

Abstract
RNA has attracted considerable attention as a target for small molecules. However, methods to identify, study, and characterize suitable RNA targets have lagged behind strategies for protein targets. One approach that has received considerable attention for protein targets has been to utilize computational analysis to investigate ligandable “pockets” on proteins that are amenable to small molecule binding. These studies have shown that selected physical properties of pockets are important parameters that govern the ability of a structure to bind to small molecules. This work describes a similar analysis to study pockets on all RNAs in the Protein Data Bank (PDB). Using parameters such as buriedness, hydrophobicity, volume, and other properties, the set of all RNAs is analyzed and compared to all proteins. Considerable overlap is observed between the properties of pockets on RNAs and proteins. Thus, many RNAs are capable of populating conformations with pockets that are likely suitable for small molecule binding. Further, principal moment of inertia (PMI) calculations reveal that liganded RNAs exist in diverse structural space, much of which overlaps with protein structural space. Taken together, these results suggest that complex folded RNAs adopt unique structures with pockets that may represent viable opportunities for small molecule targeting.
Keywords: RNA, Nucleic acid structure, Small molecule, Computational analysis, Ligandability
1. Introduction
Modern views of RNA within the cell describe a far greater functional role than a transient, unstructured message between genetic DNA and functional protein. It is now well established that RNA is utilized for many biological processes relating to gene expression,1,2 protein function,3,4 cellular homeostasis,5 and disease.5-7 RNA has been shown to adopt a wide variety of well-defined but conformationally dynamic three-dimensional structures such as hairpins,8,9 triple helices,10 G-quadruplexes,11 and pseudoknots,12,13 which are also associated with many functions of RNA. For example, structure in RNA has been linked to regulating pre-mRNA splicing7,14,15 (including alternative splicing), as well as having effects on translation efficiency.11 In conjunction with these discoveries of structure and function, RNA has been posited as a potential therapeutic target for small molecules.16 Indeed, many examples can be found of small molecules binding to RNA targets and eliciting a pharmacological response.17 However, there remains no drug that targets RNA (apart from ribosome-binding drugs18,19), despite considerable interest in the field in recent years.
While substantial effort has been focused on individual targets or classes of targets,20-23 there is a need to more broadly assess the suitability of RNA as a target for small molecules. While one intriguing recent report proposed the use of computation to investigate individual RNAs,24 a global analysis using atomic resolution structural data is lacking. Conventional wisdom states that in comparison to proteins, binding pockets on RNA are too shallow, solvent-exposed, and polar to achieve potent and selective small molecule binding. Comparatively, many approaches have been taken to both identify protein pockets which are likely to bind small molecules and to understand factors that make these pockets suitable for targeting. In these studies, the “ligandability” of a given pocket is defined as the relative ease/difficulty of developing a small molecule that can bind to the protein in vitro.25 Similarly, “druggability” is defined as the relative ease of developing a small molecule that effectively alters target activity in vivo.25-28 For clarity, we also use these definitions throughout this work, where we aim to provide insights into the ligandability of RNA.
One approach was recently undertaken to assess the ligandability of protein targets by analyzing a large data set of ligand-bound protein structures within the protein databank (PDB).26,29 Using the PocketFinder algorithm within the ICM-Molsoft software suite,30,31 the authors calculated potential small-molecule binding pockets on the surface of > 42,000 protein structures. Using metrics of volume, fraction buried, and hydrophobicity, pockets containing drug-like small molecules were characterized in a 3-dimensional property space. This data set could then be used to assess the ligandability of any given pocket based on its properties correlated with this bound pocket-space. In addition to accurately reporting on known liganded sites, this approach faithfully identifies new potential druggable sites in proteins. This approach does not depend on the orientation of the specific amino acids that make up a pocket, but rather reports on physical properties such as size, shape, polarity, and solvent accessibility that make a particular pocket suitable for small molecules. The ICM-molsoft software suite has also previously been applied to RNA modeling,32 docking,33 and virtual screening.34
Herein, we take a structure-based approach to analyze potential small-molecule binding pockets on RNA structures within the PDB. We demonstrate through this analysis that pockets on RNA structures which contain small molecule ligands occupy a similar property space to ligand-bound protein pockets. As a validation to this approach, multiple known liganded RNAs are identified and characterized. We demonstrate that although many pockets on ligand-unbound RNA structures lay outside the “ligandability” region, there are also pockets which are predicted to be potentially chemically tractable. This analysis provides important insights into the global structures that RNAs adopt with respect to small molecule targeting, and the pockets formed in individual conformations found in atomic-resolution structures throughout the PDB.
2. Results and Discussion
2.1. Assessment of ligandability in RNA
To establish whether computational methods used for proteins are also suitable to study pockets on RNA, we selected three high quality RNA structures with small molecule ligands bound. The PreQ1 riboswitch is an example of a natural RNA/ligand pair (PDB: 3Q50) (Fig. 1A).35 The FMN riboswitch bound to Ribocil is a recent prominent example of a synthetic ligand bound to an RNA that was discovered by a phenotypic screen (PDB: 5KX9) (Fig. 1B).36 Finally, the HCV IRES motif is one of the rare examples where structure-guided design has been used to optimize a synthetic ligand for RNA (PDB: 3TZR) (Fig. 1C).37 For each of these cases, we deleted the known ligand from the structure and searched for potential small molecule binding pockets using the PocketFinder algorithm in the ICM software suite (Fig. 1). We assessed each identified pocket using the parameters reported to be most important for identifying ligandable sites in proteins. In each example several pockets were found, including the ligand binding site, though in general these other pockets were smaller and had less desirable properties (Fig. S1). We calculated volume in cubic angstroms (Å3), the fraction of the pocket considered to be hydrophobic (hydrophobicity, ranging from 0 to 1), the solvent exposure of the pocket (buriedness, ranging from 0.5 for pockets that are completely solvent exposed to 1 for those completely buried), and the druglike density score (DLID) for each pocket (with unitless values typically ranging from −3 to 2). DLID has been previously used as a single metric to assess pocket ligandability and is calculated based on the location of each pocket in its three-dimensional property space and its proximity within that space to known liganded protein pockets relative to the total number of nearby pockets. More simply, if a pocket of interest has volume, buriedness, and hydrophobicity values similar to other pockets with known ligands, it is likely to be ligandable and will thus have a high DLID score.26 While a high DLID score does not guarantee success in identifying a ligand, it serves as a useful metric to evaluate pockets. Importantly, DLID has no connection to function: further effort to demonstrate a connection between binding and modulation of function would be required for a given RNA in order to consider it a worthwhile target.
Fig. 1.

A) Structures of the PreQ1 riboswitch bound to PreQ1 (yellow) (PDB: 3Q50) B) the FMN riboswitch bound to Ribocil (yellow) (PDB: 5KX9) and C) the HCV IRES motif bound to a aminobenzimidazole-based small molecule (yellow) (PDB: 3TZR) with calculated pockets shown as blue objects.
In each case, the unbiased approach faithfully identified the known ligand binding site as a suitable small molecule binding pocket. Other pockets, in addition to the known binding site, were identified on each RNA. For the PreQ1 riboswitch, we identified two pockets of interest. Both pockets have comparable hydrophobicity and buriedness, however the ligand-containing pocket is larger (160 vs 145 Å3). Importantly, the pocket with the higher DLID score contained the known ligand, validating the approach (−0.08 vs −0.30). For the FMN riboswitch, a larger and more complex RNA, five pockets were identified. The ribocil-containing pocket was the largest, but similar to other pockets in hydrophobicity and buriedness. Again, DLID for the ribocil-containing pocket was 0.70, while all other pockets were considerably lower. Two pockets were identified on the HCV IRES. In this case, the ligand-containing pocket was smaller and more hydrophobic but had comparable buriedness and DLID score. These examples validate that a computational assessment of structured RNAs can be used to successfully identify sites likely to interact with small molecules. Importantly, results for the FMN Riboswitch were comparable to those reported in a review by Weeks et al. (which proposes a similar approach using QED scores), highlighting the robustness of this approach.24
2.2. Identification and analysis of structured RNA in the PDB
Next, we sought to globally assess RNA structures using the PocketFinder algorithm. To identify RNA sequences with known, atomic resolution structural information, we queried the PDB. At the outset of this project (May 2017), the PDB contained 137,348 entries, of which 3439 contained at least one strand of RNA. To develop a list of RNAs likely to contain defined secondary/tertiary structure, we omitted short sequences of less than 12 nucleotides. We further omitted highly complex RNAs such as the ribosome, which are likely to be computationally intractable. Of the remaining RNA-containing entries, 1552 consisted of X-ray crystallographic or NMR structural information of chain length between 12 and 700 nucleotides. In the case of entries generated from NMR structural data, the lowest energy conformer was chosen for analysis. We analyzed this set of structures using the PocketFinder algorithm as described above, and calculated volume, buriedness, hydrophobicity, and DLID values for each pocket identified. We then analyzed this data set to identify RNAs in complex with known ligands. To provide additional data sets for comparison, we flagged each entry that contained small molecule ligands of > 7 heavy atoms (to omit metal ions and solvents) and developed a list of 300 ligand-containing structures (See Supplementary Fig. 1S).
2.3. Analysis of all pockets on structured RNA
In total, we evaluated all 1552 RNA structures, and calculated the properties for each identified pocket. We compared these results to the values for all proteins in the PDB as well as all liganded proteins in the PDB (Table 1, Fig. 2, Supplementary Table S2). Most of these distributions were not normal, and therefore median values were used for comparison. The pocket volume for all proteins and all RNA are similar with median values of 175 and 187 Å,3 respectively. In both cases there is a broad distribution of volumes, indicating that there is a high degree of variability of pocket size in both proteins and RNA. In terms of buriedness, median values were again similar, with proteins and RNA having values of 0.75 and 0.72 (Fig. 2A, Table 1), suggesting that pockets on a given RNA are similarly solvent exposed to protein pockets. In contrast, there is a notable difference in the hydrophobicity parameter. Here, proteins had a median value of 0.49 while RNA had a median value of 0.35 (Fig. 2 A, Table 1). Pockets on RNA are generally more polar than pockets on proteins, consistent with conventional wisdom given the highly polar nature of nucleic acids. Further difference was observed in the DLID score, which considers all three of the above parameters for a given pocket. Here, proteins had a median value of −0.84, while RNA had a median value of −1.12 (Fig. 4C, Table 1). When considering all three parameters of a given pocket it is clear that “ligandable” pockets are considerably more frequent on proteins than RNA (at least in the case of those RNAs with known atomic resolution structure). This analysis further highlights that while many RNAs do contain structural features amenable to small molecule binding, such pockets exist but are considerably less common in RNA than on proteins. Despite the less frequent occurrence of ideal pockets in RNA, many pockets do lie within acceptable ranges that are suitable for small molecule binding.
Table 1.
Properties of protein26 and RNA structures from the PDB.
| Liganded Proteins (n = 46,221) | Liganded RNAs (n = 300) | |||
|---|---|---|---|---|
| Average | Median | Average | Median | |
| Volume (Å) | 460 ± 360 | 350 | 450 ± 400 | 280 |
| Buriedness | 0.81 ± 0.09 | 0.81 | 0.78 ± 0.09 | 0.77 |
| Hydrophobicity | 0.53 ± 0.12 | 0.52 | 0.42 ± 0.10 | 0.41 |
| DLID | −0.045 ± 0.81 | 0.13 | −0.38 ± 0.72 | −0.27 |
| All Proteins (n = 291,758) | All RNAs (n = 1552) | |||
| Average | Median | Average | Median | |
| Volume (Å) | 270 ± 250 | 180 | 290 ± 330 | 190 |
| Buriedness | 0.76 ± 0.08 | 0.75 | 0.72 ± 0.08 | 0.72 |
| Hydrophobicity | 0.50 ± 0.11 | 0.49 | 0.35 ± 0.10 | 0.35 |
| DLID | −0.70 ± 0.80 | −0.84 | −1.09 ± 0.77 | −1.10 |
Fig. 2.

Scatter plots showing the distribution of ligand-occupied pockets on A) liganded RNA pockets (red) and liganded protein pockets (gray) and B) liganded RNA pockets (red), protein-bound RNA pockets (blue), and all RNA pockets (gray) in the 2-dimensional property space of hydrophobicity and buriedness. Normalized distribution of C) buriedness and D) hydrophobicity for liganded protein pockets (black) and liganded RNA pockets (red). Normalized distribution of E) buriedness and F) hydrophobicity for all RNA pockets (black), liganded RNA pockets (red), and protein-bound RNA pockets (blue).
Fig. 4.
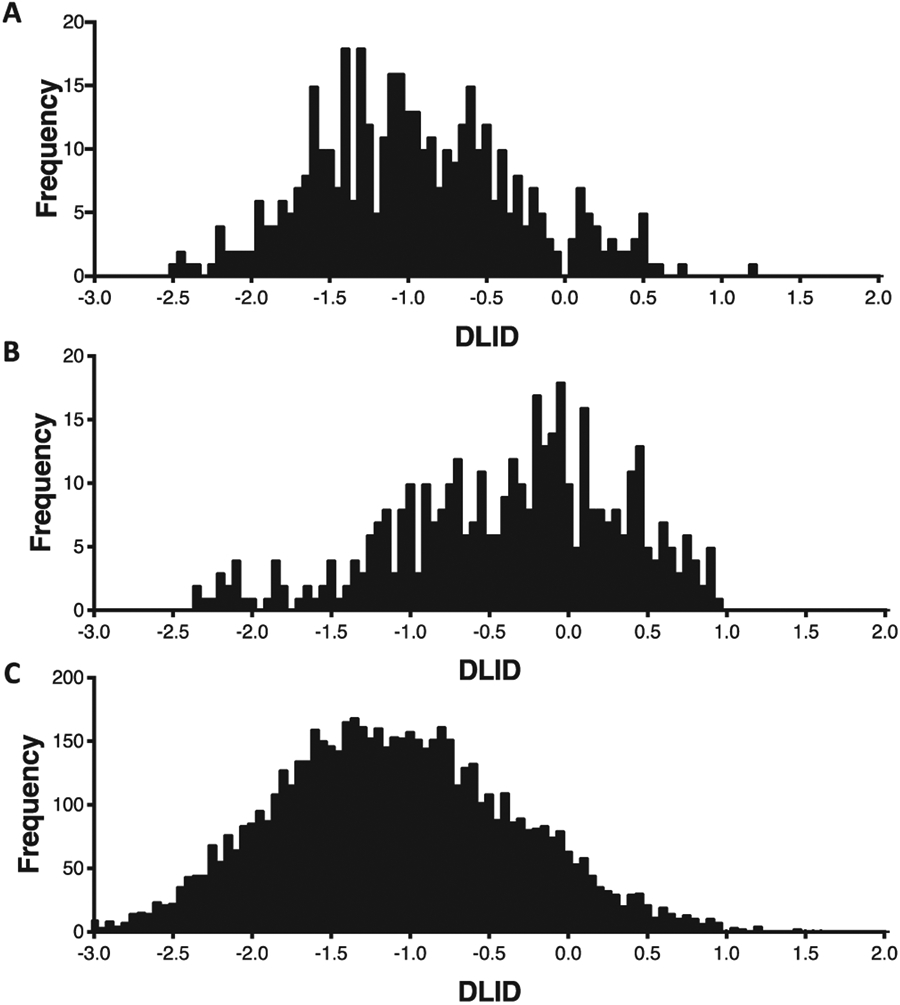
Distribution of DLID values for A) protein-bound RNA pockets, B) liganded RNA pockets, and C) all RNA pockets.
To provide further insight, we next compared liganded proteins and liganded RNAs. Of the 300 liganded RNAs, 90% had multiple pockets. Liganded pockets on proteins had median volume of 352 Å3, while liganded pockets on RNA had a median volume of 280 Å3 (Table 1). The median buriedness of liganded pockets on proteins was 0.81 and liganded pockets on RNA was 0.77 (Fig. 2C). Hydrophobicity of liganded pockets on proteins had a median of 0.52 and RNA was 0.41 (Fig. 2D). Finally, for DLID values, liganded pockets on proteins was 0.13 while RNA was −0.27 (Table 1, Fig. 4B). As can also be seen, liganded pockets on RNA are generally larger in volume, are more buried, and are more hydrophobic than unliganded pockets on RNA. This distinction likely leads to the higher DLID scores of liganded RNA pockets (Table 1, Fig. 4B and 4C). In fact, of liganded RNA, 82% of the highest DLID pockets already contained ligands, highlighting the ability of the algorithm to detect pockets known to be capable of accepting small molecules. In one specific example, the SAM-I/IV riboswitch (PDB: 4OQU) contains a pocket that the S-adenosyl-l-methionine ligand occupies (Fig. 3B).38 The pocket has a hydrophobicity of 0.51 and a buriedness of 0.88, which are properties similar to liganded protein pockets. Additionally, this pocket has a relatively high DLID score of 0.89. In a second example, the malachite green aptamer complexed with tetramethyl-rosamine (PDB: 1F1T), the pocket also has properties similar to liganded protein pockets (hydrophobicity of 0.65 and buriedness of 0.91) (Fig. 3A).39 Thus, successfully liganded pockets on both proteins and RNA have considerable overlap in terms of their physical properties, and can be readily detected with the ICM software suite.
Fig. 3.

Examples of liganded pockets with high DLID scores identified by the PocketFinder algorithm. A) The malachite green aptamer bound to tetramethyl-rosamine (yellow) (PDB: 1F1T) and B) the SAM-I/IV riboswitch bound to S-adenosyl-l-methionine (yellow) (PDB: 4OQU) with calculated pockets shown as blue objects.
In addition to ligand-bound pockets, a considerable proportion of RNA structures in the PDB are bound by proteins, and many of the pockets identified through the analysis presented here are occupied by amino acid side chains. Investigations into protein-protein interactions have revealed that they are often governed by comparatively shallow pockets.40 However, these interactions can indeed be excellent targets for small molecules that mimic the specific interactions made by side-chains. When considering the subset of protein-occupied pockets on RNA, it can be observed that these pockets tend to have comparable volumes and buriedness to all other RNA pockets. However, they are more hydrophobic (Fig. 2E and F). Importantly, protein-bound RNA pockets are enriched with higher DLID scores (Fig. 4). While the median DLID value for these pockets is not as high as known liganded RNA pockets (and is likely influenced by the buriedness component of DLID), there is already evidence that these RNAs engage in specific molecular recognition events. Studying these pockets may present compelling starting points for disrupting protein-RNA interactions with RNA-binding small molecules41,42 (in an analogy to inhibiting protein-protein interactions).43
DLID scores can be used to evaluate specific pockets for their likelihood of accepting a small ligand. Two examples are the F. nucleatum FMN riboswitch (PDB: 2YIF) and the Thermotoga maritima lysine riboswitch (PDB: 3D0X) with DLID scores of 0.97 and 0.87, respectively (Fig. 5B and C).44,45 In both cases, the indicated pocket is empty in the crystal structure but known to bind a small molecule ligand with high affinity and specificity through other studies. In another example, the HIV-1 core packaging signal RNA (PDB: 2N1Q) contains an unliganded pocket with a high DLID score of 1.46 around a pair of three-way junctions (Fig. 5A).46 This pocket is large, somewhat less hydrophobic, and has a high DLID score, suggesting that it could be suitable for binding to small molecules (though it remains unclear whether a ligand bound to this pocket would have a function). Conversely, an RNA duplex containing two G(anti).A(anti) base-pairs (PDB:157D) contains a pocket in a helical portion of the RNA with a poor DLID score of −1.87 (Fig. 5D).47 In the absence of bulges, helical structures form pockets with poor properties that are shallow and solvent exposed, in contrast that the more buried hydrophobic structures complex folds can produce. Many of the examples with highest DLID scores are associated with complex structures and binding pockets, while most of the structures with the lowest DLID score contain high proportions of helical regions.
Fig. 5.

Unliganded structures with high (desirable) and low (undesirable) DLID pockets. Structures of A) the HIV-1 core packaging signal RNA (PDB: 2N1Q), B) the F. nucleatum FMN riboswitch (PDB: 2YIF), C) the Thermotoga maritima lysine riboswitch (PDB: 3D0X), and D) an RNA duplex (PDB: 157D). In each structure, the highest DLID scoring pocket (blue) is shown with its calculated properties displayed below.
2.4. PMI calculations
Normalized Principal Momentum of Inertia (PMI) calculations can be used to provide information to describe molecular shape.10,44-52 As illustrated in Fig. 6, the corners of the graph represent the shape of fundamental geometric objects: rods, discs, and spheres.50 These measurements are normalized to eliminate the dependency of molecular weight, volume, surface area, or other descriptors. PMI calculations can be used broadly to classify collections of molecules, including both macromolecules and small molecules. For example, one previous study utilized PMI calculations to describe a relationship between protein structure and propensity for ligand binding.52 A similar approach has also been used to provide analysis of the shape of small molecule ligands for an RNA triple helix, and more broadly across a library of RNA-binding small molecules.10 Here, we report PMI calculations for the set of RNAs containing ligands in order to provide insight into the structure space occupied by these RNAs. Fig. 6A shows the normalized PMI ratios (NPRs) of selected representative RNA structures.50 The triple-helical stability element at the 3′ end of MALAT153 (PDB: 4PLX) is in the rod-like neighborhood, a quadruplex structure of an RNA aptamer against bovine prion protein is in the sphere-like neighborhood54 (PDB: 2RQJ), and telomeric RNA G-quadruplex complexed with an acridine-based ligand55 (PDB: 3MIJ) is in the disc-like neighborhood. These classifications are consistent with visual inspection of the structures. In addition to these examples, NPRs were calculated for a set of 300 liganded RNA structures containing the highest DLID scores (Fig. 6B). Here, colors and sizes of the points are reflective of the DLID score of the most druggable pocket of each RNA, while X/Y coordinates represent the NPR value. White points reflect negative DLID values, blue points reflect positive DLID values, and the size of the data point represents the magnitude of the value. There is an enrichment of RNAs in the “rod”-like region particularly with negative DLID scores. This may be due to the common RNA structural motifs of helices exhibiting coaxial stacking56 that tend to form more “rod”-like structures that are associated with inferior pockets. Liganded RNA structures with positive DLID scores are variable for their degree of “rod”-like structure, however there were no RNAs with positive DLID scores that were either strongly sphere or disc-shaped.
Fig. 6.

A) NPRs of selected ligand containing RNA structures. PBD codes in red are helixes, PDB codes in blue are quadruplexes, PDB codes in magenta are riboswitches, and black PDB codes are other structures. B) NPRs of 300 liganded RNA structures. White points are negative DLID values, blue points are positive DLID values, and the size of the data point represents the magnitude of the DLID score. C) NPRs of 1000 representative protein structures from the PDB.
In contrast to RNA, liganded proteins occupy a more diverse area of PMI space. A randomly selected set of 1000 liganded proteins have a different distribution of NPR values (Fig. 6C). Rather than being enriched in “rod”-like space, there are more proteins in the “sphere”-like space and in the hybrid (central) neighborhood of the graph. Of note, several in-depth studies have broadly investigated the “druggability” of the PDB using similar approaches.57-59 Importantly, the protein-based data set may also be biased: more globular proteins tend to be easier to crystallize.
3. Discussion
Given that > 80% of our genome is transcribed into RNA but less than 3% of these transcripts code for protein,60,61 there are far more different RNA species within the cell than proteins. While the functions of these transcripts are still being annotated, it has already been demonstrated that many of these RNAs have important, noncoding regulatory roles.5,62,63 Only a small portion of the proteome is considered druggable.64 We report here that, consistent with other studies, the transcriptome also likely contains many ligandable sites. Efforts to understand which classes of RNA are targetable with potent, selective small molecules remain in their infancy, but are necessary to fully evaluate RNA as a target for small molecules.
The study described here demonstrates that many different RNAs throughout the PDB exist in conformations that could be amenable to small molecule binding. Bulk physical properties for pockets on RNA such as buriedness and volume are, in many cases, comparable to values seen on protein targets. However, RNA pockets tend to be on average less hydrophobic than their protein counterparts, a trend that is also observed with liganded RNA pockets relative to liganded protein pockets. Lower DLID scores, likely influenced by these less hydrophobic pockets, may also be attributed to the ‘rod’-like structure that many RNAs form in stem loops, which generally provide inferior binding pockets. In contrast, higher order structures, exemplified predominantly in this study by riboswitches, provide more opportunities for complex folds that lead to pockets with superior properties. Efforts toward finding new ligands would be best spent targeting more complex RNA structures with pockets that have higher DLID scores, given that such pockets are likely to be more rare or unique throughout the transcriptome.
An argument frequently made is that nucleic acids have only four bases relative to 20 amino acids in proteins, and thus nucleic acids are unable to achieve sufficient structural diversity to generate unique binding sites. One compelling counterpoint utilizes information theory to argue that sequences with sufficient “bits” of structural information can in fact be annotated as unique (or near unique) within the transcriptome.24 Here, we present a complementary approach using NPR calculations to estimate the structural space covered by RNAs with known atomic resolution structure. Although the known set of RNA sequences with high resolution structural data to date is surprisingly small, it already covers a broad swath of structural space. Known protein structures are more enriched in sphere-like structure space than RNA (which is more enriched in rod-like space). However, it is clear that much overlap between RNA and proteins can be seen in the hybrid (central) area of the NPR graph. Further, a considerable number of the RNAs with high DLID pockets are in this space as well, demonstrating substantial overlap in structural space between the conformations made by “druggable” proteins and many liganded RNAs. Together with the pocket analysis, this suggests that many RNAs fold into structures that form pockets amenable to selective small molecule binding.
An obvious next step for this and related structure-based approaches to target RNA is to employ virtual ligand screens and related computational approaches. However, computational force fields to predict ligands for such pockets are still being developed and to date are not as effective at prospectively identifying ligands for RNA.65,66 Continued development of such force fields will be needed to implement this approach with success. Furthermore, as evident from the infrequent occurrence of RNA in the PDB, there is far less structural data to draw from when considering ligandable sites on RNA. It is even less frequent to have structural data for RNA with sufficient resolution to perform structure-guided design or virtual ligand screening. Other approaches may be needed to identify molecules for a desired RNA target (at least for now).
High-throughput screening approaches, including phenotypic and target-based screens, have been employed to identify RNA-binding molecules. Small molecule libraries have used by the Disney,67 Hargrove,10 and Schneekloth68 groups, among others,17,69 to find ligands that bind to RNA pockets. These and other RNA binding ligands have been analyzed by Lipinski rules and other metrics for “druggability”.70 However, phenotypic screens, similar to this approach, do not guarantee the identification of molecules that bind directly to RNA, and target-based screens often provide hits with poor selectivity. Hits must be carefully validated through functional assays and biophysical methods. Importantly, identifying a pocket as ligandable does not link that pocket with a function of the RNA.20 Care should be taken to study pockets that directly relate to some function of the associated RNA, for example stabilization of a fold or perturbation/stabilization of an RNA-protein interaction. Binding site accessibility is a further consideration. Additionally, the expression levels of individual RNA targets vary considerably. Thus, it will also be important to consider the expression level of a target, and how much of the target would need to be inhibited to influence a biological response.
One caveat of this approach is that it does not consider dynamics. RNA is often described as a highly dynamic biopolymer, and discussions of RNA structure generally present an ensemble of conformations.71,72 Therefore, this approach (investigating a single representative conformation) does not capture the entirety of conformational space that a given RNA can sample. However, even in the single conformations evaluated here, there is considerable evidence that many RNA structures sample conformations that contain pockets suitable for small molecule binding. A second caveat is that RNA is rarely found on its own in the cell and typically exists in complex with ribonucleoproteins. This analysis does not capture the quaternary structure sampled by such protein/RNA complexes and reports on the structure of the RNA alone. There is evidence that protein/RNA complexes could also provide excellent targets for small molecules.32,73
This work suggests that there are many highly structured RNAs capable of adopting conformations that form pockets with properties suitable for achieving a small molecule recognition event. Many of these RNAs form pockets with similar properties to their protein counterparts. However, finding compounds that selectively bind to nucleic acids and elicit a pharmacological response still remains challenging. Pockets located in RNA that form more unique, complex structures are likely better candidates for finding more selective and higher affinity compounds as they are more likely to be unique within the transcriptome24 and to accept small molecule recognition through specific interactions and molecular recognition events. Continued work in this area will shed light on the suitability of RNA as a drug target, potentially leading to mechanistically novel therapeutics for diseases with no current treatment.
4. Experimental
RNA structures were analyzed for ligandability by using the Internal Coordinate Mechanics method, or ICM (Molsoft, San Diego, CA) software. 1552 RNA structures for this analysis were chosen based on certain selection criteria to include structures of length between 12 and 700 residues per chain and containing two or fewer protein chains. These structures were analyzed by the PocketFinder algorithm contained in the ICM software suite.30,31 The algorithm generates data for identified pockets including hydrophobicity, buriedness, volume, radius, and nonsphericity.30,31 A macro was developed (see Supplemental Fig. S2) to allow application of the PocketFinder algorithm in an automated fashion to identify potential binding pockets and to flag pockets containing atoms belonging to small molecules (defined as containing eight or more non-hydrogen atoms) or protein residues.
Additional data for this structure set-including principle axes of inertia, volume and surface area of RNA structures, the number of nucleic acid residues and chains, the number of protein residues and chains, and the number of ligand molecules contained in each PDB entry-were collected utilizing ICM as well. An additional macro was developed (see Supplemental Fig. S3) to allow automated extraction of these data from the selected PDB entries.
Supplementary Material
Acknowledgments
This work was supported by the Intramural Research Program of the National Institutes of Health, Center for Cancer Research, and the National Cancer Institute (NCI), National Institutes of Health (1 ZIA BC011585 04). We thank Josh Haver for helpful discussions.
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmc.2019.04.010.
References
- 1.Holoch D, Moazed D RNA-mediated epigenetic regulation of gene expression. Nat Rev Genet 2015;16(2):71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roundtree IA, Evans ME, Pan T, He C Dynamic RNA modifications in gene expression regulation. Cell 2017;169(7):1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gerstberger S, Hafner M, Tuschl T A census of human RNA-binding proteins. Nat Rev Genet 2014;15(12):829–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kenyon JC, Prestwood LJ, Lever AML A novel combined RNA-protein interaction analysis distinguishes HIV-1 Gag protein binding sites from structural change in the viral RNA leader. Sci Rep-Uk. 2015:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmitz SU, Grote P, Herrmann BG Mechanisms of long noncoding RNA function in development and disease. Cell Mol Life Sci 2016;73(13):2491–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Faustino NA, Cooper TA Pre-mRNA splicing and human disease. Gene Dev. 2003;17(4):419–437. [DOI] [PubMed] [Google Scholar]
- 7.Hasler R, Kerick M, Mah N, et al. Alterations of pre-mRNA splicing in human inflammatory bowel disease. Eur J Cell Biol 2011;90(6-7):603–611. [DOI] [PubMed] [Google Scholar]
- 8.Yu JY, DeRuiter SL, Turner DL RNA interference by expression of short-interfering RNAs and hairpin RNAs in mammalian cells. Proc Natl Acad Sci USA 2002;99(9):6047–6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yi R, Qin Y, Macara IG, Cullen BR Exportin-5 mediates the nuclear export of premicroRNAs and short hairpin RNAs. Gene Dev. 2003;17(24):3011–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Donlic A, Morgan BS, Xu JL, Liu AQ, Roble C, Hargrove AE Discovery of small molecule ligands for MALAT1 by tuning an RNA-binding scaffold. Angew Chem Int Edit 2018;57(40):13242–13247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bugaut A, Balasubramanian S 5 '-UTR RNA G-quadruplexes: translation regulation and targeting. Nucl Acids Res. 2012;40(11):4727–4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Staple DW, Butcher SE Pseudoknots: RNA structures with diverse functions. PLoS Biol 2005;3(6):956–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rivas E, Eddy SR A dynamic programming algorithm for RNA structure prediction including pseudoknots. J Mol Biol 1999;285(5):2053–2068. [DOI] [PubMed] [Google Scholar]
- 14.Jurica MS, Moore MJ Pre-mRNA splicing: awash in a sea of proteins. Mol Cell 2003;12(1):5–14. [DOI] [PubMed] [Google Scholar]
- 15.Seino S, Bell GI Alternative splicing of human insulin-receptor messenger-RNA. Biochem Bioph Res Co. 1989;159(1):312–316. [DOI] [PubMed] [Google Scholar]
- 16.Disney MD, Yildirim I, Childs-Disney JL Methods to enable the design of bioactive small molecules targeting RNA. Org Biomol Chem. 2014;12(7):1029–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Connelly CM, Moon MH, Schneekloth JS The emerging role of RNA as a therapeutic target for small molecules. Cell Chem Biol 2016;23(9):1077–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bottger EC The ribosome as a drug target. Trends Biotechnol 2006;24(4):145–147. [DOI] [PubMed] [Google Scholar]
- 19.Hermann T Drugs targeting the ribosome. Curr Opin Struc Biol 2005;15(3):355–366. [DOI] [PubMed] [Google Scholar]
- 20.Costales MG, Haga CL, Velagapudi SP, Childs-Disney JL, Phinney DG, Disney MD Small molecule inhibition of microRNA-210 reprograms an oncogenic hypoxic circuit. J Am Chem Soc. 2017;139(9):3446–3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Osborn MF, White JD, Haley MM, DeRose VJ Platinum-RNA modifications following drug treatment in S. cerevisiae identified by click chemistry and enzymatic mapping. ACS Chem Biol 2014;9(10):2404–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang JX, Schultz PG, Johnson KA Mechanistic studies of a small-molecule modulator of SMN2 splicing. Proc Natl Acad Sci USA. 2018;115(20):E4604–E4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Velagapudi SP, Gallo SM, Disney MD Sequence-based design of bioactive small molecules that target precursor microRNAs. Nat Chem Biol 2014;10(4):291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Warner KD, Hajdin CE, Weeks KM Principles for targeting RNA with drug-like small molecules. Nat Rev Drug Discov. 2018;17(8):547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vukovic S, Huggins DJ Quantitative metrics for drug target ligandability. Drug Discov Today. 2018;23(6):1258–1266. [DOI] [PubMed] [Google Scholar]
- 26.Sheridan RP, Maiorov VN, Holloway MK, Cornell WD, Gao YD Drug-like density: a method of quantifying the “Bindability” of a protein target based on a very large set of pockets and drug-like ligands from the protein data bank. J Chem Inf Model 2010;50(11):2029–2040. [DOI] [PubMed] [Google Scholar]
- 27.Dai X, Jing RY, Guo YZ, et al. Predicting the druggability of protein-protein interactions based on sequence and structure features of active pockets. Curr Pharm Design. 2015;21 (21):3051–3061. [DOI] [PubMed] [Google Scholar]
- 28.Hussein HA, Borrel A, Geneix C, Petitjean M, Regad L, Camproux AC PockDrug-Server: a new web server for predicting pocket druggability on holo and apo proteins. Nucleic Acids Res. 2015;43(W1):W436–W442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abagyan R, Kufareva I The flexible pocketome engine for structural chemogenomics. Chemogenom: Methods Appl 2009;575:249–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.An JH, Totrov M, Abagyan R Pocketome via comprehensive identification and classification of ligand binding envelopes. Mol Cell Proteom. 2005;4(6):752–761. [DOI] [PubMed] [Google Scholar]
- 32.Palacino J, Swalley SE, Song C, et al. SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat Chem Biol 2015;11 (7):511. [DOI] [PubMed] [Google Scholar]
- 33.Stelzer AC, Frank AT, Kratz JD, et al. Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nat Chem Biol 2011;7(8):553–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Filikov AV, Mohan V, Vickers TA, et al. Identification of ligands for RNA targets via structure-based virtual screening: HIV-1 TAR. J Comput Aided Mol Des. 2000;14(6):593–610. [DOI] [PubMed] [Google Scholar]
- 35.Jenkins JL, Krucinska J, McCarty RM, Bandarian V, Wedekind JE Comparison of a preQ1 riboswitch aptamer in metabolite-bound and free states with implications for gene regulation. J Biol Chem. 2011;286(28):24626–24637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Howe JA, Xiao L, Fischmann TO, et al. Atomic resolution mechanistic studies of ribocil: a highly selective unnatural ligand mimic of the E. coli FMN riboswitch. RNA Biol 2016;13(10):946–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dibrov SM, Ding K, Brunn ND, et al. Structure of a hepatitis C virus RNA domain in complex with a translation inhibitor reveals a binding mode reminiscent of riboswitches. Proc Natl Acad Sci USA. 2012;109(14):5223–5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trausch JJ, Xu ZJ, Edwards AL, et al. Structural basis for diversity in the SAM clan of riboswitches. Proc Natl Acad Sci USA. 2014;111(18):6624–6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baugh C, Grate D, Wilson C 2.8 a crystal structure of the malachite green aptamer. J Mol Biol 2000;301 (1):117–128. [DOI] [PubMed] [Google Scholar]
- 40.Jubb H, Blundell TL, Ascher DB Flexibility and small pockets at protein-protein interfaces: new insights into druggability. Prog Biophys Mol Bio. 2015;119(1):2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bernstam VA, Gray RH, Bernstein IA Effect of microtubule-disrupting drugs on protein and rna-synthesis in physarum-polycephalum amebas. Arch Microbiol 1980;128(1):34–40. [DOI] [PubMed] [Google Scholar]
- 42.Mei HY, Cui M, Heldsinger A, et al. Inhibitors of protein-RNA complexation that target the RNA: specific recognition of human immunodeficiency virus type 1 TAR RNA by small organic molecules. Biochem-US. 1998;37(40):14204–14212. [DOI] [PubMed] [Google Scholar]
- 43.Arkin MR, Tang YY, Wells JA Small-molecule inhibitors of protein-protein interactions: progressing toward the reality. Chem Biol 2014;21 (9):1102–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vicens Q, Mondragon E, Batey RT Molecular sensing by the aptamer domain of the FMN riboswitch: a general model for ligand binding by conformational selection. Nucleic Acids Res. 2011;39(19):8586–8598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garst AD, Heroux A, Rambo RP, Batey RT Crystal structure of the lysine riboswitch regulatory mRNA element. J Biol Chem. 2008;283(33):22347–22351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keane SC, Heng X, Lu K, et al. RNA structure. Structure of the HIV-1 RNA packaging signal. Science. 2015;348(6237):917–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leonard GA, McAuley-Hecht KE, Ebel S, Lough DM, Brown T, Hunter WN Crystal and molecular structure of r(CGCGAAUUAGCG): an RNA duplex containing two G(anti).A(anti) base pairs. Structure. 1994;2(6):483–494. [DOI] [PubMed] [Google Scholar]
- 48.Wirth M, Sauer WHB Bioactive molecules: perfectly shaped for their target? Mol Inform. 2011;30(8):677–688. [DOI] [PubMed] [Google Scholar]
- 49.Silverman BD Using molecular principal axes for structural comparison: determining the tertiary changes of a FAB antibody domain induced by antigenic binding. Bmc Struct Biol 2007:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sauer WHB, Schwarz MK Molecular shape diversity of combinatorial libraries: a prerequisite for broad bioactivity. J Chem Inf Comp Sci 2003;43(3):987–1003. [DOI] [PubMed] [Google Scholar]
- 51.Aldeghi M, Malhotra S, Selwood DL, Chan AWE Two-and three-dimensional rings in drugs. Chem Biol Drug Des. 2014;83(4):450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Foote J, Raman A A relation between the principal axes of inertia and ligand binding. Proc Natl Acad Sci USA. 2000;97(3):978–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brown JA, Bulkley D, Wang J, et al. Structural insights into the stabilization of MALAT1 noncoding RNA by a bipartite triple helix. Nat Struct Mol Biol 2014;21(7):633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mashima T, Matsugami A, Nishikawa F, Nishikawa S, Katahira M Unique quadruplex structure and interaction of an RNA aptamer against bovine prion protein. Nucleic Acids Res. 2009;37(18):6249–6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Collie GW, Sparapani S, Parkinson GN, Neidle S Structural basis of telomeric RNA quadruplex-acridine ligand recognition. J Am Chem Soc. 2011;133(8):2721–2728. [DOI] [PubMed] [Google Scholar]
- 56.Hendrix DK, Brenner SE, Holbrook SR RNA structural motifs: building blocks of a modular biomolecule. Q Rev Biophys. 2005;38(3):221–243. [DOI] [PubMed] [Google Scholar]
- 57.Kellenberger E, Muller P, Schalon C, Bret G, Foata N, Rognan D sc-PDB: an annotated database of druggable binding sites from the protein data bank. J Chem Inf Model 2006;46(2):717–727. [DOI] [PubMed] [Google Scholar]
- 58.Schmidtke P, Barril X Understanding and predicting druggability. A high-throughput method for detection of drug binding sites. J Med Chem. 2010;53(15):5858–5867. [DOI] [PubMed] [Google Scholar]
- 59.Guo ZJ, Li B, Cheng LT, Zhou SG, McCammon JA, Che JW Identification of protein-ligand binding sites by the level-set variational implicit-solvent approach. J Chem Theory Comput 2015;11(2):753–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lander ES, Consortium IHGS, Linton LM, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. [DOI] [PubMed] [Google Scholar]
- 61.Smrcka AV, Hepler JR, Brown KO, Sternweis PC Regulation of polyphosphoinositide-specific phospholipase-C activity by purified Gq. Science. 1991;251(4995):804–807. [DOI] [PubMed] [Google Scholar]
- 62.Castro-Oropeza R, Melendez-Zajgla J, Maldonado V, Vazquez-Santillan K The emerging role of lncRNAs in the regulation of cancer stem cells. Cell Oncol 2018;41(6):585–603. [DOI] [PubMed] [Google Scholar]
- 63.Quinn JJ, Chang HY Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17(1):47–62. [DOI] [PubMed] [Google Scholar]
- 64.Hopkins AL, Groom CR The druggable genome. Nat Rev Drug Discov. 2002;1(9):727–730. [DOI] [PubMed] [Google Scholar]
- 65.Tan D, Piana S, Dirks RM, Shaw DE RNA force field with accuracy comparable to state-of-the-art protein force fields. Proc Natl Acad Sci USA. 2018;115(7):E1346–E1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Banas P, Hollas D, Zgarbova M, et al. Performance of molecular mechanics force fields for RNA simulations: stability of UUCG and GNRA hairpins. J Chem Theory Comput. 2010;6(12):3836–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rzuczek SG, Southern MR, Disney MD Studying a drug-like, RNA-focused small molecule library identifies compounds that inhibit RNA toxicity in myotonic dystrophy. Acs Chem Biol 2015;10(12):2706–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Abulwerdi FA, Shortridge MD, Sztuba-Solinska J, et al. Development of small molecules with a noncanonical binding mode to HIV-1 trans activation response (TAR) RNA. J Med Chem. 2016;59(24):11148–11160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thomas JR, Hergenrother PJ Targeting RNA with small molecules. Chem Rev. 2008;108(4):1171–1224. [DOI] [PubMed] [Google Scholar]
- 70.Morgan BS, Forte JE, Hargrove AE Insights into the development of chemical probes for RNA. Nucleic Acids Res. 2018;46(16):8025–8037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Al-Hashimi HM, Walter NG RNA dynamics: it is about time. Curr Opin Struc Biol. 2008;18(3):321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sponer J, Bussi G, Krepl M, et al. RNA structural dynamics as captured by molecular simulations: a comprehensive overview. Chem Rev. 2018;118(8):4177–4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sivaramakrishnan M, McCarthy KD, Campagne S, et al. Binding to SMN2 pre-mRNA-protein complex elicits specificity for small molecule splicing modifiers. Nat Commun. 2017:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.