

Supplemental Digital Content is available in the text.
Keywords: cardiomyopathy, hypertrophic; genetic testing; oligonucleotide, antisense; stem cell; transcriptome
Abstract
Background:
Transcriptome sequencing can improve genetic diagnosis of Mendelian diseases but requires access to tissue expressing disease-relevant transcripts. We explored genetic testing of hypertrophic cardiomyopathy using transcriptome sequencing of patient-specific human induced pluripotent stem cell derived cardiomyocytes (hiPSC-CMs). We also explored whether antisense oligonucleotides (AOs) could inhibit aberrant mRNA splicing in hiPSC-CMs.
Methods:
We derived hiPSC-CMs from patients with hypertrophic cardiomyopathy due to MYBPC3 splice-gain variants, or an unresolved genetic cause. We used transcriptome sequencing of hiPSC-CM RNA to identify pathogenic splicing and used AOs to inhibit this splicing.
Results:
Transcriptome sequencing of hiPSC-CMs confirmed aberrant splicing in 2 people with previously identified MYBPC3 splice-gain variants (c.1090+453C>T and c.1224-52G>A). In a patient with an unresolved genetic cause of hypertrophic cardiomyopathy following genome sequencing, transcriptome sequencing of hiPSC-CMs revealed diverse cryptic exon splicing due to an MYBPC3 c.1928-569G>T variant, and this was confirmed in cardiac tissue from an affected sibling. Antisense oligonucleotide treatment demonstrated almost complete inhibition of cryptic exon splicing in one patient-specific hiPSC-CM line.
Conclusions:
Transcriptome sequencing of patient specific hiPSC-CMs solved a previously undiagnosed genetic cause of hypertrophic cardiomyopathy and may be a useful adjunct approach to genetic testing. Antisense oligonucleotide inhibition of cryptic exon splicing is a potential future personalized therapeutic option.
Genetic testing with transcriptome sequencing is a new approach that involves sequencing all of the mRNA transcripts of a diseased tissue. A key advantage of transcriptome sequencing is that it demonstrates the outcomes of mRNA splicing; thus, it can highlight splice-disrupting intronic variants that may not have been sequenced, or considered, with DNA sequencing.1–3 A limitation of this approach is that it requires mRNA extracted from a relevant diseased tissue of the patient, which is often impossible to access without invasive or unwarranted surgery, or the resected tissue is stored in a way that does not preserve the RNA integrity.
Peripheral blood cells can be reprogrammed into induced pluripotent stem cells (iPSCs) and then differentiated into almost any cell type.4 IPSC technology could therefore overcome a key limitation of transcriptome sequencing–based genetic testing by providing convenient access to disease relevant cells of any patient. Furthermore, these patient-derived cells replicate cellular phenotypes of disease and can be considered a disease-in-a-dish, which provide patient-specific models to study disease etiology, progression, and therapeutics.5,6
Hypertrophic cardiomyopathy (HCM) is the most common genetic heart disorder, affecting up to 1 in 500 individuals, and leads to significant morbidity and mortality, including heart failure and sudden death.7 While major advances have been made in defining the genetic causes of HCM, screening of all of the known causal genes identifies a pathogenic variant in ≈40% of all HCM.8 Recently, variants within intronic regions that disrupt splicing of an established HCM gene, myosin binding protein C3 (MYBPC3 transript NM_000256.3), have emerged as an underappreciated cause of HCM.9–11 Variants causing aberrant splicing can be challenging to identify from DNA sequence context alone and usually require analysis of mRNA to confirm their clinical relevance.12 Transcriptome sequencing of patients with HCM, in which prior DNA based analysis has not revealed a molecular cause of disease, may thus be an adjunct approach to improve the genetic testing diagnostic yield. Furthermore, the emergence of deep intronic splice gain variants in an appreciable number of HCM families raises the intriguing possibility of a new future therapeutic option involving inhibition of aberrant splicing using antisense oligonucleotides (AOs).
Herein, we show how transcriptome sequencing of patient-specific human iPSC-derived cardiomyocytes (hiPSC-CMs) identifies the genetic cause of HCM. Aberrant mRNA splicing of MYBPC3 was detected with transcriptome sequencing and pinpointed deep intronic variants that activate splicing of cryptic exons. The same aberrant mRNA was confirmed in myocardial tissue of one patient’s affected sibling. We also use hiPSC-CMs to evaluate antisense oligonucleotides (AOs) inhibition of cryptic exon splicing. Transcriptome sequencing of patient-specific iPSC-derived tissue is an adjunct approach to genetic testing of families in which current strategies have not achieved a genetic diagnosis and where diseased tissue, or a suitable surrogate tissue, is not available.
Methods
Extended details of methods and computational procedures are provided in the Data Supplement.
Patients provided consent for cellular and genetic studies, which were carried out in accordance with the ethics protocol approved by the Sydney Local Health District Ethics Review Committee, Australia, and The University of Sydney, Australia. Human heart tissue was provided with approval from the University of Sydney Human Research Ethics Committee, No. 2016/923.
The Genotype-Tissue Expression Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this article were obtained from the Genotype-Tissue Expression Portal on August 27, 2019 and are publicly available at https://www.gtexportal.org/home/datasets. Raw RNA sequencing files from human heart tissue during developmental stages13 are publicly available at ArrayExpress with the accession code E-MTAB-6814 and can be accessed at https://www.ebi.ac.uk/arrayexpress/. Because of the confidential nature of some of the research materials supporting this publication, not all of the data can be made accessible to other researchers. Please contact the corresponding author for more information.
Results
Patient Clinical Characteristics and Prior Genetic Testing Outcomes
We included 3 unrelated people with HCM for transcriptome-sequencing of hiPSC-CMs, including 2 with a known genetic cause of disease and 1 with an indeterminate genetic test after genome sequencing (Figure I in the Data Supplement), as we previously reported in detail.9 DA1 is a male aged 48 years with severe nonobstructive HCM and no family history of HCM. He harbors a likely pathogenic MYBPC3 c.1224-52G>A variant that creates a new splice acceptor site resulting in a 50 bp extension of exon 14. This variant accounts for 1% of HCM in 3 large cohorts of HCM probands.11,14 SW1 is a female aged 65 years with moderate nonobstructive HCM and a family history of HCM in her father, brother, and son. She harbors a likely pathogenic MYBPC3 c.1090+453C>T variant that creates a new splice donor site resulting in activation of a 77 bp cryptic exon between exons 12 and 13. ID4 is a female aged 56 years with nonobstructive HCM. She has a family history of HCM in her sister, mother, and aunt, and an uncle had a sudden cardiac death. Her sister and mother have an implantable cardioverter defibrillator and her sister (ID2) underwent a septal myectomy from which a piece of the resected myocardial tissue was stored in liquid nitrogen within minutes of harvest. Genome sequencing identified a deep intronic MYBPC3 c.1928-569G>T variant in ID4 that creates a potential splice acceptor sequence. Analysis of RNA extracted from venous blood from ID4 and myectomy tissue from ID2 with RT-PCR showed canonical splicing of exons 20 and 21 only, and the variant was classified as benign.9 We did not establish the genetic cause of HCM in this family with genome sequencing.
Cardiac Gene Expression Levels in Accessible Tissues
We explored the expression levels of 8 established HCM genes in various accessible human tissues using the Genotype-Tissue Expression project data to assess whether any would be suitable for transcriptome sequencing (Table I in the Data Supplement). Tissues accessible using noninvasive procedures include whole blood, skin fibroblasts, skeletal muscle, and renal epithelial cells, which can be harvested from urine. Each tissue showed very low expression of <1 transcripts per million of at least one of the established HCM genes. In contrast, hiPSC-CMs of 4 unrelated individuals showed very high expression (>500 transcripts per million) of all 8 genes, suggesting transcriptome sequencing of these cells would have ample sequencing read depth to detect RNA splice junctions.
Finding Novel Splice Junctions in Patients With a Known Genetic Cause of HCM
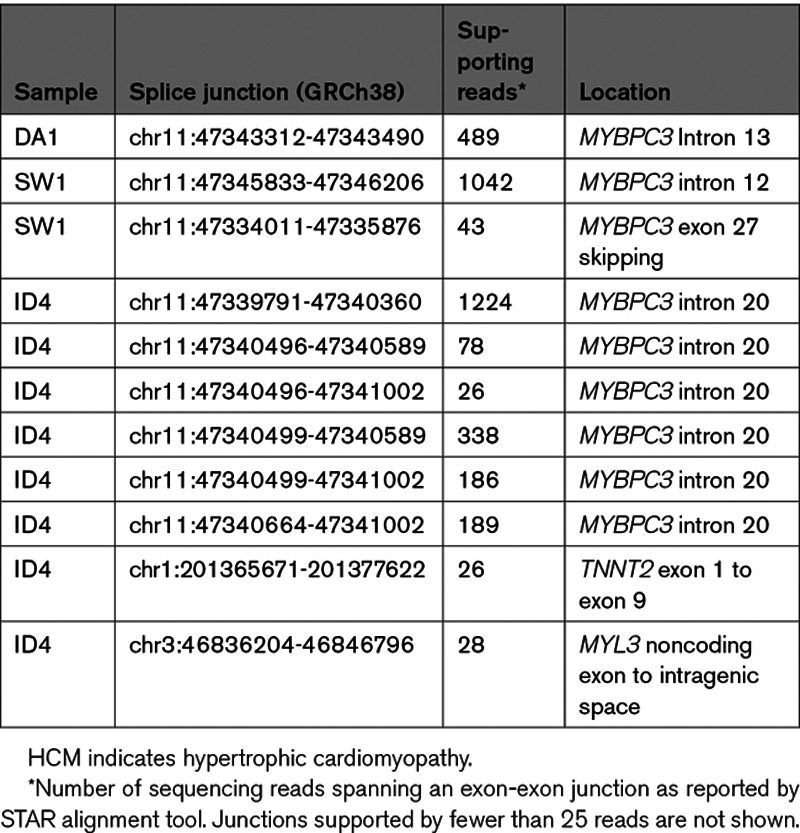
Transcriptome sequencing was performed on hiPSC-CMs of the genetically solved cases, DA1 and SW1. We extracted all splice junctions within 8 established HCM genes, which were supported by >25 sequencing reads and were absent in our splice junction reference set (Table). DA1 had only one novel splice junction, supported by 489 sequencing reads, which joins MYBPC3 exon 13 to a 50 bp extension of exon 14. This new exon has a splice acceptor site created by the previously identified c.1224-52G>A variant (Figure 1A). In SW1, we found a novel splice junction supported by 1042 sequencing reads that joins MYBPC3 exon 12 to a new 77 bp exon composed of intron 12 sequence (Figure 1B). The splice donor site of this new exon (exon 12a) is created by the previously identified c.1090+453C>T variant. Further inspection of splice junctions in MYBPC3 intron 12 of SW1 revealed a second new exon of 85 bp (exon 12b) that is flanked by canonical AG/GT splice sites in the reference sequence. We found a very low level of exon 12b splicing in both control hiPSC-CMs lines, with 59 and 7 supporting reads, but not in the GENCODE gene set. We confirmed the aberrant MYBPC3 splicing found in DA1 and SW1 with RT-PCR amplification and Sanger sequencing of mRNA extracted from hiPSC-CMs and whole blood (Figure II in the Data Supplement). We also detected a very low level of canonical MYBPC3 exon 27 skipping in hiPSC-CMs of SW1, supported by 43 sequencing reads, which was not considered to be biologically relevant.
Figure 1.
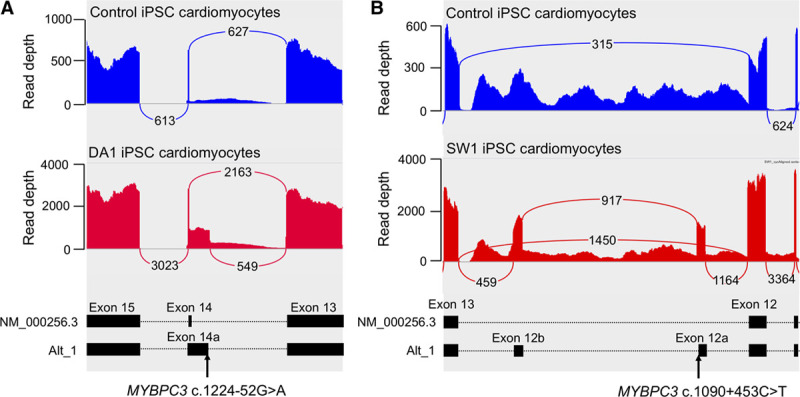
Novel MYBPC3 splice junctions detected in human induced pluripotent stem cell derived cardiomyocyte (hiPSC-CM) of DA1 and SW1. Sashimi plots show sites of aberrant MYBPC3 splice junctions in control hiPSCM-CMs (upper blue) and (A) DA1 (left red) and (B) SW1 (right red). Loops with numbers show number of sequencing reads supporting each major splice junction, as reported by Integrated Genome Viewer. Lower panels show transcript structures with exons (black boxes) and position of intronic splice gain variant.
Table.
Novel Splice Junctions in 8 Established HCM Genes
Transcriptome Sequencing of iPSC Cardiomyocytes Identifies a Cause of HCM
We next performed transcriptome sequencing on hiPSC-CMs of the genetically unsolved case, ID4, and looked for novel splice junctions in the 8 established HCM genes. We found a cluster of novel splice junctions in MYBPC3 intron 20 that corresponded to 3 cryptic exons (Table, Figure 2A). Exon 20a is composed of 138 bp of intron 20 sequence and is flanked by a novel splice donor site, created by a c.1928-569G>T variant. Splicing of exon 20a to canonical exon 21 is supported by 1224 sequencing reads. Exon 20b is a 413 bp extension of exon 20 and exon 20c is composed of 74 bp of intron 20 sequence, and both exons are flanked by canonical AG/GT splice sites in the reference sequence. The MYBPC3 c.1928-569G>T variant co-segregates with disease in affected family members ID1, ID2, ID4 and obligate carrier ID3 (Figure I in the Data Supplement). To confirm whether the same aberrant splicing of MYBPC3 occurs in the heart, we used RNA extracted from frozen septal myectomy tissue of an affected sibling, ID2. The RNA had an RNA integrity value of 3.2, indicating that it was degraded and unsuitable for transcriptome sequencing; however, it supported RT-PCR amplification using primers annealing within exon 20b and exon 21 of MYBPC3. Sanger sequencing confirmed an identical pattern of aberrant splicing as found in the proband’s hiPSC-CMs (Figure 2B), and western analysis of myectomy tissue showed ID2 had the lowest level of MYBPC3 protein when compared with 3 age- and sex-matched control hearts and 3 age-matched HCM probands without MYBPC3 truncating variants (Figure 2C and 2D). MYBPC3 transcripts containing exon 20b would result in a reading frameshift, whereas those containing the in-frame exon 20a would encode 18 new amino acids after Glu642 followed by a premature termination codon. We classified the MYBPC3 c.1928-569G>T variant as likely pathogenic using the American College of Medical Genetics and Association of Molecular Pathology guidelines for the interpretation of sequence variants.15 Our functional study of mRNA is supportive of a damaging effect on the protein (PS3), the variant is absent in gnomAD version 2.1.1 and version 3 (PM2), and it co-segregates with disease in 4 affected family members with 4 meiosis in an established disease gene (PP1). In hiPSC-CMs of ID4, we also detected very low levels of a novel splice junction between the noncoding exon 1 and exon 9 of TNNT2, and a noncoding exon of MYL3 and a downstream intragenic region, neither of which were considered to be biologically relevant (Table).
Figure 2.
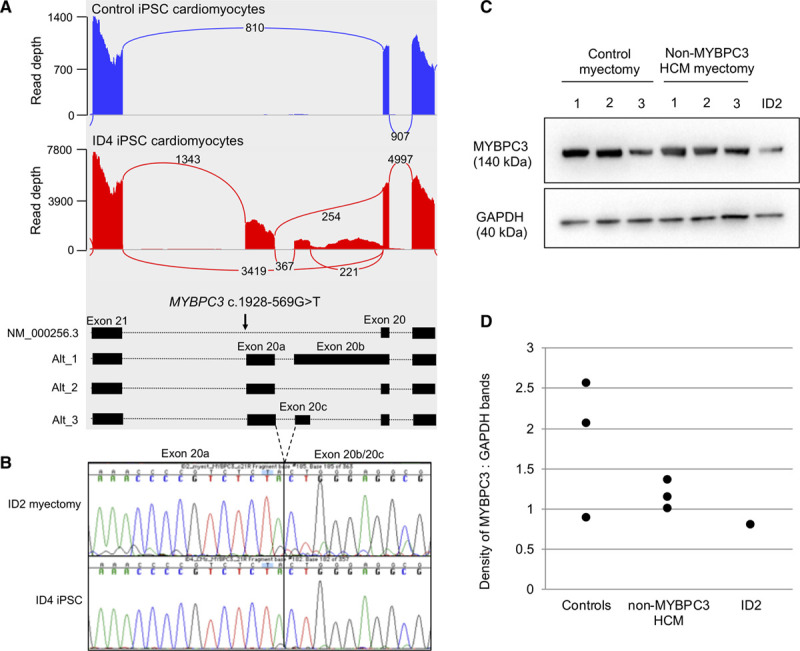
Novel MYBPC3 splice junctions detected in human induced pluripotent stem cell derived cardiomyocyte (hiPSC-CM) of ID4. A, Sashimi plots show sites of aberrant MYBPC3 splice junctions in control hiPSCM-CMs (blue) and ID4 (red) and SW1 (right red). Loops with numbers show number of sequencing reads supporting each major splice junction, as reported by IGV. Middle shows transcript structures with exons (black boxes) and position of intronic splice gain variant. B, Sanger sequencing across the novel splice junction of exon 20a and exon 20b/20c in myectomy tissue and hiPSC-CMs. C, Western blot of human heart tissue from 3 age- and sex-matched control donors, 3 age-matched HCM who do not have an MYBPC3 pathogenic variant and ID2. D, Relative band intensity of MYBPC3 compared with GAPDH.
Patient-Specific Antisense Oligonucleotide Treatment Inhibits Aberrant Splicing
MYBPC3 intronic splice-gain variants that activate splicing of cryptic exons are the underlying cause of HCM in cases DA1, SW1, and ID4. We evaluated whether AOs designed to bind across the disease-causing splice gain variants could inhibit splicing of cryptic exons and restore the natural MYBPC3 protein-coding transcript in patient-specific hiPSC-CMs. We designed up to 3 different AOs to inhibit splicing of each cryptic exon, and a standard control AO which binds to an intron of the beta-globin gene (Table II in the Data Supplement). We treated hiPSC-CMs derived from SW1 with 3 different patient-specific AOs, or control AO. RT-PCR amplification of MYBPC3 transcripts and transcriptome sequencing of RNA extracted from the hiPSC-CMs showed that each patient-specific AO inhibited splicing of exon 12a, whereas control AO had minimal effect (Figure 3). In untreated cells, or those treated with control AO, 50% and 59% of MYBPC3 transcripts contained canonical exon 12 to exon 13 splicing, respectively. Treatment of these cells with SW1_AO1, SW1_AO2, and SW1_AO3 inhibited cryptic exon 12a splicing, with up to 97% of MYBPC3 transcripts containing canonical exon 12 to exon 13 splicing. Increasing concentrations of the best performing AO (SW1_AO2) progressively reduced the levels of cryptic exon 12a splicing in MYBPC3 mRNA, as assessed with RT-PCR (Figure III in the Data Supplement). We in silico assessed off-target binding of SW1_AO2 in RefSeq transcripts and RefSeq genes and found minimal homology to established cardiac disease genes (Table III in the Data Supplement).
Figure 3.
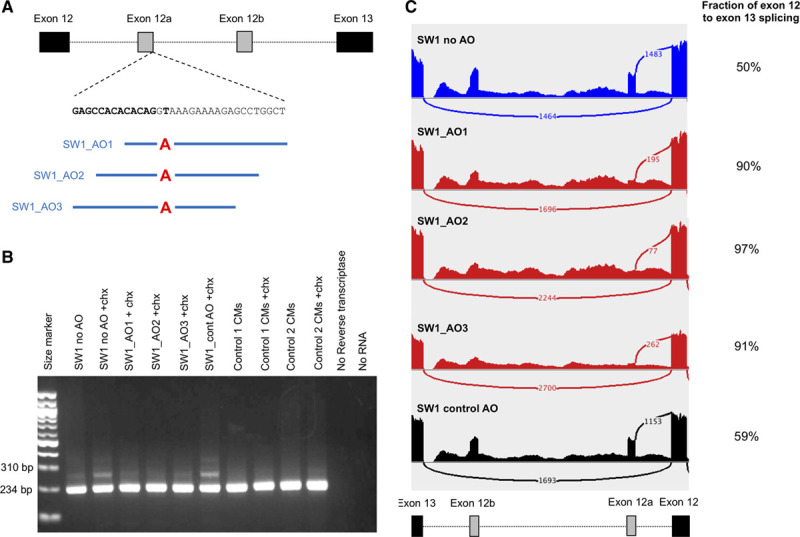
Antisense oligonucleotide treatment of SW1 human induced pluripotent stem cell–derived cardiomyocytes (hiPSC-CMs). A, Location of antisense oligonucleotides (AOs) designed to inhibit splicing of exon 12a. B, RT-PCR amplification of RNA extracted from induced pluripotent stem cell-derived cardiomyocytes of SW1 and controls. C, Sashimi plots show number of reads supporting canonical exon 12 and 13 splicing and exon 12 to cryptic exon 12a splicing in hiPSC-CMs with no AO treatment (blue), AO treatment (red), or control AO (black). Number of reads supporting canonical exon 12 to exon 13 splicing over total reads supporting exon 12 splicing are shown on the right. chx indicates cycloheximide.
We designed 2 AOs to inhibit splicing of exon 20a in hiPSC-CMs derived from case ID4, but splicing of this exon was only marginally inhibited (Figure IV in the Data Supplement). Two AOs designed to inhibit splicing of exon 14a in hiPSC-CMs derived from DA1 had no discernible effect on splicing of this exon (Figure V in the Data Supplement).
Discussion
We have explored genetic analysis of HCM with transcriptome sequencing of RNA extracted from patient-specific hiPSC-CMs. We identified aberrant mRNA splicing of MYBPC3 in 3 patients that was caused by deep intronic splice-gain variants, which are often missed with standard genetic testing of protein coding regions. The same pattern of aberrant MYBPC3 splicing found in hiPSC-CM of 1 patient was confirmed in primary heart tissue of an affected family relative. Our use of hiPSC-CMs as a source of RNA overcomes the main barrier to performing transcriptome sequencing–based genetic testing when the disease-relevant tissue is unavailable. We also showed how hiPSC-CMs can be used to explore the efficacy of patient specific AOs to inhibit cryptic exon splicing, and for 1 cell line we were able to restore the correct MYBPC3 transcript. Our work is an exploration of genetic testing with transcriptome sequencing of hiPSC-CMs and correction of MYBPC3 cryptic exon splicing with a patient-specific AO in a human HCM model.
A New Strategy for Transcriptome-Sequencing Based Genetic Testing
Although genetic testing of Mendelian diseases has improved with the widespread adoption of exome and genome sequencing, the rate at which this testing yields a genetic diagnosis ranges from 25% to 50% across many diseases.16,17 Genetic testing with transcriptome sequencing is a complimentary approach that was shown to increase the diagnostic rate for muscular and mitochondrial diseases, as it identified aberrant mRNA splicing and expression that was not found with DNA sequencing.1,3 Transcriptome sequencing requires RNA extracted from a tissue expressing relevant transcript isoforms, which is often a major barrier when such tissue, or a suitable surrogate, is not available. We showed that 4 accessible tissues do not express all 8 established HCM genes sufficiently for meaningful analysis, whereas hiPSC-CMs showed very high expression of these genes. Transcriptome sequencing of hiPSC-CMs confirmed aberrant splicing of MYBPC3 in 2 patients with HCM due to known intronic splice-gain variants and identified aberrant splicing in patient ID4 with a previously unresolved cause of HCM. The aberrant splice junctions found in ID4 guided us to an intronic splice gain variant (c.1928-569G>T) as the cause of disease. This variant was identified with prior genome sequencing but classified as benign since RT-PCR analysis of RNA extracted from blood and myectomy tissue amplified only correctly spliced products of 293 bp when using primers annealing within exons 19 and 23. Resolving the pattern of aberrant splicing with transcriptome sequencing guided us to redesign RT-PCR primers to amplify transcripts containing the additional 138 bp of cryptic exon 20a plus 413 bp of cryptic exon 20b. Confirmation of the aberrantly spliced transcripts in RNA extracted from hiPSC-CM and myectomy tissue (Figure 2) provided functional evidence of pathogenicity to support variant classification. A limitation of our study is that heart tissue was only available for 1 family member and western analysis showed a small reduction in MYBPC3 protein level that does not definitively confirm haploinsufficiency as a result of aberrant splicing. Our approach of deriving disease-relevant cells from blood, using iPSC technology, is a promising new strategy for transcriptome sequencing when a relevant tissue is otherwise not available.
Limitations of our approach include that it is costly, and time and labor intensive as it takes 3 months to derive hiPSC-CMs from blood. Nevertheless, many families have already undergone multiple genetic tests without a resolution, and transcriptome sequencing provides a new option to evaluate the clinical relevance of noncoding variants. Another limitation is that, for some genes, the transcript isoforms and splice junctions found in hiPSC-CMs may not correspond to those found in adult heart tissue. All identified variants affected splicing of MYBPC3 and genes with more numerous and complex transcript isoforms, such as titin, will be more challenging to interpret. Development of a reference set of splice junctions helped us to filter out natural alternative splicing and low-level background splicing events, leaving a manageable shortlist of candidate splice junctions for investigation. Our strategy may therefore provide a long sought-after answer for some families in which standard approaches have not achieved a genetic diagnosis.
Splice Gain Variants Enhance Transcript Diversity
We found unexpected diversity of MYBPC3 splicing in hiPSC-CMs of people with intronic splice gain variants, but not controls, including splicing of novel exons distal from the underlying splice-gain variant. Examples include splicing of cryptic exon 12b in SW1, and exons 20b and exon 20c in ID4. It is unlikely that these exons are common artifacts of hiPSC-CMs as they were unique to each patient and absent from control hiPSC-CMs. Furthermore, novel exons 20b and 20c found in hiPSC-CMs of ID4 were also found in the myocardial heart tissue of an affected sibling. Transcriptional diversity was similarly found in MYBPC3 transcripts amplified from peripheral blood lymphocyte RNA of a patient with a canonical splice site variant.18 It is possible that disruption of canonical splicing amplifies aberrant mis-splicing by impacting on splicing regulatory elements or by remodeling assembly of the spliceosome on the nascent premRNA. Although the novel exons are flanked by canonical splice sites in the reference sequence, they are not usually found in primary heart tissue and the exon sequences are not evolutionarily conserved. Our sequencing data, and data derived from long-read RNA sequencing technology,18 suggest that current predictions of mRNA splicing outcomes may be too simplistic. It is therefore important to determine the full repertoire of transcriptional diversity and the consequences on the protein sequence to improve variant interpretation as not all aberrant transcripts may contain premature stop codons that are targeted for nonsense mediated decay. We suggest that new improved approaches to demonstrate mRNA splicing outcomes are needed for accurate interpretation of the consequences of splice altering variants in MYBPC3.
Deep Intronic Splice-Gain Variants Are an Increasingly Recognized Cause of Disease
Recently, genome sequencing, targeted sequencing of the entire MYBPC3 gene, and re-analysis of intronic regions covered by exome sequencing have highlighted that MYBPC3 intronic splice-gain variants are an underappreciated cause of HCM.9–11 Identifying intronic splice-gain variants from DNA sequence context alone is challenging, and it is difficult to predict with certainty what the final outcomes of mRNA splicing will be. While computational tools that predict whether variants at canonical splice sites have an impact on splicing are commonplace,19 less focus has been given to approaches to predict deep intronic spice gain variants, which may represent a pool of missed genetic diagnoses. Any predicted splice disrupting variants must be validated using RNA analyses such as RT-PCR, minigene assay, or RNA sequencing, before clinical interpretation of pathogenicity.12 Transcriptome sequencing is one genetic testing approach that may increase the diagnostic rate by finding deep intronic splice gain variants and there is a mounting argument to include all intronic regions of MYBPC3 in DNA-based genetic testing of HCM.9–11
Splice-Redirecting Antisense Oligonucleotides Are a Potential Future Personalized Therapy
The increasing recognition of deep intronic splice gain variants opens up a possible new therapeutic approach for HCM involving AO directed inhibition of aberrant splicing. AOs bind to specific RNA regions and modulate premRNA splicing by sterically blocking the assembly of the spliceosome to the premRNA. AOs have emerged as a new clinical therapeutic for Duchenne muscular dystrophy and spinal muscular atrophy.20 We demonstrated near 100% inhibition of cryptic exon splicing in SW1 hiPSC-CMs. This restoration of full-length MYBPC3 coding transcripts would be expected to avoid chronic overactivation of nonsense-mediated decay, haploinsufficiency of MYBPC3 protein, and clearance of misfolded protein, each of which has been proposed as molecular mechanisms underlying HCM.21–23 There are limitations to this approach. We were not successful in redirecting splicing in 2 hiPSC-CM cell lines for which we had designed 2 AOs each. Additional AO designs around the target residue or at other essential splice motifs, such as the branchpoint or other splicing regulatory elements, may prove to be more successful. There is the potential for unintended off target effects, although our most successful AO had few predicted off target binding sites (Table III in the Data Supplement). hiPSC-CMs enable assessment of off target effects specific to the patient and hence development of an optimized personalized therapeutic intervention. Achieving tissue specific and sustained tissue-specific therapeutic levels of AOs is challenging; however, adenoviral vectors are emerging as a promising avenue for delivery of AOs into the heart. Systemic delivery of an adenoviral vector encoding an AO was able to modulate splicing of MYBPC3 in the heart in vivo in mice and prevented development of left ventricular hypertrophy in neonatal mice.24 More recently, AOs were shown to induce specific silencing of an MYH7 Arg403Gln missense allele in hiPSC-CMs with modest effects on hypertrophic cell size.25
Conclusions
We have explored transcriptome sequencing of patient-specific hiPSC-CMs as an adjunct genetic test for HCM. We show that patient-derived hiPSC-CMs are a suitable source of cardiac transcripts for transcriptome sequencing and validated our method in 2 patients with known deep intronic variants in MYBPC3 and a previously unsolved family with HCM. hiPSC-CMs are a useful human model for screening patient-specific AOs to inhibit aberrant splicing events, which represent a potential future therapeutic option for patients with HCM.
Acknowledgments
The Sydney Heart Bank thanks the patients and staff of Royal Prince Alfred Hospital and the Australian Red Cross Service.
Sources of Funding
Dr Bagnall is the recipient of a New South Wales Health Cardiovascular Disease Senior Scientist Grant and Investigator Development Grant. Dr Semsarian is the recipient of a National Health and Medical Research Council Practitioner Fellowship (No. 1154992) and a New South Wales Health Cardiovascular Disease Clinician Scientist Grant. Dr Ingles is the recipient of a National Health and Medical Research Council Career Development Fellowship (No. 1162929).
Disclosures
Dr Ingles receives research grant support from Myokardia, Inc. The other authors report no conflicts.
Supplemental Materials
Online Materials and Methods
Online Computational Methods
Online Figures I–VI
Online Tables I–III
Nonstandard Abbreviations and Acronyms
- iPSC
- induced pluripotent stem cells
- HCM
- hypertrophic cardiomyopathy
- hiPSC-CM
- human induced pluripotent stem cell–derived cardiomyocyte
- TPM
- transcripts per million
This manuscript was sent to Ruth McPherson, MD, PhD, Guest Editor, for review by expert referees, editorial decision, and final disposition.
For Sources of Funding and Disclosures, see page 190.
The Data Supplement is available at https://www.ahajournals.org/doi/suppl/10.1161/CIRCGEN.120.003202.
Contributor Information
Mira Holliday, Email: mholliday@hotmail.com.
Emma S. Singer, Email: e.singer@centenary.org.au.
Seakcheng Lim, Email: s.lim@centenary.org.au.
Sean Lal, Email: sean.lal@sydney.edu.au.
Jodie Ingles, Email: j.ingles@centenary.org.au.
Christopher Semsarian, Email: c.semsarian@centenary.org.au.
References
- 1.Cummings BB, Marshall JL, Tukiainen T, Lek M, Donkervoort S, Foley AR, Bolduc V, Waddell LB, Sandaradura SA, O’Grady GL, et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med. 2017; 9:eaal5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kremer LS, Bader DM, Mertes C, Kopajtich R, Pichler G, Iuso A, Haack TB, Graf E, Schwarzmayr T, Terrile C, et al. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat Commun. 2017; 8:15824. doi: 10.1038/ncomms15824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee H, Huang AY, Wang LK, Yoon AJ, Renteria G, Eskin A, Signer RH, Dorrani N, Nieves-Rodriguez S, Wan J, et al. ; Undiagnosed Diseases Network. Diagnostic utility of transcriptome sequencing for rare Mendelian diseases. Genet Med. 2020; 22:490–499. doi: 10.1038/s41436-019-0672-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Staerk J, Dawlaty MM, Gao Q, Maetzel D, Hanna J, Sommer CA, Mostoslavsky G, Jaenisch R. Reprogramming of human peripheral blood cells to induced pluripotent stem cells. Cell Stem Cell. 2010; 7:20–24. doi: 10.1016/j.stem.2010.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ross SB, Fraser ST, Semsarian C. Induced pluripotent stem cells in the inherited cardiomyopathies: From disease mechanisms to novel therapies. Trends Cardiovasc Med. 2016; 26:663–672. doi: 10.1016/j.tcm.2016.05.001 [DOI] [PubMed] [Google Scholar]
- 6.Wu JC, Garg P, Yoshida Y, Yamanaka S, Gepstein L, Hulot JS, Knollmann BC, Schwartz PJ. Towards precision medicine with human iPSCs for cardiac channelopathies. Circ Res. 2019; 125:653–658. doi: 10.1161/CIRCRESAHA.119.315209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maron BJ, Ommen SR, Semsarian C, Spirito P, Olivotto I, Maron MS. Hypertrophic cardiomyopathy: present and future, with translation into contemporary cardiovascular medicine. J Am Coll Cardiol. 2014; 64:83–99. doi: 10.1016/j.jacc.2014.05.003 [DOI] [PubMed] [Google Scholar]
- 8.Alfares AA, Kelly MA, McDermott G, Funke BH, Lebo MS, Baxter SB, Shen J, McLaughlin HM, Clark EH, Babb LJ, et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med. 2015; 17:880–888. doi: 10.1038/gim.2014.205 [DOI] [PubMed] [Google Scholar]
- 9.Bagnall RD, Ingles J, Dinger ME, Cowley MJ, Ross SB, Minoche AE, Lal S, Turner C, Colley A, Rajagopalan S, et al. Whole genome sequencing improves outcomes of genetic testing in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2018; 72:419–429. doi: 10.1016/j.jacc.2018.04.078 [DOI] [PubMed] [Google Scholar]
- 10.Janin A, Chanavat V, Rollat-Farnier PA, Bardel C, Nguyen K, Chevalier P, Eicher JC, Faivre L, Piard J, Albert E, et al. Whole MYBPC3 NGS sequencing as a molecular strategy to improve the efficiency of molecular diagnosis of patients with hypertrophic cardiomyopathy. Hum Mutat. 2020; 41:465–475. doi: 10.1002/humu.23944 [DOI] [PubMed] [Google Scholar]
- 11.Lopes LR, Barbosa P, Torrado M, Quinn E, Merino A, Ochoa JP, Jager J, Futema M, Carmo-Fonseca M, Monserrat L, et al. Cryptic splice-altering variants in MYBPC3 are a prevalent cause of hypertrophic cardiomyopathy. Circ Genom Precis Med. 2020; 13:e002905. doi: 10.1161/CIRCGEN.120.002905 [DOI] [PubMed] [Google Scholar]
- 12.Singer ES, Ingles J, Semsarian C, Bagnall RD. Key value of RNA analysis of MYBPC3 splice-site variants in hypertrophic cardiomyopathy. Circ Genom Precis Med. 2019; 12:e002368. doi: 10.1161/CIRCGEN.118.002368 [DOI] [PubMed] [Google Scholar]
- 13.Cardoso-Moreira M, Halbert J, Valloton D, Velten B, Chen C, Shao Y, Liechti A, Ascenção K, Rummel C, Ovchinnikova S, et al. Gene expression across mammalian organ development. Nature. 2019; 571:505–509. doi: 10.1038/s41586-019-1338-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harper AR, Bowman M, Hayesmoore JBG, Sage H, Salatino S, Blair E, Campbell C, Currie B, Goel A, McGuire K, et al. ; HCMR Investigators. Reevaluation of the South Asian MYBPC3Δ25bp intronic deletion in hypertrophic cardiomyopathy. Circ Genom Precis Med. 2020; 13:e002783. doi: 10.1161/CIRCGEN.119.002783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. ; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17:405–424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Retterer K, Juusola J, Cho MT, Vitazka P, Millan F, Gibellini F, Vertino-Bell A, Smaoui N, Neidich J, Monaghan KG, et al. Clinical application of whole-exome sequencing across clinical indications. Genet Med. 2016; 18:696–704. doi: 10.1038/gim.2015.148 [DOI] [PubMed] [Google Scholar]
- 17.Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013; 369:1502–1511. doi: 10.1056/NEJMoa1306555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dainis A, Tseng E, Clark TA, Hon T, Wheeler M, Ashley E. Targeted long-read RNA sequencing demonstrates transcriptional diversity driven by splice-site variation in MYBPC3. Circ Genom Precis Med. 2019; 12:e002464. doi: 10.1161/CIRCGEN.119.002464 [DOI] [PubMed] [Google Scholar]
- 19.Jian X, Boerwinkle E, Liu X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 2014; 42:13534–13544. doi: 10.1093/nar/gku1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li D, Mastaglia FL, Fletcher S, Wilton SD. Precision medicine through antisense oligonucleotide-mediated exon skipping. Trends Pharmacol Sci. 2018; 39:982–994. doi: 10.1016/j.tips.2018.09.001 [DOI] [PubMed] [Google Scholar]
- 21.Glazier AA, Thompson A, Day SM. Allelic imbalance and haploinsufficiency in MYBPC3-linked hypertrophic cardiomyopathy. Pflugers Arch. 2019; 471:781–793. doi: 10.1007/s00424-018-2226-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Helms AS, Tang VT, O’Leary TS, Friedline S, Wauchope M, Arora A, Wasserman AH, Smith ED, Lee LM, Wen XW, et al. Effects of MYBPC3 loss-of-function mutations preceding hypertrophic cardiomyopathy. JCI Insight. 2020; 5:e133782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seeger T, Shrestha R, Lam CK, Chen C, McKeithan WL, Lau E, Wnorowski A, McMullen G, Greenhaw M, Lee J, et al. A premature termination codon mutation in MYBPC3 causes hypertrophic cardiomyopathy via chronic activation of nonsense-mediated decay. Circulation. 2019; 139:799–811. doi: 10.1161/CIRCULATIONAHA.118.034624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gedicke-Hornung C, Behrens-Gawlik V, Reischmann S, Geertz B, Stimpel D, Weinberger F, Schlossarek S, Précigout G, Braren I, Eschenhagen T, et al. Rescue of cardiomyopathy through U7snRNA-mediated exon skipping in Mybpc3-targeted knock-in mice. EMBO Mol Med. 2013; 5:1128–1145. doi: 10.1002/emmm.201202168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dainis A, Zaleta-Rivera K, Ribeiro A, Chang ACH, Shang C, Lan F, Burridge PW, Liu WR, Wu JC, Chang ACY, et al. Silencing of MYH7 ameliorates disease phenotypes in human iPSC-cardiomyocytes. Physiol Genomics. 2020; 52:293–303. doi: 10.1152/physiolgenomics.00021.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ross SB, Fraser ST, Bagnall RD, Semsarian C. Peripheral blood derived induced pluripotent stem cells (iPSCs) from a female with familial hypertrophic cardiomyopathy. Stem Cell Res. 2017; 20:76–79. doi: 10.1016/j.scr.2017.02.016 [DOI] [PubMed] [Google Scholar]
- 27.Karakikes I, Ameen M, Termglinchan V, Wu JC. Human induced pluripotent stem cell-derived cardiomyocytes: insights into molecular, cellular, and functional phenotypes. Circ Res. 2015; 117:80–88. doi: 10.1161/CIRCRESAHA.117.305365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. doi: 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ingles J, Goldstein J, Thaxton C, Caleshu C, Corty EW, Crowley SB, Dougherty K, Harrison SM, McGlaughon J, Milko LV, et al. Evaluating the clinical validity of hypertrophic cardiomyopathy genes. Circ Genom Precis Med. 2019; 12:e002460. doi: 10.1161/CIRCGEN.119.002460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011; 29:24–26. doi: 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]