

Abstract
Next‐generation sequencing has resulted in an explosion of rare de novo TTN variants. The clinical interpretation of these de novo variants in patients with recessive titinopathy is very difficult. Here, we provided a useful way to identify compound heterozygous mutations with a de novo one.
Keywords: autosomal recessive, centronuclear myopathy, de novo, TTN
Next‐generation sequencing has resulted in an explosion of rare de novo TTN variants. The clinical interpretation of these de novo variants in patients with recessive titinopathy is very difficult. Here, we provided a useful way to identify compound heterozygous mutations with a de novo one.

1. INTRODUCTION
De novo mutation in an autosomal recessive disorder is a rare event, which may increase the difficulty of the interpretation of gene variants. Here, we report a case of centronuclear myopathy due to compound heterozygous mutations in TTN, including a maternal splice‐site mutation c.32312‐1G>A, and a de novo mutation c.95341C>T (p. Arg31781Ter), which has been previously reported as pathogenic mutation. Paternity was confirmed by short tandem repeats. A single nucleotide polymorphism c.95047A>G was selected, which located 390bp to the N‐terminal of the de novo mutation c.95341C>T, shared by the proband and her father. Clone and sequencing were performed which confirmed that the de novo mutation occurring on paternal chromosome. Our report provided a useful way to identify compound heterozygous mutations with a de novo one for recessive disorders.
With its 363 coding exons and a full‐length transcript of more than 100 kb, TTN(MIM#188840) encodes the largest polypeptide in nature named titin, which is an essential component of the sarcomere, spanning half the sarcomere from the Z‐disk to the M‐line. 1 , 2 , 3 TTN mutations have been reported to cause a wide spectrum of cardiomyopathy, skeletal muscle diseases, or both together. 3 The spectrum of TTN mutations varies in terms of mode of inheritance (dominant versus recessive) and location of variants. Heterozygous truncating TTN mutations have been recognized as the most common genetic cause of dilated cardiomyopathy (DCM), which are predominantly found in the A‐band region of titin. 2 While dominant heterozygous causative variants in the final exon (exon 364) cause adult‐onset tibial muscular dystrophy(TMD, 600334), 4 and variants in exon 344 cause adult‐onset hereditary myopathy with early respiratory failure (HMERF, 603689). 5 , 6 Recessive TTN mutations cause a wide range of muscle disease with or without cardiomyopathy which have been reported under different terms in the past, such as recessive early‐onset myopathy with fatal cardiomyopathy, centronuclear myopathy (CNM), core myopathy with heart disease, and arthrogryposis multiplex congenita with myopathy and others. 3 , 7 , 8 , 9 , 10 , 11 , 12
De novo mutations have been frequently identified at the molecular level in disorders with X‐linked and autosomal dominant inheritance, but rarely in human autosomal recessive disorders. Next‐generation sequencing has resulted in an explosion in the identification of new TTN variants, considering the huge size of the gene. But the clinical interpretation of these variants is still a big challenge, 13 , 14 especially when de novo variant occurs in patients with recessive titinopathy. Here, we report a case of CNM due to a de novo nonsense variant and a maternally inherited splice‐site variant in TTN.
2. CASE REPORT
A 20‐month‐old girl presented to our unit with motor developmental delay and generalized muscle weakness. There were no significant abnormal findings during pre‐ and perinatal periods. She could hold her head up until 10 months, sit without support at 14 months, roll at 17 months, and stand with support at 20 months old. No dyspnea or dysphagia was noted, and her motor skills improved slowly. When she visited us at 4 years of age, she could walk independently with unstable gait. She had never been able to run, jump, or hop. Physical examination revealed generalized limb weakness, which was more prominent in the proximal muscles. She had a positive Gowers’ sign and wadding gait. On manual muscle test, she got a grade of 3 for proximal muscles in all upper and lower extremities. Deep tendon reflexes disappeared. Spinal rigidity was noted without joint contractures, scoliosis, ptosis, or ophthalmoplegia. Her intelligence was always normal for her age. Serum creatine kinase levels and electromyographic findings were normal. Electrocardiography and ultrasonic cardiogram findings were also normal. The X‐ray studies of the whole spine indicated spinal rigidity (Figure 1), while a muscle MRI scan was refused. There was no history of delayed development, weakness, or other neuromuscular disease among the first‐degree relatives of her parents. Both of her parents had normal cardiac findings on recent electrocardiography and ultrasonic cardiogram screenings at 43(father) and 36 years (mother), respectively.
FIGURE 1.

Radiographic examination of the whole spine. Anteroposterior and lateral radiographs of the whole spine show the disappearance of physiological curvature of the neck and chest
Open muscle biopsy from the right quadriceps femoris was performed at 20 months of age. The microscopic study showed remarkable nuclear internalizations, with mildly increased fiber size variation and fibrosis, prevalence of type I fibers, mild muscle fiber‐type disproportion (diameter of type I fiber is smaller than that of type II),and irregular staining of the inter‐myofibrillar network(Figure 2). There were no significant muscle fiber necrosis, regeneration, and infiltration of inflammatory cells. The pathological findings fulfilled the criteria of CNM. 15 Additional features associated with other muscular diseases were not observed. Electron microscopic study showed marked internal nuclei with some disordered myofibers and Z‐disk streaming.
FIGURE 2.
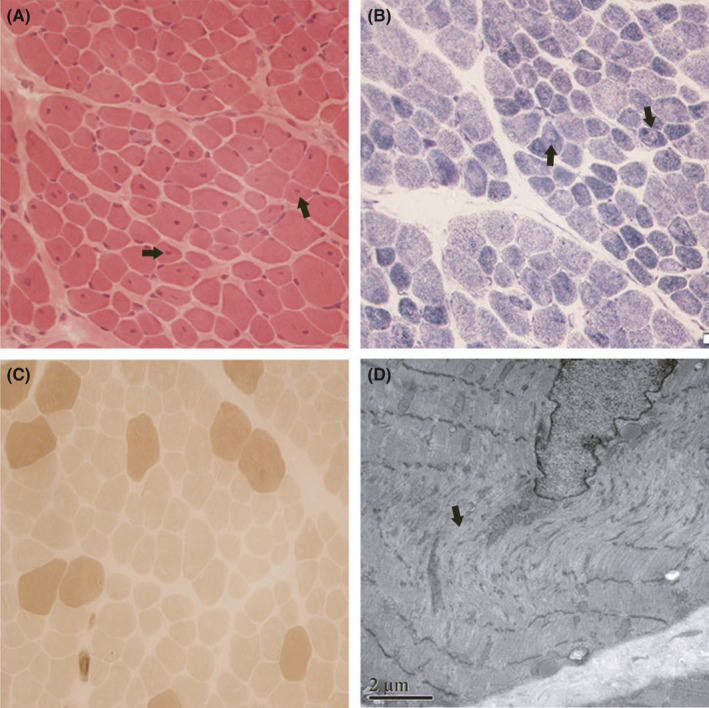
Skeletal muscle histopathological and ultrastructural pattern. A. Hematoxylin & eosin (H&E) staining of transverse muscle sections shows multiple fibers with internal and central nuclei. B. Nicotinamide adenine dinucleotide‐tetrazolium reductase (NADH‐TR) staining shows several fibers with central core‐like areas devoid of oxidative reaction. C. Adenosine triphosphatase (ATPase) staining demonstrates predominance and hypotrophy of darkly stained type I fibers. D. Electron micrograph of muscle shows sarcomere disorganization and central nuclei
Whole‐exome sequencing showed two candidate variants of TTN gene(NM_001267550): One de novo nonsense mutation c.95341C>T (p. Arg31781Ter) in exon 343 has been previously reported as pathogenic. 16 The maternal splice‐site mutation c.32312‐1G>A in intron 127 was predicted to be pathogenic (the dbscSNV_ADA_SCORE is 1.0000, dbscSNV_RF_SCORE is 0.918. Scores >0.6 are considered as splice altering) (Figure 3). 17 Parenthood was confirmed. DNA samples were short tandem repeats (STR) genotyped using Goldeneye 20A (Peoplespot). Clone and sequencing were performed to verify the de novo mutation c.95341C>T occurring on paternal chromosome. A single nucleotide polymorphism (SNP) c.95047A>G (MAF = 0.0631, reference the 1000 genome project, http://1000genomes.org), which was located 390bp to the N‐terminal of the de novo mutation c.95341C>T, was founded from whole‐exome sequencing (NGS) data. The proband and her father were heterozygosis of SNP c.95047A>G, but her mother did not carry this SNP. A 675bp fragment carrying SNP c.95047A>G and the de novo mutation c.95341C>T were amplified by primers 5′‐AAgACTgCCCTTCTCCCTTC‐3’(Forward) and 5′‐ AgTTggTggCAAACCTgAAC −3′ (Reverse), then this fragment was inserted into pEASY‐T1 vector, and the vector was transferred into DH5α cell, finally, 4 monoclones were selected and sequenced. The SNP c.95047A>G and the de novo mutation c.95341C>T were founded in one monoclone, which shown the de novo mutation c.95341C>T was occurring on paternal chromosome. Thus, the TTN mutations were identified to be compound heterogeneous. The mutation rate of the variant was 48/100 and 0/211 in proband and her father, respectively. There was no clue for mosaicism of the variant in father.
FIGURE 3.

Sanger sequence analysis for validation of exome sequencing. Sanger sequence analysis confirmed that the mutation c.95341C>T (p. Arg31781Ter) was de novo, while the mutation c.32312‐1G >A was maternally inherited
3. DISCUSSION
Titinopathies are a group of clinically heterogeneous disorders caused by TTN gene mutations, involving heart, skeletal muscle, or both. 1 , 2 , 11 The clinical presentations of titin‐related myopathies vary according to the mode of inheritance (dominant versus recessive), the specific type (nonsense, splicing, frameshift, missense, etc.), and location of the mutations. 2 , 13 , 14 , 18 Dominant titin‐related myopathies mainly include two diseases caused by mutations in specific exons: adult‐onset tibial muscular dystrophy (exon, 364) and adult‐onset hereditary myopathy with early respiratory failure (exon, 344), presented with special pattern of muscle involvement, and with no cardiomyopathy. 4 , 5 Apart from those, all other titin‐related myopathies are recessively inherited with mutations located along the whole gene, 2 , 12 and 46% of patients with recessive congenital titinopathies were noted with cardiac involvement. 11
According to the onset age, recessive titin‐related myopathies were classified as congenital form (onset within the first 12 months of life) and non‐congenital form (a childhood or later onset). 3 , 11 , 12 Patients with titin‐related congenital myopathies presented with early‐onset hypotonia and a delayed motor development, usually without ophthalmoplegia. Axial involvement has been reported to be one of the most prominent findings in patients with recessive congenital titin‐related myopathies. 11 , 12 Our patient exhibited early‐onset generalized muscle weakness with marked neck involvement and early development of spinal rigidity, without ophthalmoplegia or ptosis. Her clinical features are conformed to the presentations of recessive congenital titin‐related myopathies. Different histopathologic changes have been reported previously in congenital titinopathies including increased muscle fiber size variation, increased internalized nuclei, cores, and structural abnormalities. 7 , 9 , 11 The pathologic changes of our patient fulfilled the criteria of CNM.
Extensive genetic screening results in numerous rare variants identified in TTN, given its large size. But the interpretation of these mutations is still a great challenge. 13 , 14 Heterozygous truncating TTN mutations have been recognized as the most common genetic cause of dilated cardiomyopathy (DCM). 2 , 19 Dominant titin‐related myopathies were caused by missense mutations in specific exon(exon 364,exon 344). 4 , 5 Genotypes of recessive titin‐related myopathies are more complicated. Biallelic deleterious truncating mutations such as nonsense and frameshift variants with proven segregation in trans were considered pathogenic. 3 The interpretation of splicing variants should be careful, even the variant is in the canonical splice sites. Downstream effect of splicing variants on the messenger RNA (mRNA) and protein level should be carefully evaluated when possible. 3 , 12 , 14 , 18 While the interpretation of missense variants is tricky because the current bioinformatics tools cannot predict their functional impact effectively, and only very few missense variants have been confirmed as pathogenic. 11 , 20 , 21 The de novo mutation c.95341C>T (p. Arg31781Ter) in our patient has previously been reported to be related to dominant DCM. 16 The variant is very rare in population and is not recorded in population databases such as 1000 Genomics, ExAC, and GnomeAD. According to Orphanet database, the prevalence of familial isolated dilated cardiomyopathy is 1‐5/10 000, while the prevalence of recessive titinopathies is currently unknown. According to ACMG guidelines and the current clinical interpretation of nonsense titin variants for muscular disorders, 14 integrated with the phenotype and muscle pathology of the patient, the TTN c.95341C>T (p. Arg31781Ter) variant is classified as pathogenic. As to the maternal splicing site mutation c.32312‐1G>A, it is in the canonical splice sites. It is predicted to be splice altering with high prediction score using dbscSNV tools, observed in trans with the pathogenic mutation c.95341C>T (p. Arg31781Ter), and in line with the phenotype of congenital titinopathy. The splicing mutation c.32312‐1G>A is also classified as pathogenic. It is a pity that there was no result from mRNA or protein level to verify its precise mutant effect.
The TTN‐related cardiac risk depends on the titin isoforms caused by the mutations. Based on the presence of the N2A and N2B elements in the I‐band region, the titin isoforms were divided into three main classes: skeletal muscles express N2A isoform; cardiac muscle express N2BA and N2B isoforms. 1 , 2 As to recessive titinopathies, patients with two pathogenic mutations predicted to affect both N2BA and N2B cardiac isoforms appeared to be at higher risk of cardiac involvement. 9 , 11 , 12 In our case, TTN c.95341C>T is located in exon 343 and the A band (http://cardiodb.org/titin/titin_transcripts.php), predicted to affect the three isoforms N2A, N2BA, and N2B. The other slicing site mutation c.32312‐1G>A is located in the 127 intron and predicted to influence the expression of the I band, affecting isoforms N2BA and N2A. Though the mutant TTN c.95341C>T has previously been reported to be related to dominant DCM, it is hard to predict the cardiac risk related to the combined action of those two mutations. Long‐term follow‐up is required to assess if myocardium is involved or not in our patient.
With the advances of next‐generation sequencing, the number of rare and unique TTN variants was increased rapidly, which give rise to a great challenge related to the diagnosis of TTN‐related myopathy. While confirmed cases are important in further establishing potential genotype‐phenotype correlations. We reported a case of recessive TTN‐related CNM with one de novo mutation, which enriched the TTN phenotypic and genotypic spectrum. Additionally, we provided an elegant approach to identify compound heterozygous mutations with a de novo one for autosomal recessive disorders.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
AUTHOR CONTRIBUTIONS
HS and CX designed and performed the study. HS and ZY collected data. CX and XH wrote the draft manuscript. MY carried out molecular analysis. All authors contributed to the writing and reviewing of the manuscript, and approved the final manuscript for submission.
ETHICAL APPROVAL
Written informed consent was received from the patients. This study has been approved by the Ethics Committee of Peking University First Hospital (Beijing, China).
ACKNOWLEDGEMENTS
We thank the patient and her family. We thank Wei Wei in the Beijing Kangso Medical Inspection Co. Ltd for his technical support with the gene mutation analysis. Published with written consent of the patient.
Huang S, Ma Y, Zhang Y, Xiong H, Chang X. Centronuclear myopathy due to a de novo nonsense variant and a maternally inherited splice‐site variant in TTN: A case report. Clin Case Rep. 2021;9:e04478. 10.1002/ccr3.4478
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Tharp CA, Haywood ME, Sbaizero O, Taylor MRG, Mestroni L. The giant protein titin's role in cardiomyopathy: genetic, transcriptional, and post‐translational modifications of TTN and their contribution to cardiac disease. Front Physiol. 2019;10:1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kellermayer D, Smith JE 3rd, Granzier H. Titin mutations and muscle disease. Pflugers Arch. 2019;471(5):673‐682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Udd B. The constantly evolving spectrum of phenotypes in titinopathies ‐ will it ever stop? Curr Opin Neurol. 2020;33(5):604‐610. [DOI] [PubMed] [Google Scholar]
- 4. Hackman P, Vihola A, Haravuori H, et al. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal‐muscle protein titin. Am J Hum Genet. 2002;71(3):492‐500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Palmio J, Leonard‐Louis S, Sacconi S, et al. Expanding the importance of HMERF titinopathy: new mutations and clinical aspects. J Neurol. 2019;266(3):680‐690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pfeffer G, Elliott HR, Griffin H, et al. Titin mutation segregates with hereditary myopathy with early respiratory failure. Brain. 2012;135(Pt 6):1695‐1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chauveau C, Bonnemann CG, Julien C, et al. Recessive TTN truncating mutations define novel forms of core myopathy with heart disease. Hum Mol Genet. 2014;23(4):980‐991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chervinsky E, Khayat M, Soltsman S, Habiballa H, Elpeleg O, Shalev S. A homozygous TTN gene variant associated with lethal congenital contracture syndrome. Am J Med Genet A. 2018;176(4):1001‐1005. [DOI] [PubMed] [Google Scholar]
- 9. Ge L, Fu X, Zhang W, et al. Recessive mutations in proximal I‐band of TTN gene cause severe congenital multi‐minicore disease without cardiac involvement. Neuromuscul Disord. 2019;29(5):350‐357. [DOI] [PubMed] [Google Scholar]
- 10. Jang JY, Park Y, Jang DH, Jang JH, Ryu JS. Two novel mutations in TTN of a patient with congenital myopathy: A case report. Mol Genet Genomic Med. 2019;7(8):e866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Oates EC, Jones KJ, Donkervoort S, et al. Congenital Titinopathy: Comprehensive characterization and pathogenic insights. Ann Neurol. 2018;83(6):1105‐1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Savarese M, Vihola A, Oates EC, et al. Genotype‐phenotype correlations in recessive titinopathies. Genet Med. 2020;22(12):2029‐2040. [DOI] [PubMed] [Google Scholar]
- 13. Savarese M, Johari M, Johnson K, et al. Improved criteria for the classification of titin variants in inherited skeletal myopathies. J Neuromuscul Dis. 2020;7(2):153‐166. [DOI] [PubMed] [Google Scholar]
- 14. Savarese M, Maggi L, Vihola A, et al. Interpreting genetic variants in titin in patients with muscle disorders. JAMA Neurol. 2018;75(5):557‐565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dubowitz V, Sewry CA, Oldfors A. Muscle Biopsy. A Practical Approach, 5th edn. Edinburgh: W.B. Saunders; 2020:530. [Google Scholar]
- 16. Haas J, Frese KS, Peil B, et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J. 2015;36(18):1123‐1135. [DOI] [PubMed] [Google Scholar]
- 17. Liu X, Wu C, Li C, Boerwinkle E. dbNSFP v3.0: A one‐stop database of functional predictions and annotations for human nonsynonymous and splice‐site SNVs. Hum Mutat. 2016;37(3):235‐241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Savarese M, Jonson PH, Huovinen S, et al. The complexity of titin splicing pattern in human adult skeletal muscles. Skelet Muscle. 2018;8(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Akhtar MM, Lorenzini M, Cicerchia M, et al. Clinical phenotypes and prognosis of dilated cardiomyopathy caused by truncating variants in the TTN gene. Circ Heart Fail. 2020;13(10):e006832. [DOI] [PubMed] [Google Scholar]
- 20. Khan A, Wang R, Han S, et al. Homozygous missense variant in the TTN gene causing autosomal recessive limb‐girdle muscular dystrophy type 10. BMC Med Genet. 2019;20(1):166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Perrin A, Juntas Morales R, Rivier F, et al. The importance of an integrated genotype‐phenotype strategy to unravel the molecular bases of titinopathies. Neuromuscul Disord. 2020;30(11):877‐887. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.