

Supplemental Digital Content is available in the text.
Keywords: acute coronary syndrome, colchicine, gastrointestinal diseases, myocardial infarction, pharmacogenetics
Abstract
Background:
The randomized, placebo-controlled COLCOT (Colchicine Cardiovascular Outcomes Trial) has shown the benefits of colchicine 0.5 mg daily to lower the rate of ischemic cardiovascular events in patients with a recent myocardial infarction. Here, we conducted a post hoc pharmacogenomic study of COLCOT with the aim to identify genetic predictors of the efficacy and safety of treatment with colchicine.
Methods:
There were 1522 participants of European ancestry from the COLCOT trial available for the pharmacogenomic study of COLCOT trial. The pharmacogenomic study’s primary cardiovascular end point was defined as for the main trial, as time to first occurrence of cardiovascular death, resuscitated cardiac arrest, myocardial infarction, stroke, or urgent hospitalization for angina requiring coronary revascularization. The safety end point was time to the first report of gastrointestinal events. Patients’ DNA was genotyped using the Illumina Global Screening array followed by imputation. We performed a genome-wide association study in colchicine-treated patients.
Results:
None of the genetic variants passed the genome-wide association study significance threshold for the primary cardiovascular end point conducted in 702 patients in the colchicine arm who were compliant to medication. The genome-wide association study for gastrointestinal events was conducted in all 767 patients in the colchicine arm and found 2 significant association signals, one with lead variant rs6916345 (hazard ratio, 1.89 [95% CI, 1.52–2.35], P=7.41×10−9) in a locus which colocalizes with Crohn disease, and one with lead variant rs74795203 (hazard ratio, 2.51 [95% CI, 1.82–3.47]; P=2.70×10−8), an intronic variant in gene SEPHS1. The interaction terms between the genetic variants and treatment with colchicine versus placebo were significant.
Conclusions:
We found 2 genomic regions associated with gastrointestinal events in patients treated with colchicine. Those findings will benefit from replication to confirm that some patients may have genetic predispositions to lower tolerability of treatment with colchicine.
Inflammation plays an important role in atherosclerosis and in processes leading to and following a myocardial infarction. The COLCOT (Colchicine Cardiovascular Outcomes Trial) has recently shown the benefits of the anti-inflammatory medication colchicine in reducing the rate of ischemic cardiovascular events in 4745 patients included within 30 days after myocardial infarction.1 The study’s primary end point consisting of time to first occurrence of cardiovascular death, resuscitated cardiac arrest, nonfatal myocardial infarction, nonfatal stroke, or urgent hospitalization for angina requiring coronary revascularization was reduced by 23% by low-dose colchicine as compared to placebo after a median follow-up of 23 months.1
Considering that patients receive long-term treatment with multiple drugs after a myocardial infarction, genomics can help identify patients more or less unlikely to derive benefits to decrease polypharmacy. Given the effects of colchicine on tubulin and multiple inflammatory pathways,2,3 the identification of genes associated with clinical outcomes can provide insights into the underlying mechanisms responsible for its benefits in patients with coronary artery disease. Similarly, genes linked to adverse effects may offer clues to their pathophysiology. Here, we present the post hoc pharmacogenomic study of COLCOT in the subgroup of participants who took part in the optional genetic substudy, with the aim to identify genetic predictors of the efficacy and safety of treatment with colchicine.
Methods
The data underlying this article cannot be shared publicly to preserve the privacy of study participants; however, the data are available from the corresponding authors upon reasonable requests. The analytic methods and study materials may be made available to other researchers for purposes of reproducing the results or replicating the procedure. Summary statistics are available publicly for download and visualization via PheWEB4 at URL: http://statgen.org/pheweb/colcot. The COLCOT clinical trial was registered at URL: https://www.clinicaltrials.gov under the unique identifier NCT02551094. The study protocol was approved by the Montreal Heart Institute research ethics committee and complies with the Declaration of Helsinki. Written informed consent was obtained from all participating subjects. Full Methods are available in the Data Supplement of the article.
Results
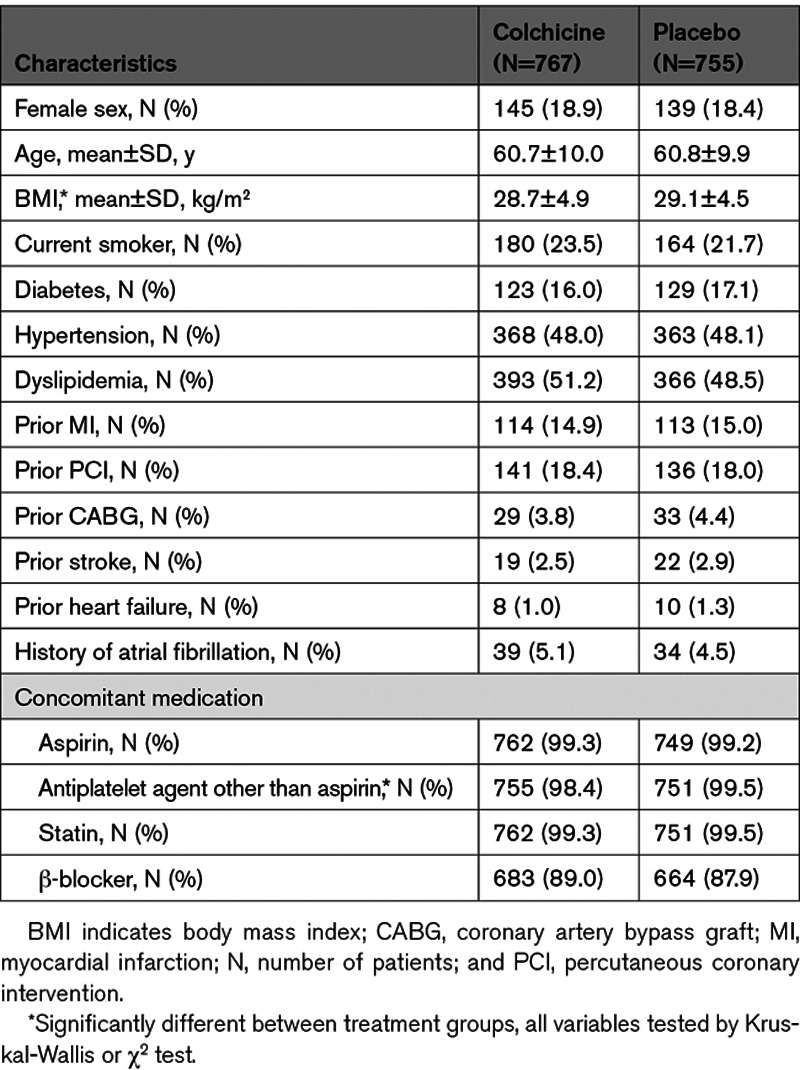
There were 1522 participants included in the pharmacogenomic analysis of COLCOT of which 767 were randomized to colchicine and 755 to placebo (Figure I in the Data Supplement). The baseline characteristics of patients according to the study treatment groups are shown in Table 1. The mean age of participants was 60.9 years and 81.3% were male. The COLCOT study primary cardiovascular end point occurred in 6.2% of patients who consented to the pharmacogenomic substudy, as compared to 6.3% of those in the main trial (P=0.86; Table I in the Data Supplement). Gastrointestinal adverse events occurred in 23.4% of the pharmacogenomic study population, as compared to 17.6% of the COLCOT trial participants (P=1.8×10−7).
Table 1.
Characteristics of the Pharmacogenomics Study Participants
Genetic Determinants of Cardiovascular Efficacy With Colchicine
The pharmacogenomic analyses of the primary cardiovascular efficacy end point were limited to the 702 participants randomized to colchicine who used the study drug with at least 80% compliance in the first 6 months of treatment. Of those, 39 patients had an event. The prespecified analysis for the ATP binding cassette subfamily B member 1 gene (ABCB1) variant rs1045642 and the CYP3A4 (cytochrome P450 family 3 subfamily A member 4) metabolizer phenotype was not associated with the primary cardiovascular efficacy end point (P=0.77 and P=0.91, respectively), and none of the tested genetic variants passed the genome-wide association study (GWAS) significance threshold (P<5×10−8; Figure IIA in the Data Supplement). However, the GWAS analysis had limited power, and negative results should be interpreted with care. The sex-stratified GWAS with 576 male participants also did not provide any GWAS-significant findings (Figure IIB in the Data Supplement), however, there was some interest for the top signal on chromosome 9 at rs10811106 (P=5.8×10−8) near the stabilizer of axonemal microtubules 1 (SAXO1) gene (also known as FAM154A), as it encodes the stabilizer of axonemal microtubules 1 (Figure IIIB in the Data Supplement).
Genetic Determinants of Gastrointestinal Adverse Events With Colchicine
There were 767 participants randomized to colchicine who were included in the genetic analyses for gastrointestinal adverse events, of those, 187 had a gastrointestinal event. The ABCB1 rs1045642 variant and the CYP3A4 metabolizer phenotype were not associated with gastrointestinal adverse events (P=0.97 and P=0.31, respectively). We found 22 genetic variants significantly associated with gastrointestinal events at 2 loci located on chromosomes 6 and 10 (Figure). The most significant association on chromosome 6 was the intergenic variant rs6916345 (P=7.41×10−9). When conditioning on rs6916345, no additional genetic variants remained significant at P<5×10−8 in the region, and rs6916345 had the highest probability of being causal by CAVIAR analysis (Data Supplement). The minor allele (A) was associated with gastrointestinal events in the colchicine group (hazard ratio [HR], 1.89 [95% CI, 1.52–2.35], P=7.41×10−9) with an estimated effect in the placebo group of HR=1.30 (95% CI, 1.04–1.62; P=0.02). The effect appeared to be mostly driven by the occurrence of diarrhea (Table II in the Data Supplement). The interaction term between rs6916345 and colchicine treatment was significant (P=2.96×10−8; Table 2). Individuals with the AA genotype represented 25% of the trial population. Gastrointestinal adverse events were reported by 36.9% of AA patients in the colchicine group compared with 18.6% in the placebo group (HR, 2.42 [95% CI, 1.57–3.72], P=5.77×10−5; Table 3). We found evidence of colocalization of the locus with Crohn disease (Material and Figure V in the Data Supplement). The risk allele (A) at rs6916345 was previously associated with Crohn disease (odds ratio, 1.07, P=3.1×10−5).5
Table 2.
Genetic Association Results of the Leading Genetic Variants Found to be Significantly Associated in the COLCOT Pharmacogenomic Study
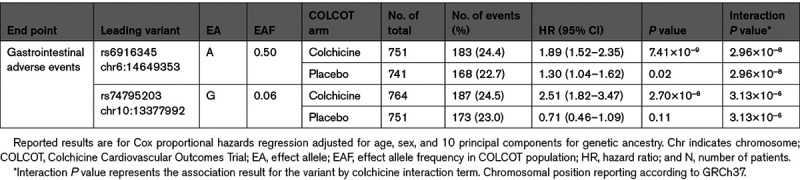
Table 3.
Effect of Colchicine on Gastrointestinal Adverse Events Compared With Placebo Stratified by Genotype Groups
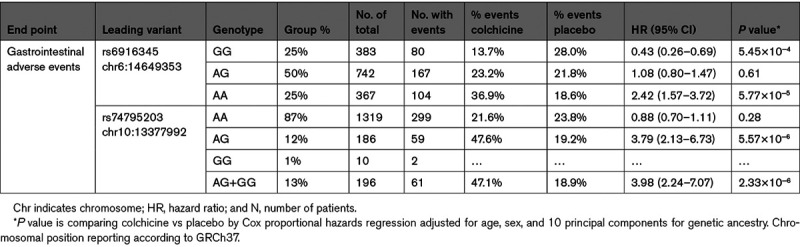
Figure.
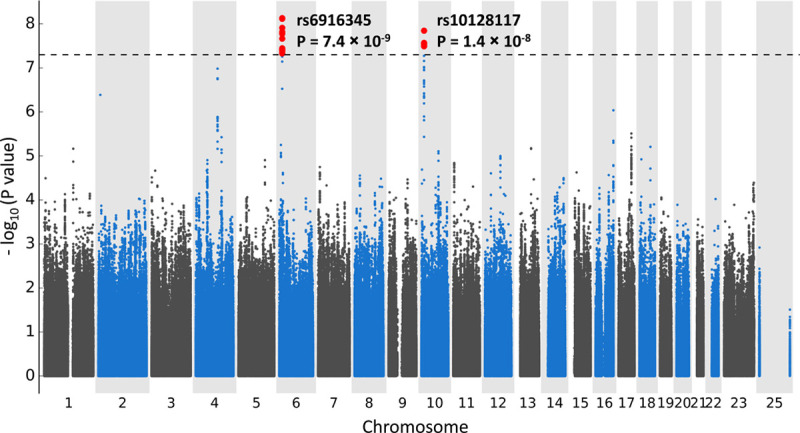
Manhattan plot for the genome-wide association study (GWAS) of gastrointestinal adverse events in COLCOT (Colchicine Cardiovascular Outcomes Trial) using Cox proportional hazards regression with 4 468 817 genetic variants of minor allele frequency ≥5% with 767 patients from the colchicine arm of COLCOT, controlling for age, sex, and principal components for genetic ancestry.
The most significant association at the chromosome 10 locus was at rs10128117 located in intron 2 of the selenophosphate synthetase 1 gene (SEPHS1). However, because this variant is triallelic and was imputed as biallelic, we report the findings based on variant, rs74795203, in strong linkage disequilibrium, located in intron 4 of the gene (Figure IVB in the Data Supplement). The G allele of variant rs74795203 was associated with gastrointestinal events with an HR of 2.51 (95% CI, 1.82–3.47; P=2.70×10−8) with an estimated effect in the placebo group of HR of 0.71 (95% CI, 0.46–1.09; P=0.11). The interaction term between rs74795203 and colchicine treatment was significant (P=3.13×10−6; Table 2). When conditioning on rs10128117 or rs74795203, no additional genetic variants remained significant at P<5×10−8. Individuals with the AG or GG genotype at rs74795203 represented 13% of the trial population. Gastrointestinal adverse events were reported by 47.1% of patients with the AG or GG genotype in the colchicine arm compared with 18.9% in the placebo arm (HR, 3.98 [95% CI, 2.24–7.07], P=2.33×10−6; Table 3). The GWAS limited to 622 male participants did not identify additional association signals.
Discussion
In this pharmacogenomic study of the randomized, placebo-controlled COLCOT trial, genetic variants were found to be associated with gastrointestinal events in patients treated with colchicine, offering insights into the biological mechanisms underlying the tolerability of treatment with colchicine. Although the signal did not reach the significance threshold, we have found an interesting genetic region on chromosome 9 in the prespecified analysis in males that is possibly associated with the cardiovascular benefits of colchicine. The locus is particularly interesting as it spans the SAXO1 gene, and it colocalizes with the expression of the HAUS augmin like complex subunit 6 (HAUS6) gene which is involved in microtubule generation from existing microtubules and in kinetochore-microtubule attachment and central spindle formation during anaphase.6 The cardiovascular event risk allele at the leading variant reduces HAUS6 expression, and it may possibly interact with the effects of colchicine on tubulin binding and microtubule polymerization. However, replication of this locus in future cardiovascular studies with colchicine is necessary.
The genome-wide analysis of gastrointestinal adverse events found 2 associated regions. The first region on chromosome 6 is particularly appealing as it colocalizes with a previously identified locus for Crohn disease.5 The risk allele of the lead variant at this locus was previously associated with Crohn disease risk and with reticulocyte counts and hemoglobin concentrations, which are common extraintestinal complication of Crohn disease. The second genetic locus on chromosome 10 overlaps the SEPHS1, which encodes an enzyme that synthesizes selenophosphate from selenide and ATP. We found evidence of colocalization of the region with expression of SEPHS1, with correlation between the gastrointestinal disorder risk allele and lower SEPHS1 gene expression.
Despite the relatively small proportion of participants who consented to take part in the pharmacogenomic substudy of COLCOT (32%), we have found significant and credible association signals predictive of gastrointestinal events with colchicine use. There may be volunteer bias in the pharmacogenomic subgroup compared with the main trial population, and we observed a lower occurrence of deaths, possibly attributable to the fact that not all patients were recruited into the pharmacogenomic substudy at the baseline visit. This may have contributed to reducing the statistical power for detecting genetic association signals with the primary cardiovascular end point which included cardiovascular death. We also noted an overrepresentation of patients who reported suffering from gastrointestinal disorders during the course of the trial from both the colchicine and the placebo arm. This could be due to correlation between patient willingness to participate and to share information on milder gastrointestinal adverse events. We do not expect that this observation had an impact on the pharmacogenomic findings with gastrointestinal events, as the 2 genetic association signals identified were strong and had strong interaction effects with colchicine treatment.
Because this study was a post hoc investigation, these results are considered as hypothesis-generating, and they will have to be replicated before using the information for clinical decision-making. Colchicine is used throughout the world for indications of gout, familial Mediterranean fever, pericarditis, and, since the COLCOT trial, for secondary cardiovascular prevention. There are other ongoing and planned clinical trials designed to assess the cardiovascular benefits of colchicine where it may be possible to replicate the findings if genetic material is collected. Reliance on observational studies and registries to conduct replication studies will become an option as the long-term use of colchicine for the prevention of secondary cardiovascular disease gains in popularity in the coming years. Shorter-term use of colchicine for the treatment of gout could provide useful data for replication of the genetic variants associated with gastrointestinal events.
In conclusion, in the present pharmacogenomic study of the COLCOT trial, we have found genetic variants associated with gastrointestinal events in patients treated with colchicine. Those findings will benefit from replication to confirm our observations that some patients may have genetic predispositions to lower tolerability of treatment with colchicine.
Acknowledgments
We acknowledge the technical support of Diane Valois and Isabelle Fillion for the genotyping work and of Yannik Couture and Sylvain Versailles for blood and DNA sample preparation. We thank the patients and staff who supported this study.
Sources of Funding
This work was supported by the Health Collaboration Acceleration Fund from the Government of Quebec (to Dr Tardif). M.-A. Legault is supported by a Frederick Banting and Charles Best Canada Graduate Scholarship Doctoral Award from the Canadian Institutes of Health Research (CIHR). Dr Tardif holds the Canada Research Chair in Personalized Medicine and the Université de Montréal endowed research chair in atherosclerosis. Dr Dubé holds the Canada Research Chair in Precision Medicine Data Analysis. Dr Berry is supported by the British Heart Foundation (RE/18/6134217). Dr de Denus holds the Université de Montréal Beaulieu-Saucier Chair in Pharmacogenomics. The funding sources had no role in study design, conduct, or analyses.
Disclosures
Dr Dubé reports grants from the Government of Quebec during the conduct of the study; personal fees from Dalcor, personal fees and other from GlaxoSmithKline, other from AstraZeneca, other from Pfizer, other from Servier, other from Sanofi, outside the submitted work; in addition, Dr Dubé has a patent Methods for Treating or Preventing Cardiovascular Disorders and Lowering Risk of Cardiovascular Events issued to Dalcor, no royalties received, a patent Genetic Markers for Predicting Responsiveness to Therapy with HDL-Raising or HDL Mimicking Agent issued to Dalcor, no royalties received, and a patent Methods for using low-dose colchicine after myocardial infarction with royalties paid to Invention assigned to the Montreal Heart Institute. Dr Tardif reports grants from the Government of Quebec, grants from Canadian Institutes of Health Research, grants from Montreal Heart Institute Foundation during the conduct of the study; grants from Amarin, grants and personal fees from Astra Zeneca, grants, personal fees and other from Dalcor, grants from Esperion, grants from Ionis, grants and personal fees from Sanofi, grants and personal fees from Servier, grants from RegenXBio, outside the submitted work; in addition, Dr Tardif has a patent Genetic markers for predicting responsiveness to therapy with HDL-raising or HDL mimicking agent pending, a patent Methods for using low-dose colchicine after myocardial infarction pending to Invention assigned to the Montreal Heart Institute, and a patent Methods of treating a coronavirus infection using Colchicine pending. Dr Waters reports personal fees from Pharmascience, outside the submitted work; Dr Kouz reports personal fees and other from Medtronic, grants, personal fees and other from Sanofi, other from Johnson & Johnson, personal fees and other from Amgen, grants, personal fees and other from Astrazeneca, grants, personal fees and other from Novartis, other from Celgene, other from Biogen, other from Gilead, other from Roche, other from Boston Scientific, personal fees and other from Bausch Health, other from GSK, personal fees and other from BMS, other from TG Therapeutics, other from Becton Dickinson, other from Spectrum Pharmaceuticals, personal fees from Merck, personal fees from Eli Lilly, personal fees from Pfizer, personal fees from Bayer, grants and personal fees from Boehringer-Ingelheim, personal fees from Servier, grants from Esperion, grants from Dalcor Pharmaceuticals, grants from Eisai, grants from Amarin Pharma, grants from Theracos, outside the submitted work; Dr Maggioni reports personal fees from Bayer, personal fees from DalCor, personal fees from Novartis, outside the submitted work; Dr Diaz reports grants from Montreal Health Innovations Coordinating Center, during the conduct of the study; grants from Dalcor, grants from Population Health Research Institute, outside the submitted work; Dr Berry reports that the University of Glasgow has received research and consultancy support for work done by CB with AstraZeneca, Abbott Vascular, DalCor, GSK, Heartflow, Menarini and Novartis. Dr Koenig reports personal fees from AstraZeneca, personal fees from Novartis, personal fees from Pfizer, personal fees from The Medicines Company, personal fees from DalCor, personal fees from Kowa, personal fees from Amgen, personal fees from Corvidia, personal fees from Daiichi-Sankyo, personal fees from Berlin-Chemie, personal fees from Sanofi, personal fees from Bristol-Myers Squibb, grants and nonfinancial support from Singulex, grants and nonfinancial support from Abbott, grants and nonfinancial support from Roche Diagnostics, grants and nonfinancial support from Beckmann, outside the submitted work; Dr Lopez-Sendon reports grants from Merk, grants from Pfizer, grants from Sanofi, grants from Amgen, grants from Boehringer Ingelheim, outside the submitted work; Dr Kiwan reports personal fees from Bayer, personal fees from Servier, personal fees from Novartis, personal fees from Astra Zeneca, personal fees from Bristol-Myers Squibb, personal fees from Roche, personal fees from Pfizer, outside the submitted work; Dr Rhainds reports personal fees from DalCor Pharmaceuticals, outside the submitted work; Dr Bouabdallaoui reports personal fees from AstraZeneca, outside the submitted work; Dr de Denus reports grants from Pfizer, AstraZeneca, Roche Molecular Science, and DalCor. Dr L’Allier reports personal fees from Pharmascience, personal fees from Philips, outside the submitted work; Dr Roubille reports grants, personal fees and nonfinancial support from Air Liquide, grants and personal fees from Abbott, personal fees from Vifor, grants and personal fees from Novartis, personal fees from Servier, personal fees from Abiomed, personal fees from Zoll, grants and personal fees from AstraZeneca, personal fees from Medtronic, personal fees from Resmed, from LVL, Eole, personal fees from Pfizer, outside the submitted work. The other authors report no conflicts.
Supplemental Materials
Online Methods and Results
Online Tables I–IV
Online Figures I–V
Nonstandard Abbreviations and Acronyms
- ABCB1
- ATP Binding Cassette Subfamily B Member 1 gene
- COLCOT
- Colchicine Cardiovascular Outcomes Trial
- CYP3A4
- cytochrome P450 family 3 subfamily A member 4
- GWAS
- genome-wide association study
- HAUS6
- HAUS augmin like complex subunit 6 gene
- HR
- hazard ratio
- SAXO1
- stabilizer of axonemal microtubules 1 gene
- SEPHS1
- selenophosphate synthetase 1 gene
For Sources of Funding and Disclosures, see page 227
The Data Supplement is available at https://www.ahajournals.org/doi/suppl/10.1161/CIRCGEN.120.003183.
Contributor Information
Marc-André Legault, Email: legaultmarc@gmail.com.
Audrey Lemaçon, Email: Audrey.lemacon@mhi-rc.org.
Louis-Philippe Lemieux Perreault, Email: louis-philippe.lemieux.perreault@statgen.org.
René Fouodjio, Email: rene.fouodjio@statgen.org.
David D. Waters, Email: David.Waters@ucsf.edu.
Simon Kouz, Email: simon.kouz@sympatico.ca.
Fausto J. Pinto, Email: fpinto@icvl.pt.
Aldo P. Maggioni, Email: maggioni@anmco.it.
Rafael Diaz, Email: rafadiaz@me.com.
Colin Berry, Email: colin.berry@glasgow.ac.uk.
Wolfgang Koenig, Email: koenig@dhm.mhn.de.
Jose Lopez-Sendon, Email: jlopezsendon@gmail.com.
Habib Gamra, Email: hgamra03@yahoo.com.
Ghassan S. Kiwan, Email: gskiwan@yahoo.ca.
Géraldine Asselin, Email: geraldine.asselin@statgen.org.
Sylvie Provost, Email: sylvie.provost@statgen.org.
Amina Barhdadi, Email: amina.barhdadi@statgen.org.
Maxine Sun, Email: maxine.sun@statgen.org.
Mariève Cossette, Email: marieve.Cossette@mhicc.org.
Lucie Blondeau, Email: Lucie.Blondeau@mhicc.org.
Ian Mongrain, Email: ian.mongrain@pharmacogenomics.ca.
Anick Dubois, Email: Anick.Dubois@cepmed.com.
David Rhainds, Email: david.rhainds@icm-mhi.org.
Nadia Bouabdallaoui, Email: nadia.bouabdallaoui@gmail.com.
Michelle Samuel, Email: michelle.samuel@mhi-rc.org.
Simon de Denus, Email: simon.kouz@sympatico.ca.
Marie-Claude Guertin, Email: marie-claude.guertin@mhicc.org.
François Roubille, Email: francois.roubille@gmail.com.
Jean-Claude Tardif, Email: jean-claude.tardif@icm-mhi.org.
References
- 1.Tardif JC, Kouz S, Waters DD, Bertrand OF, Diaz R, Maggioni AP, Pinto FJ, Ibrahim R, Gamra H, Kiwan GS, et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med. 2019; 381:2497–2505. doi: 10.1056/NEJMoa1912388 [DOI] [PubMed] [Google Scholar]
- 2.Ravelli RB, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, Knossow M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature. 2004; 428:198–202. doi: 10.1038/nature02393 [DOI] [PubMed] [Google Scholar]
- 3.Pope RM, Tschopp J. The role of interleukin-1 and the inflammasome in gout: implications for therapy. Arthritis Rheum. 2007; 56:3183–3188. doi: 10.1002/art.22938 [DOI] [PubMed] [Google Scholar]
- 4.Gagliano Taliun SA, VandeHaar P, Boughton AP, Welch RP, Taliun D, Schmidt EM, Zhou W, Nielsen JB, Willer CJ, Lee S, et al. Exploring and visualizing large-scale genetic associations by using PheWeb. Nat Genet. 2020; 52:550–552. doi: 10.1038/s41588-020-0622-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Lange KM, Moutsianas L, Lee JC, Lamb CA, Luo Y, Kennedy NA, Jostins L, Rice DL, Gutierrez-Achury J, Ji SG, et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet. 2017; 49:256–261. doi: 10.1038/ng.3760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Uehara R, Nozawa RS, Tomioka A, Petry S, Vale RD, Obuse C, Goshima G. The augmin complex plays a critical role in spindle microtubule generation for mitotic progression and cytokinesis in human cells. Proc Natl Acad Sci U S A. 2009; 106:6998–7003. doi: 10.1073/pnas.0901587106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lemieux Perreault LP, Provost S, Legault MA, Barhdadi A, Dubé MP. pyGenClean: efficient tool for genetic data clean up before association testing. Bioinformatics. 2013; 29:1704–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007; 81:559–575. doi: 10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006; 38:904–909. doi: 10.1038/ng1847 [DOI] [PubMed] [Google Scholar]
- 10.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009; 5:e1000529. doi: 10.1371/journal.pgen.1000529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet. 2012; 44:955–959. doi: 10.1038/ng.2354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delaneau O, Zagury JF, Marchini J. Improved whole-chromosome phasing for disease and population genetic studies. Nat Methods. 2013; 10:5–6. doi: 10.1038/nmeth.2307 [DOI] [PubMed] [Google Scholar]
- 13.Whirl-Carrillo M, McDonagh EM, Hebert JM, Gong L, Sangkuhl K, Thorn CF, Altman RB, Klein TE. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012; 92:414–417. doi: 10.1038/clpt.2012.96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lemieux Perreault L-P genetest. December 2019. https://github.com/pgxcentre/genetest
- 15.Hormozdiari F, Kostem E, Kang EY, Pasaniuc B, Eskin E. Identifying causal variants at loci with multiple signals of association. Genetics. 2014; 198:497–508. doi: 10.1534/genetics.114.167908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen W, Larrabee BR, Ovsyannikova IG, Kennedy RB, Haralambieva IH, Poland GA, Schaid DJ. Fine mapping causal variants with an approximate bayesian method using marginal test statistics. Genetics. 2015; 200:719–736. doi: 10.1534/genetics.115.176107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benner C, Spencer CC, Havulinna AS, Salomaa V, Ripatti S, Pirinen M. FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics. 2016; 32:1493–1501. doi: 10.1093/bioinformatics/btw018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lemaçon A, Joly Beauparlant C, Soucy P, Allen J, Easton D, Kraft P, Simard J, Droit A. VEXOR: an integrative environment for prioritization of functional variants in fine-mapping analysis. Bioinformatics. 2017; 33:1389–1391. doi: 10.1093/bioinformatics/btw826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lemaçon A, Scott-Boyer MP, Ongaro-Carcy R, Soucy P, Simard J, Droit A. DSNetwork: an integrative approach to visualize predictions of variants’ deleteriousness. Front Genet. 2019; 10:1349. doi: 10.3389/fgene.2019.01349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kamat MA, Blackshaw JA, Young R, Surendran P, Burgess S, Danesh J, Butterworth AS, Staley JR. PhenoScanner V2: an expanded tool for searching human genotype-phenotype associations. Bioinformatics. 2019; 35:4851–4853. doi: 10.1093/bioinformatics/btz469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Staley JR, Blackshaw J, Kamat MA, Ellis S, Surendran P, Sun BB, Paul DS, Freitag D, Burgess S, Danesh J, et al. PhenoScanner: a database of human genotype-phenotype associations. Bioinformatics. 2016; 32:3207–3209. doi: 10.1093/bioinformatics/btw373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, Thompson JR, Ingelsson E, Saleheen D, Erdmann J, Goldstein BA, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013; 45:25–33. doi: 10.1038/ng.2480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nelson CP, Goel A, Butterworth AS, Kanoni S, Webb TR, Marouli E, Zeng L, Ntalla I, Lai FY, Hopewell JC, et al. ; EPIC-CVD Consortium; CARDIoGRAMplusC4D; UK Biobank CardioMetabolic Consortium CHD working group. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat Genet. 2017; 49:1385–1391. doi: 10.1038/ng.3913 [DOI] [PubMed] [Google Scholar]
- 24.Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, Payne AJ, Steinthorsdottir V, Scott RA, Grarup N, et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet. 2018; 50:1505–1513. doi: 10.1038/s41588-018-0241-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Astle WJ, Elding H, Jiang T, Allen D, Ruklisa D, Mann AL, Mead D, Bouman H, Riveros-Mckay F, Kostadima MA, et al. The allelic landscape of human blood cell trait variation and links to common complex disease. Cell. 2016; 167:1415–1429.e19. doi: 10.1016/j.cell.2016.10.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, Ripke S, Lee JC, Jostins L, Shah T, et al. ; International Multiple Sclerosis Genetics Consortium; International IBD Genetics Consortium. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015; 47:979–986. doi: 10.1038/ng.3359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ligthart S, Vaez A, Võsa U, Stathopoulou MG, de Vries PS, Prins BP, Van der Most PJ, Tanaka T, Naderi E, Rose LM, et al. ; LifeLines Cohort Study; CHARGE Inflammation Working Group. Genome analyses of >200,000 Individuals Identify 58 Loci for chronic inflammation and highlight pathways that link inflammation and complex disorders. Am J Hum Genet. 2018; 103:691–706. doi: 10.1016/j.ajhg.2018.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, Plagnol V. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014; 10:e1004383. doi: 10.1371/journal.pgen.1004383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, Kang HM, Fuchsberger C, Danecek P, Sharp K, et al. ; Haplotype Reference Consortium. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016; 48:1279–1283. doi: 10.1038/ng.3643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gauderman WJ. Sample size requirements for matched case-control studies of gene-environment interaction. Stat Med. 2002; 21:35–50. doi: 10.1002/sim.973 [DOI] [PubMed] [Google Scholar]
- 31.Sáez ME, González-Pérez A, Hernández-Olasagarre B, Beà A, Moreno-Grau S, de Rojas I, Monté-Rubio G, Orellana A, Valero S, Comella JX, et al. Genome Wide Meta-Analysis identifies common genetic signatures shared by heart function and Alzheimer’s disease. Sci Rep. 2019; 9:16665. doi: 10.1038/s41598-019-52724-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ernst J, Kellis M. Chromatin-state discovery and genome annotation with ChromHMM. Nat Protoc. 2017; 12:2478–2492. doi: 10.1038/nprot.2017.124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yeh MM, Bosch DE, Daoud SS. Role of hepatocyte nuclear factor 4-alpha in gastrointestinal and liver diseases. World J Gastroenterol. 2019; 25:4074–4091. doi: 10.3748/wjg.v25.i30.4074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burdin DV, Kolobov AA, Brocker C, Soshnev AA, Samusik N, Demyanov AV, Brilloff S, Jarzebska N, Martens-Lobenhoffer J, Mieth M, et al. Diabetes-linked transcription factor HNF4α regulates metabolism of endogenous methylarginines and β-aminoisobutyric acid by controlling expression of alanine-glyoxylate aminotransferase 2. Sci Rep. 2016; 6:35503. doi: 10.1038/srep35503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Møller AM, Dalgaard LT, Ambye L, Hansen L, Schmitz O, Hansen T, Pedersen O. A novel Phe75fsdelT mutation in the hepatocyte nuclear factor-4alpha gene in a Danish pedigree with maturity-onset diabetes of the young. J Clin Endocrinol Metab. 1999; 84:367–369. doi: 10.1210/jcem.84.1.5396 [DOI] [PubMed] [Google Scholar]
- 36.Marcil V, Sinnett D, Seidman E, Boudreau F, Gendron FP, Beaulieu JF, Menard D, Lambert M, Bitton A, Sanchez R, et al. Association between genetic variants in the HNF4A gene and childhood-onset Crohn’s disease. Genes Immun. 2012; 13:556–565. doi: 10.1038/gene.2012.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilson A, Reyes E, Ofman J. Prevalence and outcomes of anemia in inflammatory bowel disease: a systematic review of the literature. Am J Med. 2004; 116(Suppl 7A):44S–49S. doi: 10.1016/j.amjmed.2003.12.011 [DOI] [PubMed] [Google Scholar]
- 38.Gentschew L, Bishop KS, Han DY, Morgan AR, Fraser AG, Lam WJ, Karunasinghe N, Campbell B, Ferguson LR. Selenium, selenoprotein genes and Crohn’s disease in a case-control population from Auckland, New Zealand. Nutrients. 2012; 4:1247–1259. doi: 10.3390/nu4091247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lauc G, Huffman JE, Pučić M, Zgaga L, Adamczyk B, Mužinić A, Novokmet M, Polašek O, Gornik O, Krištić J, et al. Loci associated with N-glycosylation of human immunoglobulin G show pleiotropy with autoimmune diseases and haematological cancers. PLoS Genet. 2013; 9:e1003225. doi: 10.1371/journal.pgen.1003225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Plomp R, Ruhaak LR, Uh HW, Reiding KR, Selman M, Houwing-Duistermaat JJ, Slagboom PE, Beekman M, Wuhrer M. Subclass-specific IgG glycosylation is associated with markers of inflammation and metabolic health. Sci Rep. 2017; 7:12325. doi: 10.1038/s41598-017-12495-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li T, DiLillo DJ, Bournazos S, Giddens JP, Ravetch JV, Wang LX. Modulating IgG effector function by Fc glycan engineering. Proc Natl Acad Sci U S A. 2017; 114:3485–3490. doi: 10.1073/pnas.1702173114 [DOI] [PMC free article] [PubMed] [Google Scholar]