

The leading form of dementia worldwide, Alzheimer’s disease (AD) is a common neurodegenerative disorder. The underlying causes of AD are not well understood, and no current treatments are preventing the onset or delay progression of the disease. Currently, most investigation is directed towards the amyloid-beta (Aβ) and tau pathologies, yet there are many other underlying processes that have been implicated to contribute directly to AD progression. One such phenomenon is glutamatergic excitotoxicity, a loss of neuromodulatory balance inducing a hyper-excitable neuronal state, leading to cell death across several brain regions (Zhang et al., 2016; Bukke et al., 2020). Glutamate is the primary excitatory neurotransmitter in the brain and is involved in many critical signaling and metabolic functions but control of the glutamatergic system requires constant moderation to avoid excitotoxicity occurring (Bukke et al., 2020). As yet, glutamatergic signaling changes that contribute to this process, or result due to this process, have not been thoroughly investigated.
Glutamate acts on a variety of receptors, traditionally categorized as ionotropic and metabotropic. Ionotropic receptors include the N-methyl-D-aspartate receptor (NMDAR), alpha-amino-3-hydroxy-5-methylisoxazole-4-propinoic acid receptor (AMPAR), and kainate (GluK) receptor classes. The metabotropic class of receptors is subdivided into three functionally distinct groups; group I are coupled with phospholipase C, while groups II and III are coupled with adenylyl cyclase. These receptor subtypes can be found on dendrites of postsynaptic cells, astrocytes, and oligodendrocytes, as well as on glial cells. The receptors are formed by multiple subunits which are classified as follows: GluN1-3 (NMDA), GluA1-4 (AMPA), GluK1-5 (kainate), and mGluR1-5 (metabotropic). Vesicular glutamate receptors (VGluTs), categorized into VGluT1 and VGluT2, are present at presynaptic neurons and are vital in maintaining vesicular glutamate concentrations. The glutamatergic system is significantly implicated in AD pathogenesis, with NMDA receptors most frequently associated with the disease, perhaps due to their ability to mediate excitotoxicity but other glutamatergic signaling components are also affected in the disease (Zhang et al., 2016; Bukke et al., 2020).
Aβ is a 4 kDa peptide product derived from the cleavage of amyloid precursor protein (APP). Cleavage of APP by alpha and gamma-secretase yields non-neurotoxic fragments, while Aβ is generated through the beta and gamma-secretase pathway. Numerous Aβ species exist, but Aβ1–40 and Aβ1–42 are the most abundant, the latter being the dominant form of Aβ in the amyloid plaques. This Aβ can further aggregate into larger polymeric structures, including oligomers, protofibrils, and amyloid fibrils with different functional properties. Amyloid plaques are formed from the assembly of insoluble amyloid fibrils, whereas amyloid oligomers are soluble and appear to exhibit much higher cytotoxicity (Bukke et al., 2020; Yeung et al., 2020a). Both amyloid plaques and soluble amyloid oligomers have been implicated in the pathogenesis of AD. Aβ is thought to be responsible for many pathogenic changes in AD, including synaptic disruption, mitochondrial and vascular dysfunction, neuroinflammation, and excitotoxicity (Zhang et al., 2016; Bukke et al., 2020).
In our recent studies, we characterized the expression changes of glutamatergic components in response to Aβ1–42exposure. We quantified expression changes in glutamatergic receptor subunits and transporters spatially within the cornu ammonis (CA)1, CA3, and dentate gyrus (DG) of the mouse hippocampus 3 and 30 days after Aβ1–42injection. These studies are the first comprehensive anatomical investigations focusing on the acute and chronic effects of Aβ1–42 on glutamate receptor and transporter expression in the hippocampus (Yeung et al., 2020a, b). We injected Aβ1–42 into the CA1 region of the mouse hippocampus as this region has been reported to be one of the earliest hippocampal regions to exhibit functional changes in AD. At the 30 day time point, we have demonstrated alterations in the expression of AMPAR subunit GluA1 and vesicular glutamate transporter (VGluT) 1 in mice injected with Aβ1–42 within the CA1 region of the hippocampus compared to naïve control and artificial cerebrospinal fluid-injected mice (Figure 1). We also demonstrate that these changes are region- and layer-specific and thereby complex, which may highlight specific compensatory mechanisms or spatial susceptibility of glutamatergic components to Aβ1–42-induced damage.
Figure 1.
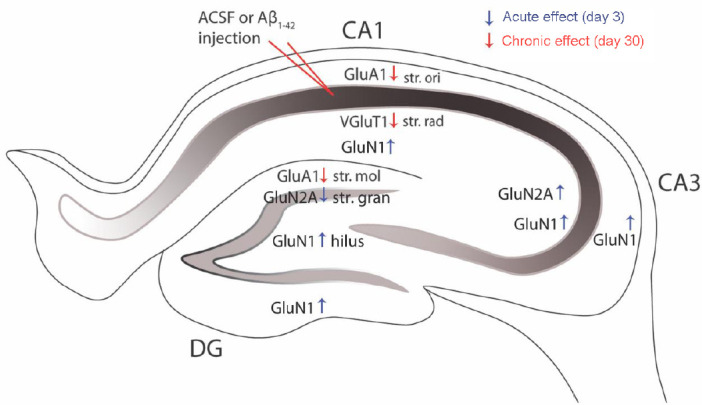
Aβ1–42-induced hippocampal glutamatergic receptor and transporter remodeling.
Aβ1–42 induces region- and layer-specific expression changes in the hippocampus of the glutamatergic receptor subunits GluA1, GluN1, GluN2A, and the transporter VGluT1, suggesting a complex and spatial vulnerability of this pathway during the development of AD neuropathology. The acute (blue arrows) and chronic (red arrows) expression changes induced by Aβ1–42 impact different glutamatergic signaling components. These glutamatergic receptor subunit and transporter expression changes have the ability to impair glutamate release, receptor activation and LTP leading to deficits in cognitive function and memory. Aβ: Amyloid-beta; ACSF: artificial cerebrospinal fluid: CA: cornu ammonis; DG: dentate gyrus; GluA1: glutamate α-amino-3-hydroxy-5-methyl-4- isoxazolepropionic acid (AMPA) receptor type subunit A1; GluN1: glutamate N-methyl-D-aspartate type subunit 1; GluN2A: glutamate N-methyl-D-aspartate type subunit 2A; str mol: startum moleculare; str. gran: stratum granulosum; str. ori: stratum oriens; str. rad: stratum radiatum; VGluT: vesicular glutamate transporter.
Examination of the acute effects of Aβ1–42 injection is relevant as physiological changes have been observed in human patients, with an increase in glutamatergic synapses observed in mildly cognitively impaired patients and subsequent reduction in AD patients, potentially reflecting changes related to the progression of the pathology (Yeung et al., 2020a). A few in vitro studies also report acute changes in receptor distribution and expression as a response to Aβ1–42exposure. A recent study demonstrated AMPAR internalization in human cultured primary neurons 4 hours after application of Aβ (Zhang et al., 2019). In another study, oligomerized Aβ induced a rapid increase in surface expression of GluA1 in hippocampal slices 30 minutes after exposure, but no changes were found in GluA2/3 expression (Whitcomb et al., 2015). We observed increased expression in the DG hilus and ventral stratum granulosum, CA3 stratum radiatum and stratum oriens, and CA1 stratum radiatum of the GluN1 subunit, and increased expression within the CA3 stratum radiatum and decreased expression within the DG stratum granulosum of the GluN2A subunit in Aβ1–42 injected mice compared to naïve control, and a similar trend was observed when compared to ACSF-injected mice (Yeung et al., 2020a). Some of these alterations in expression levels are influenced by the effect of the microinjection. We found that GluN1 expression increased within the stratum oriens and stratum pyramidale in the CA1 region of ACSF-injected mice compared to the naïve control mice (Yeung et al., 2020a). This is the limitation of the experimental design, but this model allows us to examine the acute effects of Aβ while transgenic AD mice do not provide any information about potential acute molecular and cellular changes. IImportantly, glutamate receptor subunit and transporter expression seem to be robust and resistant to Aβ1–42 at 3 days after injection (Yeung et al., 2020a). This is most likely due to compensatory changes that can balance the early pathological changes in glutamate levels and excitotoxicity and because this examination has been performed before the onset of pyramidal neuronal loss in the CA1 region. Furthermore, these observations suggest that the acute and chronic changes induced by Aβ1–42 impact different glutamatergic signaling components (Yeung et al., 2020a, b).
Glutamate levels under normal physiological conditions are finely regulated. The low concentration within the synaptic cleft is due to the action of excitatory amino acid transporters (EAATs). Impairment of EAATs has been reported in AD, with studies observing a reduction in glutamate transporter capacity with a concomitant rise in extracellular glutamate concentration. This has been partly attributed to damage by reactive oxygen species and products of lipid peroxidation (Bukke et al., 2020). Aβ-mediated release of glutamate from microglia has also been reported. The aberrant accumulation of glutamate results in glutamate excitotoxicity, with glutamate diffusing into extrasynaptic areas and activating extrasynaptic NMDARs. Overactivation of these receptors can result in excessive calcium influx, potentially disrupting the intracellular balance of calcium and other ions (Zhang et al., 2016). Indeed, stimulation of EAATs has neuroprotective effects against excitotoxicity through efficacious glutamate control (Bukke et al., 2020). VGluTs are also critical in regulating glutamate levels. The VGluTs are involved in the packaging of glutamate into vesicles prior to release into the synaptic cleft. The expression of VGluTs regulates the quantity of vesicular glutamate release (Wilson et al., 2005). The effect of Aβ on VGluT expression and function is poorly understood but a decrease of VGluT1 was observed in hippocampal cultures exposed to Aβ (Rodriguez-Moreno and Lerma, 1998) and in several regions of the AD brain (Kashani et al., 2008). These observations are in agreement with our recent finding showing VGluT1 expression decreased following Aβ treatment (Yeung et al., 2020b) suggesting that Aβ might be responsible for decreased VGluT1 expression in the AD brain.
While we observed downregulation of VGluT1 within the CA1 stratum radiatum in Aβ1–42-injected mice compared to ACSF-injected or NC mice VGluT2 levels were not affected by Aβ1–42 (Figure 1). We also found that longer exposure to Aβ1–42 is required to induce a decrease in VGluT1 expression, as no changes were observed 3 days after Aβ injection (Yeung et al., 2020a). The preferential localization of Aβ to VGluT1 positive terminals in the post-mortem human parietal cortex of AD subjects might explain why we observed alterations in VGluT1 expression while the expression of VGluT2 remained relatively robust (Sokolow et al., 2012). The expression of VGluTs is controlled via a negative feedback, with VGluT1 expression adjusted in response to calcium influx and neuronal excitability (Wilson et al., 2005). Blocking of NMDARs and AMPARs results in increased expression of VGluTs (Wilson et al., 2005), but conversely, aberrant activation of these receptors could lead to a neuroprotective decline in VGluT1 levels as observed in our study. VGluT1 decline might be a neuroprotective mechanism as this decrease in expression could reduce glutamatergic transmission but it will result in a loss of effective neuronal communication. VGluT1 heterozygous knock-out mice show impaired long-term potentiation (LTP) formation indicating that VGluT1 is critical in memory processes (Balschun et al., 2010). In addition, a decrease in VGluT1 levels not only disrupts the glutamatergic signaling in the AD brain but also increases the vulnerability to Aβ-induced neuroinflammation (Rodriguez-Perdigon et al., 2016).
As mentioned above, Aβ has a direct effect on glutamate receptor function, with many pathophysiological changes such as synaptic and neuronal loss attributed to changes in the composition and expression of glutamate receptor subunits. Synaptic loss is one of the main hallmarks of AD and has a definitive correlation with patient cognitive function, with early symptoms correlating with dysfunction of glutamatergic synapses. Studies have shown significant decreases in density of glutamatergic synapses in the cortex and the number of synapses per neuron in AD patients compared with control cases. Some glutamate receptor subunits appear to act as binding sites for Aβ, with oligomers binding to neurons expressing GluN1 and GluN2B. This binding has a direct effect on glutamate receptor expression, with interactions inducing the endocytosis of both AMPA and NMDA receptors. This is consistent with studies showing that the vulnerability of neurons to glutamate toxicity is dependent on the types of glutamate receptors expressed on the cell surface. GluN2B has been shown to mediate calcium influx and neuronal loss within rat hippocampal cultures following the injection of Aβ1–42. Furthermore, GluN2B antagonists or negative allosteric modulators provide neuroprotective effects in rodent hippocampal cultures treated with Aβ, indicating a potential for aberrant GluN2B subunit-containing NMDARs in disease. Activation of NMDARs increases Aβ1–42synthesis, resulting in a positive feedback cycle of continuing NMDAR activation and Aβ1–42accumulation. We found no significant changes in NMDA receptor subunit expression in the mouse hippocampus 3 and 30 days following Aβ1–42 administration (Yeung et al., 2020b; Yeung et al., 2020a). However, on day 3 after Aβ1–42injection, we observed a trend towards GluN1 subunit increase in the CA1, CA3, and DG regions when compared with ACSF-injected controls suggesting an early response to Aβ1–42 resulting in GluN1 subunit upregulation (Yeung et al., 2020a). Aβ-injected mice showed stronger immunostaining consisting of a more diffuse pattern within the hilus and stratum moleculare, and strong cellular staining within the stratum granulosum. Besides, Aβ-injected mice displayed increased neuronal staining within the hilar area. We also detected a nonsignificant trend towards an increase in GluN2A subunit expression in the CA3 region and decreased expression within the DG region in Aβ1–42 injected mice when compared to ACSF controls (Yeung et al., 2020a) (Figure 1). In Aβ-injected mice, we found GluN2A immunoreactivity localized closer to cellular bodies and their associated processes within the DG hilus and stratum moleculare, and an increase in diffuse immunolabeling within the stratum moleculare compared with controls. The alteration in GluN2A subunit expression is in agreement with a decrease seen in human AD cases (Zhang et al., 2016). GluN2A has been shown to reduce excitotoxic calcium transients induced by Aβ1–42, therefore a decrease in GluN2A subunit expression may render the system susceptible to pathogenic calcium fluxes triggered by Aβ.
Aβ1–42 has also been shown to interact with and disrupt normal AMPAR activity, resulting in synaptic dysregulation but also with proteins responsible for maintaining glutamate homeostasis such as uptake and release. Previous findings regarding the expression of AMPARs in AD have been controversial, with increases, decreases, and no alterations found. We reported a downregulation in GluA1 subunit expression within the stratum oriens of the CA1 region and the dorsal stratum moleculare of the DG region in the mouse hippocampus 30 days following Aβ1–42 administration and a trend toward decreased GluA1 expression has also been observed in the stratum radiatum of the CA1 region (Yeung et al., 2020b) (Figure 1). Double knockin APP/PS-1 mice also show a reduction in membrane surface GluA1 expression in the stratum radiatum of the CA1 region. In hippocampal neurons of transgenic mice overexpressing APP, and in hippocampal neurons cultured with Aβ1–42oligomers, the frequency and amplitude of AMPA-mediated excitatory postsynaptic currents are significantly decreased. These findings suggest that Aβ reduces AMPAR activation that could lead to LTP impairment. Indeed, mice carrying a variant form of GluA1 display weakened LTP generation. However, we observed no changes in AMPAR GluA2 subunit expression levels in the Aβ1–42-injected mice compared to control and ACSF-injected groups although downregulation of this subunit has been associated with excitotoxicity in a variety of disease conditions (Yeung et al., 2020b), suggesting that disrupted GluA2 subunit expression is most likely not the only factor accounting for excessive intracellular calcium influx and neuronal death. We did not examine specifically the intracellular and extracellular localization of the subunit but previous studies postulated that Aβ induces GluA2 subunit endocytosis and this is an underlying factor of Aβ-mediated synaptic loss.
In summary, the glutamatergic system is relatively robust against Aβ1–42-induced neurotoxic changes, especially during acute phases of exposure, but it is important to consider that even minor alterations in specific receptor subunit and transporter expression could lead to significant pathophysiological outcomes. Therefore, glutamatergic changes in response to Aβ warrants further research. More human studies are required to examine whether Aβ1–42 is capable of inducing the pathological mechanisms observed in AD and a better understanding of glutamatergic receptor and transporter changes in AD may lead to new pharmacological approaches to target specific components of the signaling pathway.
Footnotes
C-Editors: Zhao M, Song LP; T-Editor: Jia Y
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
References
- 1.Balschun D, Moechars D, Callaerts-Vegh Z, Vermaercke B, Van Acker N, Andries L, D’Hooge R. Vesicular glutamate transporter VGLUT1 has a role in hippocampal long-term potentiation and spatial reversal learning. Cereb Cortex. 2010;20:684–693. doi: 10.1093/cercor/bhp133. [DOI] [PubMed] [Google Scholar]
- 2.Bukke VN, Archana M, Villani R, Romano AD, Wawrzyniak A, Balawender K, Orkisz S, Beggiato S, Serviddio G, Cassano T. The dual role of glutamatergic neurotransmission in Alzheimer’s disease: from pathophysiology to pharmacotherapy. Int J Mol Sci. 2020;21:E7452. doi: 10.3390/ijms21207452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kashani A, Lepicard E, Poirel O, Videau C, David JP, Fallet-Bianco C, Simon A, Delacourte A, Giros B, Epelbaum J, Betancur C, El Mestikawy S. Loss of VGLUT1 and VGLUT2 in the prefrontal cortex is correlated with cognitive decline in Alzheimer disease. Neurobiol Aging. 2008;29:1619–1630. doi: 10.1016/j.neurobiolaging.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 4.Rodriguez-Moreno A, Lerma J. Kainate receptor modulation of GABA release involves a metabotropic function. Neuron. 1998;20:1211–1218. doi: 10.1016/s0896-6273(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 5.Rodriguez-Perdigon M, Tordera RM, Gil-Bea FJ, Gerenu G, Ramirez MJ, Solas M. Down-regulation of glutamatergic terminals (VGLUT1) driven by Abeta in Alzheimer’s disease. Hippocampus. 2016;26:1303–1312. doi: 10.1002/hipo.22607. [DOI] [PubMed] [Google Scholar]
- 6.Sokolow S, Luu SH, Nandy K, Miller CA, Vinters HV, Poon WW, Gylys KH. Preferential accumulation of amyloid-beta in presynaptic glutamatergic terminals (VGluT1 and VGluT2) in Alzheimer’s disease cortex. Neurobiol Dis. 2012;45:381–387. doi: 10.1016/j.nbd.2011.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whitcomb DJ, Hogg EL, Regan P, Piers T, Narayan P, Whitehead G, Winters BL, Kim DH, Kim E, St George-Hyslop P, Klenerman D, Collingridge GL, Jo J, Cho K. Intracellular oligomeric amyloid-beta rapidly regulates GluA1 subunit of AMPA receptor in the hippocampus. Sci Rep. 2015;5:10934. doi: 10.1038/srep10934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilson NR, Kang J, Hueske EV, Leung T, Varoqui H, Murnick JG, Erickson JD, Liu G. Presynaptic regulation of quantal size by the vesicular glutamate transporter VGLUT1. J Neurosci. 2005;25:6221–6234. doi: 10.1523/JNEUROSCI.3003-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yeung JHY, Palpagama TH, Tate WP, Peppercorn K, Waldvogel HJ, Faull RLM, Kwakowsky A. The acute effects of amyloid-beta1-42 on glutamatergic receptor and transporter expression in the mouse hippocampus. Front Neurosci. 2020a;13:1427. doi: 10.3389/fnins.2019.01427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yeung JHY, Calvo-Flores Guzman B, Palpagama HT, Ethiraj J, Zhai Y, Tate WP, Peppercorn K, Waldvogel HJ, Faull RLM, Kwakowsky A. Amyloid-beta1-42 induced glutamatergic receptor and transporter expression changes in the mouse hippocampus. J Neurochem. 2020b;155:62–80. doi: 10.1111/jnc.15099. [DOI] [PubMed] [Google Scholar]
- 11.Zhang B, Li Y, Liu JW, Liu XW, Wen W, Cui Y, Huang SM. Postsynaptic GluR2 involved in amelioration of Abeta-induced memory dysfunction by KAIXIN-San through rescuing hippocampal LTP in mice. Rejuvenation Res. 2019;22:131–137. doi: 10.1089/rej.2018.2080. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, Li P, Feng J, Wu M. Dysfunction of NMDA receptors in Alzheimer’s disease. Neurol Sci. 2016;37:1039–1047. doi: 10.1007/s10072-016-2546-5. [DOI] [PMC free article] [PubMed] [Google Scholar]