriwaki K; Nakagawa T Biological function of fucosylation in cancer biology. J. Biochem 2007, 143 (6), 725–9. [DOI] [PubMed] [Google Scholar]
- (5).Li J; Hsu HC; Mountz JD; Allen JG Unmasking Fucosylation: from Cell Adhesion to Immune System Regulation and Diseases. Cell Chem. Biol 2018, 25 (5), 499–512. [DOI] [PubMed] [Google Scholar]
- (6).Shields RL; Lai J; Keck R; O’Connell LY; Hong K; Meng YG; Weikert SH; Presta LG Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J. Biol. Chem 2002, 277 (30), 26733–40. [DOI] [PubMed] [Google Scholar]
- (7).Zeitlin L; Pettitt J; Scully C; Bohorova N; Kim D; Pauly M; Hiatt A; Ngo L; Steinkellner H; Whaley KJ; et al. Enhanced potency of a fucose-free monoclonal antibody being developed as an Ebola virus immunoprotectant. Proc. Natl. Acad. Sci. U. S. A 2011, 108 (51), 20690–20694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kapur R; Kustiawan I; Vestrheim A; Koeleman CA; Visser R; Einarsdottir HK; Porcelijn L; Jackson D; Kumpel B; Deelder AM; Blank D; Skogen B; Killie MK; Michaelsen TE; de Haas M; Rispens T; van der Schoot CE; Wuhrer M; Vidarsson G A prominent lack of IgG1-Fc fucosylation of platelet alloantibodies in pregnancy. Blood 2014, 123 (4), 471–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Li T; DiLillo DJ; Bournazos S; Giddens JP; Ravetch JV; Wang LX Modulating IgG effector function by Fc glycan engineering. Proc. Natl. Acad. Sci. U. S. A 2017, 114 (13), 3485–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Giddens JP; Lomino JV; DiLillo DJ; Ravetch JV; Wang LX Site-selective chemoenzymatic glycoengineering of Fab and Fc glycans of a therapeutic antibody. Proc. Natl. Acad. Sci. U. S. A 2018, 115 (47), 12023–12027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ferrara C; Grau S; Jager C; Sondermann P; Brunker P; Waldhauer I; Hennig M; Ruf A; Rufer AC; Stihle M; Umana P; Benz J Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcgammaRIII and antibodies lacking core fucose. Proc. Natl. Acad. Sci. U. S. A 2011, 108 (31), 12669–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Mizushima T; Yagi H; Takemoto E; Shibata-Koyama M; Isoda Y; Iida S; Masuda K; Satoh M; Kato K Structural basis for improved efficacy of therapeutic antibodies on defucosylation of their Fc glycans. Genes Cells 2011, 16 (11), 1071–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Falconer DJ; Subedi GP; Marcella AM; Barb AW Antibody Fucosylation Lowers the FcgammaRIIIa/CD16a Affinity by Limiting the Conformations Sampled by the N162-Glycan. ACS Chem. Biol 2018, 13 (8), 2179–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Li C; Zhu S; Ma C; Wang LX Designer alpha1,6-Fucosidase Mutants Enable Direct Core Fucosylation of Intact N-Glycopeptides and N-Glycoproteins. J. Am. Chem. Soc 2017, 139 (42), 15074–15087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Li C; Li T; Wang LX Chemoenzymatic Defucosylation of Therapeutic Antibodies for Enhanced Effector Functions Using Bacterial alpha-Fucosidases. Methods Mol. Biol 2018, 1827, 367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Klontz EH; Li C; Kihn K; Fields JK; Beckett D; Snyder GA; Wintrode PL; Deredge D; Wang LX; Sundberg EJ Structure and dynamics of an alpha-fucosidase reveal a mechanism for highly efficient IgG transfucosylation. Nat. Commun 2020, 11 (1), 6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Li T; Tong X; Yang Q; Giddens JP; Wang LX Glycosynthase Mutants of Endoglycosidase S2 Show Potent Trans-glycosylation Activity and Remarkably Relaxed Substrate Specificity for Antibody Glycosylation Remodeling. J. Biol. Chem 2016, 291 (32), 16508–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kaifu T; Nakamura A Polymorphisms of immunoglobulin receptors and the effects on clinical outcome in cancer immunotherapy and other immune diseases: a general review. Int. Immunol 2017, 29 (7), 319–325. [DOI] [PubMed] [Google Scholar]
- (19).Shimizu C; Mogushi K; Morioka MS; Yamamoto H; Tamura K; Fujiwara Y; Tanaka H Fc-Gamma receptor polymorphism and gene expression of peripheral blood mononuclear cells in patients with HER2-positive metastatic breast cancer receiving single-agent trastuzumab. Breast Cancer 2016, 23 (4), 624–32. [DOI] [PubMed] [Google Scholar]
- (20).Koene HR; Kleijer M; Algra J; Roos D; von dem Borne AE; de Haas M Fc gammaRIIIa-158V/F polymorphism influences the binding of IgG by natural killer cell Fc gammaRIIIa, independently of the Fc gammaRIIIa-48L/R/H phenotype. Blood 1997, 90 (3), 1109–1114. [PubMed] [Google Scholar]
- (21).Cartron G; Dacheux L; Salles G; Solal-Celigny P; Bardos P; Colombat P; Watier H Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 2002, 99 (3), 754–8. [DOI] [PubMed] [Google Scholar]
- (22).Boero S; Morabito A; Banelli B; Cardinali B; Dozin B; Lunardi G; Piccioli P; Lastraioli S; Carosio R; Salvi S; Levaggi A; Poggio F; D’Alonzo A; Romani M; Del Mastro L; Poggi A; Pistillo MP Analysis of in vitro ADCC and clinical response to trastuzumab: possible relevance of FcgammaRIIIA/FcgammaRIIA gene polymorphisms and HER-2 expression levels on breast cancer cell lines. J. Transl. Med 2015, 13, 324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Cheng ZJ; Garvin D; Paguio A; Moravec R; Engel L; Fan F; Surowy T Development of a robust reporter-based ADCC assay with frozen, thaw-and-use cells to measure Fc effector function of therapeutic antibodies. J. Immunol. Methods 2014, 414, 69–81. [DOI] [PubMed] [Google Scholar]
- (24).Li H; Sethuraman N; Stadheim TA; Zha D; Prinz B; Ballew N; Bobrowicz P; Choi B-K; Cook WJ; Cukan M; et al. Optimization of humanized IgGs in glycoengineered Pichia pastoris. Nat. Biotechnol 2006, 24 (2), 210. [DOI] [PubMed] [Google Scholar]
- (25).Huang W; Giddens J; Fan SQ; Toonstra C; Wang LX Chemoenzymatic glycoengineering of intact IgG antibodies for gain of functions. J. Am. Chem. Soc 2012, 134 (29), 12308–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Hossler P; Chumsae C; Racicot C; Ouellette D; Ibraghimov A; Serna D; Mora A; McDermott S; Labkovsky B; Scesney S; Grinnell C; Preston G; Bose S; Carrillo R Arabinosylation of recombinant human immunoglobulin-based protein therapeutics. MAbs 2017, 9 (4), 715–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Yang M; Huang J; MacKerell AD Jr. Enhanced conformational sampling using replica exchange with concurrent solute scaling and hamiltonian biasing realized in one dimension. J. Chem. Theory Comput 2015, 11 (6), 2855–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Yang M; MacKerell AD Jr. Conformational sampling of oligosaccharides using Hamiltonian replica exchange with two-dimensional dihedral biasing potentials and the weighted histogram analysis method (WHAM). J. Chem. Theory Comput 2015, 11 (2), 788–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Okeley NM; Alley SC; Anderson ME; Boursalian TE; Burke PJ; Emmerton KM; Jeffrey SC; Klussman K; Law CL; Sussman D; Toki BE; Westendorf L; Zeng W; Zhang X; Benjamin DR; Senter PD Development of orally active inhibitors of protein and cellular fucosylation. Proc. Natl. Acad. Sci. U. S. A 2013, 110 (14), 5404–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Rabuka D; Hubbard SC; Laughlin ST; Argade SP; Bertozzi CR A chemical reporter strategy to probe glycoprotein fucosylation. J. Am. Chem. Soc 2006, 128 (37), 12078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Hsu TL; Hanson SR; Kishikawa K; Wang SK; Sawa M; Wong CH Alkynyl sugar analogs for the labeling and visualization of glycoconjugates in cells. Proc. Natl. Acad. Sci. U. S. A 2007, 104 (8), 2614–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Kizuka Y; Funayama S; Shogomori H; Nakano M; Nakajima K; Oka R; Kitazume S; Yamaguchi Y; Sano M; Korekane H; Hsu TL; Lee HY; Wong CH; Taniguchi N High-Sensitivity and Low-Toxicity Fucose Probe for Glycan Imaging and Biomarker Discovery . Cell Chem. Biol 2016, 23 (7), 782–92. [DOI] [PubMed] [Google Scholar]
- (33).Sakae Y; Satoh T; Yagi H; Yanaka S; Yamaguchi T; Isoda Y; Iida S; Okamoto Y; Kato K Conformational effects of N-glycan core fucosylation of immunoglobulin G Fc region on its interaction with Fcgamma receptor IIIa. Sci. Rep 2017, 7 (1), 13780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Cumnock K; Tully T; Cornell C; Hutchinson M; Gorrell J; Skidmore K; Chen Y; Jacobson F Trisulfide modification impacts the reduction step in antibody-drug conjugation process. Bioconjugate Chem. 2013, 24 (7), 1154–60. [DOI] [PubMed] [Google Scholar]
- (35).Jo S; Kim T; Iyer VG; Im W CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem 2008, 29 (11), 1859–65. [DOI] [PubMed] [Google Scholar]
- (36).Park SJ; Lee J; Qi Y; Kern NR; Lee HS; Jo S; Joung I; Joo K; Lee J; Im W CHARMM-GUI Glycan Modeler for modeling and simulation of carbohydrates and glycoconjugates. Glycobiology 2019, 29 (4), 320–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Vanommeslaeghe K; MacKerell AD Jr. Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model 2012, 52 (12), 3144–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Vanommeslaeghe K; Raman EP; MacKerell AD Jr. Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model 2012, 52 (12), 3155–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Huang J; Rauscher S; Nawrocki G; Ran T; Feig M; de Groot BL; Grubmuller H; MacKerell AD Jr. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14 (1), 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Guvench O; Mallajosyula SS; Raman EP; Hatcher E; Vanommeslaeghe K; Foster TJ; Jamison FW 2nd; Mackerell AD Jr. CHARMM additive all-atom force field for carbohydrate derivatives and its utility in polysaccharide and carbohydrate-protein modeling. J. Chem. Theory Comput 2011, 7 (10), 3162–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Eastman P; Swails J; Chodera JD; McGibbon RT; Zhao Y; Beauchamp KA; Wang LP; Simmonett AC; Harrigan MP; Stern CD; Wiewiora RP; Brooks BR; Pande VS OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol 2017, 13 (7), e1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Wang J; Torii M; Liu H; Hart GW; Hu ZZ dbOGAP - an integrated bioinformatics resource for protein O-GlcNAcylation. BMC Bioinf. 2011, 12, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Best RB; Zhu X; Shim J; Lopes PE; Mittal J; Feig M; Mackerell AD Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J. Chem. Theory Comput 2012, 8 (9), 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.