

Abstract
Long-read transcriptome sequencing is designed to sequence full-length RNA molecules and advantageous for identifying alternative splice isoforms; however, in the absence of a reference genome, it is difficult to accurately locate splice sites because of the diversity of patterns of alternative splicing (AS). Based on long-read transcriptome data, we developed a versatile tool, IsoSplitter, to reverse-trace and validate AS gene “split sites” with the following features: (i) IsoSplitter initially invokes a modified SIM4 program to find transcript split sites; (ii) each split site is then quantified, to reveal transcript diversity, and putative isoforms are grouped into gene clusters; (iii) an optional step for aligning short reads is provided, to validate split sites by identifying unique junction reads, and revealing and quantifying tissue-specific alternative splice isoforms. We tested IsoSplitter AS prediction using data sets from multiple model and nonmodel plant species and showed that the IsoSplitter pipeline is efficient to handle different transcriptomes with high accuracy. Furthermore, we evaluated the IsoSplitter pipeline compared with that of the splice junction identification tools, Program to Assemble Spliced Alignments (PASA software needs a reference genome for AS identification) and AStrap, using data from the model plant Arabidopsis thaliana. We found that IsoSplitter determined more than twice as many AS events than AStrap analysis, and 94.13% of the IsoSplitter predicted AS events were also identified by the PASA analysis. Starting from a simple sequence file, IsoSplitter is an assembly-free tool for identification and characterization of AS. IsoSplitter is developed and implemented in Python 3.5 using the Linux platform and is freely available at https://github.com/Hengfu-Yin/IsoSplitter.
Keywords: gene structure, gene expression, alternative splicing, sequence alignment
INTRODUCTION
Alternative splicing (AS) is an evolutionarily critical characteristic of eukaryotic genes that increases proteome diversity and regulates gene expression (Baralle and Giudice 2017). A large proportion of eukaryotic genes are alternatively spliced; for example, over 95% of human genes undergo AS (Pan et al. 2008; Nilsen and Graveley 2010). AS events are also widespread in plants and AS is involved in various plant functions, including development, growth, and stress responses (Barbazuk et al. 2008). Recently, genome-wide characterizations of various plants species have shown that a large proportion of genes undergo AS, with proportions in different species, as follows: Amborella trichopoda, 70.4%; Vitis vinifera, 64.4%; Populus trichocarpa, 53.2% (Chamala et al. 2015); Arabidopsis thaliana, 60% (Marquez et al. 2012; Zhang et al. 2017); and Oryza sativa, 46.4% (Zhang et al. 2010). Based on sequence analysis of mRNA molecules and their corresponding genomic loci, AS isoforms can be categorized into groups, including those with retained introns, skipped exons, alternative 5′ or 3′ splice sites, or mutually exclusive exons (Staiger and Brown 2013). Plants and animals differ in their most common types of AS events; in plants, retained introns are the most common AS event, while skipped exons are the most common in animals (Dong et al. 2018; Slansky and Spellman 2019).
Accurate determination of AS is an important step to investigate genome characteristics and gene function. The original basic and fundamental method for discovery of AS events from sequence data is alignment of expressed sequence tag (EST) data to genomic sequences (Mironov et al. 1999; Modrek and Lee 2002). In recent years, several new methods and tools to identify AS events have been developed; however, most of them rely on the availability of a reference genome and annotation. For example, Program to Assemble Spliced Alignments (PASA) is a tool well-suited to the identification and classification of AS isoforms (Haas et al. 2008). PASA uses GMAP/BLAT to align transcripts to the genome and performs “all-versus-all” comparisons among clustered overlapping alignment assemblies to identify splicing variations (Haas et al. 2008).
With advances in sequencing technology, discovery of extensive AS events is no longer limited to model species (Wang et al. 2009). Third-generation sequencing (TGS) technologies (e.g., Pacific Biosciences and Oxford Nanopore) feature long-read sequencing. TGS platforms generate reads of 1–100 kb, which are much longer than those from second-generation sequencing (SGS) platforms (50–700 bp) (Rhoads and Au 2015; Weirather et al. 2017). Long-read transcriptomes generated using TGS have advantages for identifying gene isoforms compared with SGS, as there is no need to reconstruct the transcript variants (Weirather et al. 2017; Dong et al. 2018); however, in species where there is no high quality reference genome, accurate identification of AS sites, particularly for sequences with minor changes, remains difficult using conventional sequence similarity searches. One alternative approach is to assemble reads and generate a reference to determine AS sites by realignment. For example, the IDP-denovo tool takes both short and long reads to assemble a “pseudogenome” as the reference for AS identification (Fu et al. 2018). The use of reconstructed reference information has been shown to substantially increase efficiency for nonmodel species (Fu et al. 2018). AStrap is another software program for identifying AS isoforms without using a reference genome. AStrap first uses CD-HIT software to reduce redundancy and generate a clustered isoform file (Li and Godzik 2006; Fu et al. 2012), then performs alignments to generate an aligned sequence file using GMAP or other tools; second, Astrap identifies splice isoforms and AS types using a machine-learning model (Ji et al. 2019).
Given the copious number of isoforms present in a long-read-based transcriptome data, we were motivated to fully explore AS events without reconstructing or assembling original sequencing reads. To ensure the accuracy of sequence alignments, we adopted SIM4 algorithms (Florea et al. 1998) to identify high similarity regions for initial AS identification. As a tool designed to align cDNA to genomic DNA sequences, SIM4 determines high similarity segment pairs (HSPs) by screening 12-mers, followed by implementing the dynamic programming algorithm, and is highly accurate and efficient. Here, we implemented a reverse-tracing approach to determine AS sites (“split sites”) using long-read transcriptome data alone, via modified SIM4 alignments. We grouped potential gene isoforms and counted the occurrences of split sites to reveal transcript diversity. Further, we provide an option to validate and quantify the occurrence of split sites using short-read data sets; this feature is useful for functional evaluation of tissue-specific AS isoforms.
RESULTS
IsoSplitter design principles
IsoSplitter uses a modified SIM4 alignment algorithm (Florea et al. 1998) to handle transcriptome sequences containing diverse gene isoforms. Although transcriptomes assembled from short-read sequencing can be used, IsoSplitter is designed for use with long-read transcriptome data (e.g., the transcripts of isoform sequencing by using Pacific Biosciences Technology after clustering and removing redundancy). In a complete transcriptome, alternative gene splicing can occur in various patterns, such as exon skipping, mutually exclusive exons, or alternative donor or acceptor sites (Modrek and Lee 2002); most cases will generate a gap region at the alternative splice site: a broken point in a transcript, supported by two consecutive aligned regions of another transcript (Fig. 1); however, IsoSplitter is unable to identify AS isoforms with alternative 5′ or 3′ ends, due to the lack of a gap.
FIGURE 1.

A schematic diagram of IsoSplitter design. IsoSplittingAnchor determines the split sites of transcripts through selection using a modified SIM4 algorithm; the default parameters for HSP selection are shown and can be customized. Transcriptome sequences after removing redundancy from long-read sequencing are recommended Input files; other transcriptome collections that contain extensive gene alternative splicing isoforms are suitable for the analysis as well. When short RNA-seq reads are available, ShortReadsAligner maps the short reads and identifies junction reads and isoform-specific reads that validate and quantify the split sites. Finally, isoform expression levels are calculated based on sequence reads. Red boxes indicate output files. The prediction of AS isoforms is supported by the junction reads; and to reveal the tissue-specific expression of isoforms the isoform-specific reads are calculated into ACM for quantification. The stars indicate the split sites identified by the SIM4 alignments.
There are three major analyses implemented in the software package. (i) IsoSplitter first performs all-versus-all alignments and locates the “breaking points” of transcripts; transcripts supporting the “breaking points” are further grouped to remove redundant combinations, which generates gene clusters of transcript isoforms. (ii) IsoSplitter offers an option of aligning short reads to validate breaking points through extracting and re-indexing the associated regions. Junction reads that support the split are identified to validate the detection of AS sites in a transcript; and the number of junction reads is calculated to reveal the abundance of AS events. (iii) Furthermore, to calculate the expression levels of tissue-specific isoforms, reads that map exclusively to the split sites are counted and normalized per transcript for downstream analyses. The preferential input files are complete transcriptome sequences that include extensive variation of AS isoforms, while individual sequencing results of incomplete tissue collections and high redundancy are not suitable due to the design of all-versus-all alignment (Fig. 1).
IsoSplittingAnchor efficiently identifies AS sites based on transcriptome sequences
To evaluate the efficiency of IsoSplitter, we used transcriptome sequences from public and in-house data sources, including Arabidopsis thaliana, Populus trichocarpa, Zea mays, and Camellia japonica, for AS identification. We show that, without reference genome information, IsoSplitter can efficiently identify potential AS isoforms (Fig. 2A). We tested the performance of IsoSplitter prediction using varied data sets from different plant species (Fig. 2B). For a typical transcriptome containing 40,000–50,000 transcripts, IsoSplitter takes around 20 h to complete AS prediction, with all-versus-all SIM4 alignments using 30 CPUs on a Ubuntu platform with an Intel(R) Xeon(R) CPU E5-2630 v3 @ 2.40GHz processor and 128GB-RAM (Fig. 2C). For transcriptome over 10,000 transcripts, the prediction took a substantial longer period for prediction (Fig. 2A,C). The C. japonica transcriptome data set was generated by isoform sequencing (Hu et al. 2020), and we found the AS discovery rate (55.57%) was higher for this data set than for those of A. thaliana and P. trichocarpa (Fig. 2D). The Z. mays data set contained the highest number of transcripts, and IsoSplitter yielded the highest discovery rate (76.07%; Fig. 2D).
FIGURE 2.

Prediction of Arabidopsis AS transcripts by IsoSplitter. (A) Summary of total used and predicted numbers of transcripts for Ath, Cja, Ptr, and Zma. (B) The distribution of transcript length in different species. The total number of transcripts are displayed. (C) IsoSplitter processing time on a 30 core cluster. (D) AS event prediction rates for Ath, Cja, Ptr, and Zma. Ath, Arabidopsis thaliana; Cja, Camellia japonica; Ptr, Populus trichocarpa; Zma, Zea mays.
To investigate the accuracy of IsoSplitter, we took advantage of the AS information based on genome annotation information from A. thaliana and P. trichocarpa. Based on the gene model information of the reference genomes, the alternative gene isoforms were located; this information of AS isoforms was used for the testing of prediction accuracy. We found that, for both analyses, the majority of AS events were identified by IsoSplitter; the accuracy rates for A. thaliana and P. trichocarpa were 75.1% and 73.0%, respectively (Fig. 3A,B). To reveal the features of missed predictions, we analyzed AS isoforms that were missed by IsoSplitter in A. thaliana. First, we filtered out isoforms with different 3'UTR and 5'UTR ends, since IsoSplitter cannot detect AS transcripts without a gap. The filtered number of transcripts of Arabidopsis thaliana and Populus trichocarpa are 3682 and 4389, respectively. The remainder transcripts were evaluated for the performance of IsoSplitter prediction. Our data show that, for missed AS isoforms with a gap, gap size was a major factor for IsoSplitter prediction; small gaps <6 nt were abundant in all missed cases (Fig. 3C).
FIGURE 3.

Evaluation of the accuracy of AS prediction using IsoSplitter. (A) Venn diagram plot of recorded AS events and events predicted by IsoSplitter in Arabidopsis thaliana. (B) Venn diagram plot of recorded AS events and events predicted results by IsoSplitter in Populus trichocarpa. (C) Summary of the gap length of missed AS events in Arabidopsis thaliana; in total, 1374 events were identified from alignments containing at least one gap. The percentage of missed events of each gap length is displayed on top.
Validation and quantification of AS events using short-read data
To further investigate AS transcripts, we used high-coverage short-read data to validate and quantify AS isoforms. First, the sequences 135 bp up- and downstream of the split sites (for regions <135 bp, remainder sequences were used) were extracted and re-indexed. Next, short reads from each experiment were mapped using bowtie2 (Langmead and Salzberg 2012). We screened for two types of read: junction reads and isoform-specific reads (Fig. 1). The number of junction reads indicates the frequency of differential splicing events, while isoform-specific reads can be used to quantify unique isoforms. In this version of IsoSplitter, the identification of junction reads was based on the bowtie2; and default settings were reads with over 15 bp perfect matches next to the split site (Fig. 1). The prediction of AS isoforms is supported by the junction reads. In order to reveal the tissue-specific expression of isoforms, we provide a method to determine the expression levels of isoforms based on isoform-specific reads: Average counts per million of total reads (ACM). For an isoform with multiple split sites, the ACM is calculated as an average number of isoform-specific reads normalized to millions of total used reads. The ACM is defined by the following: ACM = (total isoform-specific reads of a transcript/split sites of the transcript) × (total mapped reads to this transcript/transcript base number) × 10−6. And the formula is listed below: (ACM = SUM(ISR)/SUM(SS) × SUM(TMR)/TL × 10−6 ISR, mapped isoform-specific reads of a transcript; SS, split-sites number; TMR, mapped reads to this transcript; TL, number of bases in the transcript). This functionality is useful for performing downstream analyses to identify differentially expressed isoforms and functional interpretation of genes involved in AS and gene regulation.
Comparisons of the IsoSplitter pipeline with other software
To investigate the use and efficiency of AS prediction, we compared the IsoSplitter pipeline with the AStrap and PASA packages using the A. thaliana transcript data set. The PASA pipeline requires a reference genome to support AS identification, while AStrap and IsoSplitter use only transcriptome sequences for prediction. We compared the runtime and memory usage between IsoSplitter, AStrap and PASA (Table 1). For the calculation of runtime and memory, AStrap needs at least three types of files: the transcriptome sequence file (FASTA); isoform cluster file (TEXT) generated by CD-HIT; aligned sequence file (GFF3) generated by GMAP or other sequence alignment tools. Therefore, runtime was the sum time of all three steps; PASA integrates the MySql database for AS identification, which greatly reduced the runtime and memory (Table 1). PASA predicted the highest number of AS events (28779), accounting for 53.46% of total transcripts (Fig. 4A). The AStrap package yielded 7083 AS events, and 6823 of those events were also identified by PASA (Fig. 4A). IsoSplitter identified 14911 AS events, comprising 6666 (44.70%) events that were identified by all prediction packages (Fig. 4A), and 7370 events that were only shared between IsoSplitter and PASA (Fig. 4A). Comparing to PASA prediction, we found that there were 14743 missed AS events (Fig. 4A). Among these missed events, we found 4779 alternative 3′ ends, 3635 alternative 5′ ends, 4788 retained introns and 1541 others (Fig. 4B). Since IsoSplitter is not designed to find alternative 3′ and 5′ ends, we evaluated the 4788 missed events of retained introns, we found that when the modification of default parameter (in this case: length ≥ 15 bp & identity ≥ 80%), 2641 more AS events that fit with a2 = a1 + 1 (Fig. 1) were found (not shown). We also revealed that the typical missing events of retained introns were due to the missing of a consecutive alignment (Fig. 4C). These results suggest that the IsoSplitter package is highly efficient for discovering AS events when no reference genome is available.
TABLE 1.
The runtime and memory comparison between IsoSplitter, AStrap, and PASA
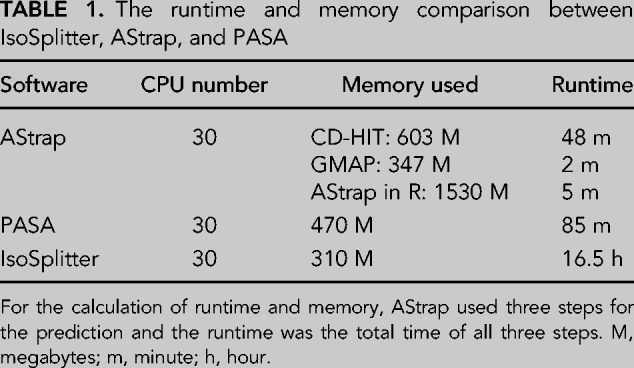
FIGURE 4.

The comparisons of prediction results using PASA, AStrap, and IsoSplitter. (A) The majority of AS events were identified by PASA; IsoSplitter predicted more AS events than Astrap. (B) Evaluation of missed AS events by IsoSplitter compared to PASA. (C) An example of missed AS event of retained intron by IsoSplitter. The two AS isoforms (NM_001340557.1 and NM_001036510.3) are from the gene model AT4G05590 in Arabidopsis thaliana. The transcripts are not supported by two consecutive aligned regions and hence were missed in the IsoSplitter prediction.
DISCUSSION
The identification and quantitation of AS isoforms is a critical step in understanding the function and regulation of eukaryotic genes. Bioinformatic tools designated for AS identification are essentially based on sequence alignments, with or without use of a reference genome (Nilsen and Graveley 2010; Liu et al. 2017; Ji et al. 2019). The SIM4 package was originally designed to align genomic DNA to mRNA sequences and is optimized for handling exon-intron boundaries (Florea et al. 1998). In the IsoSplitter pipeline, determination of a gap, or split site, is the basis for AS identification (Fig. 1), and the SIM4 algorithm is well-suited for this purpose. Using the default settings, we found that SIM4-based AS identification using complete transcriptomes is efficient (Fig. 2). We used Arabidopsis transcript sequences containing AS information to test the accuracy and efficiency of IsoSplitter and found that >70% of authentic AS events were identified by IsoSplitter in both tests; however, missing prediction rates were higher than the false-positive rate (Fig. 3). We expect that modification of the alignment parameters of IsoSplittingAnchor could alter the prediction efficiency. Using the data set of A. thaliana, we modified the parameters of SIM4 alignment for the AS prediction; we found that the less stringent parameters (e.g., length ≥ 15 bp & identity ≥ 90%) enhanced the discovery rate of AS prediction (Table 2). As we have shown in the case of retained introns (Fig. 4B,C), the prediction rate under the less stringent parameters can be highly accurate. These results indicate that the modification of parameters can be useful to increase the discovery rate for various data sets. We have manually evaluated the missed AS events in Arabidopsis with at least one gap, and showed that a large proportion (∼60%, Fig. 3C) of missing gaps were <4 bp, possibly due to the initial SIM4 scanning algorithm. The current design of IsoSplittingAnchor focuses on screening of eligible split sites and is not designed to evaluate gaps. For small sequence variations, an extra step of gap filtering could be incorporated to identify specific types of AS isoform, such as short sequence repeats, by evaluating the original alignment output file. We also identified 448 false AS sites in Arabidopsis (accounting for 2.8% of the total prediction, Fig. 3A). Evaluation of those alignments revealed that the sequences exhibited very high sequence similarity. Therefore, introduction of further stringent alignment parameters will reduce the number of false positives; however, it may also reduce total AS discovery.
TABLE 2.
The evaluation of IsoSplitter prediction by modifying the alignment parameters

We have compared the AS prediction results of IsoSplitter to those generated using PASA and AStrap. We show that IsoSplitter predicted more than twice as many AS events than AStrap analysis, which also predicts AS without using a reference genome (Fig. 4A), and that the majority (97.6%) of AS events predicted by AStrap were included in the IsoSplitter prediction (Fig. 4A). These results suggest that the IsoSplitter pipeline is efficient for AS prediction using only transcriptome data. However, we showed that the IsoSplitter analysis took a much longer runtime than PASA and AStrap (Table 1). One reason for this is because both PASA and AStrap uses the GMAP as the alignment algorithms which adopt an efficient indexing process (Zhou et al. 2009; Walenz and Florea 2011). AStrap can efficiently classify AS events into different types for further revaluation (Ji et al. 2019), and this functionality is not available with IsoSplitter. With the support of a reference genome, we find that the PASA package produces many more AS predictions than either AStrap or IsoSplitter (Fig. 4A), indicating that current IsoSplitter settings still underestimate the ubiquitous distribution of AS events.
The ability to deal with short-read RNA sequencing data sets is beneficial for AS evaluation and investigation. IsoSplitter can validate and quantify AS prediction using short-read data (Fig. 1). The current version of IsoSplitter borrows an original idea from HISAT; that is, to verify “split sites” by selecting reads that are explicitly matched to a junction site (Kim et al. 2015). This feature is improved by reindexing the split-site-associated sequences for mapping efficiency. Simultaneously, reads that mapped across split sites were extracted to reveal isoform-specific expression (Fig. 1). Based on this feature, the IsoSplitter pipeline can provide valuable information for tissue-specific analysis of isoforms. To further exploit the usage of the IsoSplitter prediction results, we developed an algorithm (ACM method) to quantify the expression level of transcripts by using only the junction reads; the ACM measure is informative to reveal the sample-specific expression of gene isoforms. Compared to the conventional quantification methods, such as, RSEM (Li and Dewey, 2011), Salmon (Patro et al. 2017), or SCUPPA2 (Trincado et al. 2018), that use different multimapping bias models for isoform quantification, the ACM methods can provide valuable information for relative estimations of sample-specific isoforms. It should be noted that, because the ACM measure is exclusively using the partial reads, the bias can be high for samples with a low sequencing depth. Further, for samples with multiple biological replicates, the differential expression of isoforms is also amenable to further analyses. In a previous experiment, we analyzed the expression of isoforms using five distinct tissues form Camellia japonica and found that tissue-specific isoforms were enriched for functions related to their tissue types (Hu et al. 2020). We conclude that the IsoSplitter pipeline is a versatile suite for AS identification and investigation and that IsoSplitter outputs can be neatly incorporated into further functional analyses of AS-related processes.
MATERIALS AND METHODS
Data and database acquisition
Transcriptomes of model and nonmodel plant species were obtained from public databases. Long-read and short-read Camellia japonica RNA sequencing data sets were described previously (Hu et al. 2020). Arabidopsis thaliana transcripts and annotation information (version 10) were from the Arabidopsis Information Resource (TAIR). Populus trichocarpa genome data sets (version 3.1) were from Phytozome (Tuskan et al. 2006). The Zea mays sequence information was obtained from MaizeGDB (Portwood et al. 2019). All transcript files are in the fasta format and detailed information of transcripts number and lengths were displayed in (Fig. 2B).
IsoSplitter supported functions
IsoSplitter was created in Python and tested under the Linux Ubuntu system. A detailed description for installation and use of the software is provided (Supplemental Data 1). IsoSplitter consists of two steps of analyses executed by IsoSplittingAnchor.py and ShortReadsAligner.py. The first script prefers a transcriptome sequence file generated by single-molecule sequencing technologies that need no transcript reconstruction. The latter analysis is optional, and takes short transcriptome sequence reads to evaluate splicing sites predicted by IsoSplittingAnchor, as well as isoform-specific expression levels.
Starting from a sequence file of transcripts containing comprehensive isoform information, IsoSplittingAnchor invokes SIM4 alignment algorithms to identify splitting sites derived from the whole long-read transcriptome. The first output file, “Breakingpoint_out,” is a tabular text file containing the sequencing ID and the location of splicing sites. The second output file, “GeneCluster,” is the results of sequence IDs that are transcript isoforms; where each cluster is a potential gene locus for alternative splice isoforms. To visualize the AS isoforms and their relationships, the script ChangeToCytoscape.py is provided to transform the prediction result to the file formats that can be evaluated and visualized by the cytoscape package (Shannon et al. 2003). ShortReadsAligner is used to evaluate and quantify splitting sites by counting eligible breaking points. This step analyzes short-read RNA sequencing data to identify “junction reads” (reads partially mapped next to break sites and exclusively split at the same location), and count the numbers of junction reads for each break site (output file, “JunctionReadsCount.txt”). The abundance of junction reads can be further analyzed for tissue-specific AS. ShortReadsAligner also generates isoform abundance results using reads that map exclusively to split sites; and these reads are normalized for each transcript to calculate the ACM values for isoform-specific expression (output file, “Average_counts_per.txt”). To track the reads of the predicted splice sites, ShortReadsMapped.sam and ACMMapped.sam that were generated by ShortReadsAligner can be directly used by samtools (Li et al. 2009) to screen split sites of interest.
Example data sets for quick testing of the IsoSplitter pipeline
We used current Arabidopsis thaliana gene transcript data (available from the TAIR website, Araport11_genes.201606.cdna.fasta.gz) to test the identification of AS sites using IsoSplitter. Further, we aligned short reads from a previous study (Li et al. 2016) to validate the prediction of split sites in the previous step. The short-reads file is available from the NCBI SRA database (Accession No: SRR3664433). For testing ShortReadsAligner.py, SRA data were first converted into fastq format and filtered for adaptor and low quality sequence reads. The following are example scripts for quick reproduction of the test results (see Supplemental Data 1 for details).
- To identify split sites:
- $ IsoSplittingAnchor Araport11_genes.201606.cdna.fasta
- To validate and quantify split sites:
- $ ShortReadsAligner Araport11_genes.201606.cdna.fasta SRR3664433.fasta Breakpoint_out.txt
or
$ ShortReadsAligner -q Araport11_genes.201606.cdna.fasta SRR3664433.fastq Breakpoint_out.txt
where breakpoint_out.txt is one of the output files of the first step, Araport11_genes.201606.cdna.fasta is the same file used in the first step, and SRR3664433.fasta/SRR3664433.fastq is sequencing data.
DATA DEPOSITION
The source code of IsoSplitter and associated test data are available at https://github.com/Hengfu-Yin/IsoSplitter.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Dr. Y.-J. Liu of the Chinese Academy of Forestry for helpful comments. We are grateful to the anonymous reviewers for critical comments and suggestions that greatly improved this work. This work is supported by Nonprofit Research Projects (CAFYBB2018ZY001-1) of the Chinese Academy of Forestry and the National Key R&D Program of China (grant no. 2019YFD1000400). We also thank the Postgraduate Research & Practice Innovation Program of Jiangsu Province for funding support.
Author contributions: All authors conceived and designed the analysis. Y.W., Z.H., and H.Y. performed the analysis; Y.W., N.Y., and H.Y. conceived and designed the bioinformatics pipeline. All authors wrote and approved the manuscript.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.077834.120.
REFERENCES
- Baralle FE, Giudice J. 2017. Alternative splicing as a regulator of development and tissue identity. Nat Rev Mol Cell Biol 18: 437–451. 10.1038/nrm.2017.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbazuk WB, Fu Y, McGinnis KM. 2008. Genome-wide analyses of alternative splicing in plants: opportunities and challenges. Genome Res 18: 1381–1392. 10.1101/gr.053678.106 [DOI] [PubMed] [Google Scholar]
- Chamala S, Feng G, Chavarro C, Barbazuk WB. 2015. Genome-wide identification of evolutionarily conserved alternative splicing events in flowering plants. Front Bioeng Biotechnol 3: 33. 10.3389/fbioe.2015.00033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong CL, He F, Berkowitz O, Liu JX, Cao PF, Tang M, Shi HC, Wang WJ, Li QL, Shen ZG, et al. 2018. Alternative splicing plays a critical role in maintaining mineral nutrient homeostasis in rice (Oryza sativa). Plant Cell 30: 2267–2285. 10.1105/tpc.18.00051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florea L, Hartzell G, Zhang Z, Rubin GM, Miller W. 1998. A computer program for aligning a cDNA sequence with a genomic DNA sequence. Genome Res 8: 967–974. 10.1101/gr.8.9.967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L, Niu B, Zhu Z, Wu S, Li W. 2012. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28: 3150–3152. 10.1093/bioinformatics/bts565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu S, Ma Y, Yao H, Xu Z, Chen S, Song J, Au KF. 2018. IDP-denovo: de novo transcriptome assembly and isoform annotation by hybrid sequencing. Bioinformatics 34: 2168–2176. 10.1093/bioinformatics/bty098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, Salzberg SL, Zhu W, Pertea M, Allen JE, Orvis J, White O, Buell CR, Wortman JR. 2008. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol 9: R7. 10.1186/gb-2008-9-1-r7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Lyu T, Yan C, Wang Y, Ye N, Fan Z, Li X, Li J, Yin H. 2020. Identification of alternatively spliced gene isoforms and novel noncoding RNAs by single-molecule long-read sequencing in Camellia. RNA Biol 17: 966–976. 10.1080/15476286.2020.1738703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji GL, Ye WB, Su YR, Chen ML, Huang GZ, Wu XH. 2019. AStrap: identification of alternative splicing from transcript sequences without a reference genome. Bioinformatics 35: 2654–2656. 10.1093/bioinformatics/bty1008 [DOI] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL. 2015. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12: 357–360. 10.1038/nmeth.3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. 2011. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12: 323. 10.1186/1471-2105-12-323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Godzik A. 2006. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22: 1658–1659. 10.1093/bioinformatics/btl158 [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map (SAM) format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Yamada M, Han X, Ohler U, Benfey PN. 2016. High-resolution expression map of the Arabidopsis root reveals alternative splicing and lincRNA regulation. Dev Cell 39: 508–522. 10.1016/j.devcel.2016.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Mei W, Soltis PS, Soltis DE, Barbazuk WB. 2017. Detecting alternatively spliced transcript isoforms from single-molecule long-read sequences without a reference genome. Mol Ecol Resour 17: 1243–1256. 10.1111/1755-0998.12670 [DOI] [PubMed] [Google Scholar]
- Marquez Y, Brown JWS, Simpson C, Barta A, Kalyna M. 2012. Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis. Genome Res 22: 1184–1195. 10.1101/gr.134106.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov AA, Fickett JW, Gelfand MS. 1999. Frequent alternative splicing of human genes. Genome Res 9: 1288–1293. 10.1101/gr.9.12.1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrek B, Lee C. 2002. A genomic view of alternative splicing. Nat Genet 30: 13–19. 10.1038/ng0102-13 [DOI] [PubMed] [Google Scholar]
- Nilsen TW, Graveley BR. 2010. Expansion of the eukaryotic proteome by alternative splicing. Nature 463: 457–463. 10.1038/nature08909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey J, Blencowe BJ. 2008. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 40: 1413–1415. 10.1038/ng.259 [DOI] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry R, Kingsford C. 2017. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14: 417–419. 10.1038/nmeth.4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portwood JL, Woodhouse MR, Cannon EK, Gardiner JM, Harper LC, Schaeffer ML, Walsh JR, Sen TZ, Cho KT, Schott DA, et al. 2019. MaizeGDB 2018: the maize multi-genome genetics and genomics database. Nucleic Acids Res 47: D1146–D1154. 10.1093/nar/gky1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoads A, Au KF. 2015. PacBio sequencing and its applications. Genomics Proteomics Bioinformatics 13: 278–289. 10.1016/j.gpb.2015.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504. 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slansky JE, Spellman PT. 2019. Alternative splicing in tumors: a path to immunogenicity? N Engl J Med 380: 877–880. 10.1056/NEJMcibr1814237 [DOI] [PubMed] [Google Scholar]
- Staiger D, Brown JW. 2013. Alternative splicing at the intersection of biological timing, development, and stress responses. Plant Cell 25: 3640–3656. 10.1105/tpc.113.113803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trincado JL, Entizne JC, Hysenaj G, Singh B, Skalic M, Elliott D, Eyras E. 2018. SUPPA2: fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol 19: 40. 10.1186/s13059-018-1417-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuskan GA, Difazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S, Salamov A. 2006. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313: 1596–1604. 10.1126/science.1128691 [DOI] [PubMed] [Google Scholar]
- Walenz B, Florea L. 2011. Sim4db and Leaff: utilities for fast batch spliced alignment and sequence indexing. Bioinformatics 13: 1869–1870. 10.1093/bioinformatics/btr285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. 2009. RNA-seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10: 57–63. 10.1038/nrg2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirather JL, Cesare MD, Wang Y, Piazza P, Sebastiano V, Wang XJ, Buck D, Au KF. 2017. Comprehensive comparison of Pacific Biosciences and Oxford Nanopore Technologies and their applications to transcriptome analysis. F1000Res 6: 100. 10.12688/f1000research.10571.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang GJ, Guo GW, Hu XD, Zhang Y, Li QY, Li RQ, Zhuang RH, Lu ZK, He ZQ, Fang XD, et al. 2010. Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res 20: 646–654. 10.1101/gr.100677.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang RX, Calixto CPG, Marquez Y, Venhuizen P, Tzioutziou NA, Guo WB, Spensley M, Entizne JC, Lewandowska D, ten Have S, et al. 2017. A high quality Arabidopsis transcriptome for accurate transcript-level analysis of alternative splicing. Nucleic Acids Res 45: 5061–5073. 10.1093/nar/gkx267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou LM, Pertea M, Delcher AL, Florea L. 2009. Sim4cc: a cross-species spliced alignment program. Nucleic Acids Res 11: e80. 10.1093/nar/gkp319 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.