

ABSTRACT
Leukocyte Nox2 is recognized to have a fundamental microbicidal function in sepsis but the specific role of Nox2 in endothelial cells (EC) remains poorly elucidated. Here, we tested the hypothesis that endothelial Nox2 participates in the pathogenesis of systemic inflammation and hypotension induced by LPS. LPS was injected intravenously in mice with Tie2-targeted deficiency or transgenic overexpression of Nox2. Mice with Tie2-targeted Nox2 deficiency had increased circulating levels of TNF-α, enhanced numbers of neutrophils trapped in lungs, and aggravated hypotension after LPS injection, as compared to control LPS-injected animals. In contrast, Tie2-driven Nox2 overexpression attenuated inflammation and prevented the hypotension induced by LPS. Because Tie2-Cre targets both EC and myeloid cells we generated bone marrow chimeric mice with Nox2 deletion restricted to leukocytes or ECs. Mice deficient in Nox2 either in leukocytes or ECs had reduced LPS-induced neutrophil trapping in the lungs and lower plasma TNF-α levels as compared to control LPS-injected mice. However, the pronounced hypotensive response to LPS was present only in mice with EC-specific Nox2 deletion. Experiments in vitro with human vein or aortic endothelial cells (HUVEC and HAEC, respectively) treated with LPS revealed that EC Nox2 controls NF-κB activation and the transcription of toll-like receptor 4 (TLR4), which is the recognition receptor for LPS. In conclusion, these results suggest that endothelial Nox2 limits NF-κB activation and TLR4 expression, which in turn attenuates the severity of hypotension and systemic inflammation induced by LPS.
Keywords: Endothelial cells, gp91phox, hypotension, NF-κB, systemic inflammation, TLR4
Abbreviations: ATB, antibiotic, BM, bone marrow, BP, blood pressure, CLP, cecal ligation and puncture, EC, endothelial cells, EDTA, ethylenediaminetetraacetic acid, ip, intraperitoneal, iv, intravenous, KO, knockout, LPS, Lipopolysaccharide, MPO, myeloperoxidase, NF-κB, nuclear factor-kappa B, NO, Nitric oxide, NOS, nitric oxide synthase, Nox2, NADPH oxidase 2, ROS, reactive oxygen species, sc, subcutaneous, Tie2, tyrosine-protein kinase receptor (Tie2∗ receptor for angiopoietin 1 and 4), TLR2, toll-like receptor 2, TLR4, toll-like receptor 4, TLR9, toll-like receptor 9, TNF-α, tumor necrosis factor-alpha, WT, wild type
INTRODUCTION
Sepsis is a life-threatening organ dysfunction caused by a dysregulated host response to infection. The systemic inflammation resulting from sepsis can be complicated by multi-organ failure and may progress to septic shock (1). Structural and functional changes of the endothelium occur in septic patients, with a positive feedback leading to production of inflammatory mediators, expression of adhesion molecules, hypotension, and hypoperfusion (1, 2). The precise mechanisms leading to endothelial dysfunction in sepsis are incompletely understood, but previous studies suggest that reactive oxygen species (ROS) are involved in its pathophysiology (3, 4).
NADPH oxidase-2 (Nox2) is a multisubunit complex based on the catalytic subunit Nox2 and dedicated to ROS production. Agonist-induced post-translational modifications in p47phox, p67phox, Rac1/2, and p40phox cytosolic subunits result in their translocation to the cell membrane where they bind to Nox2/p22phox. The assembly of cytoplasmic and membrane subunits of NADPH oxidase-2 activates electron transfer from NADPH to molecular oxygen to generate superoxide (4).
Nox2 was initially identified in neutrophils where it is fundamental for killing of phagocytized microorganisms. In addition to phagocytes, Nox2 is also expressed in other cell types where it has specific functions in redox-sensitive intracellular signaling pathways (5). For example, Nox2 limits the suppressive ability of regulatory T cells thereby aggravating T cell infiltration into the heart and cardiac remodeling induced by angiotensin II (ANG II) (6) whereas cardiomyocyte Nox2 modulates mitogen-activated protein kinase (MAPK) signaling to enhance ANG II-induced heart hypertrophy (7).
Previous studies from our laboratory and others showed that Nox2 in endothelial and other cell types participates in the pathogenesis of ANG II-induced hypertension and myocardial remodeling (6–11). However, the role of endothelial Nox2 in the setting of sepsis or in experimental models of endotoxemia remains unclear. Some studies suggest that Nox2 activation in EC is detrimental in endotoxemia because superoxide and other ROS collectively contribute to necrosis of venular EC (12) and inactivate catecholamines (13), which might aggravate hypotension and multi-organ failure. On the other hand, Nox2 could be protective in sepsis by producing superoxide, which scavenges NO and, consequently, preventing severe hypotension. In line with the latter hypothesis, many studies showed that endothelial Nox2 modulates the magnitude of endothelium-dependent relaxation by reducing NO bioactivity, both in animal and humans tissues (14–16). Additionally, Nox2 can negatively regulate TLR4 expression and NF-κB activation in leukocytes, key drivers of systemic inflammation in endotoxemia and sepsis (17–19).
TLR4, the recognition receptor for lipopolysaccharide (LPS or endotoxin), is activated upon interaction with LPS and has an important role in sepsis. Small amounts of circulating LPS (endotoxin) induce TLR4 activation and initiate intracellular signaling that results in the production of pro-inflammatory cytokines, such as TNF-α and IL-6 (20). Previous work from our group showed that animals deficient in TLR4 signaling displayed reduced mortality in a model of severe cecal ligation and puncture (CLP)-induced sepsis (21). In addition, TLR4 has been implicated in modifying blood pressure in models of endotoxemia (22, 23).
In this study, we used a variety of gene-modified mouse models to test the hypothesis that EC Nox2 modulates the systemic response to LPS and to investigate the role of TLR4 in such effects. We found that endothelial Nox2 synergizes with leukocyte Nox2 to limit the severity of systemic inflammation and hypotension induced by LPS, with endothelial Nox2 being particularly important in reducing the extent of the hypotensive response. Mechanistically, endothelial Nox2 downregulates NF-κB activation and subsequent TLR4 expression, which may contribute to reduced multi-organ failure and death.
MATERIAL AND METHODS
Reagents
LPS serotype O111:B4 from E. coli was purchased from Sigma-Aldrich (St. Louis, USA, Cat. L2630). Antibodies were: anti-gp91phox from BD Pharmigen (Franklin Lakes, USA. Cat. 611414); anti-p65 from Santa Cruz Biotechnology (Dallas, Tex. Cat. sc-372); goat anti-mouse and donkey anti-rabbit secondary antibodies conjugated, respectively, with Alexa Fluor 555 or Alexa fluor 488 from Invitrogen (Carlsbad, USA. Cat. A21422, Cat.A21206); anti-CD31 from BD Pharmigen (San Diego, Calif. Cat. 550274); anti-TLR4 from Abcam (Cambridge, UK. Cat. ab22048). Dihydroethidium (DHE) was purchased from Molecular Probes (Eugene, Ore, Cat. D1168). The peptides gp91ds-tat [H]-R-K-K-R-R-Q-R-R-R-C-S-T-R-I-R-R-Q-L-NH2 and scrambled-tat [H]-R-K-K-R-R-Q-R-R-R-C-L-R-I-T-R-Q-S-R-NH2 were synthethized by Anaspec (San Jose, Calif) (24).
Animal procedures
Experiments were performed in 6 to 8-week-old male mice on a C57BL/6 background. Male mice were used to simplify the breeding strategy as Nox2 is located on the X chromosome, and also to minimize estrogen-dependent fluctuations in response. We used C57BL/6 mice (wild type, WT), Nox2-/y (global Nox2 knockout [KO]), TLR4-/-, TLR2-/-, TLR9-/- and mice with Tie-2-targeted deletion of Nox2 (Nox2fl/flTie2Cre+ or EC/L-Nox2KO) or Tie-2-targeted overexpression of Nox2 (Nox2Tg). Global Nox2 knockout mice were obtained from Jackson Laboratories and bred locally. Mice with Tie2-targeted Nox2 overexpression or deletion were generated as described previously (9, 10, 14). Nox2fl/fl mice were used as the Nox2-replete controls for tissue-specific Nox2 KO mice. Bone marrow (BM) chimeric mice were also generated (described later) and allowed to recover for at least 4 weeks before further study (10). LPS (10 mg/kg) was administered intravenously in mice under isoflurane anesthesia.
Clinical severity scores were determined at 2-hourly intervals after LPS injection. The maximum possible score of 18 comprised the presence of the following signs: lethargy, piloerection, tremors, periorbital exudates, respiratory distress, and diarrhea, which were each assessed as mild (+), moderate (++), or severe (+++) by two independent blinded researchers.
To induce cecal ligation and puncture (CLP), mice were anesthetized intraperitoneally with ketamine (100 mg/kg) and xylazine (10 mg/kg), and, after a ligation near the ileo-cecal colic valve, a double puncture was made in the cecum using a 26-gauge needle. Some mice were treated with apocynin (200 mg/kg) or vehicle (10% Tween 20) subcutaneously 30 min before surgery. All mice were treated with antibiotics (ATB, ertapenem sodium, 30 mg/kg) i.p. 6 h after surgery and 12 hourly thereafter. Survival was evaluated up to 7 days after surgery.
All studies were conducted in accordance with institutional ethical guidelines (Ethical guidelines of the Ribeirao Preto Medical School, University of Sao Paulo, Sao Paulo, Brazil, protocols 062/2013 and 032/2011; and King's College London Biological Safety Unit) and the UK Home Office Guidance on the Operation of the Animals (Scientific Procedures) Act, 1986.
NF-κB activity.
Human umbilical vein endothelial cells (HUVEC) or human aortic endothelial cells (HAEC) were transfected with NF-κB firefly luciferase (pGL4.32[luc/NF-κB-RE/Hygro], Promega, Wis) and thymidine kinase renilla luciferase (pGL4.73[hRluc/SV40], Promega) vector plasmids using Lipofectamine LTX with Plus Reagent (Invitrogen, Carlsbad, Calif). Plasmids expressing the minimal firefly and thymidine kinase renilla promoters were used as controls. Luciferase activities in cell lysates were determined using the Dual-Glo luciferase system (Promega) in a 96-well plate luminometer (Mithras LB 940 Multimode Microplate Reader, Berthold Technologies, Calmbacher, Germany).
qRT-PCR
Standard methods were used for real-time qPCR with Syber Green reagent (ThermoFisher Scientific) on an Eppendorf Mastercycler instrument. The comparative cycle threshold method was used for quantification (fold increase = 2-ΔΔct). Primers were: murine TLR4 sense (5’-CCAAGCCTTTCAGGGAATTAA-3’), murine TLR4 antisense (5’-GCCAGGTTTTGAAGGCAAGT-5’), murine GAPDH sense (5’-CATCTTCTTGTGCAGTGC-3’), murine GAPDH antisense (5’-CGGCCAAATCCGTTCAC-3’); human GAPDH sense (5’-GGGAAGGTGAAGGTCGGA-3’); human GAPDH antisense (5’-GCAGCCCTGGTGACCAG-3’); human TLR4 sense (5’-GTCTGCAGGCGTTTTCTTCT-3’); human TLR4 antisense (5’-AAGTGAAAGCGGCAACCTTA-3’). GAPDH mRNA levels were used for normalization.
Blood pressure
Systolic blood pressure (BP) was determined by tail-cuff plethysmography using a noninvasive blood pressure system (CODA standard, Kent Scientific, San Diego, Calif). Animals were acclimatized to blood pressure measurement over 1 week prior to commencing experiments. Blood pressure measurements were determined 6 h after LPS injection (22).
Cytokine measurements
TNF-α and IL-6 levels were quantified by enzymatic-linked immunosorbent assay (ELISA) using antibodies from R&D Systems (Cat. DY410-05, Cat. DY406-05). The cytokines measured acutely after LPS injection correlate with sepsis mortality in previous clinical studies (25).
Lung neutrophil sequestration, plasma AST, and urea levels
Myeloperoxidase activity was measured as an index of neutrophil trapping in lungs as previously described (26) and modified (27). Briefly, the lungs were homogenized in a NaPO4 0.02 mol/L buffer. The erythrocytes were lysed in NaCl 0.2% buffer and neutrophils disrupted in NaPO4 buffer 0.05 mol/L with hexadecyl-trimetil-amonium bromide. Samples were diluted in NaPO4 0.08 mol/L and incubated with hydrogen peroxide 0.5 mmol/L and 3,3,5,5-tetramethyl benzidine (Sigma, St. Louis, Calif. Cat. T2885) 1.6 mmol/L.
Plasma concentrations of aspartate aminotransferase (AST) and urea were measured using commercial kits (Labtest, Brazil; Bioclin, Brazil, Cat. 109 and 27, respectively) and the absorbance measured in a Molecular Devices Spectramax 190 Laboratory Microplate spectrophotometer.
Cultures of HUVEC and HAEC
Primary HUVEC and HAEC were purchased from Life Technologies (Carlsbad, Calif) and Lonza (Cambridge, UK), respectively. Cells were cultured to passage 5 in M199 medium, with supplemental growth factors (endothelial cell growth factor 1 ng/mL, heparin 10 U/mL, thymidine 2.5 mg/mL, and human β-endothelial cell growth factor 3 μg/mL). Plates were coated with fibronectin (Sigma-Aldrich, St. Louis, USA) before seeding the cells.
Immunofluorescence
Cells or 7-μm-thick tissue sections were fixed with 4% paraformaldehyde for 30 min and permeabilized in 0.2% Triton-X for 30 min. Unspecific interactions were blocked by incubation with 2% BSA plus 1:50 goat serum solution for 30 min. Primary antibodies were used at 1:100 to 1:200 dilution. Images were acquired on a confocal microscope (TCS SP5 II, Leica) or an Olympus 1X81 inverted epifluorescence microscope.
ROS assay
Dihydroethidium (DHE) oxidation products were quantified as described previously (28). Briefly, HUVEC or HAEC were incubated with scrambled-tat or gp91ds-tat (30 μM) for 1 h, then stimulated with LPS (200 ng/mL) for 4 h. Cells were washed and incubated with DHE (100 μM, 37°C), then scraped in acetonitrile for lyophilization. Quantification of DHE products was performed by high-performance liquid chromatography (HPLC).
DHE fluorescence was used to estimate ROS production in mesenteric vessels. For this, 7-μm-thick tissue sections were incubated with 3 μM DHE in PBS with DTPA (diethylenetriamine-pentacetic acid 100 μM) for 30 min and then images obtained immediately on an Olympus 1X81 inverted epifluorescence microscope.
Statistics
Analyses were performed using GraphPad Prism software 7.03 (La Jolla, Calif). The Log-rank (Mantel-Cox) test was used to evaluate survival rates. Clinical scores and blood pressure measurements were compared using 2-way ANOVA followed by Bonferroni post-test. Mann–Whitney U test was used to assess variables with two experimental groups and Kruskal–Wallis test followed by Dunn's post-hoc correction for analyses involving >2 experimental groups. P < 0.05 was considered significant.
RESULTS
Nox2 modulates the severity of LPS-induced systemic inflammation and hypotension
To study the role of Nox2 independent of its microbicidal activity (29–31), we first compared the responses of global Nox2-deficient mice (32) and WT controls in a model of intravenous LPS injection. Experimental endotoxemia mimics features of the inflammatory response observed in sepsis (33). After LPS injection, the WT mice showed an 85% survival rate whereas global Nox2-deficient mice had a survival rate of 45% (see Figure, Supplemental Digital Content 1, at ). The global Nox2-deficient mice had higher plasma TNF-α and IL-6 levels than WT mice after LPS injection (Fig. 1, A and B). Global Nox2 deficiency also resulted in an increased number of neutrophils trapped in the lungs (Fig. 1C), lower blood pressure (Fig. 1D) and a greater degree of renal and liver damage as indexed by plasma urea (Fig. 1E) and aspartate aminotransferase (AST), respectively (Fig. 1F). Therefore, global Nox2 deficiency results in an enhanced pro-inflammatory state and evidence of organ dysfunction during the acute phase after LPS challenge.
Fig. 1.
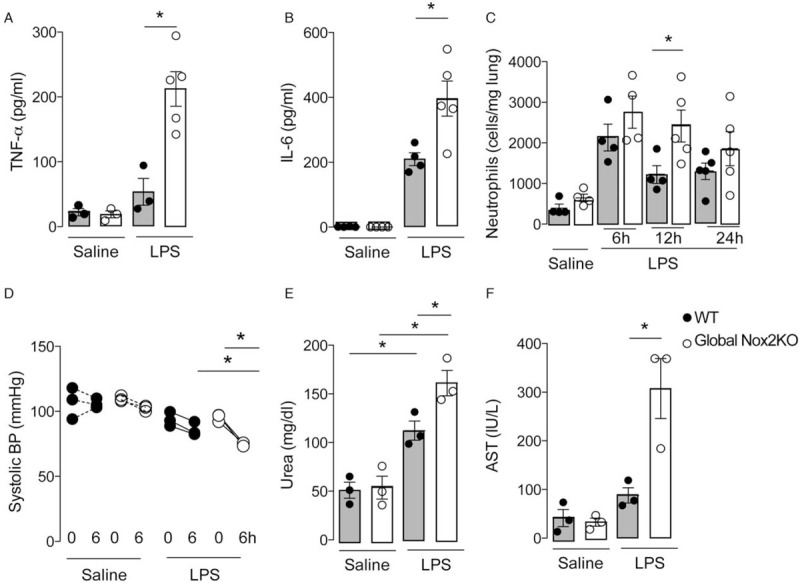
Nox2 limits LPS-induced systemic inflammation and hypotension. Wild-type (WT) and global Nox2-deficient (Global Nox2KO) mice were injected with LPS (10 mg/kg i.v.). (A, B) Plasma TNF-α and IL-6 levels (ELISA) after LPS injection; (C) lung neutrophil sequestration 6, 12, and 24 h after LPS treatment; (D) systolic blood pressure (BP) at baseline and 6 h after LPS injection; (E, F) plasma urea and aspartate aminotransferase (AST) 24 h after LPS injection. Graphs represent mean ± SEM (n = 3–5 per group). ∗P < 0.05 for indicated comparisons. 2-way ANOVA followed by Bonferroni post-test (C, D) or Kruskal–Wallis followed by Dunn's multiple comparison test (A, B, E, F).
Effects of EC Nox2 on inflammation and blood pressure
Since EC–leukocyte interaction is a key component of systemic inflammation (3), Nox2 present in either cell type might contribute to LPS-induced release of inflammatory mediators and hypotension. We evaluated the responses to LPS in mice with Tie2-targeted Nox2 deficiency in both EC and myeloid cells (EC/L-Nox2KO) (9). EC and leukocyte-specific Nox2-deficient mice expressed lower levels of Nox2 in mesenteric vessels, bone marrow neutrophils, and aorta as compared with Nox2-replete littermate controls (see Figure, Supplemental Digital Content 2A-C, at ). Additionally, a lower DHE signal indicative of ROS production was observed in EC/L-Nox2KO mice after LPS injection compared to control mice (see Figure, Supplemental Digital Content 2D, at ). LPS-treated EC/L-Nox2KO mice had higher clinical scores (Fig. 2A) and systemic inflammation than Nox2-replete control LPS-treated animals, along with a greater number of lung neutrophils (Fig. 2B), higher plasma TNF-α levels (Fig. 2C), and aggravated hypotension (Fig. 2D).
Fig. 2.
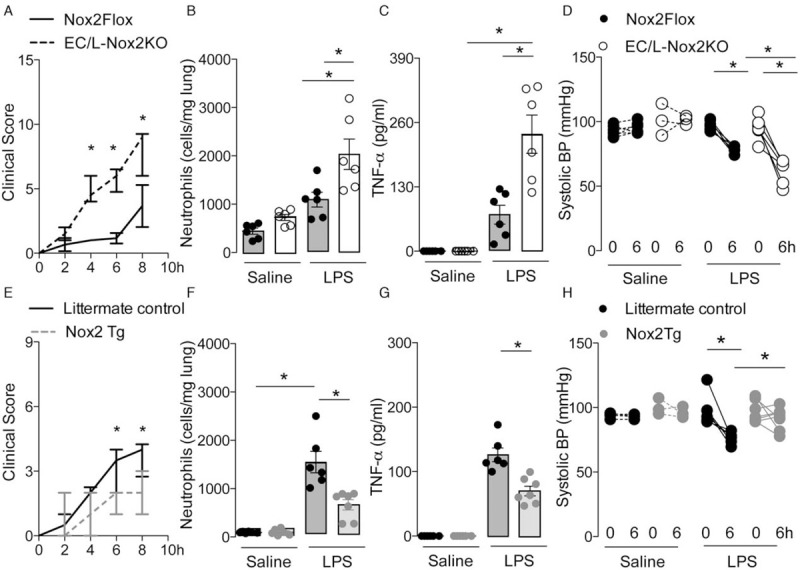
Tie2-targeted Nox2 deficiency exacerbates systemic inflammation and hypotension induced by LPS. Combined EC/L-Nox2KO, Nox2-replete (Nox2Flox), Tie2-targeted Nox2 overexpression (Nox2Tg), and littermate control mice were injected with LPS (10 mg/kg i.v.). (A, E) Median ± interquartile range of clinical scores; (B, F) mean ± SEM of neutrophil sequestration in lungs; (C, G) plasma tumor necrosis factor (TNF)-α levels, 12 h after LPS treatment. (D, H) Systolic blood pressure (BP) at baseline and 6 h after LPS injection. ∗P < 0.05 for indicated comparisons (n = 3–7 per group). 2-way ANOVA followed by Bonferroni's post-test (A, D, E, H), or Kruskal–Wallis followed by Dunn's multiple comparison test (B, C, F, G).
In contrast, the Tie2-targeted over-expression of Nox2 (Nox2Tg) (14) resulted in a phenotype opposite to that observed in EC/L-Nox2KO animals. Nox2Tg mice had lower clinical scores (Fig. 2E), less neutrophil sequestration in lungs (Fig. 2F), lower plasma TNF-α levels (Fig. 2G), and a reduced hypotensive response to LPS injection compared to littermate controls (Fig. 2H). Of note, Nox2Tg mice expressed higher levels of Nox2 in mesenteric vessels and Nox2 mRNA in aorta; and showed higher DHE signal in mesenteric vessels as compared to littermate controls under basal conditions (see Figure, Supplemental Digital Content 3, at ), as previously published (14).
To distinguish between the effects of Nox2 in EC versus leukocytes, combinations of bone marrow (BM) chimeric mice were next generated using endothelium/leukocyte-specific Nox2-deficient mice (EC/L-Nox2KO) and Nox2-replete control littermates as donors and/or recipients (Fig. 3A). Using this approach, we produced animals with selective deficiency of Nox2 solely in BM cells or in ECs (leukocyte-specific Nox2 deficiency and EC-specific Nox2 deficiency, respectively)—which was confirmed by q-PCR (see Figure, Supplemental Digital Content 4A-B, at ). Control Nox2-replete mice were given a transplant of Nox2-replete BM cells while another control was to transplant EC/L-Nox2KO mice with EC/L-Nox2KO BM. After LPS injection, leukocyte-specific Nox2-deficient chimeric mice showed an increase in ROS generation by mesenteric vessels as compared to control mice injected with saline. This response was prevented in EC-specific Nox2-deficient chimeric mice or in EC/leukocyte-specific Nox2-deficient chimeric mice (see Figure, Supplemental Digital Content 4C, at ). Consistent with the earlier results, EC/leukocyte-specific Nox2-deficient chimeric mice showed worse clinical scores (Fig. 3B), along with increased neutrophil trapping in lungs (Fig. 3C), higher plasma TNF-α levels (Fig. 3D), and aggravated hypotension as compared to Nox2-replete controls (Fig. 3E). EC-specific Nox2-deficient mice and leukocyte-specific Nox2-deficient mice showed similar lung neutrophil sequestration and plasma TNF-α levels to Nox2-replete controls (Fig. 3, C and D). Leukocyte-specific Nox2-deficient mice and EC-specific Nox2-deficient mice also had lower clinical scores than EC/leukocyte-specific Nox2-deficient mice (Fig. 3B). However, only EC-specific Nox2-deficient mice had a significant reduction in blood pressure levels as compared to control Nox2-replete mice after LPS injection (Fig. 3E).
Fig. 3.
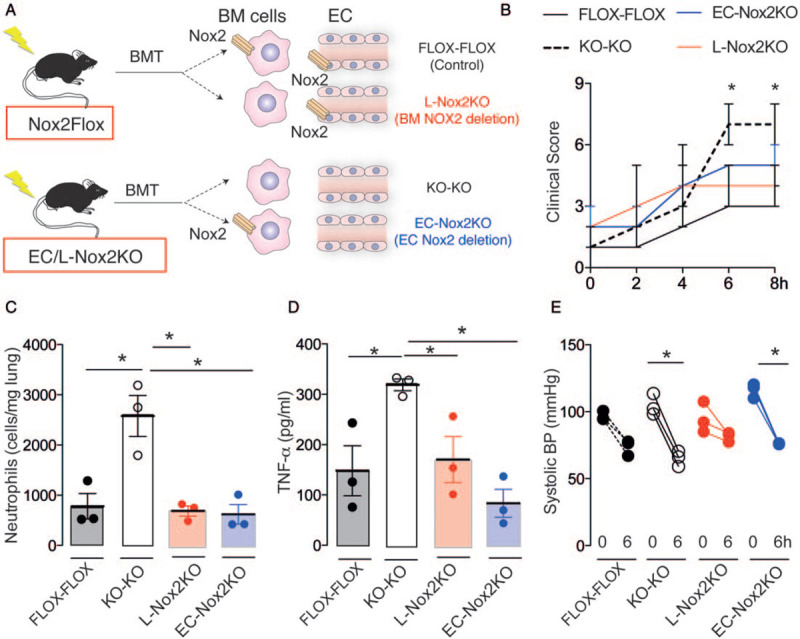
Endothelial Nox2 reduces the extent of systemic inflammation and hypotension induced by LPS. (A) Schematic depicting the generation of bone marrow (BM) chimeric mice. BMT indicates bone marrow transplantation. Nox2 was deficient solely in endothelial cells (EC) (EC-Nox2KO) or BM cells (L-Nox2KO). KO-KO represents Nox2 deficiency in both cell types and FLOX-FLOX represents irradiated Nox2-replete mice. (B) Median ± range of clinical scores; (C, D) mean ± SEM of lung neutrophil sequestration and plasma tumor necrosis-(TNF)-α levels after LPS injection; (E) systolic blood pressure (BP) under baseline conditions and 6 h after LPS injection. ∗P < 0.05 for highlighted comparisons (n = 3 per group). 2-way ANOVA followed by Bonferroni post-test (B, E), or Kruskal–Wallis followed by Dunn's multiple comparison test (C, D).
Taken together, our in vivo results indicate that both leukocyte and EC Nox2 limit systemic inflammation in response to LPS administration. However, only EC Nox2 affects the severity of hypotension induced by LPS.
Endothelial Nox2 limits NF-κB-activation and transcription of TLR4
We previously published that BM neutrophils and peritoneal macrophages deficient in Nox2 show increased expression of TLR4 compared to WT cells after in vitro LPS stimulation (17). Here, we found that after LPS injection, both global Nox2-deficient mice and animals with Tie2-targeted Nox2 deficiency (EC/L-Nox2KO) had higher TLR4 mRNA levels in aorta than WT controls (Fig. 4, A and B). Mice with Tie2-targeted Nox2 overexpression (Nox2Tg) showed lower aortic TLR4 mRNA levels (Fig. 4C). Both EC-specific Nox2-deficient and leukocyte-specific Nox2-deficient chimeric mice showed similar aortic TLR4 mRNA levels to control Nox2-replete mice injected with LPS, whereas combined EC and leukocyte-deficient mice had higher aortic TLR4 levels (Fig. 4D). These results suggest that both EC and leukocyte Nox2 contribute to the increase of TLR4 levels in aortic tissue.
Fig. 4.
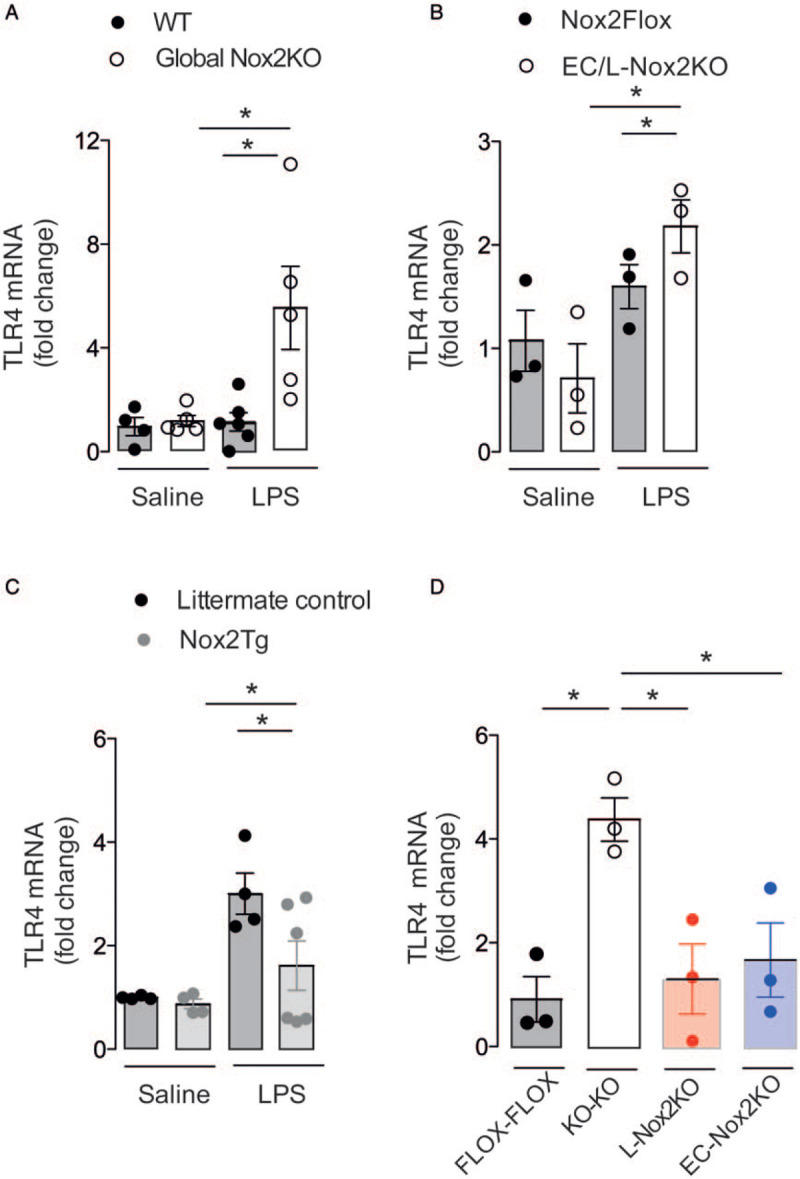
Nox2 deficiency in leukocytes and endothelial cells (EC) increases LPS-induced toll-like receptor (TLR4) expression in aorta. Levels of TLR4 mRNA in aorta 12 h after LPS injection (10 mg/kg i.v.) in global Nox2 deficient (Global Nox2KO, A), mice with Tie2-targeted Nox2 deficiency (EC/L-Nox2KO, B), mice with Tie2-targeted Nox2 overexpression (Nox2Tg, C) and chimeric mice with Nox2 deficiency solely in bone marrow cells (L-Nox2KO) or in endothelial cells (EC-Nox2KO) (D). WT indicates wild type. Graphs are mean ± SEM (n = 3–6 per group). ∗P < 0.05 for indicated comparisons. Kruskal–Wallis followed by Dunn's multiple comparison test.
To further investigate the molecular mechanism through which EC Nox2 affects TLR4 expression, we next studied HUVEC and HAEC stimulated with LPS. Pretreatment with a Nox2-specific peptide inhibitor, gp91ds-tat, led to a higher LPS-induced TLR4 expression than in cells pre-incubated with the scrambled-tat peptide control, sc-tat (Fig. 5A; See Figure, Supplemental Digital Content 5A-B, at ). HUVEC and HAEC showed increased ROS production after LPS treatment, which was prevented by gp91ds-tat but not the sc-tat peptide (see Figure, Supplemental Digital Content 5C-D, at ).
Fig. 5.
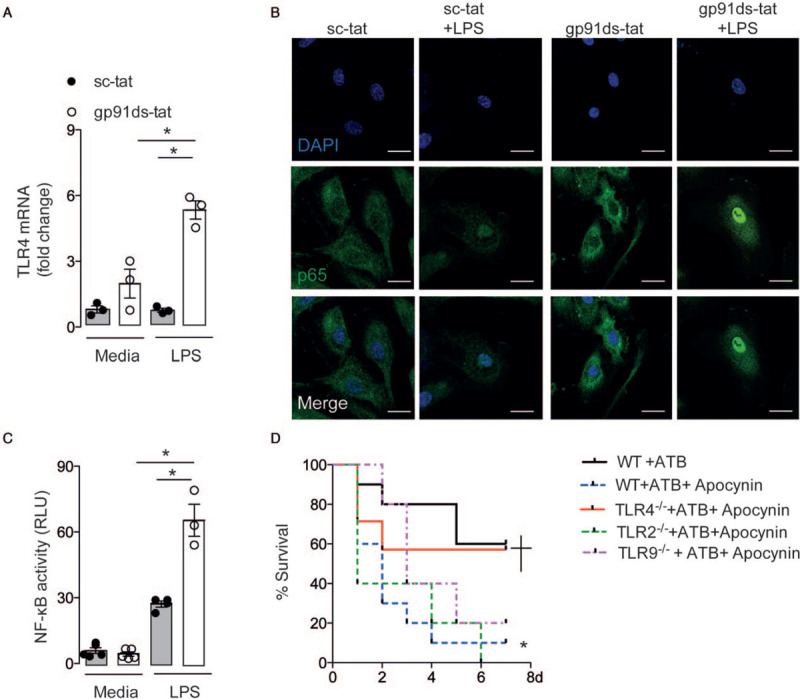
Endothelial Nox2 limits lipopolysaccharide (LPS)-induced transcription of toll-like receptor (TLR4) via nuclear factor (NF)-κB and reduces sepsis mortality. (A–D) Human umbilical vein endothelial cells (HUVEC) were pre-incubated with gp91ds-tat (30 μmol/L) or scrambled-tat (sc-tat, 30 μmol/L) and then stimulated with lipopolysaccharide (LPS, 200 ng/mL). (A) TLR4 mRNA levels 8 h after LPS stimulation. (B) Presence of p65 NF-κB subunit (green) in the nucleus (blue), 30 min after LPS stimulation. Scale bars, 25 μm. (C) Luciferase reporter assay for NF-κB activation after LPS stimulation. RLU: relative lumen units. Graphs and images represent mean ± SEM of three independent experiments performed in triplicate. ∗P < 0.05 for indicated comparisons. Kruskal–Wallis followed by Dunn's multiple comparison test. (D) Sepsis was induced by cecal ligation and puncture (CLP) in C57BL/6 wild-type (WT, n = 10) or TLR4-deficient (TLR4-/-, n = 7), or TLR2-deficient (TLR2-/-, n = 5), or TLR9-deficient (TLR9-/-, n = 5) mice. Some animals were pretreated with the Nox2 inhibitor apocynin (200 mg/kg, s.c.) All mice were treated with antibiotics (ATB, ertapenem sodium, 30 mg/kg, i.p.) after surgery. Survival was evaluated up to 7 days after surgery. ∗P < 0.05 compared to WT mice submitted to CLP and treated with ATB. +P < 0.05 compared to WT mice submitted to CLP and treated with ATB plus apocynin. Log-rank (Mantel–Cox) test.
Because TLR4 transcription is mainly regulated by NF-κB (20) and Nox2 downregulates NF-κB activity in leukocytes (19), we investigated whether endothelial Nox2 controls NF-κB activity downstream of TLR4 activation by LPS. Indeed, HUVEC and HAEC pre-incubated with gp91ds-tat showed higher nuclear levels of the p65 NF-κB subunit (Fig. 5B; See Figure, Supplemental Digital Content 5E, at ), as well as increased NF-κB activity in a luciferase reporter assay as compared to cells pre-incubated with sc-tat (Fig. 5C; see Figure, Supplemental Digital Content 5F, at ).
To further confirm the relevance of the increased TLR4 expression under Nox2-deficient conditions in vivo and using an animal model more similar to clinical polymicrobial sepsis, we submitted TLR4-deficient mice to CLP. Some mice were pretreated with an inhibitor of Nox2 (apocynin). All mice were treated with antibiotics to normalize the bacterial load between groups. Treatment with apocynin significantly decreased sepsis survival (Fig. 5D) in accordance with our previously published data (17). However, the higher mortality induced by inhibition of Nox2 was not observed in TLR4-deficient mice treated with apocynin (Fig. 5D). On the other hand, mice deficient in TLR2 or TLR9 demonstrated a higher mortality when treated with apocynin to inhibit Nox2, as compared to mice with a functional Nox2 (Fig. 5D). These results suggest a specific role of Nox2 in regulating NF-κB driven TLR4 transcription.
DISCUSSION
EC are important sources of inflammatory mediators and EC–leukocyte interactions are fundamental drivers in the pathogenesis of sepsis (2, 3). Here, we show that EC Nox2 limits systemic inflammation and hypotension induced by LPS, which may be attributed to the downregulation of NF-κB signaling and reduced expression of TLR4 (see Figure, Supplemental Digital Content 6, at ).
Global Nox2-deficient mice developed severe LPS-induced systemic inflammation, hypotension and multi-organ dysfunction as compared to control animals, in accordance with previous studies (17–19, 34). A similar phenotype was observed in mice with combined EC and leukocyte Nox2 deficiency (Tie2-targeted Nox2 KO), whereas Tie2-targeted Nox2 overexpressing mice (Nox2Tg) had reduced systemic inflammation and hypotension induced by LPS. While the Tie2-Cre approach efficiently targets EC, which abundantly express Tie2 (the receptor for angiopoietin-2) (35), leukocytes are also targeted (9). Since leukocytes participate in the pathogenesis of systemic inflammation and sepsis (36), we generated BM chimeric mice with deficiency of Nox2 solely in EC or BM cells to distinguish the relative contribution of these cell types. These experiments revealed that mice with EC-specific deficiency of Nox2 but not those with leukocyte-specific deficiency developed pronounced hypotension in response to LPS. On the other hand, both EC and leukocyte Nox2 contributed to control the overall severity of systemic inflammation. These findings corroborate the phenotype observed in global Nox2-deficient mice and EC/L-Nox2KO mice, and highlight the importance of Nox2-dependent interplay between these cell types in controlling inflammatory responses to endotoxin.
We observed that mice with combined EC and leukocyte Nox2 deficiency had higher TLR4 expression in aorta, whereas Nox2Tg mice had the opposite response after LPS injection. Because TLR4 is essential in the response to LPS, the molecular mechanism through which EC Nox2 controls LPS-induced systemic inflammation may be linked to the transcriptional regulation of TLR4. Consistent with our in vivo results, a specific Nox2-inhibitor enhanced LPS-induced transcription of TLR4 in cultured EC. Furthermore, studies in TLR4-deficient mice subjected to CLP, in which Nox2 activity was inhibited with apocynin, also supported an important role for Nox2 in regulating the response to sepsis.
NF-κB is the main transcription factor responsible for the synthesis of inflammatory mediators and TLR4 (20). Previous studies have highlighted the importance of Nox2 in downregulating NF-κB transcriptional activity in a redox-dependent manner (17–19). Similar to these previous findings in leukocytes, EC lacking Nox2 activity displayed increased NF-κB activation in response to LPS. These results help explain the overwhelming inflammatory response observed in mice with Tie2-targeted Nox2 deficiency when challenged with LPS. The effect of Nox2 deficiency in EC to aggravate LPS-induced hypotension might reflect an increased bioactivity of vasodilator NO due to reduction in Nox2-derived superoxide (37, 38). It is known that NO synthesis is induced during LPS challenge and the deficiency of Nox2-derived superoxide may facilitate an unrestrained action of NO. Although we did not directly examine this possibility, previous work from our lab showed that Nox2-overexpressing Tg mice do indeed have an increased NO–superoxide interaction which results in a decrease in NO biovailability (14). The increase in NF-κB/TLR4 signaling in the absence of EC Nox2 may also lead to an increased expression of inducible NO synthase (22, 38)—an established contributor to LPS-induced hypotension (39). Although leukocytes can also produce NO and potentially could contribute to hypotension induced by LPS, BM chimeric mice with deficiency of Nox2 solely in BM cells did not show attenuation of hypotension after LPS injection, different from the animals with EC-specific Nox2 deficiency. These results suggest that acute severe hypotension in endotoxemia is mitigated by EC Nox2 independent of Nox2 expression in leukocytes.
In contrast to the present report, some studies found a harmful rather than beneficial effect of Nox2 in specific inflammatory models or cultured EC (40, 41). These divergent findings may be due to the use of higher LPS concentrations in cultured cells. In fact, high concentrations of LPS are able to induce pronounced and non-compartmentalized ROS generation by Nox2 in EC, which may promote oxidative damage and an overstressed inflammatory response rather than modulating intracellular signaling (7, 42, 43).
We further used the CLP sepsis model to investigate whether Nox2 effects in controlling inflammatory response and hypotension were linked to TLR4 upregulation. Corroborating our previous study (17), mice lacking Nox2 activity showed higher sepsis mortality compared to those with functional Nox2. However, when TLR4 was also deficient, mortality during sepsis was similar to that in control mice. Interestingly, this effect was not observed in mice deficient in TLR2 or TLR9, which had a higher mortality than controls after Nox2 inhibition. These differences may be explained by distinct transcriptional regulation mechanisms among TLRs. Sp1/Sp3 as well as NF-κB regulate TLR2 mRNA expression (44) and TLR9 mRNA is regulated by CRE/CREB (45), whereas transcription of TLR4 mRNA is mainly induced by NF-κB (46). Interestingly, mice deficient in TLR4 treated with apocynin did not show a better survival rate than WT mice after CLP (data not shown), which is in accordance with our previous data implicating TLRs in control of infection (21, 47), which becomes irrelevant after the treatment of mice with antibiotics.
The beneficial or detrimental effects of Nox2 are likely to depend upon the context. Previous work from our group and others found that Nox2 in neutrophils and monocytes/macrophages decreases NF-κB transcriptional activity and the downstream expression of pro-inflammatory cytokines in sepsis (17, 19). The present study shows that a similar mechanism in EC acts to limit the severity of systemic inflammation and hypotension. However, in the setting of increased renin–angiotensin activation, we found that Nox2-dependent downregulation of NF-κB and FoxP3 in regulatory T cells reduces their suppressive capacity and increases pro-inflammatory cardiovascular remodeling (6). Also, myeloid cell Nox2 was found to have a modest effect on basal blood pressure through modulation of NO bioactivity (9), whereas during endotoxemia we show that endothelial Nox2 has a greater impact on the severity of hypotension. Notwithstanding the results with TLR-deficient animals subjected to an experimental model of sepsis in the current study, the effects observed in the genetic models of Nox2 deficiency in response to LPS model cannot necessarily be extrapolated to sepsis.
While we used a variety of genetic manipulations to dissect the role of EC and leukocytes, it is important to note that these approaches have potential limitations. The appropriate choice of control group for a gene-modified mouse strain is very important to minimize differences related to other factors. All the mouse strains used in the current study had been backcrossed on to a C57Bl6 background to avoid strain-related differences. We compared gene-modified animals to the relevant littermate controls in order to limit the effects of environmental changes in phenotype (48). Cell-specific Nox2-deficient animals generated using a Cre-Lox approach were compared to Flox controls, to minimize artifactual changes related to gene-targeting. BM transplantation experiments are potentially subject to off-target irradiation effects (9). To minimize this, all the controls groups also underwent irradiation and animals were allowed to recover for at least 4 weeks prior to LPS studies (49).
In conclusion, the current results identify a novel beneficial role for EC Nox2 in regulating the severity of LPS-induced systemic inflammation and hypotension by decreasing NF-κB-induced transcription of TLR4. Furthermore, our data show that leukocyte and EC Nox2 synergize to prevent overwhelming systemic inflammation during endotoxemia. Detrimental effects of increased Nox2 activity are implicated in the pathophysiology of many conditions (e.g. vascular disease and heart failure [4,8]). The current results suggest a need for some caution in the design of Nox2 inhibition strategies for such conditions. On the other hand, in patients with genetic deficiency of Nox2 (chronic granulomatous disease) the current findings that TLR4 levels are increased suggest that this pathway could be considered for targeting.
ACKNOWLEDGMENTS
The authors thank Xiaohong Zhang, Diva Amabilie, Giuliana Bertozi, Ana Katia dos Santos, Ieda Regina dos Santos, Sergio Rosa, Sidney Verissimo, Daniel Martin, Richard Thompson, Rose-Marie Minaisah, and Shana da Silva for technical assistance.
Author contributions
SCT, FQC, and AMS conceived the study, participated in experimental design, interpreted the data, and wrote the manuscript. SCT, CMS, GS, AP, and MZ carried out experimental assays. FMRL, TMC, JCA-F, CXS, AI, LRL, TM, and ACB participated in experimental design and data interpretation. AI and CXS also contributed to writing the manuscript. FQC and AMS supervised the study.
Supplementary Material
Footnotes
This research was supported by Sao Paulo Research Foundation (FAPESP) under grant agreement no 2012/24677-7 (SCT Fellowship) and 2013/08216-2 (Center for Research of Inflammatory Disease (CRID)), 2013/07937-8 (LRL as a member of CEPID Redoxoma), the European Society of Cardiology (ESC, Basic Research Fellowship), and the British Heart Foundation (BHF CH/1999001/11735).
The authors report no conflicts of interest.
Supplemental digital content is available for this article.
REFERENCES
- 1.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 315:801–810, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsiotou AG, Sakorafas GH, Anagnostopoulos G, Bramis J. Septic shock; current pathogenetic concepts from a clinical perspective. Med Sci Monitor 11:RA76–85, 2005. [PubMed] [Google Scholar]
- 3.Ince C, Mayeux PR, Nguyen T, Gomez H, Kellum JA, Ospina-Tascon GA, Hernandez G, Murray P, De Backer D, Workgroup AX. The endothelium in sepsis. Shock 45:259–270, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lassegue B, San Martin A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res 110:1364–1390, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trevelin SC, Shah AM, Lombardi G. Beyond bacterial killing: NADPH oxidase 2 is an immunomodulator. Immunol Lett 221:39–48, 2020. [DOI] [PubMed] [Google Scholar]
- 6.Emmerson A, Trevelin SC, Mongue-Din H, Becker PD, Ortiz C, Smyth LA, Peng Q, Elgueta R, Sawyer G, Ivetic A, et al. Nox2 in regulatory T cells promotes angiotensin II-induced cardiovascular remodeling. J Clin Invest 128 (7):3088–3101, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang M, Prosser BL, Bamboye MA, Gondim ANS, Santos CX, Martin D, Ghigo A, Perino A, Brewer AC, Ward CW, et al. Contractile function during angiotensin-II activation: increased Nox2 activity modulates cardiac calcium handling via phospholamban phosphorylation. J Am Coll Cardiol 66:261–272, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burgoyne JR, Mongue-Din H, Eaton P, Shah AM. Redox signaling in cardiac physiology and pathology. Circ Res 111:1091–1106, 2012. [DOI] [PubMed] [Google Scholar]
- 9.Sag CM, Schnelle M, Zhang J, Murdoch CE, Kossmann S, Protti A, Santos CXC, Sawyer G, Zhang X, Mongue-Din H, et al. Distinct regulatory effects of myeloid cell and endothelial cell NAPDH oxidase 2 on blood pressure. Circulation 135:2163–2177, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murdoch CE, Chaubey S, Zeng L, Yu B, Ivetic A, Walker SJ, Vanhoutte D, Heymans S, Grieve DJ, Cave AC, et al. Endothelial NADPH oxidase-2 promotes interstitial cardiac fibrosis and diastolic dysfunction through proinflammatory effects and endothelial-mesenchymal transition. J Am Coll Cardiol 63:2734–2741, 2014. [DOI] [PubMed] [Google Scholar]
- 11.Bendall JK, Rinze R, Adlam D, Tatham AL, de Bono J, Wilson N, Volpi E, Channon KM. Endothelial Nox2 overexpression potentiates vascular oxidative stress and hemodynamic response to angiotensin II: studies in endothelial-targeted Nox2 transgenic mice. Circ Res 100:1016–1025, 2007. [DOI] [PubMed] [Google Scholar]
- 12.Simon F, Fernandez R. Early lipopolysaccharide-induced reactive oxygen species production evokes necrotic cell death in human umbilical vein endothelial cells. J Hypertens 27:1202–1216, 2009. [DOI] [PubMed] [Google Scholar]
- 13.Macarthur H, Westfall TC, Riley DP, Misko TP, Salvemini D. Inactivation of catecholamines by superoxide gives new insights on the pathogenesis of septic shock. Proc Nat Acad Sci U S A 97:9753–9758, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murdoch CE, Alom-Ruiz SP, Wang M, Zhang M, Walker S, Yu B, Brewer A, Shah AM. Role of endothelial Nox2 NADPH oxidase in angiotensin II-induced hypertension and vasomotor dysfunction. Basic Res Cardiol 106:527–538, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Violi F, Sanguigni V, Carnevale R, Plebani A, Rossi P, Finocchi A, Pignata C, De Mattia D, Martire B, Pietrogrande MC, et al. Hereditary deficiency of gp91(phox) is associated with enhanced arterial dilatation: results of a multicenter study. Circulation 120:1616–1622, 2009. [DOI] [PubMed] [Google Scholar]
- 16.Gryglewski RJ, Palmer RM, Moncada S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature 320:454–456, 1986. [DOI] [PubMed] [Google Scholar]
- 17.Trevelin SC, Dos Santos CX, Ferreira RG, de Sa Lima L, Silva RL, Scavone C, Curi R, Alves-Filho JC, Cunha TM, Roxo-Junior P, et al. Apocynin and Nox2 regulate NF-kappaB by modifying thioredoxin-1 redox-state. Sci Rep 6:34581, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han W, Li H, Cai J, Gleaves LA, Polosukhin VV, Segal BH, Yull FE, Blackwell TS. NADPH oxidase limits lipopolysaccharide-induced lung inflammation and injury in mice through reduction-oxidation regulation of NF-kappaB activity. J Immunol 190:4786–4794, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singel KL, Segal BH. NOX2-dependent regulation of inflammation. Clin Sci 130:479–490, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med 13:460–469, 2007. [DOI] [PubMed] [Google Scholar]
- 21.Alves-Filho JC, de Freitas A, Russo M, Cunha FQ. Toll-like receptor 4 signaling leads to neutrophil migration impairment in polymicrobial sepsis. Crit Care Med 34:461–470, 2006. [DOI] [PubMed] [Google Scholar]
- 22.Ehrentraut S, Frede S, Stapel H, Mengden T, Grohe C, Fandrey J, Meyer R, Baumgarten G. Antagonism of lipopolysaccharide-induced blood pressure attenuation and vascular contractility. Arterioscler Thromb Vasc Biol 27:2170–2176, 2007. [DOI] [PubMed] [Google Scholar]
- 23.Nunes KP, de Oliveira AA, Lima VV, Webb RC. Toll-like receptor 4 and blood pressure: lessons from animal studies. Front Physiol 10:655, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(-) and systolic blood pressure in mice. Circ Res 89:408–414, 2001. [DOI] [PubMed] [Google Scholar]
- 25.Matsumoto H, Ogura H, Shimizu K, Ikeda M, Hirose T, Matsuura H, Kang S, Takahashi K, Tanaka T, Shimazu T. The clinical importance of a cytokine network in the acute phase of sepsis. Sci Rep 8:13995, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suzuki K, Ota H, Sasagawa S, Sakatani T, Fujikura T. Assay method for myeloperoxidase in human polymorphonuclear leukocytes. Anal Biochem 132:345–352, 1983. [DOI] [PubMed] [Google Scholar]
- 27.Grisham MB, Benoit JN, Granger DN. Assessment of leukocyte involvement during ischemia and reperfusion of intestine. Method Enzymol 186:729–742, 1990. [DOI] [PubMed] [Google Scholar]
- 28.Laurindo FR, Fernandes DC, Santos CX. Assessment of superoxide production and NADPH oxidase activity by HPLC analysis of dihydroethidium oxidation products. Method Enzymol 441:237–260, 2008. [DOI] [PubMed] [Google Scholar]
- 29.Berendes H, Bridges RA, Good RA. A fatal granulomatosus of childhood: the clinical study of a new syndrome. Minnesota Med 40:309–312, 1957. [PubMed] [Google Scholar]
- 30.Holmes B, Quie PG, Windhorst DB, Good RA. Fatal granulomatous disease of childhood. An inborn abnormality of phagocytic function. Lancet 1:1225–1228, 1966. [DOI] [PubMed] [Google Scholar]
- 31.Cooper MR, DeChatelet LR, McCall CE, LaVia MF, Spurr CL, Baehner RL. Complete deficiency of leukocyte glucose-6-phosphate dehydrogenase with defective bactericidal activity. J Clin Invest 51:769–778, 1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pollock JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J, Orkin SH, Doerschuk CM, Dinauer MC. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet 9:202–209, 1995. [DOI] [PubMed] [Google Scholar]
- 33.Kamisoglu K, Haimovich B, Calvano SE, Coyle SM, Corbett SA, Langley RJ, Kingsmore SF, Androulakis IP. Human metabolic response to systemic inflammation: assessment of the concordance between experimental endotoxemia and clinical cases of sepsis/SIRS. Crit Care 19:71, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whitmore LC, Hilkin BM, Goss KL, Wahle EM, Colaizy TT, Boggiatto PM, Varga SM, Miller FJ, Moreland JG. NOX2 protects against prolonged inflammation, lung injury, and mortality following systemic insults. J Inn Immunity 5:565–580, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang Y, Harrington A, Yang X, Friesel RE, Liaw L. The contribution of the Tie2+ lineage to primitive and definitive hematopoietic cells. Genesis 48:563–567, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hotchkiss RS, Moldawer LL, Opal SM, Reinhart K, Turnbull IR, Vincent JL. Sepsis and septic shock. Nature Rev Dis Primers 2:16045, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Henke N, Schmidt-Ullrich R, Dechend R, Park JK, Qadri F, Wellner M, Obst M, Gross V, Dietz R, Luft FC, et al. Vascular endothelial cell-specific NF-kappaB suppression attenuates hypertension-induced renal damage. Circ Res 101:268–276, 2007. [DOI] [PubMed] [Google Scholar]
- 38.Heo SK, Yun HJ, Noh EK, Park WH, Park SD. LPS induces inflammatory responses in human aortic vascular smooth muscle cells via Toll-like receptor 4 expression and nitric oxide production. Immunol Lett 120:57–64, 2008. [DOI] [PubMed] [Google Scholar]
- 39.Levy B, Collin S, Sennoun N, Ducrocq N, Kimmoun A, Asfar P, Perez P, Meziani F. Vascular hyporesponsiveness to vasopressors in septic shock: from bench to bedside. Intens Care Med 36:2019–2029, 2010. [DOI] [PubMed] [Google Scholar]
- 40.Weissmann N, Sydykov A, Kalwa H, Storch U, Fuchs B, Schnitzler M, Brandes RP, Grimminger F, Meissner M, Freichel M, et al. Activation of TRPC6 channels is essential for lung ischaemia-reperfusion induced oedema in mice. Nature Commun 3:649, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gandhirajan RK, Meng S, Chandramoorthy HC, Mallilankaraman K, Mancarella S, Gao H, Razmpour R, Yang XF, Houser SR, Chen J, et al. Blockade of NOX2 and STIM1 signaling limits lipopolysaccharide-induced vascular inflammation. J Clin Invest 123:887–902, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Joseph LC, Kokkinaki D, Valenti MC, Kim GJ, Barca E, Tomar D, Hoffman NE, Subramanyam P, Colecraft HM, Hirano M, et al. Inhibition of NADPH oxidase 2 (NOX2) prevents sepsis-induced cardiomyopathy by improving calcium handling and mitochondrial function. JCI Insight 2 (17):e94248, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Toda N, Tanabe S, Nakanishi S. Nitric oxide-mediated coronary flow regulation in patients with coronary artery disease: recent advances. Int J Angiol 20:121–134, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang T, Lafuse WP, Zwilling BS. NFkappaB and Sp1 elements are necessary for maximal transcription of toll-like receptor 2 induced by Mycobacterium avium. J Immunol 167:6924–6932, 2001. [DOI] [PubMed] [Google Scholar]
- 45.Tout I, Gomes M, Ainouze M, Marotel M, Pecoul T, Durantel D, Vaccarella S, Dubois B, Loustaud-Ratti V, Walzer T, et al. Hepatitis B virus blocks the CRE/CREB complex and prevents TLR9 transcription and function in human B cells. J Immunol 201:2331–2344, 2018. [DOI] [PubMed] [Google Scholar]
- 46.Nomura F, Akashi S, Sakao Y, Sato S, Kawai T, Matsumoto M, Nakanishi K, Kimoto M, Miyake K, Takeda K, et al. Cutting edge: endotoxin tolerance in mouse peritoneal macrophages correlates with down-regulation of surface toll-like receptor 4 expression. J J Immunol 164:3476–3479, 2000. [DOI] [PubMed] [Google Scholar]
- 47.Trevelin SC, Alves-Filho JC, Sonego F, Turato W, Nascimento DC, Souto FO, Cunha TM, Gazzinelli RT, Cunha FQ. Toll-like receptor 9 activation in neutrophils impairs chemotaxis and reduces sepsis outcome. Crit Care Med 40:2631–2637, 2012. [DOI] [PubMed] [Google Scholar]
- 48.Robertson SJ, Lemire P, Maughan H, Goethel A, Turpin W, Bedrani L, Guttman DS, Croitoru K, Girardin SE, Philpott DJ. Comparison of co-housing and littermate methods for microbiota standardization in mouse models. Cell Rep 2019; 27: 1910-1919.e2. [DOI] [PubMed] [Google Scholar]
- 49.Hu Y, Davison F, Ludewig B, Erdel M, Mayr M, Url M, Dietrich H, Xu Q. Smooth muscle cells in transplant atherosclerotic lesions are originated from recipients, but not bone marrow progenitor cells. Circulation 106:1834–1839, 2002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.