

Abstract
The stability and aggregation of NIST monoclonal antibody (NISTmAb) were investigated by hydrogen/deuterium exchange mass spectrometry (HDX‐MS), differential scanning calorimetry (DSC), and nano‐differential scanning fluorimetry (nanoDSF). NISTmAb was prepared in eight formulations at four different pHs (pH 5, 6, 7, and 8) in the presence and absence of 150 mM NaCl and analyzed by the three methods. The HDX‐MS results showed that NISTmAb is more conformationally stable at a pH near its isoelectric point (pI) in the presence of NaCl than a pH far from its pI in the absence of NaCl. The stabilization effects were global and not localized. The midpoint temperature of protein thermal unfolding transition results also showed the CH2 domain of the protein is more conformationally stable at a pH near its pI. On the other hand, the onset of aggregation temperature results showed that NISTmAb is less prone to aggregate at a pH far from its pI, particularly in the absence of NaCl. These seemingly contradicting results, higher conformational stability yet higher aggregation propensity near the pI than far away from the pI, can be explained by intramolecular and intermolecular electrostatic repulsion using Lumry‐Eyring model, which separates folding/unfolding equilibrium and aggregation event. The further a pH from the pI, the higher the net charge of the protein. The higher net charge leads to greater intramolecular and intermolecular electrostatic repulsions. The greater intramolecular electrostatic repulsion destabilizes the protein and the greater intermolecular electrostatic repulsion prevents aggregation of the protein molecules at pH far from the pI.
Keywords: differential scanning calorimetry, differential scanning fluorimetry, electrostatic repulsion, hydrogen/deuterium exchange, isoelectric point, mass spectrometry, NISTmAb, protein aggregation, protein stability
Abbreviations
- CH
heavy chain constant domain
- CL
light chain constant domain
- DSC
differential scanning calorimetry
- DSF
differential scanning fluorimetry
- Fab
antigen binding fragment
- Fc
crystallizable fragment
- FPXIII
protease from Aspergillus saitoi
- HEPES
4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid
- LC
liquid chromatography
- mAb
monoclonal antibody
- MS
mass spectrometry
- MS/MS
tandem mass spectrometry
- NaOH
sodium hydroxide
- pf
protection factor
- pI
isoelectric point
- T agg
onset of aggregation temperature
- TCEP
tris(2‐carboxyethyl)phosphine hydrochloride
- TFA
trifluoroacetic acid
- T m
midpoint temperature of protein thermal unfolding transition
- type XIII
HDX, hydrogen/deuterium exchange
- VH
heavy chain variable domain
- VL
light chain variable domain
1. INTRODUCTION
Therapeutic monoclonal antibodies (mAbs) are the largest sector in the biotherapeutic space and the market is expected to near US$200 billion in worldwide sales by 2022. 1 While mAbs and their derivatives are expected to be the focus of the biotherapeutic industry and vast improvements have been made in characterization, manufacturing, and storage of mAbs, there are still many technical challenges. 2 One of the major challenges for therapeutic proteins is to maintain structural integrity, especially their higher order structure, throughout production and storage. A battery of biophysical assays is performed during development to assure structural integrity. However, it is possible for individual therapeutic assay results to pass with little understanding as to the inter‐relatability and connectivity of the data.
An IgG antibody, the most common scaffold used for antibody‐based therapeutics, consists of two heavy chain and two light chains connected by disulfide bonds. One heavy chain includes one variable domain (VH) and three constant domains (CH1, CH2, and CH3) while one light chain includes one variable domain (VL) and one constant domain (CL). One mAb has two antibody‐binding fragments (Fabs) and one crystallizable fragment (Fc). One Fab is made up of one each of VH, VL, CH1, and CL domains with two variable domains packed together and two constant domains packed together, whereas one Fc is formed by two closely packed CH2 domains and two closely packed CH3 domains.
The NIST monoclonal antibody (NISTmAb) reference material, RM8671, is a recombinant humanized IgG1κ expressed in murine suspension culture. 3 NISTmAb is available from NIST and is intended for investigating biochemical and biophysical attributes of mAbs as well as for evaluating the biochemical and biophysical methods for mAbs. Extensive biochemical and biophysical characterizations of this material is available. 4 , 5 , 6
Hydrogen/deuterium exchange mass spectrometry (HDX‐MS) is an analytical tool to study protein's dynamic properties. 7 , 8 , 9 HDX‐MS employs the unique combination of high‐resolution (in a few amino acid residues), quick turnaround time (in days), and wide applicability to scientific questions. Although X‐ray crystallography and nuclear magnetic resonance (NMR) have high resolution, they have limited applicability and the time requirements, which are often inhibitory to addressing questions in an expeditious fashion in drug discovery. While the majority of other commonly accessible biophysical tools, such as circular dichroism (CD) and ultraviolet (UV), have quick turnaround time, they do not have the resolution to provide detailed information into a molecule's structure and structural perturbations upon change in solution environment.
HDX‐MS has been used to study the change in the dynamic properties of mAbs after various perturbations. One type of perturbation is covalent modifications of mAbs themselves, such as amino acid mutations, 10 deglycosylation, 11 oxidation, 12 , 13 Fc glycan variations, 14 disulfide bond isoforms, 15 and the presence of extra light chains. 16 In these studies, the location of the modifications is known prior to the HDX‐MS analysis. HDX‐MS localized the changes in the dynamic properties of the mAbs near the modifications to understand the consequences of the modifications. The other type of perturbation is the change in the environment of a mAb as a result of formulation, including salts, 17 excipients, 18 , 19 as well as the conditions the mAb is exposed to, such as thermal stress 20 and dimerization. 21 In these studies, the presence and the location of the changes in HDX‐MS patterns are unknown prior to the analysis. The HDX‐MS changes observed in these studies tend to be subtler than those observed in the covalent modifications of mAbs.
In this article, we studied the stability and aggregation propensity of NISTmAb at four different pHs (5, 6, 7, and 8) with or without NaCl using HDX‐MS, differential scanning calorimetry (DSC) and nano‐differential scanning fluorimetry (nanoDSF). HDX‐MS and DSC are used for determining free energy of folding (ΔG) and midpoint temperatures of thermal unfolding (T ms) of individual domains, respectively. NanoDSF is used for determining both T m as well as the onset of aggregation temperature (T agg) of NISTmAb. The data were interpreted cohesively by intra‐ and inter‐molecular electrostatic repulsions using the Lumry‐Eyring model.
2. EXPERIMENTAL
2.1. Materials
All reagents were obtained from Sigma‐Aldrich (St. Louis, MO) except the following items: Pierce premium grade TCEP‐HCl was purchased from Thermo Fisher Scientific (Waltham, MA). Urea was purchased from VWR (Radnor, PA). Water and acetonitrile were HPLC grade from Honeywell (Charlotte, NC).
2.2. Preparation of formulated NISTmAb
The NISTmAb sample (Table S1 in Supplemental Information) was first dialyzed into a low‐ionic strength buffer (5 mM Acetate/5 mM Histidine pH 6.2) using a Slide‐A‐Lyzer MINI dialysis device, 10 K MWCO, 2 ml (Thermo Fisher Scientific). After determining post‐dialysis concentration, protein was diluted fivefold or higher into each of the test buffers for DSF and HDX‐MS to a final concentration of 1 mg/ml. For DSC, protein was further dialyzed into test buffers using a fresh Slide‐A‐Lyzer MINI dialysis membranes. DSC experiments setup at 0.5–1 mg/ml (Table 1). All protein concentrations were determined using the Lunatic instrument (Unchained Labs; Pleasanton, CA) in triplicates.
TABLE 1.
T m and T agg of NISTmAb measured by nanoDSF and DSC in eight different formulations (°C) a
| 10 mM buffer | pH | 150 mM NaCl | T agg b DSF | T m1 b DSF (CH2) | T m1 b DSC (CH2) | T m2 b DSC (CH3) | T m3 b DSC (fab) | T m c DSC fab |
|---|---|---|---|---|---|---|---|---|
| Acetate | 5.0 | − | >95.0 | 67.0 | 67.4 | 85.0 | 93.3 | 93.4 |
| Acetate | 5.0 | + | 89.4 | 63.3 | 64.1 | 84.7 | 92.6 | 92.5 |
| Histidine | 6.0 | − | >95.0 | 67.5 | 69.4 (69.2) d | 85.0 (83.1) d | 93.0 (93.4) d | 93.2 |
| Histidine | 6.0 | + | 88.8 | 64.9 | 66.8 (67.0) d | 84.8 (81.4) d | 92.7 (92.3) d | 92.4 |
| Phosphate | 7.0 | − | 81.4 | 70.2 | 70.7 | 84.9 | 88.7 | 89.1 |
| Phosphate | 7.0 | + | 84.6 | 69.3 | 69.9 | 84.2 | 88.7 | 89.2 |
| HEPES | 8.0 | − | 82.3 | 70.5 | 69.8 | 84.6 | 88.3 | 88.7 |
| HEPES | 8.0 | + | 83.9 | 69.4 | 70.3 | 84.0 | 88.2 | 88.3 |
Abbreviations: DSC, differential scanning calorimetry; DSF, differential scanning fluorimetry; Fab, antigen binding fragment; HEPES, 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid.
Values are averages of duplicate or triplicate measurements.
Please see Figures S3–S5 for the profiles of DSF and DSC data.
T m of the isolated Fab domain of NISTmAb.
The values in parenthesis are measure in 25 mM histidine instead of 10 mM histidine. 5
The NIST Fab was generated using the Pierce Fab Preparation Kit (Thermo Fisher Scientific) and the protocol was followed as per manufacturer's suggestion, with one modification—Fab purification was done using HiTrap MabSleect SuRe (GE Life Sciences; Marlborough, MA) to capture digested/undigested Fc as per the manufacturer's protocol. After measuring protein concentrations of flow‐through (Fab alone), fractions were pooled and dialyzed into the low‐ionic strength buffer as indicated for NISTmAb above. Further sample prep followed similar protocol as NISTmAb.
2.3. Conformational stability determination by DSC
DSC experiments were performed using a MicroCal Auto VP‐capillary DSC system (GE Healthcare; Chicago, IL) in which temperature differences between the reference and sample cell were continuously measured and converted to power units. Samples were heated from 25 to 110°C at a rate of 1°C/min. A pre‐scan time of 15 min and a filtering period of 10 s were used for each run. DSC measurements were made at a NISTmAb concentration of 0.5–1 mg/ml in each of the eight buffer conditions in duplicates, with a preceding buffer background scan taken before the sample scans. Data analysis was performed using MicroCal Origin 7 software and fitted with the “non‐two‐state” function analysis model.
2.4. Conformational and colloidal stability determination by NanoDSF
The thermal unfolding of NISTmAb in different buffers was determined by nanoDSF on the Prometheus NT.Plex instrument (NanoTemper Technologies; Munchen, Germany). Measurements were made by loading samples into Standard Capillaries (NanoTemper Technologies) from a 384 well sample plate. Duplicate or triplicate runs were performed. Thermal unfolding was monitored in a 1°C/min thermal ramp from 20 to 95°C. Thermal inflection temperatures (T ms) and T agg were determined automatically by PR. ThermControl Software (NanoTemper Technologies) and further analyzed using the PR.Stability Analysis Software (NanoTemper Technologies).
2.5. On‐exchange experiment for HDX‐MS
The on‐exchange reaction was initiated by mixing 10 μl of formulated NISTmAb with 10 μl of D2O (D2O with 150 mM NaCl was used when the formulation has 150 mM NaCl). The reaction mixture was incubated for 15, 50, 150, 500, 1,500, 5,000, 15,000, 50,000, and 150,000 s at 23 C (Table S2). All time points were duplicated. The on‐exchanged solution was quenched by the addition of 30 μl of chilled 8 M urea, 1 M TCEP, pH 3.0 (pH was adjusted with aqueous sodium hydroxide [NaOH]) and immediately analyzed.
2.6. Non‐deuterated experiment for HDX‐MS
A non‐deuterated sample was prepared by mixing 10 μl of NISTmAb and 10 μl of H2O. An aliquot of 20 μl of non‐deuterated sample was mixed with 30 μl of 8 M urea, 1 M TCEP, pH 3.0 and immediately analyzed.
2.7. Fully deuterated experiment for HDX‐MS
A fully deuterated sample was prepared by incubating a mixture of 25 μl of NISTmAb and 25 μl of 100 mM TCEP in D2O at 55°C for 2 hr. After the fully deuterated sample was cooled down to 23°C, a 30‐μl aliquot of 8 M urea, 1 M TCEP, pH 3.0 was added to 20 ul of the fully deuterated sample and immediately analyzed.
2.8. General procedure for HDX‐MS data acquisition
HDX‐MS analysis was performed using an automated HDx3 system (LEAP Technologies, Morrisville, NC) system analogous to previously described 9 , 22 except the protease column was placed outside of the cold box.
The columns and pump configuration was as follows: protease, pepsin/protease type XIII (protease from Aspergillus saitoi, type XIII) column (wt/wt, 1:1; 2.1 × 30 mm2) (NovaBioAssays Inc., Woburn, MA); 23 trap column, Acquity UPLC BEH C18 VanGuard Pre‐column (Waters, Milford, MA); analytical column, Accucore Vanquish C18 (2.1 × 100 mm2, 1.5 μm) (Thermo Fisher Scientific); and LC pump, Vanquish (Thermo Fisher Scientific). The loading pump (from the protease column to the trap column) was set at 600 μl/min with 1% acetonitrile, 0.1% formic acid in water. The gradient pump (from the trap column to the analytical column) was set from 8 to 33% acetonitrile in 0.1% aqueous formic acid in 20 min at 100 μl/min.
2.9. MS data acquisition
Mass spectrometric analyses were carried out using an LTQ Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific) with the capillary temperature at 275°C, resolution 120,000, and mass range (m/z) 300–1,500.
2.10. HDX‐MS data extraction
BioPharma Finder 2.0 (Thermo Fisher Scientific) was used for the peptide identification of non‐deuterated samples prior to the HDX experiments. HDExaminer version 2.4 (Sierra Analytics, Modesto, CA) was used to extract centroid values from the MS raw data files for the HDX experiments.
The deuteration level (D%) at each segment at each time point was calculated using Equation (1) in Excel.
| (1) |
where M(ON) is the centroid value of on‐exchanged experiment, M(ND) is the centroid value of non‐deuterated experiment, and M(FD) is the centroid value of fully deuterated experiment. 7 In this equation, the number of deuterium incorporated in on‐exchanged experiment was divided by the number of deuterium incorporated in fully deuterated experiment.
The deuteration level (D%) of sub‐localized segment was calculated using Equation (2).
| (2) |
where a indicates the centroid value of the larger segment used for sub‐localization (e.g., segment 1–10) and b indicates the centroid value of the smaller segment used for sub‐localization (e.g., segment 1–9). In this equation, the number of the deuterium incorporated at the sub‐localized segment (e.g., residue 10) after on‐exchanged experiment was divided by the number of deuterium incorporated at the sub‐localized segment after fully deuterated experiment.
All experimental exchange times were converted into those at pH 7 and 23°C (Figure 2, Figure S2, and Table S2). 24 , 25 , 26
FIGURE 2.
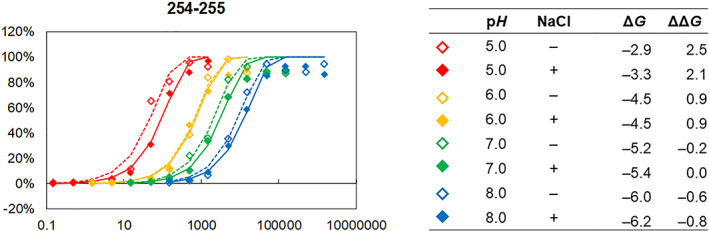
A representative deuterium buildup curve (segment 254–255 from peptide 252–255). All exchange times are converted to pH 7, 23°C equivalent using Table S2. The deuteration level (D%) at each time point at each condition is indicated with a colored diamond. The eight sets of deuterium buildup curves for this segment were fitted with eight sets of Equation (3) with a common stretch factor, B (eight k's and one B were optimized at the same time). The optimized B for segment 254–255 was 0.91. ΔG is the free energy change for each formulation compared with unstructured polyalanine using Equation (4a). ΔΔG is the relative free energy in each formulation compared with that for pH 7, 150 mM NaCl obtained by Equation (54b)
2.11. HDX‐MS data analysis
Excel was used to further process and present the data. All HDX data were fitted with stretched exponential curve (Equation (3)), 27
| (3) |
where k is rate factor which becomes larger when HDX rates in the segment is faster and B is stretch factor which becomes larger when the HDX curve is steeper.
For the curve fitting of eight formulations for each segment, a common B was used, because the deuterium buildup slopes of various formulations were very similar for each segment (Figure 2 and Figure S2).
A protection factor (pf), between two stretched exponential curves were calculated by Equation (4a) (Stretched exponential Analysis in Supporting Information).
| (4a) |
where a and b indicate two different sets of stretched exponential curves.
Equation (5) can convert a pf to the free energy change between the two states when EX2 is applicable. 9 , 26 , 28 Using Equations (4a) and (5), the current HDX data can be compared with intrinsic exchange rates to yield the free energy change from unstructured state (ΔG in Figure 2 and Table 2). ΔG in this study was obtained by comparing with the calculated exchange rate of unstructured polyalanine. 29
TABLE 2.
Average folding free energy, ΔG, and average relative free energy, ΔΔG, of NISTmAb in eight different formulations (kcal/mol)
| pH | 150 mM NaCl | mAb | Fab | VH–VL | CH1–CL | CH2 | CH3 | |
|---|---|---|---|---|---|---|---|---|
| 5.0 | − | −4.1 | −4.3 | −4.8 | −3.8 | −3.1 | −4.8 | |
| 5.0 | + | −4.6 | −4.7 | −5.1 | −4.4 | −3.8 | −5.1 | |
| 6.0 | − | −5.3 | −5.5 | −5.9 | −5.1 | −4.6 | −5.9 | |
| ΔG | 6.0 | + | −5.5 | −5.6 | −6.0 | −5.2 | −4.9 | −5.8 |
| 7.0 | − | −5.7 | −5.8 | −6.3 | −5.3 | −5.5 | −6.0 | |
| 7.0 | + | −5.9 | −6.0 | −6.5 | −5.6 | −5.6 | −6.1 | |
| 8.0 | − | −6.5 | −6.6 | −7.0 | −6.2 | −6.4 | −6.8 | |
| 8.0 | + | −6.7 | −6.8 | −7.2 | −6.3 | −6.5 | −6.9 | |
| 5.0 | − | +1.9 | +1.8 | +1.7 | +1.8 | +2.5 | +1.3 | |
| 5.0 | + | +1.4 | +1.3 | +1.4 | +1.3 | +1.9 | +1.1 | |
| 6.0 | − | +0.6 | +0.6 | +0.6 | +0.5 | +1.1 | +0.2 | |
| ΔΔG | 6.0 | + | +0.5 | +0.5 | +0.5 | +0.4 | +0.7 | +0.3 |
| 7.0 | − | +0.2 | +0.3 | +0.2 | +0.3 | +0.1 | +0.1 | |
| 7.0 | + | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | |
| 8.0 | − | −0.6 | −0.5 | −0.5 | −0.6 | −0.8 | −0.6 | |
| 8.0 | + | −0.8 | −0.7 | −0.7 | −0.7 | −0.9 | −0.7 |
Note: Please see Figure 2 for the definitions of ΔG and ΔΔG.
Abbreviations: CH, heavy chain constant domain; CL, light chain constant domain; Fab, antigen binding fragment; mAb, monoclonal antibody; VH, heavy chain variable domain; VL, light chain variable domain.
The pf between two different formulations for one segment is simply,
| (4b) |
| (5) |
where a and b indicate two different formulations. In this case, the log portion of Equation (4a) is canceled out to become Equation (4b), because a common B is used. Equations (4b) and (5) can yield the free energy change between the two formulations (ΔΔG in Figures 1 and 2).
FIGURE 1.
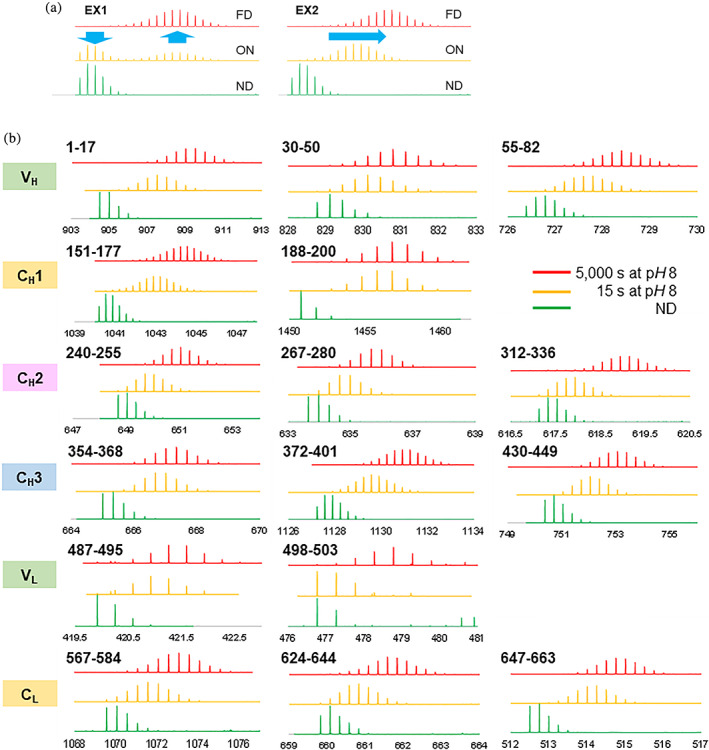
Isotope envelopes of peptides in HDX‐MS analysis. (a) Imaginary isotope envelopes of non‐deuterated, on‐exchanged, and fully deuterated isotope envelope in EX1 mechanism and EX2 mechanism. (b) Observed isotope envelopes of non‐deuterated, 15‐s on‐exchanged, and 5,000‐s on‐exchanged experiments in various segments at pH 8 in 90% D2O
3. RESULTS
3.1. Digestion and sub‐localization of NISTmAb
The sequence coverage of NISTmAb was 94% when quenched with 8 M urea, 1 M TCEP, pH 3.0 and digested with pepsin/protease type XIII mixed bed column (= 626/663; Figure S1). This sequence coverage is good enough to assess the folding free energy of entire NISTmAb. There are more peptides covering the constant regions (CH1, CH2, CH3, and CL) of both heavy and light chains than the variable regions (VH and VL).
NISTmAb was dissected into 140 individual segments (Figure 3 and Figure S2) by using small peptic fragments and a sub‐localization strategy (see Section 2) to achieve approximately four residues per segment resolution on average. Additional 101 segments were also analyzed to check the data consistency (Table S3, S4 and Figure S2). The resolution of the constant domains was higher than that of the variable domains in general, because there are more smaller peptides and overlapping peptides covering the constant domains (Figure S1).
FIGURE 3.
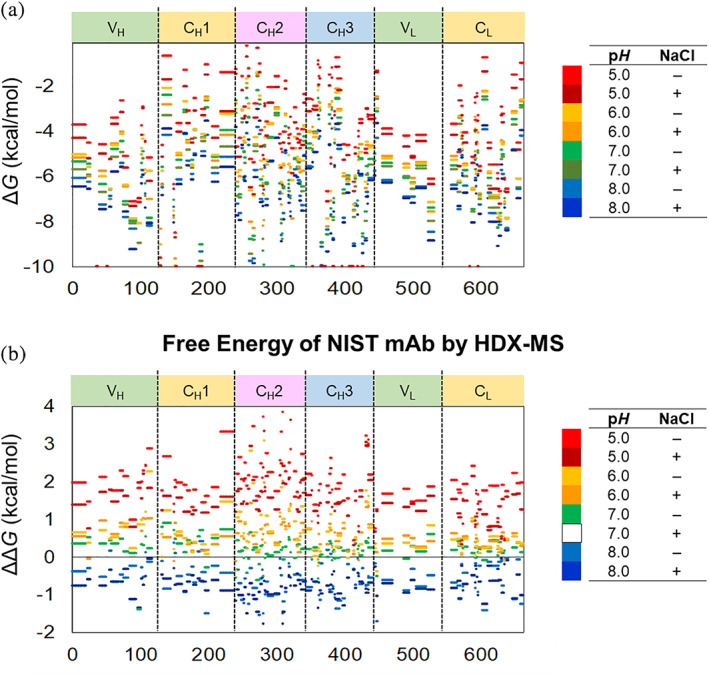
(a) The folding free energy and (b) the relative free energy of NISTmAb segments in eight formulations. Each colored line indicates the free energy change in the segment spanning the corresponding residues. Please see Figure 2 for the definitions of ΔG and ΔΔG
3.2. Confirming EX2 mechanism for NISTmAb
Prior to HDX analysis of NISTmab in various formulations, it is checked if the backbone amide hydrogens of NISTmAb exchange via EX1 mechanism or via EX2 mechanism. In slightly different experimental conditions, NISTmAb was incubated in 20 mM Tris, 150 mM NaCl, pH 8 in 90% D2O. If a protein exchanges via EX1 mechanism, a partially deuterated isotope envelope should be bimodal and the ratio of the low‐m/z lobe and the high‐m/z lobe should increase with a longer exchange time (Figure 1a). 30 , 31 One the other hand, if a protein exchanges via EX2 mechanism, a partially deuterated isotope envelope should be monomodal and the envelope should shift toward right with a longer exchange time (Figure 1a). The reaction mechanism was checked at only pH 8, the highest pH used in the following formulation study. Because the higher the pH is, the more likely to exchange via EX1, the backbone amide hydrogens of NISTmAb exchange via EX2 at all pHs used if they exchange via EX2 at pH 8.
The backbone amide hydrogens in all six NISTmAb domains exchange via EX2 mechanism at pH 8. The isotope envelopes of non‐deuterated, 15‐s on‐exchanged, and 5,000‐s on‐exchanged experiments from 16 different segments from all six NISTmAb domains were monitored (Figure 1b). All of the partially deuterated (15‐s on‐exchanged) isotope envelopes were monomodal, indicating that all segments exchanged via EX2 mechanism. This result ensures that the conversion of HDX‐MS data to the free energy described in HDX‐MS Data Analysis section is valid.
3.3. Fitting HDX‐MS data with stretched exponential curves
A set of eight deuterium buildup curves of a NISTmAb segment was fitted with eight stretched exponential curves simultaneously with a common stretch factor, B (Figure 2 and Figure S2). In this analysis, eight rate factors, k's, and one stretch factor, B, were optimized using a set of eight deuterium buildup curves for each segment. It was clear from a visual inspection that the slopes of the eight‐deuterium buildup curves for each segment are very similar (Figure S2). This approximation simplifies the calculation of pfs among the formulations and relative free energy changes (ΔΔG).
3.4. Folding free energy (ΔG ) of NISTmAb by HDX‐MS analysis
Most segments of NISTmAb exchange at significantly slower rates than unstructured polyalanine at all eight conditions, indicating NISTmAb is well‐structured at all eight formulation conditions (Figure 3a). NISTmAb does not contain a long patch of disordered structure, which should appear as −1 kcal/mol or higher in folding free energy. 29
The HDX data show that the CH3 domain and the VH–VL domain are more stable than the CH2 domain and the CH1–CL domain (Table 2, Figures 3a and 4a). This is consistent with the observation that CH2 domains usually have the lowest T m among the IgG domains for many IgGs. 5 Approximately 50 residues did not exchange at all in the time window employed in this study and about 40% of these very stable residues are located in the CH3 domain. The folding free energy of these segments were not possible to calculate and were estimated as < −10 kcal/mol (Figure 3a and Table S3). 32 , 33
FIGURE 4.
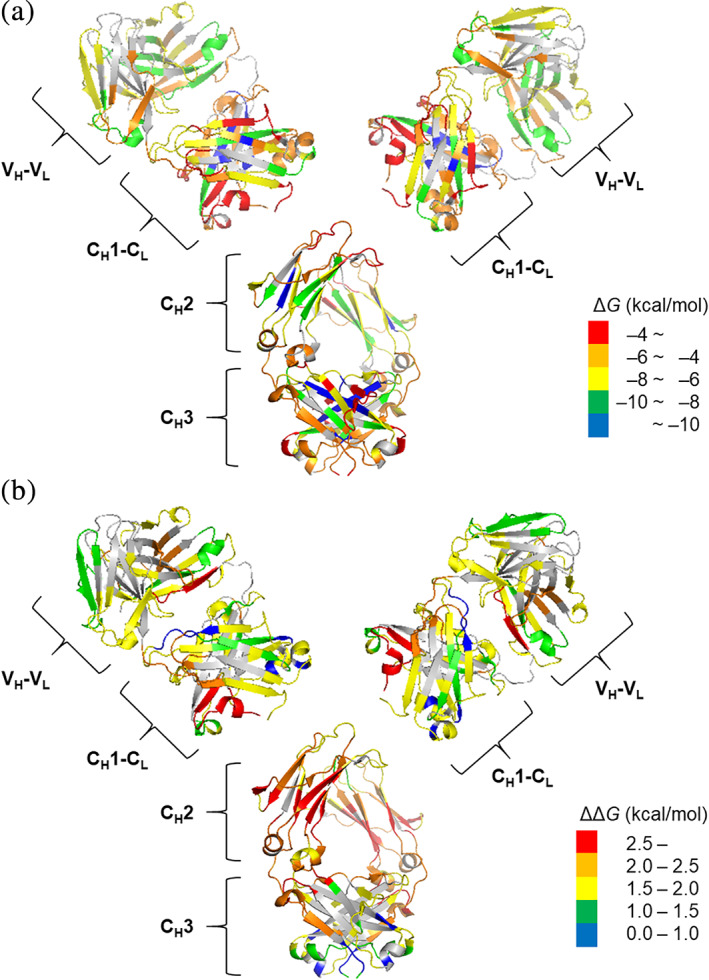
The folding free energy and the relative free energy of NISTmAb segments overlaid on X‐ray crystal structures (5K8A for Fab and 5VGP for Fc). (a) The folding free energy (ΔG) at pH 7.0, 150 mM NaCl. (b) The relative free energy (ΔΔG) at pH 5.0, 0 mM NaCl compared with pH 7.0, 150 mM NaCl
3.5. Relative free energy (ΔΔG ) of NISTmAb by HDX‐MS analysis
The relative free energy of NISTmAb decreases in the order of pH 5, 0 mM NaCl > pH 5, 150 mM NaCl > pH 6, 0 mM NaCl > pH 6, 150 mM NaCl > pH 7, 0 mM NaCl > pH 7, 150 mM NaCl > pH 8, 0 mM NaCl > pH 8, 150 mM NaCl (Table 2). This trend is almost uniform throughout the molecule (Figures 3b and 4b, Table S4). All deuterium buildup curves from the eight formulations should be on top of each other in Figure 2 and Figure S2 when the difference in intrinsic exchange rates at various pHs is considered (Table S2) and if the dynamic properties of the protein stay the same in those formulations. 24 , 25 , 26 In most cases here, however, this is not observed. The HDX rates were slower at a higher pH and in the presence of NaCl (Figure 2 and Figure S2), indicating that NISTmAb has lower free energy (= is more stable) at higher pH and in the presence of NaCl.
3.6. Thermal stability ( T m) of NISTmAb by DSC
NISTmAb exhibits three distinct transitions around 70, 85, and 90°C, which, based on IgG structure and enthalpy change values, can be assigned to CH2, CH3, and Fab domain unfolding transitions, respectively (Table 1 and Figure 5b). The thermal stability properties of NISTmAb have been studied by DSC in histidine buffer systems 5 and have been known to exhibit a favorable stability profile, especially in terms of Fab stability. Our data are consistent with these observations, and further expand the data to additional buffer conditions.
FIGURE 5.
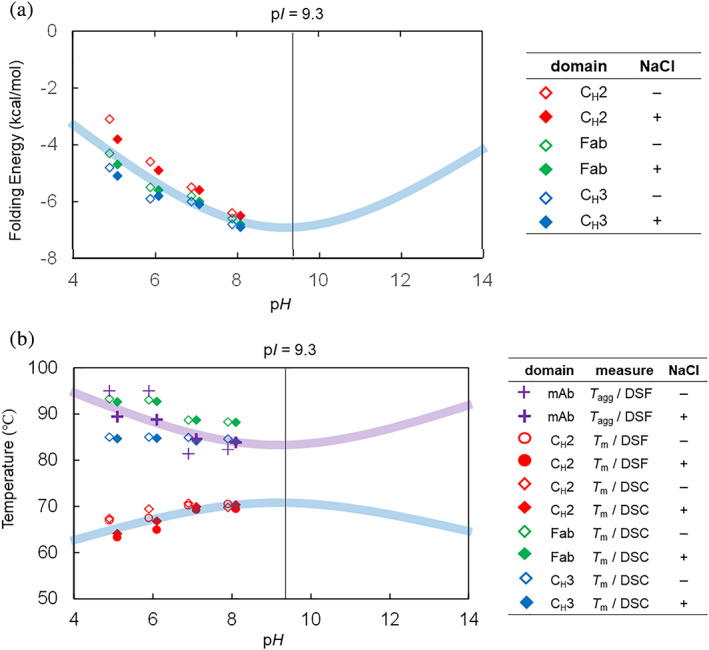
(a) Average folding free energy and (b) T ms and T aggs of NISTmAb domains at pH 5–8. Light blue curves are the trend expected for the HDX‐MS determined folding free energy and the T m from intramolecular electrostatic repulsion as depicted in Figure 6. Light purple curve is the trend expected for the T agg from intermolecular electrostatic repulsion as depicted in Figure 6. The pHs of plots are slightly shifted, because the data points overlapped
The T m1 of NISTmAb increases with pH in general, indicating the CH2 domain is more stable at higher pH (Table 1 and Figure 5b). On the other hand, the T m3 of NISTmAb decreases at a higher pH. The T m3 of NISTmAb matches very well with the T m of isolated Fab domain of NISTmAb, confirming that the T m3 of NISTmAb describes the thermal stability of the Fab (Table 1). The T m2 of NISTmAb appears almost insensitive to pH (Table 1 and Figure 5b).
3.7. Thermal stability ( T m) of NISTmAb by DSF
NanoDSF data of NISTmAb shows one clearly defined first thermal transition (T m1) corresponding to the CH2 domain. The other two thermal transitions were however not well defined, due to the unusually high‐Fab unfolding transition of NISTmAb and instrument limitations (upper limit of 95°C).
NanoDSF data shows that the higher the pH is, the higher the T m1 of NISTmAb was in the pH range employed in this study (Table 1 and Figure 5b). This is in accordance with the observation that NISTmAb is more stable at a higher pH by HDX‐MS, because a higher T m implies more conformationally stable protein.
The T m1 of NISTmAb was slightly higher in the absence of NaCl (Table 1). This agrees with the T m behaviors of NISTmAb determined with DSC by others and us. 5 On the other hand, this disagreed with the NaCl effects on relative free energy observed by HDX‐MS analysis.
3.8. Aggregation ( T agg) of NISTmAb by DSF
T agg of NISTmAb was the highest at lower pH and in the absence of NaCl (Table 1 and Figure 5b). T agg, is a direct measure of colloidal stability. NISTmAb did not show any detectable aggregation even at 95°C (the upper limit of the instrument) at pH 5 or 6 in the absence of NaCl. The addition of NaCl lowered the T agg down to a measurable range of approximately 89°C at both pH 5 and 6. T agg of NISTmAb at a higher pH (pH 7 or 8) was between 81 and 85°C, significantly lower than that at pH 5 or 6 (Table 1 and Figure 5b).
4. DISCUSSION
4.1. Global effects of pH on the conformational stability of NISTmAb
It has been previously established that a protein should be conformationally most stable at a pH near its isoelectric point (pI). 34 , 35 The net charge of a protein increases as the pH of the protein solution moves away from its pI (Figure S6) and the higher net charge leads to greater intramolecular electrostatic repulsion (Figure 6a, purple arrows). The increased intramolecular electrostatic repulsion destabilizes the folded protein, because the charge density on the folded molecule is higher than that on the unfolded molecule. It may be worth pointing out that most intrinsically disordered proteins have high‐net charges, presumably due to intramolecular electrostatic repulsion. 36 In the free energy diagram, a protein's folding well is shallower when the pH of a protein solution is further from the protein's pI, due to greater intramolecular electrostatic repulsion (Figure 6b, purple arrow).
FIGURE 6.
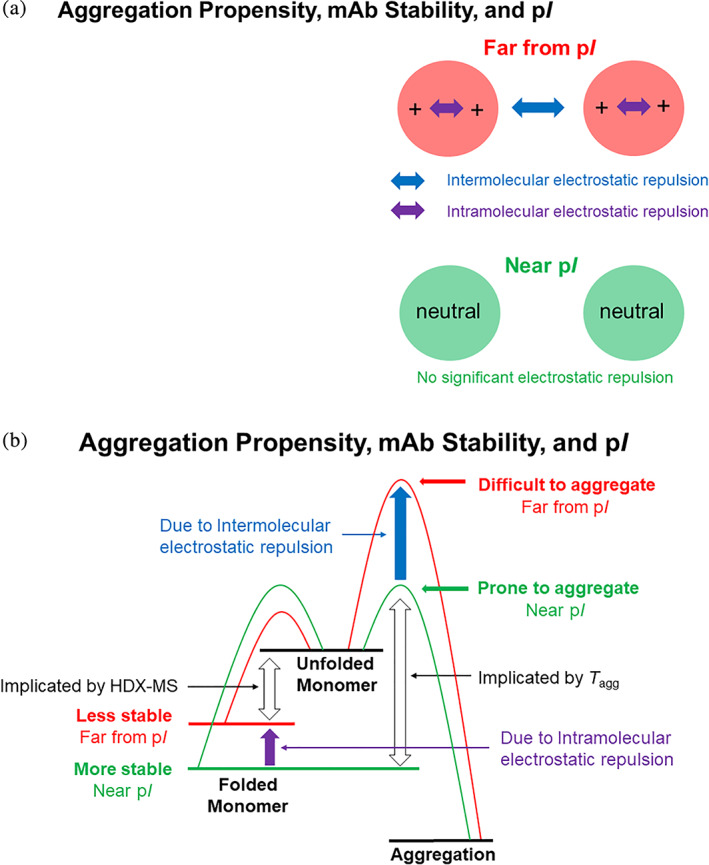
(a) Simplified electrostatic repulsion and (b) simplified free energy diagram of aggregation pathway according to Lumry‐Eyring model for a protein near and far from its pI. In the Lumry‐Eyring model, folded monomer unfolds reversibly and then the unfolded monomer aggregates irreversibly. The aggregation step is usually expected to be the rate‐determining step in a non‐accelerated storage condition 54
HDX‐MS results showed that NISTmAb is more stable at a pH near its pI than at a pH far from its pI (Table 2 and Figure 3). Since the pI of NISTmAb is 9.3, 37 the net charge of a protein molecule is lower at a higher pH between pH 5 and 8 (Figure S6) and intramolecular electrostatic repulsion should be smaller at a higher pH. Therefore, when the pH is higher, the free energy should be lower (the protein should be more stable) in the pH range (Figure 5b). Indeed, the relative free energy of NISTmAb calculated by HDX‐MS analysis showed a lower free energy at a higher pH (Table 2 and Figure 3).
4.2. Global effects of NaCl on the conformational stability of NISTmAb
The higher conformational stability of NISTmAb in the presence of NaCl observed in HDX‐MS analysis stems from the charge screening effects of NaCl (Table 2 and Figure 3b). 32 The presence of NaCl may decrease the free energy of a protein by shielding the intramolecular electrostatic repulsion. In the current study, the presence of NaCl decreased approximately 0.5 kcal/mol per residue of the free energy at pH 5, while the effects were 0.1–0.2 kcal/mol per residue at pH 6–8 (Table 2). NISTmAb has more intramolecular electrostatic repulsion at pH 5 than at higher pHs, and thus the stabilization due to the charge screening effects is larger at pH 5.
4.3. Global effects of pH on the aggregation of NISTmAb
It is generally accepted that a protein is least soluble near its pI in the absence of neutral salts 38 , 39 and that working a protein near its pI is risky because the protein is prone to aggregation. 40 The presence of net charge in the protein molecules at a pH far from its pI generates intermolecular electrostatic repulsion (Figure 6a, blue arrow) and reduces the chance of collision and thus aggregation. 41 , 42 On the other hand, the lack of intermolecular repulsion near its pI allows the protein molecules to collide and aggregate more easily. In the free energy diagram, the energy barrier to aggregation is higher when the protein is at pH far from its pI (Figure 6b, blue arrow).
The T agg is customarily used in protein formulation studies. 43 T agg of NISTmAb was lower at a higher pH (Table 1 and Figure 5b). This observation is consistent with the consensus that a protein is more prone to aggregation near its pI (9.3 for NISTmAb). 37 Consistent with this theory and with the goal of preventing aggregation in solution, NISTmAb is formulated in histidine buffer around pH ~ 6.3, which is far from its pI. 6
4.4. Global effects of NaCl on the aggregation of NISTmAb
T agg of NISTmAb was the highest at a lower pH in the absence of NaCl (Table 1). This is because NISTmAb molecules have the greater intermolecular electrostatic repulsion at a lower pH in the absence of NaCl. A lower pH increases the net charge on each molecule (Figure S6) and intermolecular electrostatic repulsion. The absence of NaCl means no shielding effects to decrease the intermolecular electrostatic repulsion. 29
4.5. Average folding free energy of each domain of NISTmAb
Average folding free energies (ΔG) of CH1–CL and CH2 domains calculated from the HDX‐MS data were higher (less stable) than those of VH–VL and CH3 domains between pH 5 and 8 (Table 2 and Figures 3a and 4a). This observation is mostly consistent with the thermal stability of NISTmAb by DSC (Table 1). The DSC results showed that the CH2 has the lowest thermal stability and Fab has the highest thermal stability at all pHs. Both HDX‐MS and DSC results showed that CH2 domain is the least stable domain of NISTmAb. Although the DSC data indicated that the Fab domain is more stable than CH3 domains, the folding free energies of the two domains determined by HDX‐MS are very close.
4.6. Average relative free energy of each domain of NISTmAb
Average relative free energy (ΔΔG) calculated from the HDX‐MS data showed very similar trend among all domains with higher pH and the presence of NaCl lowering the free energy of each domain (Table 2 and Figures 3b and 5a). The coarse uniformity of relative free energy throughout the molecule suggests that non‐localized, global forces are contributing to the relative free energy among these formulations (vide supra). Among the domains, the CH2 domain is the most sensitive to the formulation change with 2.5 kcal/mol less stable at pH 5 without NaCl than at pH 7 with NaCl (Table 2 and Figures 4b and 5a). Domain specific net charge calculations at different pHs on molecular operating environment (MOE) 44 molecular modeling interface using NISTmAb structural data (PDB ID 5VGP for CH2 and PDB ID 5K8A for Fab) showed a steep decline of net charge with pH increase for the CH2 domain while the changes for the Fab domain were only modest (Figure S6 in Supplemental Information). These are consistent with an earlier work, which showed Fc domain being more pH sensitive than Fab domain. 45 , 46
4.7. Aggregation mechanism of NISTmAb and electrostatic repulsion
Protein aggregation is a complicated phenomenon with various possible pathways and influencing factors. 47 , 48 , 49 While many protein aggregation models were devised to untangle the situation, 42 , 50 , 51 , 52 , 53 , 54 most of them are variations of Lumry‐Eyring model which is based on the following simple scheme: 50 , 51
In this model, irreversible protein aggregation involves two steps: (i) reversible partial or full unfolding of the folded monomer and (ii) irreversible aggregation of partially or fully unfolded monomer. The free energy diagram of this scheme is shown in Figure 6b.
The Lumry‐Eyring model can provide a cohesive view for seemingly contradicting results that NISTmAb is conformationally more stable (the folding free energy is lower) yet is more prone to aggregation (the T agg is lower) near its pI. A virtue of the Lumry‐Eyring model is that it dissects the aggregation process into two steps. In the first step where unfolding event occurs within a molecule, intramolecular electrostatic repulsion plays a major role. Here intramolecular electrostatic repulsion, which is greater at a pH far from the pI destabilizes the protein (Figure 6, purple arrows). The folding free energy is expected to be the lowest near the pI where little intramolecular electrostatic repulsion is expected (Figure 6b). In the second step where the aggregation event occurs between the molecules, intermolecular electrostatic repulsion plays a major role. Here intermolecular electrostatic repulsion, which is also greater at a pH far from the pI, prevents the aggregation of the protein molecules (Figure 6, blue arrows). The T agg is expected to be the lowest near the pI where little intermolecular electrostatic repulsion is expected (Figure 6b). In this simplistic view, intramolecular and intermolecular electrostatic repulsions work at two different steps of aggregation process.
4.8. Interpretation of DSC data
At a glance, the model described above cannot explain the DSC data which show that T m2 and T m3 decrease as the pH increases while T m1 increases as the pH increases (Figure 5b). It is usually assumed that a higher T m value indicates a protein with lower folding free energy (a more stable protein). 55 Therefore, the DSC data suggest that CH3 (implicated by T m2) and Fab (implicated by T m3) are less stable at a higher pH while CH2 (implicated by T m1) is more stable at a higher pH. However, according to the intramolecular electrostatic repulsion consideration described above and the HDX‐MS data, all three domains should be more stable at a higher pH due to decreasing intramolecular electrostatic repulsion (Figure 4).
Thermal stability and folding free energy of a protein can correlate only when the folding/unfolding event is a two‐state equilibrium. 55 In the case of NISTmAb, it is reasonable to approximate that T m1 is primarily monitoring folding/unfolding equilibrium of CH2 domain, since T m1 is significantly lower than T agg in all conditions employed. Indeed, T m1 is generally high at a higher pH, indicating CH2 domain is more stable at a higher pH as the intramolecular electrostatic repulsion consideration and the HDX‐MS data predict (Figure 5b). On the other hand, T agg is lower than T m3 in most cases and lower than T m2 in some cases, especially at pH 7 or 8 (Table 1 and Figure 5b). In such cases, T m3 and T m2 may reflect an irreversible cascade to the aggregation in addition to the folding/unfolding equilibria of the CH3 and Fab domains. Therefore, T m3 and T m2 may not indicate the conformational stability (folding/unfolding equilibria) at ambient temperature and may not agree with the intramolecular electrostatic repulsion consideration and the HDX‐MS data.
5. CONCLUSION
The biophysical characterization and developability assays for biopharmaceuticals are critical for lead selection and the prediction of optimal manufacturing properties. An important property in these characterizations is the conformational and colloidal stability of the biomolecule, as evidenced by T ms and T agg. However, understanding the meaning of the conformational and colloidal stability measurements at elevated temperatures is not an easy task despite their relatively simple outputs. Complicated events appear to happen behind their simple outputs and extrapolation is required to translate high‐temperature folding/unfolding equilibrium by T m or high‐temperature aggregation kinetics by T agg to ambient temperature properties. 55 We used HDX‐MS, which can measure folding free energy of an analyte protein at room temperature at high‐resolution, to shed light to untangle the issues and understand the meaning of the measurements. The emerged picture was quite simple: The protein is more stable (the folding free energy is lower) near the pI and less stable (the folding free energy is higher) far away from the pI throughout the molecule. At the same time, the protein is more prone to aggregation (the T agg is lower) near the pI and less prone to aggregation (the T agg is higher) far away from the pI. Lumry‐Eyring model which separates folding/unfolding equilibrium and aggregation event can cohesively explain these seemingly contradicting results. 50 , 51 Near the pI, the net charge of the protein molecule is low. The low‐net charge leads to weak intramolecular and intermolecular electrostatic repulsions which in turn result in higher conformational stability 34 , 35 and higher aggregation propensity, respectively. 41 , 42 On the other hand, far from the pI, the net charge of the protein molecule is high. The high‐net charge leads to strong intramolecular electrostatic repulsion which results in low‐conformational stability 34 , 35 and strong intermolecular electrostatic repulsion which results in low‐aggregation propensity. 41 , 42 Also, T ms may not be a good indicator for conformational stability at ambient temperature when T ms are close to T agg. 42 When T ms are similar to T agg, the folding/unfolding equilibrium significantly deviates from the two‐state model at the elevated temperature with additional pathway to aggregation. This can explain the unexpected lower T m2 and T m3 near the pI than far away from the pI.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
AUTHOR CONTRIBUTIONS
Mehabaw Derebe: Formal analysis; methodology; writing‐original draft. Sathya Venkataramani: Project administration; supervision. Jennifer F. Nemeth‐Seay: Conceptualization; project administration; supervision; writing‐review & editing.
Supporting information
Appendix S1: Supplementary Information
ACKNOWLEDGMENTS
The authors thank Stephen J. Coales of LEAP Technologies for his technical support.
Hamuro Y, Derebe MG, Venkataramani S, Nemeth JF. The effects of intramolecular and intermolecular electrostatic repulsions on the stability and aggregation of NISTmAb revealed by HDX‐MS, DSC, and nanoDSF . Protein Science. 2021;30:1686–1700. 10.1002/pro.4129
REFERENCES
- 1. Grilo AL, Mantalaris A. The increasingly human and profitable monoclonal antibody market. Trends Biotechnol. 2019;37:9–16. [DOI] [PubMed] [Google Scholar]
- 2. Sumit Goswami S, Wang W, Arakawa T, Ohtake S. Developments and challenges for mAb‐based therapeutics. Antibodies. 2013;2:452–500. [Google Scholar]
- 3. Karageorgos I, Gallagher ES, Galvin C, Gallagher DT, Hudgens JW. Biophysical characterization and structure of the fab fragment from the NIST reference antibody, RM8671. Biologicals. 2017;50:27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schiel JE, Davis DL, Borisov OV. State‐of‐the‐art and emerging technologies for therapeutic monoclonal antibody characterization Volume 2. Biopharmaceutical characterization. The NISTmAb Case Study. Washington D.C: American Chemical Society, 2015. [Google Scholar]
- 5. Gokarn Y, Agarwal S, Arthur K, et al. Chapter 6, Biophysical techniques for characterizing the higher order structure and interactions of monoclonal antibodies. In state‐of‐the‐art and emerging technologies for therapeutic monoclonal antibody characterization volume 2. Biopharmaceutical characterization: The NISTmAb Case Study. Washington D.C.: ACS Symposium Series, American Chemical Society, 2015. [Google Scholar]
- 6. Saro D, Baker A, Hepler R, et al. Chapter 7, Developability Assessment of a Proposed NIST Monoclonal Antibody. In State‐of‐the‐Art and Emerging Technologies for Therapeutic Monoclonal Antibody Characterization Volume 2. Washington D.C.: Biopharmaceutical Characterization: The NISTmAb Case Study, ACS Symposium Series, American Chemical Society, 2015. [Google Scholar]
- 7. Zhang Z, Smith DL. Determination of amide hydrogen exchange by mass spectrometry: A new tool for protein structure elucidation. Protein Sci. 1993;2:522–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Engen JR, Smith DL. Investigating protein structure and dynamics by hydrogen exchange MS. Anal Chem. 2001;73:256A–265A. [DOI] [PubMed] [Google Scholar]
- 9. Hamuro Y, Coales SJ, Southern MR, Nemeth‐Cawley JF, Stranz DD, Griffin PR. Rapid analysis of protein structure and dynamics by hydrogen/deuterium exchange mass spectrometry. J Biomol Tech. 2003;14:171–182. [PMC free article] [PubMed] [Google Scholar]
- 10. Majumdar R, Esfandiary R, Bishop SM, et al. Correlations between changes in conformational dynamics and physical stability in a mutant IgG1 mAb engineered for extended serum half‐life. MAbs. 2015;7:84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Houde D, Arndt J, Domeier W, Berkowitz S, Engen JR. Characterization of IgG1 conformation and conformational dynamics by hydrogen/deuterium exchange mass spectrometry. Anal Chem. 2009;81:2644–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Houde D, Peng Y, Berkowitz SA, Engen JR. Post‐translational modifications differentially affects IgG1 conformation and receptor binding. Mol Cell Proteomics. 2010;9:1716–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang A, Hu P, MacGregor P, et al. Understanding the conformational impact of chemical modifications on monoclonal antibodies with diverse sequence variation using hydrogen/deuterium exchange mass spectrometry and structural modeling. Anal Chem. 2014;86:3468–3475. [DOI] [PubMed] [Google Scholar]
- 14. Fang J, Richardson J, Du Z, Zhang Z. Effect of fc‐glycan structure on the conformational stability of IgG revealed by hydrogen/deuterium exchange and limited proteolysis. Biochemistry. 2016;55:860–868. [DOI] [PubMed] [Google Scholar]
- 15. Zhang J, Fang J, Chou RY‐T, Bondarenko PV, Zhang Z. Conformational difference in human IgG2 disulfide isoforms revealed by hydrogen/deuterium exchange mass spectrometry. Biochemistry. 2015;54:1956–1962. [DOI] [PubMed] [Google Scholar]
- 16. Yan Y, Wei H, Jusuf S, et al. Mapping the binding interface in a noncovalent size variant of a monoclonal antibody using native mass spectrometry, hydrogen–deuterium exchange mass spectrometry, and computational analysis. J Pharm Sci. 2017;106:3222–3229. [DOI] [PubMed] [Google Scholar]
- 17. Majumdar R, Manikwar P, Hickey JM, et al. Effects of salts from the Hofmeister series on the conformational stability, aggregation propensity, and local flexibility of an IgG1 monoclonal antibody. Biochemistry. 2013;52:3376–3389. [DOI] [PubMed] [Google Scholar]
- 18. Mamikwar P, Majumdar R, Hickey JM, et al. Correlating excipient effects on conformational and storage stability of an IgG1 monoclonal antibody with local dynamics as measured by hydrogen/deuterium‐exchange mass spectrometry. J Pharm Sci. 2013;102:2136–2151. [DOI] [PubMed] [Google Scholar]
- 19. Toth RT, Pace SE, Mills BJ, et al. Evaluation of hydrogen exchange mass spectrometry as a stability‐indicating method for formulation excipient screening for an IgG4 monoclonal antibody. J Pharm Sci. 2017;107:1009–1019. [DOI] [PubMed] [Google Scholar]
- 20. Zhang A, Singh SK, Shirts MR, Kumar S, Fernandez EJ. Distinct aggregation mechanisms of monoclonal antibody under thermal and freeze‐thaw stresses revealed by hydrogen exchange. Pharm Res. 2012;29:236–250. [DOI] [PubMed] [Google Scholar]
- 21. Iacob RE, Bou‐Assaf GM, Makowski L, Engen JR, Berkowitz SA, Houde D. Investigating monoclonal antibody aggregation using a combination of H/DX‐MS and other biophysical measurements. J Pharm Sci. 2013;102:4315–4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hamuro Y, Coales SJ. Optimization of feasibility stage for hydrogen/deuterium exchange mass spectrometry. J Am Soc Mass Spectrom. 2018;29:623–629. [DOI] [PubMed] [Google Scholar]
- 23. Hamuro Y, Zhang T. High‐resolution HDX‐MS of cytochrome c using pepsin/fungal protease type XIII mixed bed column. J Am Soc Mass Spectrom. 2019;30:227–234. [DOI] [PubMed] [Google Scholar]
- 24. Coales SJ, Yen ES, Lee JE, Ma A, Morrow JA, Hamuro Y. Expansion of time window for mass spectrometric measurement of amide hydrogen/deuterium exchange reactions. Rapid Commun Mass Spectrom. 2010;24:3585–3592. [DOI] [PubMed] [Google Scholar]
- 25. Hamuro Y. Determination of equine cytochrome c backbone amide hydrogen/deuterium exchange rates by mass spectrometry using a wider time window and isotope envelope. J Am Soc Mass Spectrom. 2017;28:486–497. [DOI] [PubMed] [Google Scholar]
- 26. Hamuro Y. Tutorial: Chemistry of hydrogen exchange mass spectrometry. J Am Soc Mass Spectrom. 2021;32:133–151. [DOI] [PubMed] [Google Scholar]
- 27. Chetty PS, Mayne L, Lund‐Katz S, Stranz D, Englander SW, Phillips MC. Helical structure and stability in human apolipoprotein A‐I by hydrogen exchange and mass spectrometry. Proc Natl Acad Sci U S A. 2009;106:19005–19010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Walters BT. Empirical method to accurately determine peptide‐averaged protection factors from hydrogen exchange MS data. Anal Chem. 2017;89:1049–1053. [DOI] [PubMed] [Google Scholar]
- 29. Bai Y, Milne JS, Mayne LC, Englander SW. Primary structure effects on peptide group hydrogen exchange. Proteins. 1993;17:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Weis DD, Wales TE, Engen JR. Identification and characterization of EX1 kinetics in H/D exchange mass spectrometry by peak width analysis. J Am Soc Mass Spectrom. 2006;17:1498–1509. [DOI] [PubMed] [Google Scholar]
- 31. Mayne L. Hydrogen exchange mass spectrometry. Methods Enzymol. 2016;566:335–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Englander SW, Mayne L. The case for defined protein folding pathways. Proc Natl Acad Sci U S A. 2014;111:15873–15880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Englander SW, Mayne L. The nature of protein folding pathways. Proc Natl Acad Sci U S A. 2017;114:8253–8258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dill KA. Dominant forces in protein folding. Biochemistry. 1990;29:7133–7155. [DOI] [PubMed] [Google Scholar]
- 35. Lindman S, Xue W‐F, Szczepankiewicz O, Bauer MC, Nilsson H, Linse S. Salting the charged surface: pH and salt dependence of protein G B1 stability. Biophys J. 2006;90:2911–2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41:415–427. [DOI] [PubMed] [Google Scholar]
- 37. Michels DA, Ip AY, Dillon TM, et al. Chapter 5, Separation methods and orthogonal techniques. In state‐of‐the‐art and emerging technologies for therapeutic monoclonal antibody characterization volume 2. Biopharmaceutical characterization: The NISTmAb Case Study. Washington D.C.: American Chemical Society, 2015. [Google Scholar]
- 38. Cohn EJ, Edsall JT. Proteins, amino acids and peptides. New York: Hafner Publishing Co, 1943. [Google Scholar]
- 39. Pace CN, Grimsley GR, Scholtz JM. Protein ionizable groups: pK values and their contribution to protein stability and solubility. J Biol Chem. 2009;284:13285–13289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Golovanov AP, Hautbergue GM, Wilson SA, Lian L‐Y. A simple method for improving protein solubility and long‐term stability. J Am Chem Soc. 2004;126:8933–8939. [DOI] [PubMed] [Google Scholar]
- 41. Roberts CJ. Therapeutic protein aggregation: Mechanisms, design, and control. Trends Biotechnol. 2014;32:372–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chakroun N, Hilton D, Ahmad SS, Platt GW, Dalby PA. Mapping the aggregation kinetics of a therapeutic antibody fragment. Mol Pharm. 2016;13:307–319. [DOI] [PubMed] [Google Scholar]
- 43. Brader ML, Estey T, Bai S, et al. Examination of thermal unfolding and aggregation profiles of a series of developable therapeutic monoclonal antibodies. Mol Pharm. 2015;12:1005–1017. [DOI] [PubMed] [Google Scholar]
- 44. Molecular Operating Environment (MOE), 2019.01: Chemical Computing Coup ULC, 1010 Sherbooke St., West, Suite #910, Montreal, QC, Canada, H2A 2R7, 2021.
- 45. Vermeer AW, Norde W. The thermal stability of immunoglobulin: Unfolding and aggregation of a multidomain protein. Biophys J. 2000;78:394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wu H, Kroe‐Barrett R, Singh S, Robinson AS, Roberts CJ. Competing aggregation pathways for monoclonal antibodies. FEBS Lett. 2014;588:936–941. [DOI] [PubMed] [Google Scholar]
- 47. Philo JS, Arakawa T. Mechanisms of protein aggregation. Curr Pharm Biotechnol. 2009;10:348–351. [DOI] [PubMed] [Google Scholar]
- 48. Wang W, Nema S, Teagarden D. Protein aggregation – Pathways and influencing factors. Int J Pharm. 2010;390:89–99. [DOI] [PubMed] [Google Scholar]
- 49. Roberts CJ, Das TK, Sahin R. Predicting solution aggregation rates for therapeutic proteins: Approaches and challenges. Int J Pharm. 2011;418:318–333. [DOI] [PubMed] [Google Scholar]
- 50. Lumry R, Eyring H. Conformation changes of proteins. J Phys Chem. 1954;58:110–120. [Google Scholar]
- 51. Sanchez‐Ruiz JM. Theoretical analysis of Lumry‐Eyring models in differential scanning calorimetry. Biophys J. 1992;61:921–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Roberts CJ. Kinetics of irreversible protein aggregation: Analysis of extended Lumry‐Eyring and implications for predicting protein shelf life. J Phys Chem. 2003;107:1194–1207. [Google Scholar]
- 53. Roberts CJ. Non‐native protein aggregation kinetics. Biotechnol Bioeng. 2007;98:927–938. [DOI] [PubMed] [Google Scholar]
- 54. Saluja A, Sadineni V, Mungikar A, et al. Significance of unfolding thermodynamics for predicting aggregation kinetics: A case study on high concentration solutions of a multi‐domain protein. Pharm Res. 2014;31:1575–1587. [DOI] [PubMed] [Google Scholar]
- 55. Rees DC, Robertson AD. Some thermodynamic implications for the thermostability of proteins. Protein Sci. 2001;10:1187–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supplementary Information