

Supplemental Digital Content is available in the text.
Keywords: arrhythmogenic right ventricular cardiomyopathy, desmosome, genetic screening, genomics, sudden cardiac death
Abstract
Background:
Genomic screening holds great promise for presymptomatic identification of hidden disease, and prevention of dramatic events, including sudden cardiac death associated with arrhythmogenic cardiomyopathy (ACM). Herein, we present findings from clinical follow-up of carriers of ACM-associated pathogenic/likely pathogenic desmosome variants ascertained through genomic screening.
Methods:
Of 64 548 eligible participants in Geisinger MyCode Genomic Screening and Counseling program (2015–present), 92 individuals (0.14%) identified with pathogenic/likely pathogenic desmosome variants by clinical laboratory testing were referred for evaluation. We reviewed preresult medical history, patient-reported family history, and diagnostic testing results to assess both arrhythmogenic right ventricular cardiomyopathy and left-dominant ACM.
Results:
One carrier had a prior diagnosis of dilated cardiomyopathy with arrhythmia; no other related diagnoses or diagnostic family history criteria were reported. Fifty-nine carriers (64%) had diagnostic testing in follow-up. Excluding the variant, 21/59 carriers satisfied at least one arrhythmogenic right ventricular cardiomyopathy task force criterion, 11 (52%) of whom harbored DSP variants, but only 5 exhibited multiple criteria. Six (10%) carriers demonstrated evidence of left-dominant ACM, including high rates of atypical late gadolinium enhancement by magnetic resonance imaging and nonsustained ventricular tachycardia. Two individuals received new cardiomyopathy diagnoses and received defibrillators for primary prevention.
Conclusions:
Genomic screening for pathogenic/likely pathogenic variants in desmosome genes can uncover both left- and right-dominant ACM. Findings of overt cardiomyopathy were limited but were most common in DSP-variant carriers and notably absent in PKP2-variant carriers. Consideration of the pathogenic/likely pathogenic variant as a major criterion for diagnosis is inappropriate in the setting of genomic screening.
An important aspiration of genomic medicine, as a cornerstone of the vision for precision medicine, is genetically informed, individualized risk stratification enabling early action for disease prevention or mitigation.1,2 The potential benefit of such foresight is perhaps most profound for heritable diseases characterized by a quiescent phase culminating in a dramatic or fatal presentation that is potentially avoidable via earlier detection and intervention. This premise underlies the current recommendations from the American College of Medical Genetics and Genomics for reporting secondary findings from clinical sequencing for specific genes and conditions, such as arrhythmogenic right ventricular (RV) cardiomyopathy (ARVC).3 ARVC—a specific form of arrhythmogenic cardiomyopathy (ACM)4 frequently linked with variants in desmosome genes—is associated with potentially deadly arrhythmias, with sudden cardiac death (SCD) reported as the presenting symptom in up to 23% of index cases,5 particularly in young adults.6,7 Early risk stratification based on genomic findings is, therefore, a promising potential paradigm for prevention of SCD in this setting through medical intervention or other means of mitigation.
Early efforts exploring the feasibility of this genomics-first paradigm to ACM diagnosis have identified significant potential challenges. One study estimated disease penetrance in the absence of symptoms or family history at ≈6%8—based in part on the population prevalence of loss-of-function desmosome variants (0.25%), which have the strongest association with ACM.9 Consistent with these low penetrance estimates, we have previously reviewed electronic health records (EHR) of desmosome-variant carriers bioinformatically identified through our institutional DNA sequenced biobank (MyCode) and have found generally weak or absent disease associations.8,10 These studies have been limited, however, by reliance on retrospective EHR data. This limitation is particularly important given both the complexity of ARVC/ACM diagnostic criteria11,12 and the inability to assess disease status for patients without sufficient EHR data, especially those with no prior indication for cardiac evaluation.
Through MyCode, many of these variant carriers have been notified of pathogenic (P) or likely pathogenic (LP) variants confirmed through a College of American Pathologists–accredited/Clinical Laboratory Improvement Amendments–certified laboratory and referred for genetic counseling and diagnostic evaluation, often by cardiologists with expertise in this area. Such follow-up allows for more detailed phenotypic assessment than prior studies, inclusive of detailed family history collection and disease-specific testing in many instances. Hence, we herein report clinical findings from the first 92 patients identified with P/LP desmosome variants. Our hypothesis was that appropriate clinical evaluation of desmosome-variant carriers would uncover otherwise masked or sub-clinical disease.
Methods
A full description of the methods used in this study is available in the Data Supplement. Institutional review board approval was obtained for this study; all individuals previously provided informed consent to participate in the MyCode Community Health Initiative (MyCode). The data that support the findings of this study will not be made available.13–18
Results
Patient and Variant Details
At the time of this analysis, 64 548 participants were consented in the Geisinger MyCode project and eligible for genomic screening. From this cohort, 92 individuals (0.14%) from 85 distinct families were identified with a clinical laboratory-confirmed P/LP variant in desmoplakin (DSP; n=35), plakophilin-2 (PKP2; n=24), desmocollin-2 (DSC2; n=24), or desmoglein-2 (DSG2; n=9); Figure 1. No patient was aware of his/her genetic variant before the result disclosure. The median (interquartile range) age at the time of result disclosure was 56 (38–65) years and 73% were female (Table 1). Collectively, 29 patients’ variants were classified as pathogenic and 63 were classified as LP. Postdisclosure, one LP variant was reclassified to uncertain significance, and 2 LP variants were reclassified to pathogenic. Details of observed variants are provided in Table 2; we note that all reported variants are putatively truncating (null), which have strong or very strong evidence for pathogenicity by current standards and guidelines.
Figure 1.
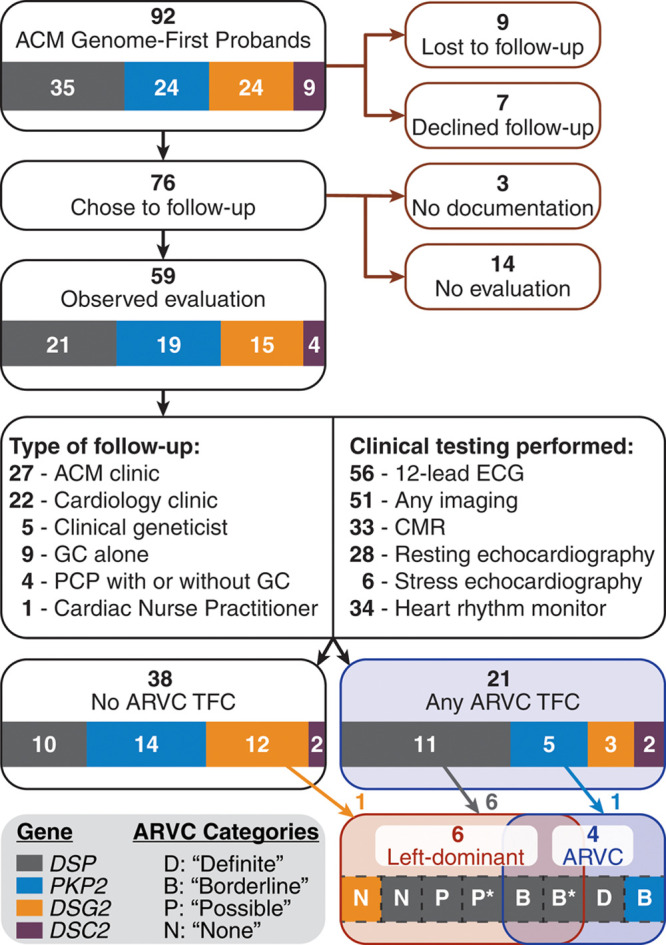
Summary of postdisclosure clinical course for desmosome-variant carriers identified genomics-first through MyCode. ACM indicates arrhythmogenic cardiomyopathy; ARVC, arrhythmogenic right ventricular cardiomyopathy; CMR, cardiac magnetic resonance; GC, genetic counselor; PCP, primary care provider; and TFC, task force criteria. *Denotes the 2 individuals with new cardiomyopathy diagnoses and implanted defibrillators.
Table 1.
Demographics and Past Medical History of Desmosome-Variant Carriers Before the Genomic Result Disclosure vs Eligible Noncarriers From MyCode

Table 2.
Observed Pathogenic or Likely Pathogenic Variants (N=58) in Desmosome Genes Identified From Genomic Screening and Clinical Confirmation Within MyCode

Personal Medical History and Family History
None of the carriers of a desmosome variant had an existing diagnosis of ARVC. One carrier of a DSP variant had a history of nonischemic, left ventricular noncompaction dilated cardiomyopathy with significant ventricular ectopy diagnosed before age 50. Another DSP-variant carrier had an existing diagnosis of pulmonary sarcoidosis with presumed cardiac involvement based on clinical and imaging criteria and noted ventricular ectopy. The clinical presentation in both of these patients is consistent with ACM based on current guidelines,4 although the contribution of the genetic variant to the cardiac findings in the setting of sarcoidosis is unclear.
The past medical history of the variant carriers, in aggregate, was similar to the 64 456 screening-eligible noncarriers from MyCode, inclusive of cardiomyopathy and heart failure diagnoses, and prescription of β-blocker and antiarrhythmic medications (Table 1). There was, however, a significantly higher proportion of preexisting ventricular tachycardia diagnoses in the carrier group (7% versus 1%, P=0.018; Table 1). We note that the relatively high rates of some findings are consistent with the characteristics of MyCode as a health care-system-based population.
Family history details are summarized in Table 3. Individuals carrying the same variant and self-reporting as relatives were grouped as a family (n=85) for this analysis. No family history of ACM/ARVC, ventricular tachycardia, or ventricular fibrillation was reported. Sudden cardiac arrest/death was reported in 10 families (12%), but none was due to suspected ACM. Implantable cardioverter-defibrillator implantation was reported in 4 (5%) families and heart failure in 19 (22%). Besides the P/LP variant, no family history ARVC Task Force Criteria (TFC) were observed.
Table 3.
Self-Reported Family History From Unique Desmosome-Variant-Carrying Families

Postdisclosure Course and Diagnostic Findings
The postdisclosure clinical course is summarized in Figure 1. Nine carriers were lost to follow-up at the time of result, while 7 carriers declined any follow-up based on the disclosure. Of the 76 carriers who reported an intent to follow-up from the initial disclosure, 17 had either no observed diagnostic evaluation at least 6 months postdisclosure or followed-up externally to the Geisinger system. Hence, 59 carriers (64%) had at least some observed diagnostic evaluation based on the genomic finding (ie, 12-lead ECG, imaging, or ambulatory rhythm monitoring), and thus form the basis of our retrospective assessment. All 3 diagnostic evaluations were completed for 33 of these carriers (56%; Figure I in the Data Supplement). Demographics (age, sex, ethnicity, race) were not significantly different between the groups with or without observed follow-up.
All carriers, with 2 exceptions, completed a resting 12-lead ECG during follow-up. One additional patient’s ECG was paced and, therefore, not scored. From the remaining 56, 4 carriers (7%) satisfied major ARVC repolarization criteria, while 8 (14%) had minor ARVC repolarization criteria. One of these 8 had T-wave inversions exclusively in leads V3–V6. Finally, 2 carriers (3%) had terminal activation delay (minor ARVC conduction criteria; Figure 2; Table 4).
Figure 2.
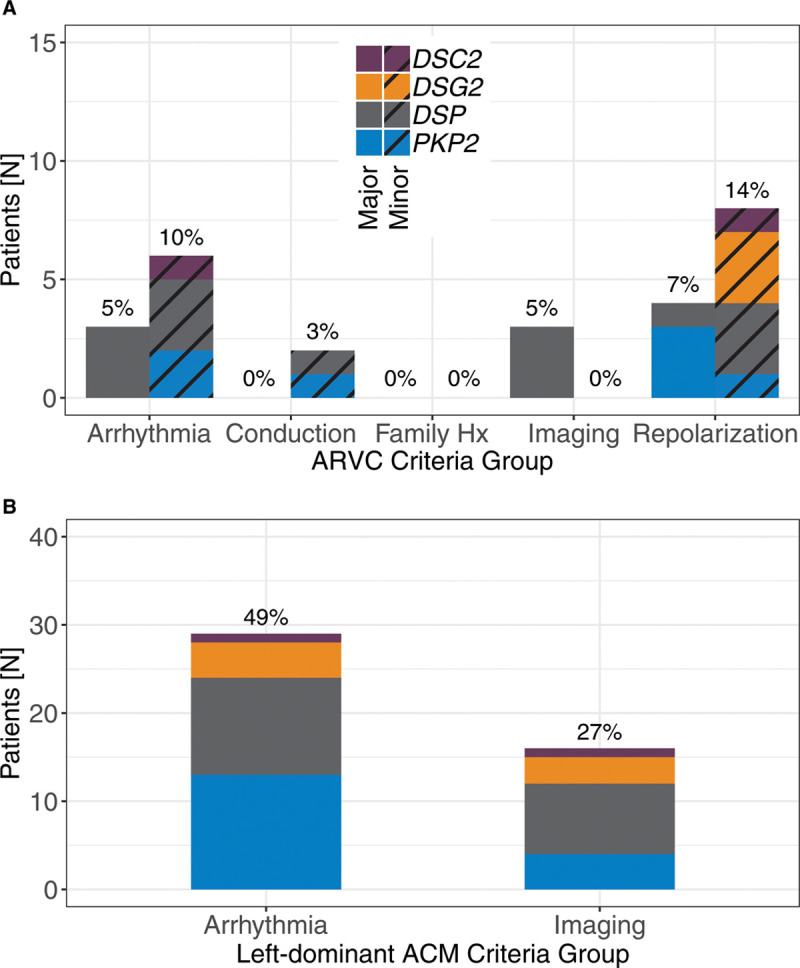
Number of patients (and percentage of 59 with follow-up) satisfying diagnostic criteria by group. A, Diagnostic arrhythmogenic right ventricular cardiomyopathy (ARVC) task force criteria groups, separated by major and minor criteria. B, Left-dominant criteria groups. Imaging findings included reduced ejection fraction or atypical late gadolinium enhancement in the left ventricle. Of note, the majority of arrhythmia findings (15/29; 52%) were in carriers with only other cardiac arrhythmias. ACM indicates arrhythmogenic cardiomyopathy; and Hx, history.
Table 4.
Imaging, ECG, and Arrhythmia Characteristics of Desmosome-Variant Carriers

Imaging studies were completed for 51 carriers (86%), inclusive of cardiac magnetic resonance (CMR) and echocardiography (resting or stress; Table II in the Data Supplement). Three carriers (5%) satisfied major imaging TFC (2 based on echocardiography and one based on CMR); no minor imaging criteria were observed (Figure 2). Of note, 15 carriers had global RV dysfunction/dilation without regional abnormalities. In addition, 10 carriers (17%) had findings of atypical LV late gadolinium enhancement (LGE), predominantly in the midwall or subepicardium of septal (10 carriers), inferior (4), inferolateral (3), anterolateral (2), or anterior (2) segments (Figure 3, Table 4). Four of these carriers had no evidence of arrhythmias or ECG criteria, although other nondiagnostic imaging abnormalities (mild LV or RV dysfunction, moderate RV dilation, mild septal thickening) were present in 3 of the 5. Finally, 5 individuals had reduced (<50%) LV ejection fraction (LVEF).
Figure 3.
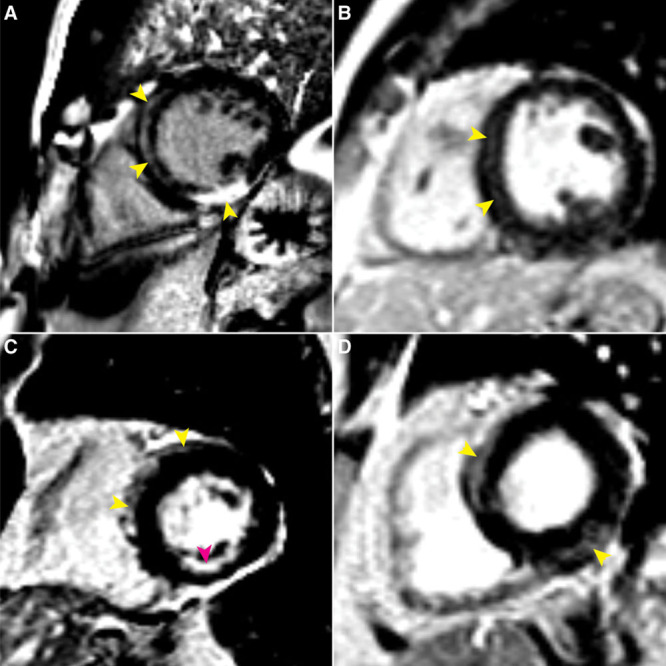
Features of atypical late gadolinium-enhanced (LGE) cardiac magnetic resonance imaging of 4 desmosome-variant carriers. Yellow arrowheads denote atypical LGE locations in the left ventricle (LV). A, Marked atypical LV LGE in the 57 y-old male DSP-variant carrier subsequently diagnosed with arrhythmogenic right ventricular cardiomyopathy (ARVC). B, Septal midwall LGE in a 35 y-old male PKP2-variant carrier. C, Septal and anterior LGE in the subepicardium of a 72 y-old female DSP-variant carrier. Subendocardial enhancement in the inferior LV wall possibly due to prior infarct also visible (magenta arrowhead). D, Atypical midwall LGE in the basal to mid interventricular septum, as well as basal inferolateral segment in a 59 y-old female DSP-variant carrier.
Ambulatory rhythm monitoring (Holter monitor or Zio Patch) was completed for 34 (58%) carriers. Episodes of nonsustained VT with left bundle branch morphology and superior axis (major ARVC arrhythmia criterion) were observed in 3 carriers (5%), while 6 carriers (10%) had >500 premature ventricular contractions in a 24-hour period (minor ARVC arrhythmia criterion; Figure 2, Table 4). In 4 other cases, episodes of nonsustained VT were observed with other/unknown morphology.
Diagnostic Results for ARVC
In total, 21 of the 59 unique carriers (36%) satisfied at least one of the ARVC TFC from the diagnostic evaluations above. As seen in Figure 2A, the majority of patients with diagnostic criteria had variants in DSP (11/21; 52%), particularly arrhythmia and imaging findings. Conversely, few criteria were noted for DSG2- (3/15; 20%) or DSC2- (2/4, 50%) variant carriers, and all were minor criteria. PKP2-carriers also had few criteria (5/19; 26%). Comparing these gene-specific rates showed no statistical differences.
The translation of these observed criteria into an ARVC diagnosis is dependent on the choice of how to incorporate the P/LP variant—a major criterion according to the 2010 TFC—as demonstrated in Figure 4. Strict inclusion of the variant as a major criterion yielded a high degree of purported diagnoses for ARVC, with 10/59 individuals (17%) qualifying for definite diagnosis, 11 (19%) satisfying criteria for a borderline diagnosis, and the remaining 38 (64%) having at least a possible diagnosis based on the variant alone. However, recognizing that a variant-based ascertainment may require a different diagnostic weighting for the variant than in the traditional symptoms-based or family history-based presentation, we additionally considered alternative weighting options. At the opposite extreme, not counting the variant toward diagnostic criteria yielded one individual (2%) with a definite diagnosis, 3 (5%) borderline diagnoses, 6 (10%) possible diagnoses, and the remaining 49 (83%) with no diagnosis.
Figure 4.
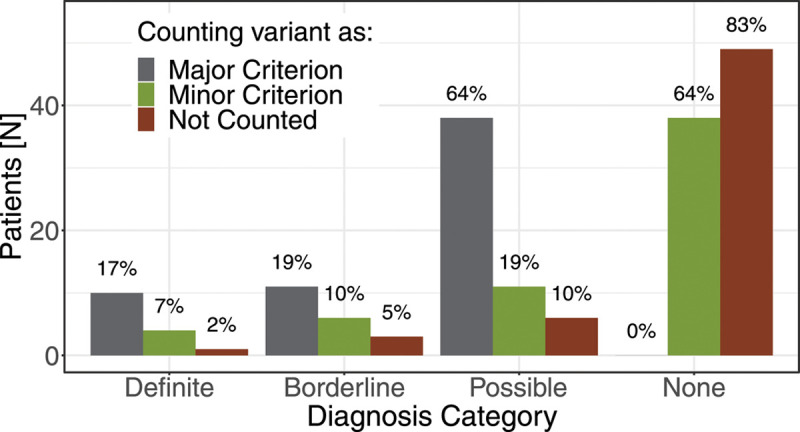
Distributions of arrhythmogenic right ventricular cardiomyopathy diagnostic categories according to the 2010 Task Force Criteria, modulated by the weight applied to the pathogenic/likely pathogenic variant.
To date, one patient from this cohort has been diagnosed with ARVC (with bi-ventricular involvement)—one of the borderline diagnosis cases from the variant-exclusive summary above. This was a 57-year old male with a LP DSP variant (NM_004415.2:c.1141-2A>T), who met major imaging criteria by CMR (RV dyskinesia and RV end-diastolic volume, 117 mL/m2) and minor arrhythmia criteria (1219 ventricular ecotopies on 24-hour Holter monitor). Imaging also revealed a LVEF of 45% and regions of mid-myocardial and subepicardial LGE, including near transmural enhancement of the mid inferior wall of the LV. None of these findings was known before patient evaluation following the genomic result disclosure. He subsequently underwent a dual-chamber defibrillator implant for primary prevention of SCD.
The individual with the definite diagnosis from the variant-exclusive summary was a 38-year old male with a LP DSP variant (NM_004415.2:c.273delT, p.Glu92fs), who met major criteria for both imaging (RV akinesia and fractional area change, 14% by echocardiography) and repolarization (T-wave inversions V1–V4 in the absence of complete right bundle branch block). However, this case was diagnostically confounded by the presence of chronic thromboembolic pulmonary hypertension—to which these findings have been attributed—precluding a diagnosis of ARVC, although the potential contribution of the genetic variant to the development of these secondary cardiac findings is unclear.
Left-Dominant Phenotype
Left-dominant criteria findings (arrhythmia or LV structure/function abnormality) are summarized in Figure 2B. Of note, the arrhythmia criteria for this purpose were broadly inclusive of observed VT as well as past diagnostic history of cardiac arrhythmias (see Methods). Six carriers (10%) satisfied both criteria (5 with a variant in DSP, 1 in DSG2), including the 1 ARVC-diagnosed case from above (Figure 1). Of the other 5 carriers, 4 had a postresult CMR, all of which revealed atypical LV LGE in mid-myocardial or subepicardial regions of the basal to mid-LV, including the septum. Reduced (<50%) LVEF was observed in 3 of 5. Regarding arrhythmia findings, there was no history of ventricular fibrillation or cardiac arrest, but 4 of the 5 had history of (predisclosure or postdisclosure) nonsustained ventricular tachycardia, and 3 had an existing diagnosis of cardiac arrhythmia (including atrial arrhythmias, Table I in the Data Supplement). Of note, the existing ARVC TFC demonstrated poor sensitivity for identifying these patients with left-dominant phenotypes as only 2 of 6 cases had findings sufficient for a borderline diagnosis, with another 2 having possible diagnoses (Figure 1). Finally, though suggestive, the differences in observed criteria by gene were not statistically significant (eg, DSP [24%] versus PKP2 [0%]; P=0.25).
Upon follow-up, one patient with a DSP variant (NM_004415.2:c.699G>A, p.Trp233X) received new diagnoses of nonischemic dilated cardiomyopathy and heart failure in conjunction with this genomic finding. This was a 59-year old male with significant arrhythmia burden (nonsustained VT and >10 000 PVCs (burden of 12%; multi-focal) observed via prior 24-hour Holter monitor) and postdisclosure LVEF of 29%. He has since received an implantable cardioverter-defibrillator. The other 2 patients with reduced LVEF were the 2 DSP-variant carriers (NM_004415.2:c.7469delA, p.Tyr2490LeufsX17; NM_004415.2:c.3133C>T, p.Arg1045X) noted with preexisting ACM diagnoses (dilated cardiomyopathy/left ventricular noncompaction and sarcoidosis).
Discussion
In this first retrospective evaluation of a clinical program aimed at follow-up of P/LP desmosome-variant carriers identified by genomic screening, we found that genomics-first ascertainment indeed resulted in new diagnoses of ACM. These diagnoses represented both ARVC—the primary phenotype expected with desmosome variants—and left-dominant ACM, a newer and still evolving disease entity. Moreover, we observed both new diagnoses of previously unrecognized cardiomyopathy, as well as refined classification of existing diagnoses. However, it is important to recognize that the overall diagnostic yield, particularly for ARVC, was low. Thus, a conservative approach to diagnosis and invasive intervention based on genomic screening is warranted.
Diagnostic Considerations for ARVC
Of the carriers with postdisclosure follow-up, 36% satisfied at least 1 of the 2010 ARVC TFC, independent of the genetic variant. However, the findings represented only isolated criteria (major or minor) for all but 5 carriers (8%) who met multiple criteria. A formal diagnosis of ARVC has so far been made in one of those 5 cases. We have previously observed that isolated TFC from secondary review of clinical testing are comparable in frequency between desmosome-variant carriers and noncarriers.8,10 It is, therefore, reasonable to expect that findings of isolated criteria through genomic result-directed testing are also unlikely to carry diagnostic significance. This understanding requires that genetic variants identified by population or secondary screening not be counted as a major criterion; otherwise, finding another isolated major or minor criterion yields a definite or borderline diagnosis, respectively, as illustrated by Figure 4. Instead, counting the variant only as a minor criterion, or not counting it at all, resulted in diagnostic outcomes that were better aligned with the clinical impression in this cohort—that most carriers did not have overt evidence of ACM. Continued surveillance, particularly of individuals with possible or borderline diagnoses, will provide further clarification on the most appropriate strategy in this regard.
This low yield of definitive ARVC is consistent with our prior work,8,10 even in the new context of informed clinical evaluation following the genomic result disclosure. However, this analysis adds support to these prior findings as, in addition to the exclusive focus on postresult clinical evaluations, we reported findings from a detailed family history analysis. These family histories were generally unremarkable given the lack of reported ACM/ARVC and low or expected rates of other nonspecific characteristics, such as heart failure and SCD; that is, no independent family history criteria suggestive of a diagnosis. These data, therefore, support prior conclusions, including previous theoretical estimates of genomics-first ARVC penetrance (6%).8
Left-Dominant ACM
The collection of postdisclosure diagnostic data, particularly CMR images with LGE, enabled an assessment of left-dominant ACM prevalence from genomics-first ascertainment for the first time. In general, the findings were similar to ARVC in that only 10% of variant carriers met the specified criteria for possible left-dominant disease. However, the burden of overt disease was higher for the LV than the RV as 3 carriers had clinically recognized cardiomyopathy in conjunction with reduced LVEF, 2 of which preceded the variant disclosure.
The frequency of LGE—found in 30% of the carriers who underwent CMR evaluation—was another novel finding of this analysis. In 5 carriers, the LGE was accompanied by arrhythmia criteria satisfying the left-dominant phenotype definition, but similar LGE findings in the absence of noted arrhythmias were identified in 5 additional desmosome-variant carriers (10/59, 17% in total). In both situations, the diagnostic and prognostic significance of these findings remains largely unclear. However, a study of clinically acquired CMR data found that of 409 patients without ischemic disease, nonischemic LGE findings were present in only 19 (5%), suggesting a much higher frequency in our cohort.19 Although LGE has historically not been included in diagnostic criteria for ARVC, more recent studies have provided evidence for its inclusion, presumably in combination with other criteria, particularly for left-dominant ACM.12,20–22 Longitudinal follow-up of these carriers will help clarify the prognostic significance of nonspecific imaging findings, including LGE and isolated global RV dilation/dysfunction. Our analysis also highlights the need for greater awareness and consensus guidelines for diagnosis of left-dominant disease.
Gene-Specific Observations
Our observed genotype-phenotype associations are noteworthy in 2 regards. First, none of the PKP2-variant carriers in this cohort had a demonstrable phenotype. As the most commonly affected gene in ARVC,23 this specific lack of PKP2 association is likely a main contributor to the low number of observed ARVC cases. The explanation for this result is unclear, but may relate to survival bias, or the influence of secondary modifying factors, such as exercise or lack thereof. The link between vigorous exercise/athletics and ARVC has the strongest supporting evidence in PKP2-variant carriers,24,25 and the median body mass index in our PKP2-variant carriers was 35 (32 for all desmosome-variant carriers), suggesting a lack of such activities based on this rate of obesity. Additionally, increasing evidence suggests a mediating role of other cellular proteins, such as the RYR2 (ryanodine receptor 2) and integrin β1D,26,27 in the setting of PKP2/desmosome disruption leading to ARVC, so it is also possible that secondary genetic variation may be moderating penetrance in these individuals as well.
Conversely, the most notable phenotypes from our analysis were observed in DSP-variant carriers. These represented both the majority of the left-dominant ACM phenotypes and also perhaps explains the LV involvement in the patient with diagnosed ARVC. The tendency for LV involvement in association with DSP variants has been well documented,28 and DSP is frequently cited as a risk gene both for dilated cardiomyopathy as well as ACM.4,29,30 Yet, the unique attributes of the DSP-specific phenotype are rapidly evolving, as exemplified by the recent work of Smith et al.20 Findings in many of our DSP-variant carriers are largely consistent with this study, including a high prevalence of LGE, frequently without overt systolic dysfunction, frequent PVCs, and poor sensitivity of the ARVC TFC.20 Our findings further underscore the need for a comprehensive bi-ventricular evaluation—inclusive of CMR with LGE—following genomics-first identification of DSP variants.
It is important to note, however, that while visually striking, the apparent differences in gene penetrance were not statistically significant in this analysis. It is most likely that the study was under-powered (≈40%) to detect such differences (≈25%) based on the sample sizes. Therefore, these specific findings should be interpreted with caution and warrant replication in future analyses.
Implications for Genomics-First Screening of Desmosome Genes
This study represents the first report of new ACM diagnoses following genomics-first ascertainment and is thus an important proof-of-concept. The value of genomic screening efforts for ACM-associated variants is also highlighted by the novel discovery of otherwise unknown disease, with subsequent interventions in 2 cases (ie, defibrillator implantation) to prevent potentially severe outcomes. Moreover, with surveillance and follow-up of the other borderline or possible diagnoses for both phenotypes, the low diagnostic yield we observed may increase over time.
Additionally, the genomics-first identification of LV-dominant disease is an important finding of this work considering that American College of Medical Genetics and Genomics guidelines for screening desmosome genes focus exclusively on ARVC. Instead, these data demonstrate that the potential for either left- or right-dominant disease presentations in connection with these genes must be appreciated in the clinical assessment, particularly for carriers of DSP and DSG2 variants.28 Future revisions of the American College of Medical Genetics and Genomics secondary findings recommendations should, therefore, focus on associations with ACM instead of the more specific ARVC.
Finally, an important question to consider is whether desmosome variants should remain included in initiatives of genomic screening or disclosure of secondary findings. Having a high rate of apparent nonpenetrance is problematic, particularly given the costs associated with unnecessary diagnostic evaluations and the potential burden of anxiety associated with result disclosure. Yet, the potential benefits of uncovering unrecognized disease—which we have now shown to be possible—are substantial. Therefore, a critical focus of future study will be to develop additional criteria (eg, history of vigorous exercise/athletics) to improve the precision of risk assessment given the finding of a desmosome variant, potentially leveraging our developing understanding of gene-phenotype associations. Such refinements will guide development of risk management recommendations that help reduce the cost of population-level desmosome screening (both financial and psychological) and improve its effectiveness in the long-term.
Limitations
Due to heterogeneous clinical testing, some patients were incompletely evaluated, even within the subset of 59 carriers analyzed for diagnostic criteria. Therefore, phenotypes and diagnostic findings may be underestimated (Figure I in the Data Supplement). Efforts are ongoing to continue engaging and longitudinally evaluating this cohort with more standardized testing procedures to further refine these insights.
This cohort has a relatively high median age compared with typical disease onset,31 so as mentioned above, there is some degree of survival bias in this group. Our family history assessment provides some confidence that survival bias is not masking early SCD in these families. Yet, evaluation of phenotypes in younger individuals, for example, through ongoing cascade testing, may uncover additional disease, despite an unaffected proband.
The history of exercise/athletics in this cohort is as yet unknown, which very likely influences disease expressivity and progression.24 Future work will better establish this connection in genome-first ascertainment.
Finally, it is also important to recognize that this experience is from a single healthcare system cohort, which is susceptible to other potential sources of ascertainment bias, including female predominance. Ultimately, replication in another cohort will be critical to confirm these findings.
Conclusions
Genomics-based identification of individuals at risk for ACM can uncover new evidence of disease as well as sub-clinical abnormalities. This represents an important proof-of-concept for a genomics-first approach to ACM, although the likelihood of newly discovered disease, at least in the short-term following result disclosure, appears relatively low. We observed an equal, if not greater likelihood of left-sided disease as right-sided disease, demonstrating the importance of bi-ventricular clinical follow-up of these genomic findings, although consensus guidelines for left-dominant ACM are needed to facilitate a formal diagnosis and provide greater awareness of that phenotype. Relatedly, DSP-variant carriers constituted the majority of patients observed with clinical criteria, particularly left ventricular disease, while only one PKP2-variant carrier had a demonstrable ARVC phenotype. Hence, the penetrance associated with DSP variants may be higher than PKP2, but future work is needed to confirm. Finally, the consideration of a P/LP variant as a major criterion for ARVC diagnosis is not appropriate in the setting of genomic screening; it should at most be regarded as a minor criterion.
Acknowledgments
We gratefully acknowledge the data brokerage assistance of Dustin Hartzel, services of Geisinger’s MyCode genetic counseling team and providers, participation of MyCode participants, sequencing efforts of Regeneron Genetics Center, and the contributions of Drs Michael F. Murray and Kandamurugu Manickam.
Sources of Funding
Research reported in this publication was supported by the National Heart, Lung, And Blood Institute of the National Institutes of Health under Award Number R01HL141901. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosures
Dr Calkins is a consultant for Medtronic Inc and St. Jude Medical/Abbott. Dr Calkins receives research support from Boston Scientific Corp and Dr James and C. Tichnell receive salary support from this grant. Dr James has received a lecture fee from Abbott. Dr Fornwalt is a consultant for the Novartis Cardiovascular Data Science Advisory Board. The other authors report no conflicts.
Supplemental Materials
Online Methods
Online Tables I and II
Online Figure I
Nonstandard Abbreviations and Acronyms
- ACM
- arrhythmogenic cardiomyopathy
- ARVC
- arrhythmogenic right ventricular cardiomyopathy
- CMR
- cardiac magnetic resonance
- DSC2
- desmocollin 2
- DSG2
- desmoglein 2
- DSP
- desmoplakin
- EHR
- electronic health record
- LGE
- late gadolinium enhancement
- LP
- likely pathogenic
- LVEF
- left ventricular ejection fraction
- P
- pathogenic
- PKP2
- plakophilin 2
- RYR2
- ryanodine receptor 2
- SCD
- sudden cardiac death
- TFC
- 2010 Task force criteria
E.D. Carruth and D. Beer contributed equally.
The Data Supplement is available at https://www.ahajournals.org/doi/suppl/10.1161/CIRCGEN.120.003302.
For Sources of Funding and Disclosures, see page 211.
Contributor Information
Eric D. Carruth, Email: ecarruth@geisinger.edu.
Dominik Beer, Email: dominikbeer@gmail.com.
Amro Alsaid, Email: aalsaid@geisinger.edu.
Marci L.B. Schwartz, Email: mlbschwartz@geisinger.edu.
Megan McMinn, Email: mnbetts@geisinger.edu.
Melissa A. Kelly, Email: makelly2@geisinger.edu.
Adam H. Buchanan, Email: ahbuchanan@geisinger.edu.
Christopher D. Nevius, Email: cdnevius@geisinger.edu.
Hugh Calkins, Email: hcalkins@jhmi.edu.
Cynthia A. James, Email: cjames7@jhmi.edu.
Brittney Murray, Email: bdye1@jhmi.edu.
Crystal Tichnell, Email: ctichne1@jhmi.edu.
Martin E. Matsumura, Email: mmatsumura@geisinger.edu.
H. Lester Kirchner, Email: hlkirchner@geisinger.edu.
Brandon K. Fornwalt, Email: bkf@gatech.edu.
Amy C. Sturm, Email: asturm@geisinger.edu.
References
- 1.Antman EM, Loscalzo J. Precision medicine in cardiology. Nat Rev Cardiol. 2016; 13:591–602. doi: 10.1038/nrcardio.2016.101 [DOI] [PubMed] [Google Scholar]
- 2.Biesecker LG. Opportunities and challenges for the integration of massively parallel genomic sequencing into clinical practice: lessons from the ClinSeq project. Genet Med. 2012; 14:393–398. doi: 10.1038/gim.2011.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, Herman GE, Hufnagel SB, Klein TE, Korf BR, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017; 19:249–255. doi: 10.1038/gim.2016.190 [DOI] [PubMed] [Google Scholar]
- 4.Towbin JA, McKenna WJ, Abrams DJ, Ackerman MJ, Calkins H, Darrieux FCC, Daubert JP, de Chillou C, DePasquale EC, Desai MY, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm. 2019; 16:e301–e372. doi: 10.1016/j.hrthm.2019.05.007 [DOI] [PubMed] [Google Scholar]
- 5.Dalal D, Nasir K, Bomma C, Prakasa K, Tandri H, Piccini J, Roguin A, Tichnell C, James C, Russell SD, et al. Arrhythmogenic right ventricular dysplasia: a United States experience. Circulation. 2005; 112:3823–3832. doi: 10.1161/CIRCULATIONAHA.105.542266 [DOI] [PubMed] [Google Scholar]
- 6.Maron BJ, Haas TS, Duncanson ER, Garberich RF, Baker AM, Mackey-Bojack S. Comparison of the frequency of sudden cardiovascular deaths in young competitive athletes versus nonathletes: should we really screen only athletes? Am J Cardiol. 2016; 117:1339–1341. doi: 10.1016/j.amjcard.2016.01.026 [DOI] [PubMed] [Google Scholar]
- 7.Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988; 318:129–133. doi: 10.1056/NEJM198801213180301 [DOI] [PubMed] [Google Scholar]
- 8.Carruth ED, Young W, Beer D, James CA, Calkins H, Jing L, Raghunath S, Hartzel DN, Leader JB, Kirchner HL, et al. Prevalence and electronic health record-based phenotype of loss-of-function genetic variants in arrhythmogenic right ventricular cardiomyopathy-associated genes. Circ Genom Precis Med. 2019; 12:e002579. doi: 10.1161/CIRCGEN.119.002579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, Mazzarotto F, Blair E, Seller A, Taylor JC, et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med. 2017; 19:192–203. doi: 10.1038/gim.2016.90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haggerty CM, James CA, Calkins H, Tichnell C, Leader JB, Hartzel DN, Nevius CD, Pendergrass SA, Person TN, Schwartz M, et al. Electronic health record phenotype in subjects with genetic variants associated with arrhythmogenic right ventricular cardiomyopathy: a study of 30,716 subjects with exome sequencing. Genet Med. 2017; 19:1245–1252. doi: 10.1038/gim.2017.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MG, Daubert JP, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010; 121:1533–1541. doi: 10.1161/CIRCULATIONAHA.108.840827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sen-Chowdhry S, Syrris P, Prasad SK, Hughes SE, Merrifield R, Ward D, Pennell DJ, McKenna WJ. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol. 2008; 52:2175–2187. doi: 10.1016/j.jacc.2008.09.019 [DOI] [PubMed] [Google Scholar]
- 13.Carey DJ, Fetterolf SN, Davis FD, Faucett WA, Kirchner HL, Mirshahi U, Murray MF, Smelser DT, Gerhard GS, Ledbetter DH. The Geisinger MyCode community health initiative: an electronic health record-linked biobank for precision medicine research. Genet Med. 2016; 18:906–913. doi: 10.1038/gim.2015.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dewey FE, Murray MF, Overton JD, Habegger L, Leader JB, Fetterolf SN, O’Dushlaine C, Van Hout C V, Staples J, Gonzaga-Jauregui C, et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science. 2016; 354:aaf6814. [DOI] [PubMed] [Google Scholar]
- 15.Staples J, Maxwell EK, Gosalia N, Gonzaga-Jauregui C, Snyder C, Hawes A, Penn J, Ulloa R, Bai X, Lopez AE, et al. Profiling and leveraging relatedness in a precision medicine cohort of 92,455 exomes. Am J Hum Genet. 2018; 102:874–889. doi: 10.1016/j.ajhg.2018.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwartz MLB, McCormick CZ, Lazzeri AL, Lindbuchler DM, Hallquist MLG, Manickam K, Buchanan AH, Rahm AK, Giovanni MA, Frisbie L, et al. A model for genome-first care: returning secondary genomic findings to participants and their healthcare providers in a large research cohort. Am J Hum Genet. 2018; 103:328–337. doi: 10.1016/j.ajhg.2018.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. ; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17:405–424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995; 57:289–300 [Google Scholar]
- 19.Hunold P, Schlosser T, Vogt FM, Eggebrecht H, Schmermund A, Bruder O, Schüler WO, Barkhausen J. Myocardial late enhancement in contrast-enhanced cardiac MRI: distinction between infarction acar and non–infarction-related disease. Am J Roentgenol. 2005; 184:1420–1426 [DOI] [PubMed] [Google Scholar]
- 20.Smith ED, Lakdawala NK, Papoutsidakis N, Aubert G, Mazzanti A, McCanta AC, Agarwal PP, Arscott P, Dellefave-Castillo LM, Vorovich EE, et al. Desmoplakin cardiomyopathy, a fibrotic and inflammatory form of cardiomyopathy distinct from typical dilated or arrhythmogenic right ventricular cardiomyopathy. Circulation. 2020; 141:1872–1884. doi: 10.1161/CIRCULATIONAHA.119.044934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corrado D, Marra MP, Zorzi A, Beffagna G, Cipriani A, Lazzari M De, Migliore F, Pilichou K, Rampazzo A, Rigato I, et al. Diagnosis of arrhythmogenic cardiomyopathy: the Padua criteria. Int J Cardiol. 2020; 319:106–114 [DOI] [PubMed] [Google Scholar]
- 22.DeWitt ES, Chandler SF, Hylind RJ, Beausejour Ladouceur V, Blume ED, VanderPluym C, Powell AJ, Fynn-Thompson F, Roberts AE, Sanders SP, et al. Phenotypic manifestations of arrhythmogenic cardiomyopathy in children and adolescents. J Am Coll Cardiol. 2019; 74:346–358. doi: 10.1016/j.jacc.2019.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Groeneweg JA, Bhonsale A, James CA, te Riele AS, Dooijes D, Tichnell C, Murray B, Wiesfeld AC, Sawant AC, Kassamali B, et al. Clinical presentation, long-term follow-up, and outcomes of 1001 arrhythmogenic right ventricular dysplasia/cardiomyopathy patients and family members. Circ Cardiovasc Genet. 2015; 8:437–446. doi: 10.1161/CIRCGENETICS.114.001003 [DOI] [PubMed] [Google Scholar]
- 24.James CA, Bhonsale A, Tichnell C, Murray B, Russell SD, Tandri H, Tedford RJ, Judge DP, Calkins H. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol. 2013; 62:1290–1297. doi: 10.1016/j.jacc.2013.06.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lie ØH, Rootwelt-Norberg C, Dejgaard LA, Leren IS, Stokke MK, Edvardsen T, Haugaa KH. Prediction of life-threatening ventricular arrhythmia in patients with arrhythmogenic cardiomyopathy: a primary prevention cohort study. JACC Cardiovasc Imaging. 2018; 11:1377–1386. doi: 10.1016/j.jcmg.2018.05.017 [DOI] [PubMed] [Google Scholar]
- 26.Delmar M, Alvarado FJ, Valdivia HH. Desmosome-dyad crosstalk: an arrhythmogenic axis in arrhythmogenic right ventricular cardiomyopathy. Circulation. 2020; 141:1494–1497. doi: 10.1161/CIRCULATIONAHA.120.046020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Li C, Shi L, Chen X, Cui C, Huang J, Chen B, Hall DD, Pan Z, Lu M, et al. Integrin β1D deficiency-mediated RyR2 dysfunction contributes to catecholamine-sensitive ventricular tachycardia in arrhythmogenic right ventricular cardiomyopathy. Circulation. 2020; 141:1477–1493. doi: 10.1161/CIRCULATIONAHA.119.043504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bhonsale A, Groeneweg JA, James CA, Dooijes D, Tichnell C, Jongbloed JD, Murray B, te Riele AS, van den Berg MP, Bikker H, et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur Heart J. 2015; 36:847–855. doi: 10.1093/eurheartj/ehu509 [DOI] [PubMed] [Google Scholar]
- 29.Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013; 10:531–547. doi: 10.1038/nrcardio.2013.105 [DOI] [PubMed] [Google Scholar]
- 30.Haas J, Frese KS, Peil B, Kloos W, Keller A, Nietsch R, Feng Z, Müller S, Kayvanpour E, Vogel B, et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J. 2015; 36:1123–135a. doi: 10.1093/eurheartj/ehu301 [DOI] [PubMed] [Google Scholar]
- 31.Bhonsale A, Te Riele ASJM, Sawant AC, Groeneweg JA, James CA, Murray B, Tichnell C, Mast TP, van der Pols MJ, Cramer MJM, et al. Cardiac phenotype and long-term prognosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia patients with late presentation. Heart Rhythm. 2017; 14:883–891. doi: 10.1016/j.hrthm.2017.02.013 [DOI] [PubMed] [Google Scholar]