

ABSTRACT
Here, we identified a novel class of compounds which demonstrated good antiviral activity against dengue and Zika virus infection. These derivatives constitute intermediates in the synthesis of indole (ervatamine-silicine) alkaloids and share a tetracyclic structure, with an indole and a piperidine fused to a seven-membered carbocyclic ring. Structure-activity relationship studies indicated the importance of substituent at position C-6 and especially the presence of a benzyl ester for the activity and cytotoxicity of the molecules. In addition, the stereochemistry at C-7 and C-8, as well as the presence of an oxazolidine ring, influenced the potency of the compounds. Mechanism of action studies with two analogues of this family (compounds 22 and trans-14) showed that this class of molecules can suppress viral infection during the later stages of the replication cycle (RNA replication/assembly). Moreover, a cell-dependent antiviral profile of the compounds against several Zika strains was observed, possibly implying the involvement of a cellular factor(s) in the activity of the molecules. Sequencing of compound-resistant Zika mutants revealed a single nonsynonymous amino acid mutation (aspartic acid to histidine) at the beginning of the predicted transmembrane domain 1 of NS4B protein, which plays a vital role in the formation of the viral replication complex. To conclude, our study provides detailed information on a new class of NS4B-associated inhibitors and strengthens the importance of identifying host-virus interactions in order to tackle flavivirus infections.
KEYWORDS: NS4B protein, SAR studies, Zika virus, cross resistance, dengue virus, indole alkaloids, mechanisms of action
INTRODUCTION
Flaviviruses (belonging to the family Flaviviridae) are small enveloped viruses with a positive-sense single-stranded RNA. Their genome is approximately 11 kb and encodes a single polyprotein, which is posttranslationally processed by several viral and host enzymes into three structural proteins (capsid, envelope, and premembrane) and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) (1). Two of the most important human pathogens in this family are dengue virus (DENV) and Zika virus (ZIKV), which are mainly spread through Aedes mosquitoes in many regions across the world (such as the Americas, Africa, Southeast Asia, and the Pacific Islands) (2, 3). Additionally, more transmission routes have been reported, such as through sexual contact and from mother to child (4, 5).
DENV, a prevailing arbovirus, is found in tropical and subtropical areas and it is responsible for more than 300 million infections per year (6). In most cases, the virus can cause flu-like illness symptoms, such as headache and fever. However, under extreme conditions, DENV has been associated with severe dengue disease, which can lead to shock syndrome and death (7, 8). There are four antigenically different but closely related serotypes (DENV1, DENV2, DENV3, and DENV4), which have been found to cocirculate with other flaviviruses, including ZIKV (9).
ZIKV was first isolated from rhesus macaques in the 1950s, and it was responsible for three main outbreaks: in Southeast Asia (Yap Islands, 2007), the Pacific Islands (French Polynesia, 2013), and South America (Brazil, 2015). During that period, ZIKV was considered a Public Health Emergency of International Concern by the World Health Organization (WHO) (10). Although the virus generally causes asymptomatic infections, it has been correlated with neurological disorders, including microcephaly and the autoimmune disease Guillain-Barré syndrome (11, 12).
Currently, an approved vaccine against DENV (Dengvaxia) has been shown to efficiently enhance immune responses in people who were previously infected by the virus. However, important limitations have been raised due to its efficacy and side effects in those with no signs of previous viral infection (13, 14). On the other hand, no ZIKV vaccines are available at the moment. As previously mentioned, the fact that ZIKV infection mostly occurs in areas where DENV is endemic makes vaccine development even more complex due to serological cross-reactivity between the two viruses (15).
Therefore, development of an effective treatment against both flaviviruses is of utmost importance. During the last years, the research community has made serious efforts to identify potent antiviral molecules against both DENV and ZIKV. These compounds can target either viral proteins (direct acting) or cellular factors (host acting) to tackle the infection. Screening assays with a number of repurposed Food and Drug Administration-approved drugs have revealed analogues targeting several viral proteins (e.g., sofosbuvir against NS5 and castanospermine against envelope protein) (16, 17). Despite the promising outcomes, important limitations have to be considered with regard to their use, since occurrence of mutations in the viral genome is common, possibly leading to the generation of resistant strains, which might evade the compounds’ pressure. On the other hand, host factor-targeting molecules are able to address the problem of a low genetic barrier to resistance (18). In particular, several compounds targeting cellular factors have been shown to exhibit antiviral activity against DENV and ZIKV, including chloroquine, bortezomib, and lovastatin. However, these molecules did not result in a significant improvement in patient symptoms when evaluated in clinical trials (19, 20). Taking all the above into consideration, it is evident that development and characterization of novel molecules will be crucial in the fight against these two emerging flaviviruses.
In the present study, we identified and characterized a novel class of indole derivatives, having in common an oxazolopiperidone moiety, with pronounced antiviral activity (in the low-micromolar range) against DENV and ZIKV. Following a number of in vitro antiviral assays, we showed that our compounds possibly interfere with the viral RNA replication and assembly processes. Moreover, selection and sequencing analysis of compound-resistant virus mutants revealed a single nonsynonymous amino acid mutation, located at the beginning of predicted transmembrane domain 1 (pTMD1) of the ZIKV NS4B protein. This motif possesses a dual role, as it is involved in the formation of viral replication complex system and antagonizes host interferon (IFN) responses (21). Finally, a cell-dependent inhibition profile indicated the possible involvement of cellular factors in the activity of the molecules.
RESULTS
Cellular cytotoxicity and antiviral activity of indole alkaloid derivatives against DENV and ZIKV replication in several susceptible host cell lines.
The synthesis of this series of molecules is described in the supplemental material. As shown in Fig. 1, all analogues are synthetic intermediates of ervatamine-silicine indole alkaloids. To evaluate the antiviral activity of the compounds, African green monkey kidney cells (Vero) were infected with DENV2 NGC and ZIKV MR766 prototype strains in the presence of these molecules. Cellular cytotoxicity was assessed in parallel. Based on the results from Table 1, we could divide the compounds into three distinct groups based on their antiviral activity and cytotoxic profile, as follows. (i) Compounds 22, 20, trans-14, and cis-12 exhibited good activity against DENV2 (EC50, 0.3 to 5.9 μΜ) but no or limited (36.2 ± 6.5 μΜ) inhibition of ZIKV. (ii) In contrast, compounds trans-13, cis-13, and cis-11, lacking the benzyloxy substituent at C-6 (Fig. 1, red circles), were active against both DENV (1.1 to 10.9 μΜ) and ZIKV (3.3 to 7.2 μΜ) but showed more pronounced cytotoxicity (CC50 < 40 μΜ) in our evaluation. (iii) Finally, cis-10 and trans-10 derivatives displayed good antiviral activity against both viruses (50% effective concentrations [EC50], 0.4 to 2.7 μΜ and 0.2 to 12 μΜ for DENV and ZIKV, respectively) with no associated cytotoxicity. Similar results were observed for compound 24, an analogue without the sulfonamide group in the indole nitrogen moiety (Fig. 1, blue circle). In addition, analogue 21, which lacks the oxazolidine ring (Fig. 1, green circle), was less active against DENV (37.1 ± 6.0 μΜ) than the other molecules. Furthermore, molecules 15a and 15b (de-ethyl analogues of cis-12 and trans-14, respectively), bearing a methyl ester at C-6 (instead of a larger benzyl ester group) (Fig. 1, yellow circles), showed more pronounced cytotoxicity and decreased antiviral activity against DENV than their analogues. Moreover, stereochemistry at C-7 seemed to affect the cytotoxicity levels between compounds 15a (50% cytotoxic concentration [CC50] >100 μΜ) and 15b (CC50: 41.8 ± 9.8 μΜ) (Fig. 1, blue arrows). Finally, compounds cis-10 and cis-12 displayed C-7/C-8-cis and C-8/C-8a-trans relative configurations (Fig. 1, orange arrows), although with opposite absolute stereochemistry, which seemed to be beneficial for the antiviral activity.
FIG 1.

Structure of indole alkaloid derivatives. Chemical groups and sites crucial for the enhancement of antiviral activity are marked with circles and arrows.
TABLE 1.
Antiviral activity, cellular cytotoxicity, and selectivity index of a novel series of compounds in Vero cellsa
| Compound | EC50 (μΜ)b |
CC50 (μΜ)c | SId |
||
|---|---|---|---|---|---|
| DENV2 | ZIKV | DENV2 | ZIKV | ||
| 15a | 19.4 ± 7.5 | 40 ± 3.8 | 41.8 ± 9.8 | 2.1 | 1 |
| 15b | 18.6 ± 3.5 | 12.1 ± 1.7 | >100 | 5.4 | 8.3 |
| 20 | 5.9 ± 3.9 | >100 | >100 | >17 | >1 |
| 21 | 37.1 ± 6.0 | 4.0 ± 2.0 | >100 | >2.7 | >25 |
| 22 | 2.8 ± 2.8 | >100 | >100 | >35.7 | >1 |
| 24 | 0.4 ± 0.2 | 0.2 ± 0.1 | 80 ± 35 | 200 | 400 |
| cis−10 | 1.0 ± 0.2 | 0.8 ± 0.1 | >100 | >100 | >125 |
| trans−10 | 2.7 ± 0.9 | 12 ± 3.8 | >100 | >37 | >8.3 |
| cis−11 | 10.9 ± 1.7 | 7.2 ± 1.4 | 27.7 ± 9.4 | 2.5 | 3.8 |
| cis−12 | 0.3 ± 0.1 | 36.2 ± 6.5 | >100 | >333 | >2.7 |
| cis−13 | 1.1 ± 0.2 | 3.3 ± 0.5 | 5.8 ± 0.1 | 5.3 | 1.8 |
| trans−13 | 4.0 ± 2.8 | 6.5 ± 0.2 | 9.2 | 2.3 | 1.4 |
| trans−14 | 2.8 ± 1.9 | >100 | >100 | >35.7 | >1 |
The laboratory-adapted viral strains DENV2 NGC and ZIKV MR766 were used.
EC50, effective concentration of the compound able to inhibit viral replication by 50%, as assessed by qRT-PCR of supernatants at 72 h p.i.
CC50, concentration of the compound required to reduce cell viability by 50%, as assessed by MTS analysis of cell lysates at 72 h p.i.
SI, selectivity index (CC50/EC50).
To obtain more knowledge of this class of indole derivatives, two analogues (compounds 22 and trans-14) were chosen for further analysis. This choice was made based on their good selectivity index and the absence of solubility issues that could hamper the experiments. Table 2 shows the antiviral activity and cellular cytotoxicity of compounds 22 and trans-14 against the prototype DENV2 NGC strain, evaluated in different cells. These molecules showed no cellular cytotoxicity in all evaluated cells lines (CC50 >100). Both analogues inhibited viral replication in the low-micromolar range; However, a slightly different antiviral profile was observed in the five susceptible cell lines. Specifically, compounds 22 and trans-14 were more potent in baby hamster kidney (BHK; EC50, 0.08 to 0.15 μΜ), placenta choriocarcinoma (Jeg3; EC50, 0.3 to 1.0 μΜ), lung epithelial adenocarcinoma (A549; EC50, 0.03 to 0.04 μΜ) and hepatocarcinoma (Huh; EC50, 0.1 to 0.4 μΜ) cells than in Vero (EC50, 2.8 μΜ) cells.
TABLE 2.
Antiviral activity, cytotoxicity, and selectivity index profiles of compounds 22 and trans-14 against the laboratory strain DENV2 NGC, evaluated in five susceptible cell linesa
| Compound | Vero |
BHK |
Huh |
Jeg3 |
A549 |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EC50 | CC50 | SI | EC50 | CC50 | SI | EC50 | CC50 | SI | EC50 | CC50 | SI | EC50 | CC50 | SI | |
| 22 | 2.8 ± 2.8 | >100 | >35.7 | 0.15 ± 0.08 | >100 | >667 | 0.4 ± 0.1 | >100 | >250 | 1.0 ± 0.4 | >100 | >100 | 0.03 ± 0.02 | >100 | >3,000 |
| trans-14 | 2.8 ±1.9 | >100 | >35.7 | 0.08 ± 0.04 | >100 | >1,250 | 0.1 ± 0.08 | >100 | >1,000 | 0.3 ± 0.1 | >100 | >330 | 0.04 ± 0.02 | >100 | >2,500 |
Data are averages ± SD of at least three independent experiments. EC50s, CC50s, and SIs for the compounds in Vero cells are from Table 1.
To determine their broad-spectrum activity, compounds 22 and trans-14 were tested against a number of different laboratory and clinical strains of DENV in Vero cells. As shown in Table 3, both molecules presented slightly stronger activity against the clinical isolates (EC50, 0.6 to 1.9 μΜ) than the laboratory strains (EC50, 2.2 to 6.0 μΜ). Moreover, we also assessed their antiviral activity against three ZIKV strains (the laboratory strain MR766 and the clinical isolates PRVAVC59 and FLR) in several susceptible cell lines (Table 4). Surprisingly, neither molecule was found to be active against any of the tested viral strains in Vero cells (EC50, >100 μΜ). Subsequently, compounds 22 and trans-14 were evaluated in human-derived cell lines (Huh, Jeg3, and A549) against all ZIKV strains. Remarkably, both compounds were active in the low-micromolar range against the tested ZIKV strains in Jeg3 cells (EC50, 0.2 to 1.2 μΜ) and A549 cells (EC50, 0.1 to 0.6 μΜ) but not in Huh cells (EC50, >100 μM), strongly indicating a possible cell type-dependent antiviral profile.
TABLE 3.
Antiviral activity of 22 and trans-14 against several DENV laboratory and clinical strains in Vero cells
| Compound | EC50 (μM)a |
|||||||
|---|---|---|---|---|---|---|---|---|
| Laboratory strains |
Clinical strains |
|||||||
| DENV1 Djibouti | DENV2 nGC | DENV3 H87 | DENV4 dak | DENV1 | DENV2 | DENV3 | DENV4 | |
| 22 | 2.2 ± 0.4 | 2.8 ± 2.8 | 3.2 ± 0.5 | 2.7 ± 1.1 | 0.6 ± 0.1 | 0.6 ± 0.4 | 0.8 ± 0.5 | 0.8 ± 0.6 |
| trans-14 | 6.0 ± 5.7 | 2.8 ± 1.9 | 3.2 ± 1.0 | 2.9 ± 1.3 | 1.1 ± 0.2 | 1.3 ± 0.3 | 0.8 ± 0.4 | 1.9 ± 1.7 |
Data are averages ± SD from at least three independent experiments. EC50s against the DENV2 NGC laboratory strain in Vero cells are from Table 1.
TABLE 4.
Antiviral activities of compounds 22 and trans-14 against several ZIKV laboratory and clinical strains evaluated in four susceptible cell lines
| Compound | EC50 (μM)a |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Vero |
Huh |
Jeg3 |
A549 |
|||||||||
| MR766 | PR | FLR | MR766 | PR | FLR | MR766 | PR | FLR | MR766 | PR | FLR | |
| 22 | >100 | >100 | >100 | >100 | >100 | >100 | 0.2 ± 0.1 | 0.2 ± 0.1 | 0.9 ± 0.6 | 0.1 ± 0.05 | 0.2 ± 0.1 | 0.1 ± 0.04 |
| trans-14 | >100 | >100 | >100 | >100 | >100 | >100 | 0.2 ± 0.05 | 0.7 ± 0.3 | 1.2 ± 1.0 | 0.2 ± 0.1 | 0.6 ± 0.4 | 0.2 ± 0.1 |
Data are averages ± SD from at least three independent experiments. EC50s against ZIKV MR766 in Vero cells are from Table 1.
Based on our findings shown in Table 4, we also assessed the activity of compound 22 against the prototype ZIKV MR766 strain using immunofluorescence staining for the presence of viral envelope protein. Specifically, two cell lines were used (A549 and Vero), as striking differences in antiviral activity levels were observed. As shown in Fig. 2, incubation of A549 cells with high concentrations (10 μΜ and 50 μΜ) of compound 22 (Fig. 2, left, panels E and F) led to full inhibition of viral infection, whereas lower concentrations (0.4 μΜ and 2 μΜ) of the compound (Fig. 2, left, panels C and D) resulted in decreased levels of viral infection. In contrast, treatment of Vero cells with the compound was not able to block virus replication, even at the highest tested concentration of 50 μΜ (Fig. 2, right, panels C to F). In both experiments, negative (noninfected, dimethyl sulfoxide [DMSO]-treated cells without compound 22) (Fig. 2, panels A) and positive controls (virus-infected cells without compound 22) (Fig. 2, panels B) were included.
FIG 2.

Detection of ZIKV envelope protein in the presence of compound 22 using immunofluorescence staining in A549 and Vero cells. Both A549 and Vero cells were exposed to the ZIKV MR766 strain at an MOI of 0.2 and incubated with several concentrations of compound 22. Fluorescence images were acquired 72 h postinfection. (A) Cell control; (B) virus control; (C to F) 0.4 μM, 2 μM, 10 μM, and 50 μM compound 22.
Compounds 22 and trans-14 interfere with DENV RNA replication/assembly.
To gain more insight into the mechanism of action of compounds 22 and trans-14, time-of-drug-addition and subgenomic replicon assays were performed. In the first set of assays, we treated Vero cells with compounds 22 and trans-14 at a concentration of 9 μΜ (approximately 3-fold the EC50) at different time points pre- and postinfection (Fig. 3A). Time zero corresponds to the time at which cells were exposed to the virus. As shown in Fig. 3A, both analogues were able to inhibit virus replication at later stages (between 8 h [P < 0.0001] and 12 h [P = 0.005]). At 12 h postinfection (p.i.), an increase in viral load was observed. To assess these findings, established inhibitors like dextran sulfate (DS; 8,000 Da average molecular weight) and 7-deaza-2′-C-methyladenosine (7DMA) were included in the study. Specifically, DS (an entry inhibitor) inhibited viral infection during the early stages (−2 h and 0 h) (P = 0.0002). On the other hand, the nucleoside analogue 7DMA (a replication inhibitor) presented a profile similar to that of our molecules, suppressing viral replication even 12 h p.i. (P = 0.0012). To consolidate these results, we proceeded with the subgenomic replicon assay. Specifically, serial dilutions of compounds 22 and trans-14 (100μΜ, 20μΜ, 4μΜ, and 0.8μΜ) were added to BHK/DENV cells. As observed in Fig. 3B, incubation of cells with both compounds led to a decrease in luciferase activity (blue), which corresponded to inhibition of viral replication and a possible interaction with the nonstructural proteins (statistical analysis is provided in the supplemental material). Data for 7DMA were similar to those obtained with our molecules. In parallel, no cytotoxic effects were noticed at all tested concentrations (Fig. 3B, red).
FIG 3.
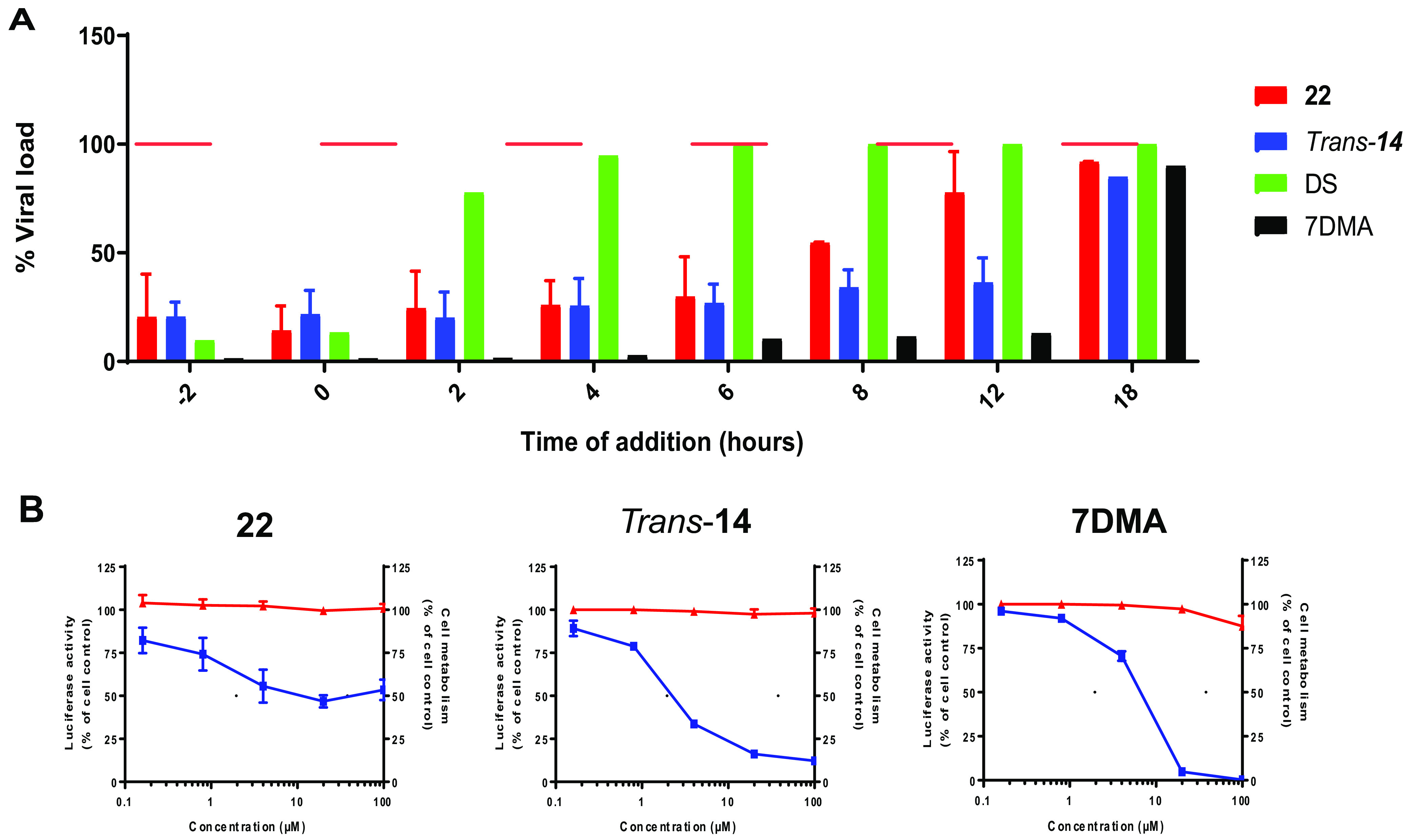
Compounds interfere with the viral RNA replication/assembly. (A) Treatment of Vero cells with a 9 μΜ concentration of compounds 22 (red) and trans-14 (blue) shows inhibition of DENV infection even at later stages (8 h to 12 h p.i.), as assessed by qRT-PCR of cell lysates 20 h p.i. The viral polymerase inhibitor 7-deaza-2′-C-methyladenosine (7DMA) (black) presents a similar profile, as it suppresses the infection at 12 h p.i. On the other hand, the entry inhibitor dextran sulfate (DS) (green) inhibits virus replication at the early stages (−2 h to 0 h). Horizontal red lines represent virus controls. (B) Compounds 22 and trans-14 were incubated with BHK/DENV cells at different concentrations. A decrease in the luciferase activity was observed for both compounds, as well as the viral inhibitor 7DMA (blue), as assessed by luminescence. These results indicate an interaction with the nonstructural proteins or the replication complex. Incubation of compounds 22 and trans-14 shows no cellular cytotoxicity at the tested concentrations (red). All data are averages from two independent experiments.
Compounds do not interact with intact viral particles.
Next, we investigated whether these compounds are able to directly target viral particles. Specifically, two in vitro assays were performed, ultrafiltration and virus inactivation. In the first assay, DENV2 stock was incubated with compound 22 (50μΜ, 10μΜ, and 2μΜ) for 1 h, followed by filtration to remove the unbound compound. If the compound does not strongly interact with the viral particles, it appears in the flowthrough (3.5 × 102 PFU/ml; P = 0.0002). A plaque assay was used to further evaluate the presence of viable virus in the fraction retained on the filter. As shown in Fig. 4A, inoculation of BHK cells treated with different concentrations of compound 22 resulted in the formation of viral plaques, implying no suppression of viral infection (2 μΜ compound 22, 1.3 × 103 PFU/ml [P = 0.57]; 10 μΜ compound 22, 5 × 102 PFU/ml [P = 0.051]; 50 μΜ compound 22, 2.5 × 102 PFU/ml [P = 0.007]). On the other hand, incubation of BHK with epigallocatechin gallate (EGCG; 1 mg/ml; positive control) showed a complete inhibition of viral infection (P < 0.0001). A virus control was also included to evaluate the significance of our results (5.5 × 102 PFU/ml) (supplemental material). Moreover, we confirmed these findings by a virus inactivation assay, where viral stock was incubated with a 10 μΜ concentration of compound 22, followed by dilution of the sample to reach a nonactive (suboptimal) concentration of the compound. As indicated in Fig. 4B, compound 22 did not inhibit the viral infection process (2.5 × 104 PFU/ml; P = 0.56). Comparable results were also observed for the virus control (3.5 × 104 PFU/ml), suggesting that compound 22 did not interact with the intact viral particles.
FIG 4.

Compounds do not interact with the intact viral particles. (A) Compound 22 at 50μΜ, 10μΜ, and 2μΜ, as well as the positive control epigallocatechin gallate (EGCG) (1 mg/ml), was mixed with DENV2 stock and passed through a filter. Incubation of compound 22-treated samples on BHK cells resulted in no inhibition of viral infection in each tested condition (formation of viral plaques). Inoculation of cells with EGCG-treated sample demonstrated full inhibition of viral infection. (B) Compound 22 T 10 μΜ was mixed with DENV2 viral stock and further diluted 100 times to infect BHK cells. No suppression of viral infection was noticed in the compound-treated sample, as assessed by the plaque assay. Both positive (virus-infected cells) and negative (noninfected, DMSO-treated cells) controls were included in the assays.
Sequencing analysis of compound-resistant viral mutants and cross-resistance assay.
To identify the potential target protein, we proceeded with the selection of resistant viruses by subcultivating ZIKV strain MR766 in the presence of increasing concentrations of compounds 22 and trans-14 in A549 cells. Two passages (P1 and P16) were selected for whole-genome sequencing using MinION technology (accession numbers are provided in “Data availability” below). Comparative analysis across the ZIKV genomes at P16 revealed a single substitution at nucleotide position 6984 (from G to C), which corresponded to a nonsynonymous amino acid change from aspartic acid (D) to histidine (H) for both viral mutants obtained with compounds 22 and trans-14 (Fig. 5A). This mutation is located at amino acid position 31 of the NS4B protein. Based on the topology of DENV2 and Japanese encephalitis virus (JEV) NS4B (22, 23) that position is located at the beginning of one of its predicted transmembrane domains, pTMD1.
FIG 5.

Sequencing analysis of the viruses used in the cross-resistance assay. (A) NS4B membrane topology and alignment of amino acid sequence at the N-terminal site of the protein in compound 22- and trans-14-resistant virus mutants compared to virus control. An aspartic acid-to-histidine (D31H) nonsynonymous mutation is located at the pTMD1 domain and marked in orange. Passage 16 (P16) of each virus strain was used in the assay. (B) Antiviral activity of several derivatives against the viruses used above (wild-type ZIKV MR766 and compound 22- and trans-14-resistant virus mutants) was evaluated by qRT-PCR in the supernatant of A549 cells at 72 h p.i. Fold shifts in EC50 are indicated in parentheses, while major findings on the activity of some analogues against the trans-14-resistant mutant and wild-type virus are highlighted in yellow.
To verify that these indole alkaloids target the same protein, we evaluated the antiviral activity of compounds 20, 22, 24, trans-14, cis-10, and trans-10 against wild-type ZIKV MR766 and trans-14- and 22-resistant virus mutants (Fig. 5B). Although all derivatives presented high inhibitory activity against the wild-type ZIKV strain MR766 (EC50, 0.1 to 4.2 μΜ), a complete loss of activity against the compound 22-resistant mutant (EC50 > 100 μΜ) was observed. However, assessment of activity against the compound trans-14 resistant strain revealed differences among the molecules. Interestingly, we observed that only cis-10 (EC50, 1.4 ± 0.5 μΜ) and trans-10 (EC50, 2.4 ± 1.8 μΜ) analogues retained their inhibitory activity, with EC50s being quite comparable to those observed for the wild-type strain (Fig. 5B, yellow). DS was used as a reference compound in this assay, since it suppresses viral infection by means of a completely different mode of action.
Compounds 22 and trans-14 constitute specific inhibitors of DENV and ZIKV.
In order to address the specificity of our molecules, we evaluated the antiviral activity of compounds 22 and trans-14 against viruses of different families. Specifically, HIV-1 (NL4.3), HIV-2 (ROD), influenza A virus (H1N1 and H3N2), influenza B virus, and human coronavirus (229E) were tested. Interestingly, the tested derivatives were devoid of any antiviral activity against these viruses (data not shown), indicating that our molecules are highly selective against DENV and ZIKV.
DISCUSSION
Despite our growing interest in curbing the public health threat posed by DENV and ZIKV, there are no approved antiviral drugs available at the moment (24). In the present study, we screened a number of molecules from a library of chemically diverse compounds for their activity against both DENV and ZIKV. A novel class of indole alkaloids was found to be active in the low-micromolar range against the prototype DENV2 NGC strain in several virus-susceptible cell lines. Interestingly, striking differences in antiviral activity against the prototype ZIKV MR766 strain were observed among the tested cell lines, suggesting a possible cell-dependent inhibition profile.
To map the essential modifications required in the structure of this class for the antiviral activity against DENV and ZIKV, structure-activity relationship (SAR) studies were performed. In particular, we demonstrate the importance of substituent and its size at position C-6 for both activity and cytotoxicity of the molecules. In addition, the presence of an oxazolidine ring seems to be crucial for the potency of the compounds. Finally, stereochemistry at C-7 and C-8 has an impact on the inhibitory activity and cellular cytotoxicity of the analogues (Fig. 1 and Table 1).
To gain more insight into the antiviral activity and mechanism of action of these indole alkaloid derivatives, the analogues 22 and trans-14 were selected for further analysis, based on their selectivity index and absence of solubility issues even at the highest concentrations. As mentioned above, these molecules presented broad-spectrum activity against all DENV serotypes, with slightly higher potency against the clinical isolates (EC50, 0.6 to 1.9 μΜ) than the laboratory strains (EC50, 2.2 to 6.0 μΜ). Interestingly, a cell-dependent antiviral profile against ZIKV strains was noticed, since compounds 22 and trans-14 were able to inhibit infection in human A549 (EC50, 0.1 to 0.6 μΜ) and Jeg3 (EC50, 0.2 to 1.2 μΜ) cells but not in human Huh and monkey Vero cells (EC50 > 100 μΜ). We speculate that even though ZIKV and DENV belong to the same virus family and share high sequence similarity, they present differences in the pathways they follow to infect human- and non-human-derived cells (25). Our findings further highlight the importance of cell line selection in the development and evaluation of new antiviral compounds, strengthening the idea that antiviral screening assays should not be restricted to a single type of susceptible cells (26).
Additionally, time-of-drug-addition and subgenomic replicon assays revealed that our compounds act during viral RNA replication/assembly. Despite the fact that compounds 22 and trans-14 belong to the same family of molecules, sharing some common features, such as the oxazolopiperidone moiety, minor differences were noticed in their antiviral profiles. This divergence can be explained either by the fact that these analogues target a different protein or a different site on the same viral protein. To address this question, compound resistance selection experiments were performed in A549 cells. The choice of this cell line was based on the following three criteria: (i) high ZIKV replication kinetics, (ii) a clearly visible cytopathogenic effect, and (iii) no cytotoxicity or solubility issues at higher tested concentrations of the molecules (27). Whole-genome sequencing and comparative analysis between compound-resistant virus mutants and the wild-type virus indicated a single nonsynonymous amino acid change at position 31 of NS4B in both compound 22- and trans-14-resistant viral strains. This substitution led to replacement of the negatively charged aspartic acid with the partially protonated histidine.
Notably, we further demonstrated that our compounds do not interact with the intact viral particles (Fig. 4), possibly suggesting the involvement of a cellular factor(s) associated with the NS4B protein. NS4B is an integral endoplasmic reticulum (ER) membrane protein, which consists of five domains (pTMD1, pTMD2, TMD3, TMD4, and TMD5) (Fig. 5A) (28). This nonstructural protein plays a vital role in viral RNA replication by interacting with the endoplasmic reticulum membrane protein complex (EMC), in which a large number of host proteins reside (23, 29). Our single point mutation (D31H) is located at the beginning of the pTMD1 domain (ER lumen), which has also been implicated in IFN antagonism host responses (30). The fact that compounds 22 and trans-14 do not exhibit anti-ZIKV activity in Vero cells, an IFN-I-defective cell line (31), could suggest a possible interference of our molecules within the interferon pathway. However, a similar antiviral profile was observed in IFN-competent Huh cells (32), indicating that compounds 22 and trans-14 are associated with a different function of NS4B. Given that the topology of NS4B of the closely related dengue virus and the discrimination of its different domains (pTMDs and TMDs) have been described by Miller et al. (28), we can use this proposed model to specify the topology of the single mutation in the compound-resistant strains (Fig. 5A). Several reports have shown that position 31 of NS4B protein is characterized by a high degree of conservation not only among ZIKV strains (33) but also between different flaviviruses, such as DENV and yellow fever virus (23), proposing it as a potential vulnerable point for these members.
In addition, a phenotypic resistance assessment was performed in order to determine whether the viral mutants that we developed are cross resistant to several derivatives of this family (Fig. 5B). Surprisingly, we found that only two analogues, cis-10 and trans-10, were active against the trans-14-resistant mutant, whereas no activity was noticed against 22-resistant mutant for all the tested molecules. These findings were interesting, since no qualitative differences on the amino acid level were observed between the 22- and trans-14-resistant mutants. In order to obtain more information, we implemented variant calling by using wild-type ZIKV MR766 (passage 16) as a reference. Notably, the frequency of variations at nucleotide position 6984 differed slightly between the two resistant mutants, thus highly reflecting differences in their population/sample (see the supplemental material). Based on these elements, we hypothesize that the varying rate of occurrence of these mutations could possibly explain the deviations on the inhibitory activity of some analogues against these two mutants. In addition, the growth of viral stocks (passage 16) in the absence of compound selection demonstrated an intrinsic fitness advantage of the 22-resistant mutant compared to the trans-14-resistant mutant (see the supplemental material). This may account for the different behavior of these viruses in response to compounds cis-10 and trans-10.
The molecules presented in this study constitute intermediates in the enantioselective synthesis of silicine alkaloids, having a distinct stereochemistry. The large family of indole alkaloids has been found to possess extremely broad pharmacological activities and thus can be used for treatment of viral infections. Numerous studies have reported the antiviral activity of several indole alkaloid derivatives against a number of viruses, such as HIV (34), influenza virus (35), and DENV (36).
To summarize, we have developed and determined the antiviral activity profiles of a new series of indole alkaloid derivatives against DENV and ZIKV. These molecules exhibited promising antiviral activity against all DENV serotypes in a number of susceptible host cell lines. On the other hand, a cell-dependent inhibitory activity against ZIKV strains was observed, while no activity against a number of other viruses, including HIV, influenza virus, and coronavirus, was noticed, which identifies them as specific inhibitors against these two flaviviruses. Mode-of-action studies and whole-genome sequencing analysis suggested a possible involvement of a host factor(s) associated with the pTMD1 domain of ZIKV NS4B, a protein with crucial role in the viral replication complex. To address this, further studies are ongoing in order to elucidate the exact molecular mechanism of these compounds. To the best of our knowledge, we describe for the first time a novel class of indole alkaloid derivatives indirectly targeting the multifunctional ZIKV NS4B protein, and more specifically the pTMD1 domain, thus opening new horizons in the virus-host interaction network. In-depth analysis and identification of cellular factors able to interact with these viral proteins might be of utmost importance to unravel new mechanisms that flaviviruses use during their replication cycle. Finally, the antiviral activity of these compounds against other members of flaviviruses needs to be evaluated to potentially promote them as pan-flavivirus inhibitors.
MATERIALS AND METHODS
Cells and viruses.
Different cell lines were used to evaluate the antiviral activity and cellular cytotoxicity of the tested molecules. Hepatocarcinoma (Huh; ATCC) and baby hamster kidney cells (BHK; kindly provided by M. Diamond, Washington University, USA) were grown in Dulbecco’s modified Eagle medium (1× DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS; HyClone, USA), 1 mM sodium pyruvate (Gibco), 0.01 M HEPES (Gibco) and 1× nonessential amino acids. Placenta choriocarcinoma cells (Jeg-3; ATCC) were grown in 10% FBS, 1 mM sodium pyruvate, and 1× nonessential amino acids. Finally, lung epithelial adenocarcinoma cells (A549; ATCC) and African green monkey kidney cells (Vero; ATCC) were grown in Eagle’s minimum essential medium (1× MEM-Rega3, Gibco, Belgium) supplemented with 8% FBS, 1 mM sodium bicarbonate (Gibco), 2 mM l-glutamine (Gibco), and 1× nonessential amino acids (100×, stock concentration; Gibco). Vero cells were used for the initial screening and lead identification of our hits. All cell lines were maintained at 37°C in a humidified 5% CO2 incubator.
In addition, a number of laboratory and clinical strains of both DENV and ZIKV were used in this study. Details about the strains and the propagation and titration of viral stocks were previously described (37).
Evaluation of antiviral activity and cellular viability.
The MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium; Promega, Leiden, The Netherlands] method was used to assess cell viability in the presence of our molecules. Each cell line was seeded at 104 cells/well in a 96-well plate (Corning, USA). After 24 h, cells were incubated with serial dilutions (5-fold) of each compound (100μΜ, 20μΜ, 4μΜ, and 0.8μΜ) for 72 h. Medium was replaced by 0.2% MTS diluted in phosphate-buffered saline (PBS) and incubated for additional 3 h at 37°C. The CC50s (the concentration at which the compound is able to reduce cell viability by 50%) were determined by measuring the optical density at 490 nm (SpectraMax Plus 384; Molecular Devices).
Regarding the antiviral assay, all cell lines were seeded at the same density as previously described. Briefly, 5-fold serial dilutions of compounds were added in the presence of each viral strain at a multiplicity of infection (MOI) of 1 for DENV and of 0.2 for ZIKV. Cells were washed 2 h p.i. and further incubated with fresh medium containing the appropriate concentration of compounds. Supernatant was collected at 72 h p.i., and the virus yield was determined with a Cells Direct one-step quantitative reverse transcription-PCR (qRT-PCR) kit (Thermo Fisher), according to the manufacturer’s instructions. Primers, probes, qRT-PCR conditions, and analysis of the results were reported previously (37). All compounds were dissolved in dimethyl sulfoxide (DMSO). The final concentration of DMSO in all tested conditions was 0.01%.
Immunofluorescence assay.
Vero and A549 cells (104 cells/well) were seeded in black 96-well plates (Falcon, Germany). The next day, cells were infected with the prototype ZIKV strain (MR766) at an MOI of 0.2 in the presence of serial dilutions of compound 22 (50 μM, 10 μM, 2 μM, and 0.4 μM) at 37°C for 2 h. Cells were washed with PBS and incubated with cell culture infection medium for 72 h. Later, they were fixed with 2% aqueous paraformaldehyde (PFA; Sigma-Aldrich, USA) at room temperature and permeabilized with 0.1% Triton X-100 (Sigma-Aldrich, USA). Bovine serum albumin (BSA; 1%; Cell Signaling Technology, The Netherlands) diluted in PBS was used to block the fixed cells for 1 h. Cells were further incubated with the primary 4G2 pan-flavivirus antibody (Merck Millipore, Belgium) overnight at 4°C. Goat anti-mouse IgG Alexa Fluor 488 (Invitrogen) was used as a secondary antibody, while the nucleus was stained with DAPI (4′,6-diamidino-2-phenylindole; Invitrogen). Fluorescence microscopy was performed on a Zeiss Axiovert 200M inverted microscope (Germany) using a 10× enhanced green fluorescent protein objective.
Time of drug addition assay.
To define at which stage of viral replication our compounds are active, Vero (104 cells/well) were infected with DENV2 strain NGC at an MOI of 1 for 2 h. Then, cells were washed twice with PBS and treated with an active concentration of each molecule (3-fold higher than the corresponding EC50s) at different time points pre- and postinfection (−2, 0, 2, 4, 6, 8, 12, and 18 h). Cell lysates were collected 20 h p.i., and the viral RNA levels were measured by qRT-PCR. Two reference compounds were used in this assay at 10 μΜ: dextran sulfate (DS; 8,000 Da), an established entry/fusion inhibitor, and 7-deaza-2′-C-methyladenosine (7DMA), a viral polymerase inhibitor.
Subgenomic replicon assay.
To assess any interaction with the nonstructural proteins (NS) or the replication complex, a subgenomic replicon assay was performed. Briefly, BHK cells stably transfected with the DENV2 replicon (encodes the firefly luciferase expression cassette instead of prM and E proteins, as well as NS proteins) were seeded in DMEM–2% FBS supplemented with 3 μg/ml puromycin (Sigma-Aldrich, USA) in a white-bottom 96-well plate (Nunclon Delta Surface; Thermo Fisher Scientific). Serial dilutions of compounds 22 and trans-14 were added to the cells (100μΜ, 20μΜ, 4μΜ, 0.8μΜ, and 0.16μΜ) for 72 h. Cells were further washed twice, the Renilla-Glo luciferase assay reagent (Thermo Fisher Scientific) was added to each well and the luminescence was measured at 490 nm (Tecan Spark 10M multimode microplate reader; BioExpress). 7DMA was also included as a control, while cytotoxicity of the molecules was determined in parallel, using the MTS method.
Ultrafiltration and virus inactivation assay.
To evaluate whether compound 22 can directly interact with DENV2 strain NGC, ultrafiltration and virus inactivation assays were performed. In the first assay, viral stock (104 PFU) was incubated with compound 22 at different concentrations (50 μM, 10 μM, and 2 μM) for 1 h and subsequently passed through a 10-kDa-cutoff concentrator filter unit (Vivaspin 500; Vivaproducts) to allow nonbinding compound to permeate. The fraction still present on the filter (200 μl) was serially diluted (10-fold) in cell culture medium (2 ml in total) and subsequently added (1 ml/condition) to BHK cells for a plaque assay to evaluate its infectivity/inhibition (each condition was tested in duplicate). Epigallocatechin gallate (EGCG; 1 mg/ml) was used as a positive control to evaluate the assay’s specificity.
In the virus inactivation assay, viral stock was incubated with 10 μΜ compound 22 for 1 h. Samples were diluted 100 times to obtain a concentration of the compound below the EC50s and used to infect Vero cells. Virus infectivity/inhibition was assessed by plaque reduction assay. Finally, positive (viral stock without the pressure of any compound) and negative controls (DMSO-treated cells) were also included.
In vitro selection of compound-resistant ZIKV mutants.
Compound-resistant viral strains were obtained through subsequent passaging of the prototype ZIKV MR766 strain in the presence of increasing concentrations of compound 22 or trans-14. For the selection procedure, A549 (105 cells/24-well plate) were used. More specifically, we started by incubating A549 cells with ZIKV MR766 in the presence of compound 22 or trans-14 at 0.1μΜ and 0.2μΜ, respectively. Three days postinfection, cytopathic effect (CPE) was observed, and the supernatant/condition (200 μl) was used to infect fresh susceptible cells by maintaining the same concentration of the compounds. When CPE was noticed for the second time, we increased the concentration of each molecule. In total, 16 passages (P1 to P16) were carried out for each compound (2μΜ final concentration). In order to exclude any spontaneous mutations that can naturally occur, A549 cells were inoculated with ZIKV MR766 in the absence of our molecules (virus control). Approximately 500 μl of each passage/condition was kept at −80°C for sequencing analysis.
RNA purification and metagenomic library preparation.
Viruses from two passages (P1 and P16) were sequenced using a metagenomic protocol. To prepare for sequencing, total RNA was extracted from 140 μl of the supernatant of six different samples (3 conditions per passage) using the QIAamp viral RNA kit (Qiagen). Carrier RNA was replaced by linear polyacrylamide, and an intermediate on-column DNase treatment was performed using an RNase-free DNase set (Qiagen) to remove any residual DNA fragments. Samples were eluted twice in RNase-free water (Qiagen) with 60 μl and 40 μl, respectively. RNA was further subjected to random reverse transcription and amplification using the sequence-independent single-primer amplification (SISPA) approach, as detailed in reference 38.
MinION nanopore sequencing and data analysis.
MinION sequencing libraries were generated from 200 ng of cDNA for each sample using the 1D ligation sequencing kit (SQK-LSK109) and the native barcoding expansion 1-12 kit (EXP-NBD104;j Oxford Nanopore Technologies). Libraries were loaded onto a Flo-Min 106 flow cell and run for 48 h with live base-calling enabled using high-accuracy base-calling.
Raw reads were demultiplexed using qcat v1.1.0 (https://github.com/nanoporetech/qcat) and SISPA primers from both the 5′ and 3′ ends were trimmed using seqtk v1.3 (https://github.com/lh3/seqtk). Minimap2 v2.17 (https://github.com/lh3/minimap2) was utilized to map the reads against the prototype ZIKV MR766 strain (GenBank accession number MK105975), and the resulting alignments were visually assessed with Tablet v1.19.09.03 (39). To calculate coverage depth and compute sequencing statistics, SAMtools v1.7 (40) and SeqKit v0.12.1 (41) were employed. Majority consensus was called at a minimum depth of 20× and 60% support fraction, with any base location not fulfilling the depth and support fraction assigned an N IUPAC ambiguity code. Finally, multiple-sequence alignments were generated on both nucleotide and amino acid levels with Aliview (42).
Cross-resistance assay.
To identify whether different analogues of this family (Table 1) share the same target protein, a cross-resistance assay was performed. Briefly, A549 (104 cells/well) were incubated with compounds 22, 20, 24, trans-14, cis-10, and trans-10 at 5-fold serial dilutions (100 μΜ to 0.8 μΜ) and subsequently exposed to the wild-type ZIKV strain MR766 or the compound 22- or trans-14-resistant viral strains (passage 16). Supernatant was collected 72 h p.i., and viral RNA (vRNA) levels were measured by qRT-PCR analysis. DS was included as a reference compound in this assay.
Statistical analysis.
One-way analysis of variance (ANOVA) followed by a correction posttest (Bonferroni) was used to evaluate significant differences for Fig. 3A and B and 4A. An unpaired t test was performed for Fig. 4B. Significance was calculated based on P values.
Data availability.
GenBank accession numbers for viral strains used in this study are as follows: AF298808.1, DENV serotype 1 Djibouti D1/H/IMTSSA/98/606 strain (43); M29095.1, DENV serotype 2 NGC strain (44); M93130.1, DENV serotype 3 H87 strain (45); MF004387.1, DENV serotype 4 Dak strain; MK105975.1, ZIKV strain MR766; MH916806.1, ZIKV strain PRVAVC59; and KX087102.2, ZIKV strain FLR. ZIKV genomic sequences generated in this study have been deposited in GenBank under the following accession numbers: MW143017 (compound 22-resistant mutant passage 1), MW143018 (compound trans-14-resistant mutant passage 1), MW143019 (wild-type virus passage 1), MW143020 (compound 22-resistant mutant passage 16), MW143021 (compound trans-14-resistant mutant passage 16), and MW143022 (wild-type virus passage 16).
ACKNOWLEDGMENTS
We thank Leentje Persoons, Cindy Heens, Daisy Ceusters, Sandra Claes, and Eric Fonteyn for their excellent technical assistance. We also acknowledge the networking contribution from the COST Action CM1407.
Financial support from the MINECO/FEDER and MICIU/FEDER, Spain (projects CTQ2015-65384-R and RTI2018-093974-B-I00) is gratefully acknowledged. P.L. acknowledges support by the Research Foundation—Flanders (Fonds voor Wetenschappelijk Onderzoek—Vlaanderen; G066215N, G0D5117N and G0B9317N) and the European Research Council under the European Union’s Horizon 2020 research and innovation program (grant agreement no. 725422-ReservoirDOCS), as well as funding from the Wellcome Trust through project 206298/Z/17/Z.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Bollati M, Alvarez K, Assenberg R, Baronti C, Canard B, Cook S, Coutard B, Decroly E, de Lamballerie X, Gould EA, Grard G, Grimes JM, Hilgenfeld R, Jansson AM, Malet H, Mancini EJ, Mastrangelo E, Mattevi A, Milani M, Moureau G, Neyts J, Owens RJ, Ren J, Selisko B, Speroni S, Steuber H, Stuart DI, Unge T, Bolognesi M. 2010. Structure and functionality in flavivirus NS-proteins: perspectives for drug design. Antiviral Res 87:125–148. 10.1016/j.antiviral.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kazmi SS, Ali W, Bibi N, Nouroz F. 2020. A review on Zika virus outbreak, epidemiology, transmission and infection dynamics. J Biol Res (Thessalon) 27:5. 10.1186/s40709-020-00115-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rezende HR, Romano CM, Claro IM, Caleiro GS, Sabino EC, Felix AC, Bissoli J, Hill J, Faria NR, da Silva TCC, Brioschi Santos AP, Junior CC, Vicente CR. 2020. First report of Aedes albopictus infected by dengue and Zika virus in a rural outbreak in Brazil. PLoS One 15:e0229847. 10.1371/journal.pone.0229847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferdousi T, Cohnstaedt LW, McVey DS, Scoglio CM. 2019. Understanding the survival of Zika virus in a vector interconnected sexual contact network. Sci Rep 9:7253. 10.1038/s41598-019-43651-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blohm GM, Lednicky JA, Márquez M, White SK, Loeb JC, Pacheco CA, Nolan DJ, Paisie T, Salemi M, Rodriguez-Morales AJ, Morris JG, Jr, Pulliam JRC, Paniz-Mondolfi AE. 2018. Evidence for mother-to-child transmission of Zika virus through breast milk. Clin Infect Dis 66:1120–1121. 10.1093/cid/cix968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Begum F, Das S, Mukherjee D, Mal S, Ray U. 2019. Insight into the tropism of dengue virus in humans. Viruses 11:1136. 10.3390/v11121136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pang X, Zhang R, Cheng G. 2017. Progress towards understanding the pathogenesis of dengue hemorrhagic fever. Virol Sin 32:16–22. 10.1007/s12250-016-3855-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hottz ED, Oliveira MF, Nunes PCG, Nogueira RMR, Valls-de-Souza R, Da Poian AT, Weyrich AS, Zimmerman GA, Bozza PT, Bozza FA. 2013. Dengue induces platelet activation, mitochondrial dysfunction and cell death through mechanisms that involve DC-SIGN and caspases. J Thromb Haemost 11:951–962. 10.1111/jth.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borchering RK, Huang AT, Mier-y-Teran-Romero L, Rojas DP, Rodriguez-Barraquer I, Katzelnick LC, Martinez SD, King GD, Cinkovich ST, Lessler J, Cummings DAT. 2019. Impacts of Zika emergence in Latin America on endemic dengue transmission. Nat Commun 10:5730. 10.1038/s41467-019-13628-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baud D, Gubler DJ, Schaub B, Lanteri MC, Musso D. 2017. An update on Zika virus infection. Lancet 390:2099–2109. 10.1016/S0140-6736(17)31450-2. [DOI] [PubMed] [Google Scholar]
- 11.Rather IA, Lone JB, Bajpai VK, Park YH. 2017. Zika virus infection during pregnancy and congenital abnormalities. Front Microbiol 8:581. 10.3389/fmicb.2017.00581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leung C. 2020. A lesson learnt from the emergence of Zika virus: what flaviviruses can trigger Guillain-Barré syndrome? J Med Virol 92:2938–2945. 10.1002/jmv.25717. [DOI] [PubMed] [Google Scholar]
- 13.Thomas SJ, Yoon IK. 2019. A review of Dengvaxia: development to deployment. Hum Vaccin Immunother 15:2295–2314. 10.1080/21645515.2019.1658503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Halstead SB. 2017. Dengvaxia sensitizes seronegatives to vaccine enhanced disease regardless of age. Vaccine 35:6355–6358. 10.1016/j.vaccine.2017.09.089. [DOI] [PubMed] [Google Scholar]
- 15.Ngono AE, Shresta S. 2019. Cross-reactive T cell immunity to dengue and Zika viruses: new insights into vaccine development. Front Immunol 10:1316. 10.3389/fimmu.2019.01316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sacramento CQ, De Melo GR, De Freitas CS, Rocha N, Hoelz LVB, Miranda M, Fintelman-Rodrigues N, Marttorelli A, Ferreira AC, Barbosa-Lima G, Abrantes JL, Vieira YR, Bastos MM, Volotao EM, Nunes EP, Tschoeke DA, Leomil L, Loiola EC, Trindade P, Rehen SK, Bozza FA, Bozza PT, Boechat N, Thompson FL, de Filippis AMB, Bruning K, Souza TML. 2017. The clinically approved antiviral drug sofosbuvir inhibits Zika virus replication. Sci Rep 7:40920. 10.1038/srep40920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whitby K, Pierson TC, Geiss B, Lane K, Engle M, Zhou Y, Doms RW, Diamond MS. 2005. Castanospermine, a potent inhibitor of dengue virus infection in vitro and in vivo. J Virol 79:8698–8706. 10.1128/JVI.79.14.8698-8706.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Plummer E, Buck MD, Sanchez M, Greenbaum JA, Turner J, Grewal R, Klose B, Sampath A, Warfield KL, Peters B, Ramstedt U, Shresta S. 2015. Dengue virus evolution under a host-targeted antiviral. J Virol 89:5592–5601. 10.1128/JVI.00028-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Low JGH, Ooi EE, Vasudevan SG. 2017. Current status of dengue therapeutics research and development. J Infect Dis 215:S96–S102. 10.1093/infdis/jiw423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choy MM, Zhang SL, Costa VV, Tan HC, Horrevorts S, Ooi EE. 2015. Proteasome inhibition suppresses dengue virus egress in antibody dependent infection. PLoS Negl Trop Dis 9:e0004058. 10.1371/journal.pntd.0004058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castillo Ramirez JA, Urcuqui-Inchima S. 2015. Dengue virus control of type I IFN responses: a history of manipulation and control. J Interferon Cytokine Res 35:421–430. 10.1089/jir.2014.0129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zou J, Xie X, Lee LT, Chandrasekaran R, Reynaud A, Yap L, Wang QY, Dong H, Kang C, Yuan Z, Lescar J, Shi PY. 2014. Dimerization of flavivirus NS4B protein. J Virol 88:3379–3391. 10.1128/JVI.02782-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zmurko J, Neyts J, Dallmeier K. 2015. Flaviviral NS4b, chameleon and jack-in-the-box roles in viral replication and pathogenesis, and a molecular target for antiviral intervention. Rev Med Virol 25:205–223. 10.1002/rmv.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barrows NJ, Campos RK, Liao K, Reddisiva K, Soto-Acosta R, Yeh SC, Schott-Lerner G, Pompon J, Sessions OM, Bradrick SS, Garcia-Blanco MA. 2018. Biochemistry and molecular biology of flaviviruses. Chem Rev 118:4448–4482. 10.1021/acs.chemrev.7b00719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brault JB, Khou C, Basset J, Coquand L, Fraisier V, Frenkiel MP, Goud B, Manuguerra JC, Pardigon N, Baffet AD. 2016. Comparative analysis between flaviviruses reveals specific neural stem cell tropism for Zika virus in the mouse developing neocortex. EBioMedicine 10:71–76. 10.1016/j.ebiom.2016.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Franco EJ, Rodriquez JL, Pomeroy JJ, Hanrahan KC, Brown AN. 2018. The effectiveness of antiviral agents with broad-spectrum activity against chikungunya virus varies between host cell lines. Antivir Chem Chemother 26:2040206618807580. 10.1177/2040206618807580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Himmelsbach K, Hildt E. 2018. Identification of various cell culture models for the study of Zika virus. World J Virol 7:10–20. 10.5501/wjv.v7.i1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller S, Sparacio S, Bartenschlager R. 2006. Subcellular localization and membrane topology of the dengue virus type 2 non-structural protein 4B. J Biol Chem 281:8854–8863. 10.1074/jbc.M512697200. [DOI] [PubMed] [Google Scholar]
- 29.Mohd Ropidi MI, Khazali AS, Nor Rashid N, Yusof R. 2020. Endoplasmic reticulum: a focal point of Zika virus infection. J Biomed Sci 27:27. 10.1186/s12929-020-0618-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xie X, Zou J, Wang QY, Shi PY. 2015. Targeting dengue virus NS4B protein for drug discovery. Antiviral Res 118:39–45. 10.1016/j.antiviral.2015.03.007. [DOI] [PubMed] [Google Scholar]
- 31.van Cleef KWR, Overheul GJ, Thomassen MC, Kaptein SJF, Davidson AD, Jacobs M, Neyts J, van Kuppeveld FJM, van Rij RP. 2013. Identification of a new dengue virus inhibitor that targets the viral NS4B protein and restricts genomic RNA replication. Antiviral Res 99:165–171. 10.1016/j.antiviral.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 32.Zhang T, Lin R-T, Li Y, Douglas SD, Maxcey C, Ho C, Lai J-P, Wang Y-J, Wan Q, Ho W-Z. 2005. Hepatitis C virus inhibits intracellullar interferon alpha expression in human hepatic cell lines. Hepatology 42:819–827. 10.1002/hep.20854. [DOI] [PubMed] [Google Scholar]
- 33.Sironi M, Forni D, Clerici M, Cagliani R. 2016. Nonstructural proteins are preferential positive selection targets in Zika virus and related flaviviruses. PLoS Negl Trop Dis 10:e0004978. 10.1371/journal.pntd.0004978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jassim SAA, Naji MA. 2003. Novel antiviral agents: a medicinal plant perspective. J Appl Microbiol 95:412–427. 10.1046/j.1365-2672.2003.02026.x. [DOI] [PubMed] [Google Scholar]
- 35.Moradi MT, Karimi A, Lorigooini Z. 2018. Alkaloids as the natural anti-influenza virus agents: a systematic review. Toxin Rev 37:11–18. 10.1080/15569543.2017.1323338. [DOI] [Google Scholar]
- 36.Hishiki T, Kato F, Tajima S, Toume K, Umezaki M, Takasaki T, Miura T. 2017. Hirsutine, an indole alkaloid of uncaria rhynchophylla, inhibits late step in dengue virus lifecycle. Front Microbiol 8:1674. 10.3389/fmicb.2017.01674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fikatas A, Vervaeke P, Martínez-Guald B, Martí-Marí O, Noppen S, Meyen E, Camarasa MJ, San-Felix A, Pannecouque C, Schols D. 2020. Tryptophan trimers and tetramers inhibit dengue and Zika virus replication by interfering with viral attachment processes. Antimicrob Agents Chemother 64:e02130-19. 10.1128/AAC.02130-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kafetzopoulou LE, Efthymiadis K, Lewandowski K, Crook A, Carter D, Osborne J, Aarons E, Hewson R, Hiscox JA, Carroll MW, Vipond R, Pullan ST. 2018. Assessment of metagenomic Nanopore and Illumina sequencing for recovering whole genome sequences of Chikungunya and dengue viruses directly from clinical samples. Eurosurveillance 23:1800228. 10.2807/1560-7917.ES.2018.23.50.1800228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Milne I, Stephen G, Bayer M, Cock PJA, Pritchard L, Cardle L, Shaw PD, Marshall D. 2013. Using Tablet for visual exploration of second-generation sequencing data. Brief Bioinform 14:193–202. 10.1093/bib/bbs012. [DOI] [PubMed] [Google Scholar]
- 40.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shen W, Le S, Li Y, Hu F. 2016. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One 11:e0163962. 10.1371/journal.pone.0163962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Larsson A. 2014. AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30:3276–3278. 10.1093/bioinformatics/btu531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tolou HJG, Couissinier-Paris P, Durand J-P, Mercier V, de Pina J-J, de Micco P, Billoir F, Charrel RN, de Lamballerie X. 2001. Evidence for recombination in natural populations of dengue virus type 1 based on the analysis of complete genome sequences. J Gen Virol 82:1283–1290. 10.1099/0022-1317-82-6-1283. [DOI] [PubMed] [Google Scholar]
- 44.Irie K, Mohan PM, Sasaguri Y, Putnak R, Padmanabhan R. 1989. Sequence analysis of cloned dengue virus type 2 genome (New Guinea-C strain). Gene 75:197–211. 10.1016/0378-1119(89)90266-7. [DOI] [PubMed] [Google Scholar]
- 45.Osatomi K, Sumiyoshi H. 1990. Complete nucleotide sequence of dengue type 3 virus genome RNA. Virology 176:643–647. 10.1016/0042-6822(90)90037-R. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download AAC02349-20_Supp_S1_seq4.pdf, PDF file, 3.0 MB (3MB, pdf)
Data Availability Statement
GenBank accession numbers for viral strains used in this study are as follows: AF298808.1, DENV serotype 1 Djibouti D1/H/IMTSSA/98/606 strain (43); M29095.1, DENV serotype 2 NGC strain (44); M93130.1, DENV serotype 3 H87 strain (45); MF004387.1, DENV serotype 4 Dak strain; MK105975.1, ZIKV strain MR766; MH916806.1, ZIKV strain PRVAVC59; and KX087102.2, ZIKV strain FLR. ZIKV genomic sequences generated in this study have been deposited in GenBank under the following accession numbers: MW143017 (compound 22-resistant mutant passage 1), MW143018 (compound trans-14-resistant mutant passage 1), MW143019 (wild-type virus passage 1), MW143020 (compound 22-resistant mutant passage 16), MW143021 (compound trans-14-resistant mutant passage 16), and MW143022 (wild-type virus passage 16).