

Supplemental Digital Content is available in the text.
Keywords: cadherins, cardiomyopathy, mutation, sudden cardiac death, tachycardia
Abstract
Background:
Arrhythmogenic cardiomyopathy (ACM) is an inherited cardiac disease characterized by fibrofatty replacement of the right and left ventricle, often causing ventricular dysfunction and life-threatening arrhythmias. Variants in desmosomal genes account for up to 60% of cases. Our objective was to establish the prevalence and clinical features of ACM stemming from pathogenic variants in the nondesmosomal cadherin 2 (CDH2), a novel genetic substrate of ACM.
Methods:
A cohort of 500 unrelated patients with a definite diagnosis of ACM and no disease-causing variants in the main ACM genes was assembled. Genetic screening of CDH2 was performed through next-generation or Sanger sequencing. Whenever possible, cascade screening was initiated in the families of CDH2-positive probands, and clinical evaluation was performed.
Results:
Genetic screening of CDH2 led to the identification of 7 rare variants: 5, identified in 6 probands, were classified as pathogenic or likely pathogenic. The previously established p.D407N pathogenic variant was detected in 2 additional probands. Probands and family members with pathogenic/likely pathogenic variants in CDH2 were clinically evaluated, and along with previously published cases, altogether contributed to the identification of gene-specific features (13 cases from this cohort and 11 previously published, for a total of 9 probands and 15 family members). Ventricular arrhythmic events occurred in most CDH2-positive subjects (20/24, 83%), while the occurrence of heart failure was rare (2/24, 8.3%). Among probands, sustained ventricular tachycardia and sudden cardiac death occurred in 5/9 (56%).
Conclusions:
In this worldwide cohort of previously genotype-negative ACM patients, the prevalence of probands with CDH2 pathogenic/likely pathogenic variants was 1.2% (6/500). Our data show that this cohort of CDH2-ACM patients has a high incidence of ventricular arrhythmias, while evolution toward heart failure is rare.
Arrhythmogenic right ventricular (RV) cardiomyopathy is an inherited cardiac disease characterized by fibrofatty replacement of the RV, accompanied by electrical instability predisposing to ventricular arrhythmias and sudden cardiac death.1,2 The major form of the disease (arrhythmogenic RV cardiomyopathy) is characterized by a predominant RV involvement. However, given the evidence of forms with biventricular or predominant left ventricular involvement,3 the term arrhythmogenic cardiomyopathy (ACM) is now preferred.4,5
ACM is mainly caused by variants in genes encoding proteins of the desmosome (PKP2, DSP, DSG2, DSC2, and JUP), a specialized junction involved in cell adhesion.5,6 Desmosomes, together with the fascia adherens junctions and the gap junctions, are located at the intercalated discs, which maintain a tight connection between cardiomyocytes and allow cellular communication. In mammals, these 3 functional structures are found in close proximity7 and form a hybrid zone that operates as a single functional network known as the area composita.8,9 Nowadays, the idea of ACM being a disease whose functional basis extends beyond the desmosome to the wider zone of the intercalated discs is gaining interest.10,11 In a previous study by Mayosi et al,12 we identified clinically actionable variants in the CDH2 gene, encoding cadherin 2, a major structural component of fascia adherens junctions, as a novel genetic substrate of ACM.13 The concept that the genetic basis of ACM extends to nondesmosomal protein components of the intercalated disc has been further demonstrated by 2 other studies linking the ACM phenotype to variants in the CTNNA314 and TJP1 genes.15
These initial discovery studies12,14,15 highlighted the potential contribution of components of the intercalated disc to the pathogenesis of ACM, thus widening its genetic basis. However, these findings require validation in large ACM patient cohorts to allow correct estimates of their relative contribution, prevalence, and clinical characteristics. To this end, we have collected the largest available worldwide population of previously genotype-negative ACM probands (n=500) to validate the contribution of CDH2 variants to ACM and to establish a reliable prevalence. As a second step, we performed, whenever possible, cascade screening in the families of CDH2 positive probands. Finally, to identify gene-specific features, we analyzed all CDH2-positive probands and affected family members from this and previously published studies,12,13 for a total of 9 probands and 15 family members.
Methods
All data, analytical methods, and study materials supporting this study are available from the corresponding author on reasonable request for purposes of reproducing the results or replicating the procedures. The study was approved by the local Ethics institutional Review Boards, and the participants gave informed consent. A full description of the methods is provided in the Data Supplement.
Results
Study Population
Most of the 500 patients were White (n=416; 83%), followed by Han Chinese (n=77; 15%) and Black (n=2). In 5 cases, ethnicity was unknown. Males were 59% and females 41%.
Genetic Findings in CDH2
We identified 7 rare genetic variants (minor allele frequency <0.0001) in CDH2, 6 missense and 1 frameshift, in 8 unrelated subjects (Tables 1 and 2; Figure 1A). Among these, one variant was classified as pathogenic (p.D407N), 4 as likely pathogenic (p.P205L, p.N261D, p.N658Kfs*3, and p.P679A), and 2 as variants of unknown significance (Table 1). Cascade family screening was feasible in 4 CDH2-proband’s families (p.P205L, p.N261D, p.D407N, and p.P679A), for a total of 12 subjects (7 variant positive and 5 variant negative). An additional family member, affected by ACM, died suddenly before genetic testing could be performed.
Table 1.
Genetic Variants in the CDH2 Gene Identified in the ACM Cohort
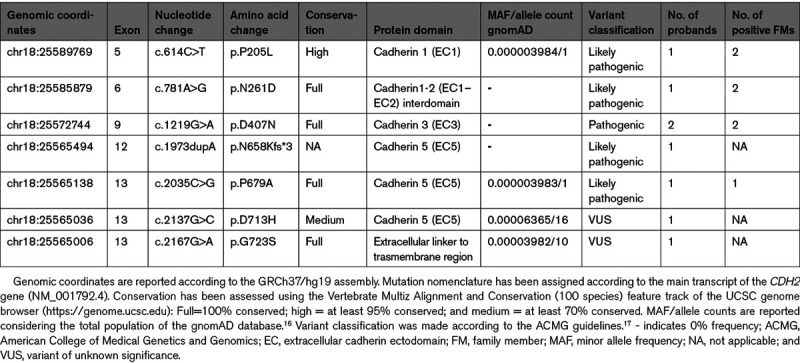
Table 2.
Task Force Diagnostic Criteria for ACM of the 6 Index Cases With Pathogenic/Likely Pathogenic Variants in CDH2
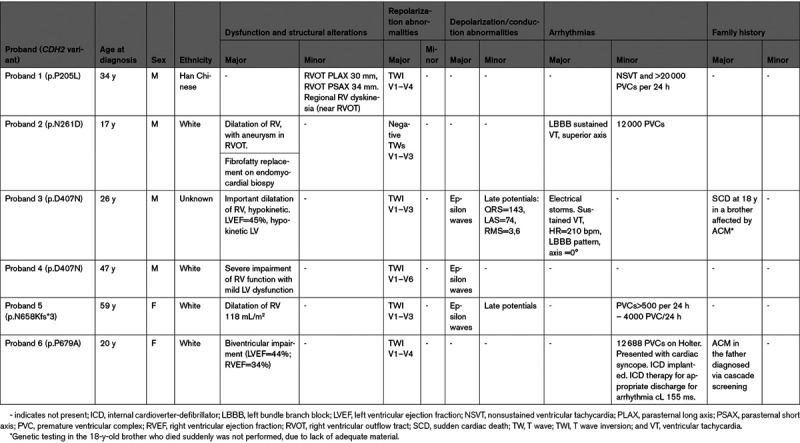
Figure 1.
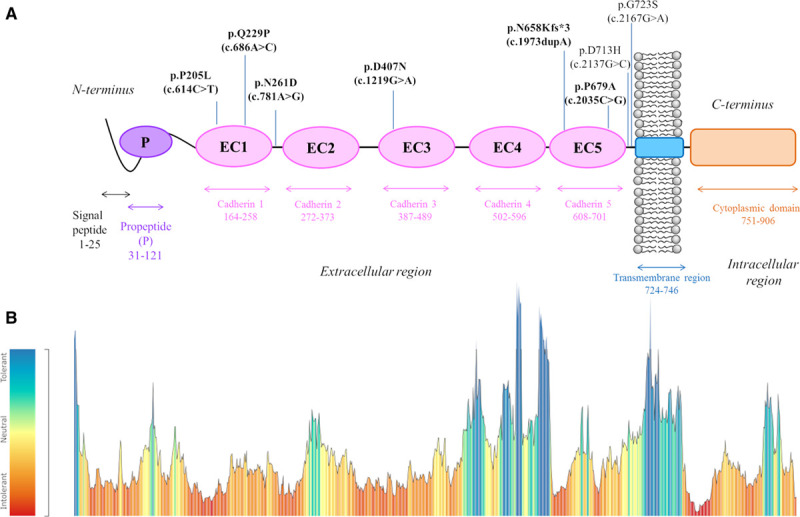
Schematic representation of the distinct domains of cadherin 2 protein and their respective genetic variant tolerance landscape. A, The mature protein is formed by 5 extracellular repeats (EC1–EC5 [ectodomain 1–ectodomain 5]), a single-pass transmembrane region and a cytoplasmic C-terminal domain, while the N-terminal propeptide (P), following the signal peptide, is removed by proteolysis (Pfam database EMBL-EBI).18 The length/boundaries of each domain are indicated with double-headed arrows just below the protein. All arrhythmogenic cardiomyopathy (ACM)-associated CDH2 genetic variants so far identified12,13 are depicted above the protein according to their position. Mutation nomenclature has been assigned according to the CDH2 main transcript (NM_001792.4), with the amino acid change (p.) reported first and the corresponding nucleotide change (c.) in brackets. Pathogenic/likely pathogenic variants are shown in bold. B, CDH2 tolerance landscape to missense variation, generated by MetaDome software analysis.19 The tolerance score is computed by the MetaDome software using the gnomAD population,16 as a missense over synonymous variant count ratio, calculated in a sliding window manner, for each position. The tolerance color-coded legend is reported on the left. The pathogenic/likely pathogenic missense variants identified in the ACM patient cohort all reside in red/orange regions (ie, the most intolerant to missense variation).
The p.D407N pathogenic variant, previously identified in 5 affected subjects of 2 independent studies,12,13 was also detected in 2/500 unrelated subjects comprising this study cohort. Both probands satisfied major ACM diagnostic criteria, such as structural dysfunction (important dilatation of RV) and depolarization/repolarization abnormalities (Table 2). One of the 2 p.D407N probands, symptomatic for arrhythmic events, had 3 affected relatives, one brother who died suddenly at age 18 years, another brother and a nephew (both carrying the p.D407N variant) with RV involvement and frequent premature ventricular complexes (PVCs) despite β-blocker therapy.
The p.N658Kfs*3 frameshift variant, resulting from a nucleotide duplication that putatively results in premature termination of the protein, is the first non missense variant thus far described in CDH2-associated ACM. The patient with this likely pathogenic variant is a female, diagnosed at 59 years, with dilatation of the RV, accompanied by depolarization/repolarization abnormalities and frequent PVCs (>500 per 24 hours, Table 2).
The other 3 variants classified as likely pathogenic (p.P205L, p.N261D, and p.P679A) are ultrarare missense variants (0 or 1 allele in gnomAD), located in the extracellular region of the protein (Figure 1A), with evidence in support of pathogenicity coming also from genotype-phenotype correlation data in the respective patient families (Figure 2). The p.N261D variant affects the Ca2+-binding site in the EC1–EC2 (ectodomain 1–ectodomain 2) interdomain that plays a key role in cadherin-cadherin adhesion.20 This variant was identified in a proband with sustained ventricular tachycardia (VT), implanted with an internal cardioverter-defibrillator at age 18 years, in one affected brother with left bundle branch block, nonsustained VTs and very frequent PVCs (40 000/24 hours), and in a cousin with signs of noncompaction and an episode of heart failure triggered by supra-VT (Figure 2). The p.P679A positive individual is a female who presented with syncope at age 20 years and who eventually developed biventricular structural impairment (Table 2). The variant was also present in her affected father, while it was absent in her unaffected sibling (Figure 2). The p.P205L variant resides in the EC1 domain that, together with EC2, is considered an essential structural element for cadherin-mediated intercellular adhesion.21 This variant was identified in a male proband who presented with repolarization abnormalities and nonsustained VTs. Cascade family screening also identified the variant in the affected father, while it was absent in 3 unaffected family members. A brother, positive for the same variant, was only partially investigated, and at present he cannot be clinically classified (Table 2; Figure 2).
Figure 2.
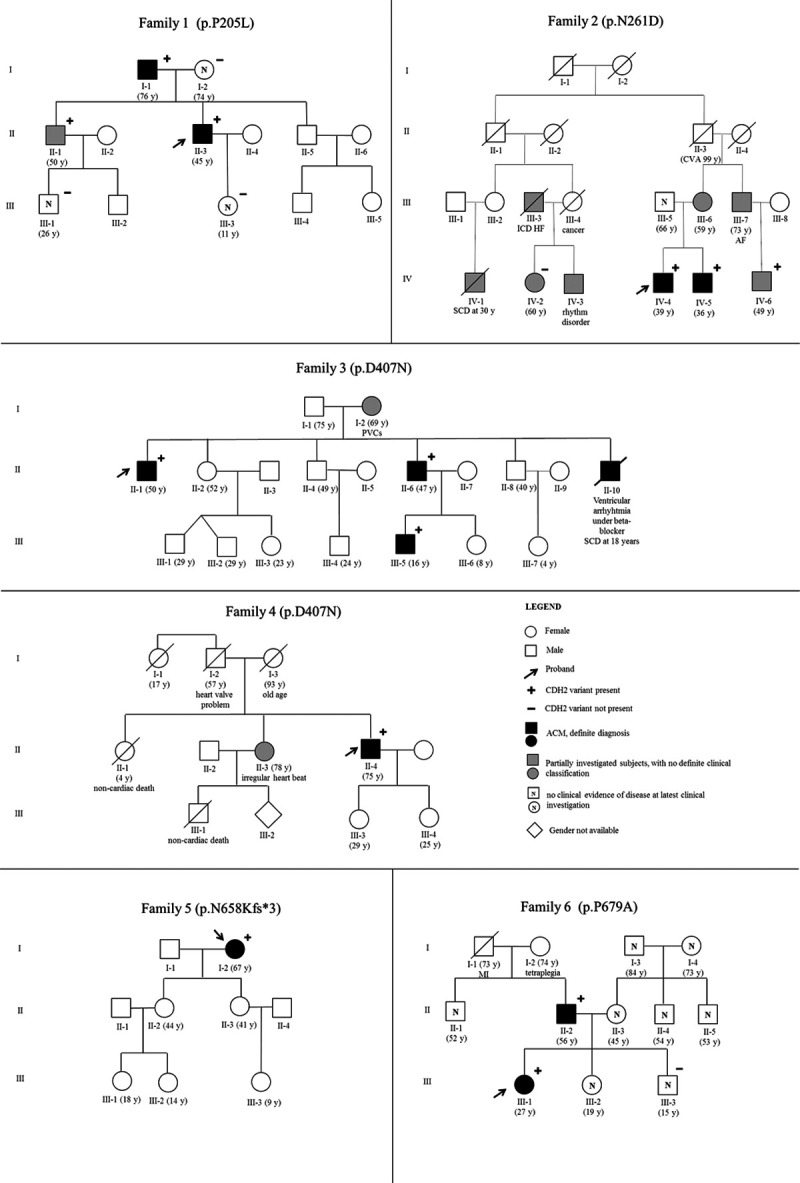
Pedigrees of the 6 families segregating the pathogenic/likely pathogenic variants identified in CDH2. The proband is indicated by an arrow. Black-filled symbols indicate affected subjects with a definite arrhythmogenic cardiomyopathy (ACM) diagnosis, while gray-colored symbols stand for partially investigated subjects for whom a complete clinical picture is not available. White symbols marked up with N correspond to subjects with no clinical evidence of disease at latest clinical investigation. + indicates mutation-positive subjects; − indicates mutation-negative subjects. CVA indicates cerebrovascular accident; HF, heart failure; MI, myocardial infarction; and PVC, premature ventricular contraction.
Finally, 2 rare variants were classified as variants of unknown significance due to lack of a high degree of conservation of the affected residue, few clinical information available and the presence of the variant in exome/genome datasets, albeit with minor allele frequencies much lower than the disease prevalence (Table 1).22
Pathogenic/likely pathogenic variants in other genes associated with arrhythmogenic and dilated cardiomyopathy were identified in only 3 probands not carrying any CDH2 variant (Table I in the Data Supplement).
In conclusion, 7 rare variants in CDH2 were identified in 8 out of 500 ACM unrelated patients (1.6% [95% CI, 0.5%–2.7%]) who lack a pathogenic/likely pathogenic variant in the major ACM genes. Considering only the 6 patients with American College of Medical Genetics and Genomics-graded pathogenic/likely pathogenic variants, the prevalence of CDH2 clinically actionable variants in this ACM cohort is 1.2% (6/500; 95% CI, 0.2%–2.2%).
CDH2 Genetic Variability and Variant Location
Since CDH2 is a novel human disease gene and to aid the eventual classification of genetic variants, it could be useful to evaluate its natural genetic variability. To this end, we took advantage of the publicly available exome/genome datasets including >140 000 subjects.
CDH2 is a gene extremely intolerant to loss-of-function variation, as indicated by the constraint metrics in the gnomAD database, with a high discrepancy between the number of expected (n=41) and observed (n=6) loss-of-function variants.
At the same time, the CDH2 gene, as many other genes, seems to be more tolerant to missense variation, with 74% of the theoretically expected variants being actually observed in the gnomAD population.16 However, the number of subjects with rare (minor allele frequency <0.0001) and potentially clinically relevant missense variants differs significantly between the ACM cohort (pathogenic/likely pathogenic, 5/500, 1.0%) and the gnomAD population (potentially pathogenic, 300/140 000, 0.2%; P=0.0010). This difference becomes even more striking when considering only the ultrarare (minor allele frequency <0.000005) and potentially clinically relevant missense variants (pathogenic/likely pathogenic, 5/500, 1.0% in ACM cases versus potentially pathogenic, 59/140 000, 0.04% in gnomAD; P<0.0001).
Regarding the localization within the protein of the pathogenic/likely pathogenic amino acid substitutions identified in our patient cohort, they exclusively reside in the extracellular region of cadherin 2 that appears intolerant to missense variation according to the computational analysis by MetaDome (Figure 1B).19 Specifically, most of these variants cluster in the head EC1–EC3 domains.
CDH2-Associated ACM Clinical Features
To identify gene-specific clinical features, we considered all probands with CDH2 pathogenic and likely pathogenic variants of the present (n=6) and of the 2 previously published studies (n=3)12,13 along with their 15 affected family members with complete clinical information (7 from this and 8 from previously published studies), for a total of 24 patients with CDH2-associated ACM (Table 3).
Table 3.
Arrhythmogenic Features of CDH2-Related ACM Patients so far Described
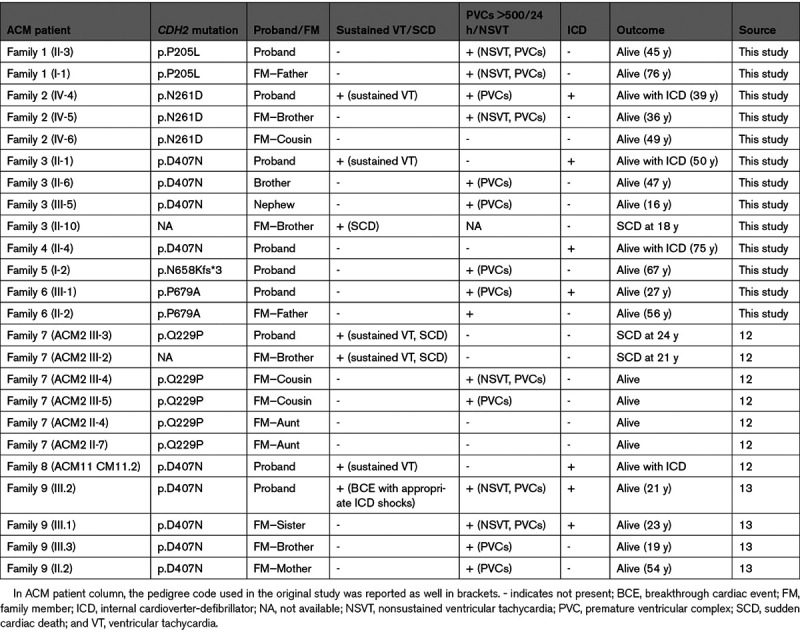
Sustained VTs and sudden cardiac death occurred in 5/9 probands (56%). All except one of the remaining index cases had frequent PVCs and nonsustained VTs, with an internal cardioverter-defibrillator implanted in 6 (67%) (Table 3). The only proband with no spontaneous arrhythmias had VT induction at the EP study (electrophysiological) and recurrent episodes of heart failure. None of the other probands had heart failure episodes and none underwent a heart transplant during a mean follow-up period of 13.5±8.5 years. Regarding the structural alterations observed, all probands had RV involvement, with a biventricular involvement observed in 3 (33%).
Among the 15 clinically affected CDH2-positive family members, 2 suffered sudden cardiac death and 10 had frequent PVCs and nonsustained VTs (Table 3). Left ventricular involvement was present in 4 genotype-positive family members and in only one of them heart failure was reported during supra-VT.
Discussion
The present report provides the first assessment of the prevalence of CDH2 clinically actionable variants in ACM and establishes its potential clinical features. CDH2 encodes cadherin 2, also known as N-cadherin, the main protein component of fascia adherens junctions at the intercalated disc. The main novel finding of this study is that pathogenic/likely pathogenic variants in CDH2 are responsible for 1.2% of genetically elusive ACM cases (95% CI, 0.2%–2.2%). Second, the CDH2-p.D407N, found in 9 patients with ACM appears to be the most common variant associated with CDH2-mediated ACM.
In addition, this study shows the presence of 2 rather specific clinical features for this cohort of CDH2-ACM. One is that left ventricular involvement was present in one-third of patients, with a rare evolution toward heart failure despite adequate follow-up period. The second is that the majority of CDH2-positive ACM patients have a ventricular arrhythmic phenotype (20/24, 83%), an observation with both prognostic and therapeutic implications.
Prevalence of CDH2 Variants in ACM and Clinical Implications
After the initial discovery of CDH2 variants as a novel genetic substrate of ACM,12 a finding soon after replicated in another study,13 the need of estimating the contribution of CDH2 variants to ACM pathogenesis in a large patient cohort emerged. Therefore, we have collected a cohort of 500 genetically negative ACM patients and performed genetic analysis of the CDH2 gene. The prevalence of CDH2 clinically actionable variants identified in this cohort is 1.2% (95% CI, 0.2%–2.2%), similar to our original estimation in the discovery study.12 The yield of genetic testing in ACM is estimated to be close to 60%,23,24 and therefore, a significant number of patients remains genetically elusive. Although the prevalence of CDH2 variants in ACM is low, this finding is not without potentially relevant clinical implications.
Indeed, the thorough clinical evaluation of those patients with CDH2-ACM enabled the identification of few gene-specific features. Specifically, some of the pathogenic variants so far identified, such as the p.Q229P12 and the original p.D407N,12,13 seem to be associated with a severe arrhythmic phenotype, with recurrent sustained VT and sudden cardiac death. It seems reasonable to postulate that the sudden deaths which occurred in these families have been arrhythmic in nature also because all these cases were clinically affected by ACM. This is consistent with the fact that most of probands and affected family members with CDH2 pathogenic/likely pathogenic variants had an arrhythmic phenotype (20/24, 83%), while the evolution toward heart failure appeared to be rare despite adequate follow-up.
These observations matter in terms of risk stratification and clinical management. Accordingly, CDH2 genetic screening should be considered in patients with a definite diagnosis of ACM but without significant variants in major desmosomal genes. The identification of the disease-causing variant has clinical consequences for probands and family members alike. Given that the disease progression and arrhythmic risk for ACM patients are mostly linked to physical activity,1,5 the early identification of family members with subclinical or silent forms is important to establish preventive strategies that might have a favorable impact on the natural history of the disease.
Another issue to be considered is that if the concept that the CDH2-associated ACM has a predominantly arrhythmogenic profile will be validated—similar to other genetic forms of ACM, such as those linked to lamin A (LMNA), filamin C (FLNC), and phospholamban (PLN)4—specific recommendations for internal cardioverter-defibrillator implantation might ensue.25 Clearly, all these observations are based on a limited number of patients. Further research and wider ascertainment strategies should provide more robust data for accurate genetic counseling of patients with CDH2 variants.
Mechanistic Implications of CDH2 Variants and Electrical Impairment in ACM
At present, the exact mechanism by which CDH2 variants may affect cadherin 2 protein function and foster the clinical development of ACM is unknown; however, current knowledge allows discussing possible hypotheses.
The only non missense CDH2 variant identified in this study, the p.N658Kfs*3, is expected to produce an aberrant mRNA transcript likely to be degraded by nonsense-mediated RNA decay26 without being translated into protein, leading to loss-of-function by haploinsufficiency. The haploinsufficiency mechanism has been described previously for truncating variants in proteins primarily involved in cell adhesion, such as the desmosomal cadherin desmocollin 2 (DSC2)27 and other components of the desmosome.28,29 Cadherin 2 (or N-cadherin) haploinsufficiency, in particular, has been mimicked in vivo in a cardiac-specific mouse model (heterozygous conditional knockout) where it decreases connexin 43 plaque size leading to an increased susceptibility to ventricular arrhythmias.30 Connexin 43 is the main structural component of gap junctions at the intercalated disc, and its reduction results in slow conduction, favoring arrhythmias by reentry.31 Furthermore, in these mice, also the expression of the zonula occludens 1, another protein of the intercalated disc recently implicated in ACM pathogenesis,15 was significantly reduced,30 potentially concurring to the destabilization of gap junctions.
CDH2-encoded cadherin 2 (N-cadherin) is part of a protein cluster at the intercalated disc that also includes the cardiac sodium channel Nav1.532 and the ability of these 2 proteins to influence each other has been demonstrated in induced pluripotent stem cell-derived cardiomyocytes of a patient with ACM,33 suggesting another potential mechanism of arrhythmogenesis.
The majority of pathogenic or likely pathogenic variants identified so far in CDH2 are missense variants. A reasonable hypothesis for their mechanism of action could be that since they all reside in the extracellular EC domains of the protein (Figure 1), these variants may adversely affect normal adhesive function. Such a mechanism was recently demonstrated in vitro for CDH2 missense variants associated to the agenesis of corpus callosum, axon pathfinding, cardiac, ocular, and genital defects syndrome, a multisystemic disorder characterized by developmental delay and intellectual disability, ocular and genital defects and congenital heart abnormalities.34 In this condition, mutant cadherins with missense variants in EC4–EC5 showed a defect in cell-cell adhesion, with consequences both on self-binding and trans-binding with wild-type cadherin 2 proteins.34 Another multisystemic disorder recently associated to genetic variants in CDH2 is the Peters anomaly, mainly characterized by corneal abnormalities.35 The reason why some CDH2 variants may create these multisystemic disorders while others result in a pure ACM phenotype is currently unknown; further studies are needed to unravel the functional effect of specific CDH2 variants and the molecular mechanisms leading from cadherin 2 dysfunction to different phenotypes.
Limitations
One limitation, intrinsic to the nature of collection of data from multiple sources, is the limited number of family members that could favor an ascertainment bias towards the more severe end of the spectrum of the disease. The family members effectively evaluated (CDH2-variant positive, with available clinical data) are only part of all other potential carriers. The evaluation of further CDH2-positive patients should reflect more precisely the different phenotypic variability present in this disease subtype and may modulate the severity of disease or the specific clinical features emerged so far.
This implies that the severity of CDH2 variants, as well as their association with other cardiac phenotypes beyond ACM, may need to be refined once a greater number of positive patients will be available.
Conclusions
The contribution of CDH2 clinically actionable variants to ACM is small but significant. Indeed, this rare genetic form appears to be associated in our cohort with a high arrhythmogenic profile and, therefore, its evaluation may be reasonable in patients affected by ACM in whom a disease-causing variant has not been identified. Whether or not the identification of disease-causing variants in this gene should modify the management of an otherwise still asymptomatic patient, as already happens for other rare genetic subtypes, such as LMNA and FLNC, remains to be determined.
We recognize the inherent ascertainment biases present in this type of study; however, current data suggest that there is a high incidence of ventricular arrhythmias in CDH2-ACM patients, with few evolving to heart failure.
Acknowledgments
We thank Pinuccia De Tomasi for expert editorial support. We thank Emmanuelle Bourcereau and Aurélie Thollet, the members of the National Referral Centre for Inherited Cardiac Arrhythmias of Nantes and its associated competence centers as well as the biological resource centre for biobanking (CHU Nantes, Hôtel Dieu, Centre de ressources biologiques [CRB], Nantes, F-44093, France [BRIF: BB-0033-00040]). We thank Jade Violleau and Stéphanie Bonnaud for their expert technical assistance and we are most grateful to the Genomics and Bioinformatics Core Facility of Nantes (GenoBiRD, Biogenouest) for its technical support. We thank Rob Zwart and Alex V. Postma for the genetic analyses performed in the Netherlands. Finally, the authors are grateful to the patients and families who agreed to participate in the research. Additional Information: Coauthor Bongani M. Mayosi, MD, PhD, died July 27, 2018.
Sources of Funding
The coordinating center was supported by the University of Milano Bicocca (2018-ATE-0359), by funds of the Italian Ministry of Health to the IRCCS Istituto Auxologico Italiano (CDH2VAL 26C722 2017_10_24_01; CARDGEN 26A502 2021_01_26_06), and partially by grant ERA-CVD JTC-2018-026 “Electromechanical presages of sudden cardiac death in the young: integrating imaging, modeling, and genetics for patient stratification”. The study was further supported by the Baylor-Hopkins Center for Mendelian Genomics (2UM1HG006542; United States). The Johns Hopkins ARVD/C Program is supported by the Leonie-Wild Foundation, the Dr Francis P. Chiaramonte Private Foundation, the Leyla Erkan Family Fund for ARVD Research, the Dr Satish, Rupal, and Robin Shah ARVD Fund at Johns Hopkins, the Bogle Foundation, the Healing Hearts Foundation, the Campanella family, the Patrick J. Harrison Family, the Peter French Memorial Foundation, and the Wilmerding Endowments. This work was also supported by the Fondation pour la Recherche Médicale (Equipe FRM DEQ20140329545), the Agence Nationale de la Recherche (ANR-14-CE10-0001-01), the H2020-MSCA-IF-2014 Program of the European Commission (RISTRAD-661617), the Regional Council of Pays-de-la-Loire (Etoile montante: REGIOCARD) in France, by the Netherlands Cardiovascular Research Initiative, supported by the Dutch Heart Foundation (CVON2012-10, CVON2018-30 PREDICT2, and CVON2015-12 eDETECT projects), and by a National Health and Medical Research Council (NHMRC) Practitioner Fellowship (no. 1154992) in Australia.
Disclosures
Dr Calkins has an investigator-initiated research grant from Boston Scientific Corp and Dr James receives salary support from this grant. The other authors report no conflicts.
Supplemental Materials
Online Materials and Methods
Online Tables I–II
Nonstandard Abbreviations and Acronyms
- ACM
- arrhythmogenic cardiomyopathy
- EC
- ectodomain
- EP
- electrophysiological study
- PVC
- premature ventricular complexes
- RV
- right ventricle
- VT
- ventricular tachycardia
This manuscript was sent to Ruth McPherson, MD, PhD, Guest Editor, for review by expert referees, editorial decision, and final disposition.
For Sources of Funding and Disclosures, see page 167.
B.M. Mayosi, P.J. Schwartz, and L. Crotti are joint senior authors.
The Data Supplement is available at https://www.ahajournals.org/doi/suppl/10.1161/CIRCGEN.120.003097.
This manuscript is dedicated to the memory of Professor Bongani M. Mayosi (28 January 1967 – 27 July 2018), Professor of Cardiology and Dean of the Faculty of Health Sciences at the University of Cape Town, South Africa. This study was inspired by him and made possible because of his relentless and successful search for the genetic cause of ACM in a family that he had personally followed for 20 years. He is sorely missed.
Contributor Information
Alice Ghidoni, Email: a.ghidoni@auxologico.it.
Perry M. Elliott, Email: perry.elliott@ucl.ac.uk.
Petros Syrris, Email: p.syrris@ucl.ac.uk.
Hugh Calkins, Email: hcalkins@jhmi.edu.
Cynthia A. James, Email: cjames7@jhmi.edu.
Daniel P. Judge, Email: judged@musc.edu.
Brittney Murray, Email: bdye1@jhmi.edu.
Julien Barc, Email: Julien.Barc@univ-nantes.fr.
Vincent Probst, Email: vincent.probst@univ-nantes.fr.
Jean Jacques Schott, Email: jean-jacques.schott@univ-nantes.fr.
Jiang-Ping Song, Email: fwsongjiangping@126.com.
Richard N.W. Hauer, Email: r.n.w.hauer@umcutrecht.nl.
Edgar T. Hoorntje, Email: e.t.hoorntje@umcg.nl.
J. Peter van Tintelen, Email: j.p.vantintelen-3@umcutrecht.nl.
Eric Schulze-Bahr, Email: eric.schulze-bahr@ukmuenster.de.
Robert M. Hamilton, Email: robert.hamilton@sickkids.ca.
Kirti Mittal, Email: kirtigenetics@gmail.com.
Christopher Semsarian, Email: c.semsarian@centenary.org.au.
Elijah R. Behr, Email: ebehr@sgul.ac.uk.
Michael J. Ackerman, Email: ackerman.michael@mayo.edu.
Cristina Basso, Email: cristina.basso@unipd.it.
Gianfranco Parati, Email: gianfranco.parati@unimib.it.
Davide Gentilini, Email: gentilini.davide@gmail.com.
Maria-Christina Kotta, Email: m.kotta@auxologico.it.
Bongani M. Mayosi, Email: mayosibonganim@yahoo.com.
References
- 1.Corrado D, Link MS, Calkins H. Arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2017; 376:61–72. doi: 10.1056/NEJMra1509267 [DOI] [PubMed] [Google Scholar]
- 2.Corrado D, van Tintelen PJ, McKenna WJ, Hauer RNW, Anastastakis A, Asimaki A, Basso C, Bauce B, Brunckhorst C, Bucciarelli-Ducci C, et al. Arrhythmogenic right ventricular cardiomyopathy: evaluation of the current diagnostic criteria and differential diagnosis. Eur Heart J. 2020; 41:1414–1429. doi: 10.1093/eurheartj/ehz669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Castelletti S, Vischer AS, Syrris P, Crotti L, Spazzolini C, Ghidoni A, Parati G, Jenkins S, Kotta MC, McKenna WJ, et al. Desmoplakin missense and non-missense mutations in arrhythmogenic right ventricular cardiomyopathy: Genotype-phenotype correlation. Int J Cardiol. 2017; 249:268–273. doi: 10.1016/j.ijcard.2017.05.018 [DOI] [PubMed] [Google Scholar]
- 4.Towbin JA, McKenna WJ, Abrams DJ, Ackerman MJ, Calkins H, Darrieux FCC, Daubert JP, de Chillou C, DePasquale EC, Desai MY, et al. 2019 HRS Expert Consensus Statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm. 2019; 16:e301–e372. doi: 10.1016/j.hrthm.2019.05.007 [DOI] [PubMed] [Google Scholar]
- 5.Corrado D, Basso C, Judge DP. Arrhythmogenic cardiomyopathy. Circ Res. 2017; 121:784–802. doi: 10.1161/CIRCRESAHA.117.309345 [DOI] [PubMed] [Google Scholar]
- 6.Delmar M, McKenna WJ. The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res. 2010; 107:700–714. doi: 10.1161/CIRCRESAHA.110.223412 [DOI] [PubMed] [Google Scholar]
- 7.Kline CF, Mohler PJ. Evolving form to fit function: cardiomyocyte intercalated disc and transverse-tubule membranes. Curr Top Membr. 2013; 72:121–158. doi: 10.1016/B978-0-12-417027-8.00004-0 [DOI] [PubMed] [Google Scholar]
- 8.Franke WW, Borrmann CM, Grund C, Pieperhoff S. The area composita of adhering junctions connecting heart muscle cells of vertebrates. I. Molecular definition in intercalated disks of cardiomyocytes by immunoelectron microscopy of desmosomal proteins. Eur J Cell Biol. 2006; 85:69–82. doi: 10.1016/j.ejcb.2005.11.003 [DOI] [PubMed] [Google Scholar]
- 9.Borrmann CM, Grund C, Kuhn C, Hofmann I, Pieperhoff S, Franke WW. The area composita of adhering junctions connecting heart muscle cells of vertebrates. II. Colocalizations of desmosomal and fascia adhaerens molecules in the intercalated disk. Eur J Cell Biol. 2006; 85:469–485. doi: 10.1016/j.ejcb.2006.02.009 [DOI] [PubMed] [Google Scholar]
- 10.Calore M, Lorenzon A, De Bortoli M, Poloni G, Rampazzo A. Arrhythmogenic cardiomyopathy: a disease of intercalated discs. Cell Tissue Res. 2015; 360:491–500. doi: 10.1007/s00441-014-2015-5 [DOI] [PubMed] [Google Scholar]
- 11.Zhao G, Qiu Y, Zhang HM, Yang D. Intercalated discs: cellular adhesion and signaling in heart health and diseases. Heart Fail Rev. 2019; 24:115–132. doi: 10.1007/s10741-018-9743-7 [DOI] [PubMed] [Google Scholar]
- 12.Mayosi BM, Fish M, Shaboodien G, Mastantuono E, Kraus S, Wieland T, Kotta MC, Chin A, Laing N, Ntusi NBA, et al. Identification of cadherin 2 (CDH2) mutations in arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Genet. 2017; 10:e001605. doi: 10.1161/CIRCGENETICS.116.001605 [DOI] [PubMed] [Google Scholar]
- 13.Turkowski KL, Tester DJ, Bos JM, Haugaa KH, Ackerman MJ. Whole exome sequencing with genomic triangulation implicates CDH2-encoded N-cadherin as a novel pathogenic substrate for arrhythmogenic cardiomyopathy. Congenit Heart Dis. 2017; 12:226–235. doi: 10.1111/chd.12462 [DOI] [PubMed] [Google Scholar]
- 14.van Hengel J, Calore M, Bauce B, Dazzo E, Mazzotti E, De Bortoli M, Lorenzon A, Li Mura IE, Beffagna G, Rigato I, et al. Mutations in the area composita protein αT-catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2013; 34:201–210. doi: 10.1093/eurheartj/ehs373 [DOI] [PubMed] [Google Scholar]
- 15.De Bortoli M, Postma AV, Poloni G, Calore M, Minervini G, Mazzotti E, Rigato I, Ebert M, Lorenzon A, Vazza G, et al. Whole-exome sequencing identifies pathogenic variants in TJP1 gene associated with arrhythmogenic cardiomyopathy. Circ Genom Precis Med. 2018; 11:e002123. doi: 10.1161/CIRCGEN.118.002123 [DOI] [PubMed] [Google Scholar]
- 16.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. ; Exome Aggregation Consortium. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016; 536:285–291. doi: 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. ; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17:405–424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, Qureshi M, Richardson LJ, Salazar GA, Smart A, et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019; 47(D1):D427–D432. doi: 10.1093/nar/gky995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wiel L, Baakman C, Gilissen D, Veltman JA, Vriend G, Gilissen C. MetaDome: Pathogenicity analysis of genetic variants through aggregation of homologous human protein domains. Hum Mutat. 2019; 40:1030–1038. doi: 10.1002/humu.23798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shapiro L, Weis WI. Structure and biochemistry of cadherins and catenins. Cold Spring Harb Perspect Biol. 2009; 1:a003053. doi: 10.1101/cshperspect.a003053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shan W, Yagita Y, Wang Z, Koch A, Fex Svenningsen A, Gruzglin E, Pedraza L, Colman DR. The minimal essential unit for cadherin-mediated intercellular adhesion comprises extracellular domains 1 and 2. J Biol Chem. 2004; 279:55914–55923. doi: 10.1074/jbc.M407827200 [DOI] [PubMed] [Google Scholar]
- 22.Pilichou K, Thiene G, Bauce B, Rigato I, Lazzarini E, Migliore F, Perazzolo Marra M, Rizzo S, Zorzi A, Daliento L, et al. Arrhythmogenic cardiomyopathy. Orphanet J Rare Dis. 2016; 11:33. doi: 10.1186/s13023-016-0407-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, Camm AJ, Ellinor PT, Gollob M, Hamilton R, et al. ; Heart Rhythm Society (HRS); European Heart Rhythm Association (EHRA). HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies. Europace. 2011; 13:1077–1109. doi: 10.1093/europace/eur245 [DOI] [PubMed] [Google Scholar]
- 24.Groeneweg JA, Bhonsale A, James CA, te Riele AS, Dooijes D, Tichnell C, Murray B, Wiesfeld AC, Sawant AC, Kassamali B, et al. Clinical presentation, long-term follow-up, and outcomes of 1001 arrhythmogenic right ventricular dysplasia/cardiomyopathy patients and family members. Circ Cardiovasc Genet. 2015; 8:437–446. doi: 10.1161/CIRCGENETICS.114.001003 [DOI] [PubMed] [Google Scholar]
- 25.Rella V, Parati G, Crotti L. Sudden cardiac death in children affected by cardiomyopathies: an update on risk factors and indications at transvenous or subcutaneous implantable defibrillators. Front Pediatr. 2020; 8:139. doi: 10.3389/fped.2020.00139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hug N, Longman D, Cáceres JF. Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 2016; 44:1483–1495. doi: 10.1093/nar/gkw010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gehmlich K, Syrris P, Peskett E, Evans A, Ehler E, Asimaki A, Anastasakis A, Tsatsopoulou A, Vouliotis AI, Stefanadis C, et al. Mechanistic insights into arrhythmogenic right ventricular cardiomyopathy caused by desmocollin-2 mutations. Cardiovasc Res. 2011; 90:77–87. doi: 10.1093/cvr/cvq353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rasmussen TB, Hansen J, Nissen PH, Palmfeldt J, Dalager S, Jensen UB, Kim WY, Heickendorff L, Mølgaard H, Jensen HK, et al. Protein expression studies of desmoplakin mutations in cardiomyopathy patients reveal different molecular disease mechanisms. Clin Genet. 2013; 84:20–30. doi: 10.1111/cge.12056 [DOI] [PubMed] [Google Scholar]
- 29.Rasmussen TB, Nissen PH, Palmfeldt J, Gehmlich K, Dalager S, Jensen UB, Kim WY, Heickendorff L, Mølgaard H, Jensen HK, et al. Truncating plakophilin-2 mutations in arrhythmogenic cardiomyopathy are associated with protein haploinsufficiency in both myocardium and epidermis. Circ Cardiovasc Genet. 2014; 7:230–240. doi: 10.1161/CIRCGENETICS.113.000338 [DOI] [PubMed] [Google Scholar]
- 30.Li J, Levin MD, Xiong Y, Petrenko N, Patel VV, Radice GL. N-cadherin haploinsufficiency affects cardiac gap junctions and arrhythmic susceptibility. J Mol Cell Cardiol. 2008; 44:597–606. doi: 10.1016/j.yjmcc.2007.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Severs NJ, Bruce AF, Dupont E, Rothery S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc Res. 2008; 80:9–19. doi: 10.1093/cvr/cvn133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leo-Macias A, Agullo-Pascual E, Sanchez-Alonso JL, Keegan S, Lin X, Arcos T, Liang FX, Korchev YE, Gorelik J, Fenyo D, et al. Nanoscale visualization of functional adhesion/excitability nodes at the intercalated disc. Nat Commun. 2016; 7:10342. doi: 10.1038/ncomms10342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Te Riele AS, Agullo-Pascual E, James CA, Leo-Macias A, Cerrone M, Zhang M, Lin X, Lin B, Sobreira NL, Amat-Alarcon N, et al. Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non-canonical mechanisms for disease pathogenesis. Cardiovasc Res. 2017; 113:102–111. doi: 10.1093/cvr/cvw234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Accogli A, Calabretta S, St-Onge J, Boudrahem-Addour N, Dionne-Laporte A, Joset P, Azzarello-Burri S, Rauch A, Krier J, Fieg E, et al. ; Undiagnosed Diseases Network. de novo pathogenic variants in N-cadherin cause a syndromic neurodevelopmental disorder with corpus collosum, axon, cardiac, ocular, and genital defects. Am J Hum Genet. 2019; 105:854–868. doi: 10.1016/j.ajhg.2019.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reis LM, Houssin NS, Zamora C, Abdul-Rahman O, Kalish JM, Zackai EH, Plageman TF, Jr, Semina EV. Novel variants in CDH2 are associated with a new syndrome including Peters anomaly. Clin Genet. 2020; 97:502–508. doi: 10.1111/cge.13660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MG, Daubert JP, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010; 121:1533–1541. doi: 10.1161/CIRCULATIONAHA.108.840827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gollob MH, Blier L, Brugada R, Champagne J, Chauhan V, Connors S, Gardner M, Green MS, Gow R, Hamilton R, et al. Recommendations for the use of genetic testing in the clinical evaluation of inherited cardiac arrhythmias associated with sudden cardiac death: Canadian Cardiovascular Society/Canadian Heart Rhythm Society joint position paper. Can J Cardiol. 2011; 27:232–245. doi: 10.1016/j.cjca.2010.12.078 [DOI] [PubMed] [Google Scholar]
- 38.Hershberger RE, Givertz MM, Ho CY, Judge DP, Kantor PF, McBride KL, Morales A, Taylor MRG, Vatta M, Ware SM; ACMG Professional Practice and Guidelines Committee. Genetic evaluation of cardiomyopathy: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2018; 20:899–909. doi: 10.1038/s41436-018-0039-z [DOI] [PubMed] [Google Scholar]
- 39.den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, Roux AF, Smith T, Antonarakis SE, Taschner PE. HGVS recommendations for the description of sequence variants: 2016 Update. Hum Mutat. 2016; 37:564–569. doi: 10.1002/humu.22981 [DOI] [PubMed] [Google Scholar]
- 40.Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR; 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015; 526:68–74. doi: 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seattle, WA: Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP). Accessed January 8, 2020. http://evs.gs.washington.edu/EVS/. [Google Scholar]
- 42.Ghosh R, Oak N, Plon SE. Evaluation of in silico algorithms for use with ACMG/AMP clinical variant interpretation guidelines. Genome Biol. 2017; 18:225. doi: 10.1186/s13059-017-1353-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019; 47(D1):D886–D894. doi: 10.1093/nar/gky1016 [DOI] [PMC free article] [PubMed] [Google Scholar]