

Abstract
Hsp104, a yeast protein disaggregase, can be potentiated via numerous missense mutations at disparate locations throughout the coiled‐coil middle domain (MD). Potentiated Hsp104 variants can counter the toxicity and misfolding of TDP‐43, FUS, and α‐synuclein, proteins which are implicated in neurodegenerative disorders. However, potentiated MD variants typically exhibit off‐target toxicity. Further, it has remained confounding how numerous degenerate mutations confer potentiation, hampering engineering of therapeutic Hsp104 variants. Here, we sought to comprehensively define the key drivers of Hsp104 potentiation. Using scanning mutagenesis, we iteratively studied the effects of modulation at each position in the Hsp104 MD. Screening this library to identify enhanced variants reveals that missense mutations at 26% of positions in the MD yield variants that counter FUS toxicity. Modulation of the helix 2–helix 3/4 MD interface potentiates Hsp104, whereas mutations in the analogous helix 1–2 interface do not. Surprisingly, we find that there is a higher likelihood of enhancing Hsp104 activity against human disease substrates than impairing Hsp104 native function. We find that single mutations can broadly destabilize the MD structure and lead to functional potentiation, suggesting this may be a common mechanism conferring Hsp104 potentiation. Using this approach, we have demonstrated that modulation of the MD can yield engineered variants with decreased off‐target effects.
Keywords: alpha‐synuclein, amyloid, fused in sarcoma, Hsp104, protein disaggregase, protein misfolding, TAR DNA‐binding protein 43 (TDP‐43)
1. INTRODUCTION
Protein misfolding underpins numerous neurodegenerative disorders including amyotrophic lateral sclerosis (ALS) and Parkinson's disease (PD). 1 , 2 , 3 Protein misfolding is also implicated in certain cardiovascular disorders and some cancers. In ALS, several different proteins, including TAR DNA‐binding protein 43 (TDP‐43) and FUS, have been found to accumulate in degenerating motor neurons. 1 , 2 Similarly, α‐synuclein (α‐syn) accumulations in Lewy bodies are the pathological hallmark of PD. 3 The genes encoding TDP‐43 and FUS are essential, 4 thus therapeutic strategies aimed at decreasing TDP‐43 and FUS expression levels or promoting their degradation may be ineffective. 5 , 6 , 7 An interesting alternative approach would be to reactivate misfolded proteins to their native folds and functions, which would simultaneously counter a possible loss‐ or gain‐of‐function mechanism. 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13
While commonly implicated in disease, amyloid formation is not invariably deleterious. For instance, yeast harness beneficial prions for adaptive purposes. 14 , 15 Beneficial prions are regulated in yeast by Hsp104, a hexameric AAA+ protein that is comprised of an N‐terminal domain, nucleotide‐binding domain 1 (NBD1), a coiled‐coil middle domain (MD), NBD2, and a short C‐terminal domain. 16 , 17 Hsp104 regulates both yeast prions and the diverse pool of proteins that aggregate following exposure to environmental stress. 18 , 19 Because Hsp104 can potently remodel yeast prions, it was hypothesized that it may also remodel amyloid conformers implicated in human disease. Indeed, studies have demonstrated that Hsp104 can dissolve amyloid and pre‐amyloid forms of α‐syn, Aβ, polyglutamine, and other proteins. 20 , 21 Hsp104 has also been found to suppress protein misfolding and mitigate neurodegeneration in animal models of disease. 20 , 22 , 23 However, this activity is limited and requires very high concentrations of Hsp104. 8 , 20 , 21 Further, Hsp104 is inactive against TDP‐43 and FUS. 8
To pursue the development of therapeutic protein disaggregases, potentiated variants of Hsp104 have been developed that confer a therapeutic gain‐of‐function. 8 , 9 , 24 , 25 These potentiated variants robustly disaggregate TDP‐43, FUS, α‐syn, and other substrates. 8 , 9 , 11 , 12 , 13 , 24 , 26 These Hsp104 variants suppress the toxicity and mislocalization of TDP‐43, FUS, and α‐syn in yeast under conditions where Hsp104 WT is ineffective. 8 Potentiated Hsp104 variants can dissolve preformed aggregates and fibrils of TDP‐43, FUS, and α‐syn, reverse the aggregation of FUS in mammalian cells, and suppress dopaminergic neurodegeneration in Caenorhabditis elegans. 8 , 27 Numerous missense mutations have been uncovered that potentiate Hsp104 via introduction of single point mutations at disparate positions throughout all four helices of the coiled‐coil MD. 8 , 24 , 28 Potentiated variants have also been identified harboring missense mutations in NBD1, and a single potentiated variant harboring an NBD2 missense mutation has also been identified. 8 , 12 It is puzzling that both conservative and nonconservative mutations to the MD at seemingly disparate positions all confer a therapeutic gain‐of‐function. 8 , 24 While potentiated Hsp104 variants may hold great therapeutic promise, they can exhibit off‐target toxicity, which is likely due to limited substrate specificity. 8 , 9 , 29 It is therefore important to comprehensively understand the drivers of Hsp104 potentiation so that these insights can be applied to precisely engineer Hsp104 variants without off‐target effects.
While many studies have probed the basis for Hsp104 potentiation, the specific mechanistic drivers that enhance Hsp104 activity have remained confounding. 8 , 9 , 12 , 13 , 24 , 27 , 28 , 30 Structural, genetic, and biochemical studies have suggested that the coiled‐coil MD is an autoinhibitory domain, whereby potentiation can be conferred by breaking both intra‐ and inter‐molecular MD‐MD contacts, as well as contacts between the MD and NBD1. 12 , 16 , 28 , 31 Studies linking potentiation to more specific structural changes have been challenging, which is likely due to the highly dynamic nature of the MD, which is typically poorly resolved in otherwise high resolution cryo‐EM structures. 16 , 26 , 31 , 32 , 33 Recent work employing hydrogen exchange mass spectrometry (HX MS) to study potentiated Hsp104 variants has suggested that potentiating mutations can make interactions within the MD and in the MD‐NBD1 interface more dynamic, thereby speeding ADP dissociation from NBD1. 34 While this study has provided more insight linking Hsp104 potentiation to structural changes, 34 key questions remain about the specific mechanistic drivers of potentiation. In order to achieve the ultimate goal of being able to engineer potentiated Hsp104 variants with fine‐tuned activities, it is essential that we comprehensively understand the drivers of potentiation. To gain new insights into the mechanistic basis for enhanced Hsp104 activity, we sought to comprehensively explore the MD via a scanning mutagenesis approach. We hypothesized that, by introducing mutations at each position throughout the MD, trends dictating Hsp104 potentiation might be uncovered. We introduced single point mutations to valine at each position in the 128 amino acid‐long coiled‐coil MD and screened this library in high throughput using yeast‐based growth assays to identify potentiated variants that suppress FUS, TDP‐43, and/or α‐syn toxicity. We also screened these variants for retention of native Hsp104 activity in a thermotolerance assay and find that these generic mutations to valine are twice as likely to confer potentiation as to inactivate the protein. Our studies reveal that modulation of a key interface in the Hsp104 MD is a driver of Hsp104 potentiation. We also studied the changes in secondary structure accompanying potentiation and found that introduction of potentiating mutations broadly disrupts the secondary structure of the coiled‐coil MD. We then performed additional mutagenesis studies to validate our hypotheses. Our studies have provided key new insights that improve our understanding of the requirements for Hsp104 potentiation. Importantly, we have demonstrated that modulation of the MD can yield potentiated Hsp104 variants with diminished off‐target effects.
2. RESULTS
2.1. Valine scanning mutagenesis of the Hsp104 middle domain reveals numerous variants suppress FUS toxicity
Although the activity of Hsp104 is limited against human disease‐associated proteins, a diverse range of single missense mutations have been found to potentiate Hsp104. 8 , 20 , 24 While numerous enhanced variants have been successfully identified, it remains unclear how so many seemingly disparate mutations can confer potentiation, and what specific molecular changes drive this enhanced activity. To comprehensively and methodically define the molecular changes that drive potentiation, we adopted an iterative scanning mutagenesis approach coupled with screening using yeast models of proteinopathy. Using site‐directed mutagenesis, we iteratively constructed a library of MD variants with every position from residue P411 to Q538 mutated to valine. We then screened these variants in yeast models where expression of FUS, TDP‐43, or α‐syn elicits robust toxicity. 35 , 36 , 37 , 38 We first coexpressed this library of variants with FUS in Δhsp104 yeast. Here, both FUS and Hsp104 expression are under the control of a galactose‐inducible promoter. We screened for toxicity suppressors by plating the yeast in duplicate onto both glucose and galactose‐supplemented media (Figure 1, Figure S1). Toxicity suppression was scored on a scale from 0 to 6 based on the average number of spots from three replicates. Here Hsp104WT was scored at 0.5, and so variants scoring above 0.5 were identified as potentiated. We find that 24 variants from this 128 variant library suppress FUS toxicity, with a score of 1.0 or greater (Figure 1, Figure S1, Table 1). These residues are dispersed throughout the MD, but primarily centered around the end of helix 2 and helix 3 (Figure 1a). The strength of rescue ranged considerably, with Hsp104E432V, Hsp104R465V, Hsp104K480V, Hsp104A503V, Hsp104Y507V, and Hsp104G532V conferring the strongest rescue, with scores of 3.5 each, and Hsp104K451V and Hsp104A479V conferring the weakest rescue, with scores of 1 each (Figure 1b). The remaining Hsp104 variants did not rescue FUS toxicity (Figure S1). Using immunoblotting, we confirmed expression of all 128 variants, and that potentiation was not simply due to consistently enhanced Hsp104 expression or impaired FUS expression (Figure 1c). Several of the strains harboring potentiated variants appeared to display decreased expression of both Hsp104 and FUS (Figure 1c). Potentiated Hsp104 variants are typically expressed at lower levels than Hsp104WT, and decreased Hsp104 expression often correlates with decreased expression of the disease‐associated substrate. 8 , 9 , 12 , 24 Indeed, we find that five of the six variants conferring a rescue of FUS toxicity of 3.5 show impaired expression of Hsp104 and FUS (Figure 1b,c). To quantitatively assess these differences, for a subset of strains we performed three independent replicate blots and measured expression of FUS and Hsp104. We included strains with Hsp104 variants that both strongly and more modestly suppressed FUS toxicity. For comparison we also included Hsp104Q420V, which does not rescue FUS toxicity. While FUS expression levels were generally decreased for the potentiated variants relative to the strain harboring Hsp104WT, there were no statistically significant changes in expression level of FUS. Hsp104 expression levels were also generally impaired for the potentiated variants. Statistically significant decreased expression was observed for strains expressing Hsp104A437V, Hsp104K480V, Hsp104R495V, and Hsp104G532V (Figure 1d). Interestingly, these variants suppress toxicity with scores ranging from 1.5 to 3.5, indicating that Hsp104 expression level does not directly correlate with level of potentiation. Furthermore, Hsp104R465V rescues FUS toxicity with a score of 3.5, yet in this strain Hsp104 and FUS are expressed at similar levels to a strain expressing Hsp104WT. Together, these results indicate that potentiation is not invariably coupled to decreased expression of Hsp104 and that potentiated Hsp104 variants do not uniformly impair FUS expression (Figure 1b‐d). Instead, differences in expression are likely due to other properties of the Hsp104 variants. In sum, these studies suggest that Hsp104R465V may be an ideal candidate to study for possible therapeutic applications.
FIGURE 1.
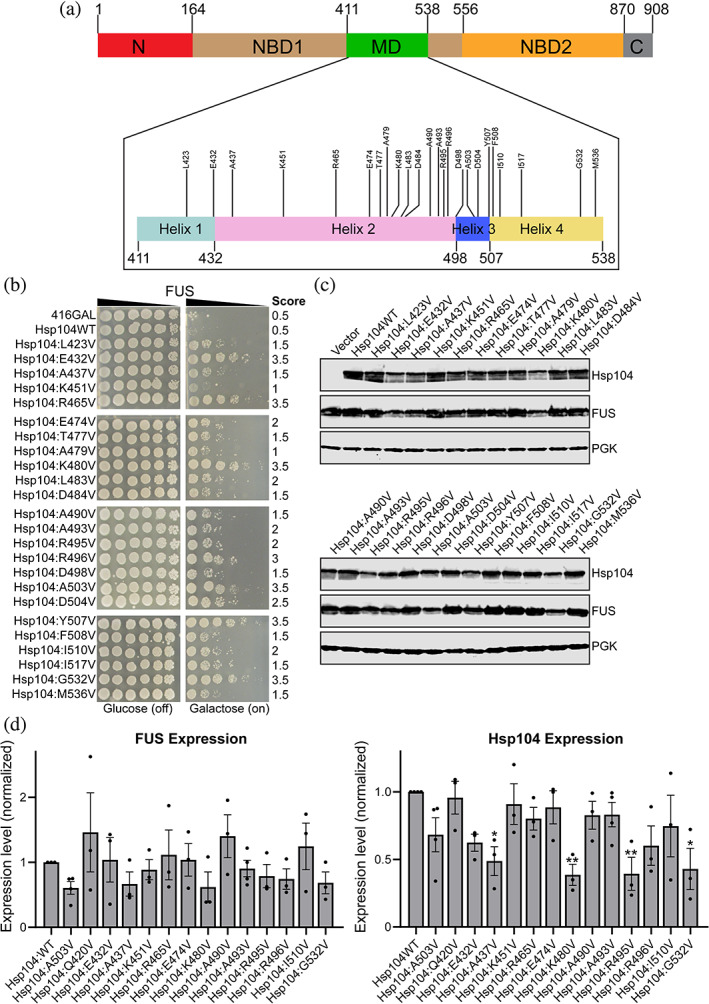
Scanning mutagenesis of the Hsp104 middle domain reveals Hsp104 variants that suppress FUS toxicity. (a) Domain map of Hsp104 shows the location of the MD (green) and its four helices (inset). Variants that suppress FUS toxicity are shown in the inset of the MD. (b) w303aΔhsp104‐pAG‐303GAL‐FUS yeast were transformed with Hsp104 variants or vector. Strains were serially diluted five‐fold and spotted in duplicate onto glucose (non‐inducing, left) or galactose (inducing, right) media. Scores are based on number of spots from an average of three replicates. Only variants that suppress FUS toxicity are shown, see Figure S1 for the full set of spotting assays. (c) Strains in (b) were induced for 5 hr, lysed, and immunoblotted for Hsp104, FUS, and 3‐phosphogylcerate kinase (PGK; loading control). (d) Quantification of FUS (left) and Hsp104 (right) immunoblots for selected strains. Values were normalized to the 3‐PGK loading control and are expressed relative to Hsp104WT. Expression of FUS and Hsp104 levels were compared to the strain expressing FUS and Hsp104WT using a one‐way ANOVA with a Dunnett's multiple comparisons test (N ≥ 3, individual data points shown as dots, bars show mean ± SEM, all p values >.05 except where denoted: *p < .05, **p < .01)
TABLE 1.
Potentiating Hsp104 mutations
| Potentiating mutations (valine) | Potentiating mutations (non‐valine) | Source |
|---|---|---|
| L423V | — | This paper |
| — | V426L/G | Jackrel et al., 2014 6 , 8 , 9 , 25 |
| E432V | — | This paper |
| A437V | A437W | This paper and Jackrel et al., 2014 6 , 8 , 9 , 25 |
| K451V | K451E | This paper and Jackrel et al., 2015 5 , 24 |
| R465V | R465G | This paper and Jackrel et al., 2015 5 , 24 |
| — | Y466S | Jackrel et al., 2015 5 , 24 |
| — | E469D | Jackrel et al., 2015 5 , 24 |
| — | K470Q | Jackrel et al., 2015 5 , 24 |
| E474V | — | This paper and Jackrel et al., 2015 5 , 24 |
| T477V | — | This paper |
| A479V | — | This paper |
| K480V | K480E | This paper and Jackrel et al., 2015 5 , 24 |
| L483V | L483S | This paper and Jackrel et al., 2015 5 , 24 |
| D484V | — | This paper |
| A490V | — | This paper |
| A493V | A493T | This paper and Jackrel et al., 2015 5 , 24 |
| R495V | R495D/E/M/N | This paper and Tariq et al., 2019 12 |
| R496V | — | This paper |
| D498V | — | Jackrel et al., 2014 |
| — | T499D | Tariq et al., 2018 13 |
| — | A502F | This paper |
| A503V | All amino acids | Jackrel et al., 2014 6 , 8 , 9 , 25 |
| D504V | — | Jackrel et al., 2014 6 , 8 , 9 , 25 |
| Y507V | Y507A/C/D | Jackrel et al., 2014 6 , 8 , 9 , 25 |
| F508V | — | This paper |
| I510V | — | This paper |
| — | P511A | Jackrel et al., 2015 5 , 24 |
| — | I513F | Jackrel et al., 2015 5 , 24 |
| I517V | — | This paper |
| G532V | G532S | This paper and Jackrel et al., 2015 5 , 24 |
| — | S535D | Tariq et al., 2018 13 |
| M536V | M536K | This paper and Jackrel et al., 2015 5 , 24 |
Note: The mutations that potentiate Hsp104 activity, leading to a rescue of TDP‐43, FUS, and/or α‐syn toxicity.
2.2. Potentiated MD variants suppress TDP‐43 toxicity
We next screened the 128 variant library for suppression of TDP‐43 toxicity to assess differences in substrate specificity. Eleven variants suppressed TDP‐43 toxicity: Hsp104E432V, Hsp104R465V, Hsp104E474V, Hsp104K480V, Hsp104R495V, Hsp104R496V, Hsp104A503V, Hsp104D504V, Hsp104Y507V, Hsp104I510V, and Hsp104G532V, all of which had a score of 2 or higher (Figure 2a, Figure S2, Table 1). All 11 of these variants that rescue TDP‐43 toxicity also rescue FUS toxicity. The remaining 117 variants did not rescue TDP‐43 toxicity (Figure S2), including 13 variants that rescue FUS but not TDP‐43 toxicity. These differences suggest that there are distinct and more stringent requirements for rescue of TDP‐43 toxicity than FUS toxicity. Immunoblotting of these strains indicated that expression of the potentiated variants was generally decreased as compared to Hsp104WT. Quantitation of expression of TDP‐43 in these strains was uniformly consistent relative to the strain expressing Hsp104WT, with the exception of the strain expressing Hsp104A493V, which displayed increased expression of TDP‐43. While expression levels of potentiated Hsp104 variants were generally decreased relative to Hsp104WT, these trends were not statistically significant. Additionally, as is the case with co‐expression of FUS, the Hsp104R465V strain again displayed expression of TDP‐43 and Hsp104 that was just modestly impaired, despite its robust potentiation (Figure 2c). Surprisingly, while Hsp104A437V does not confer a strong rescue of TDP‐43 toxicity, it is expressed at the lower level characteristic of the robustly potentiated variants, similar to observations for the strain co‐expressing Hsp104A437V and FUS. However, Hsp104A437W has been reported to be a robustly potentiated variant. 8 These differences provide further evidence that potentiation is not directly linked to protein expression levels.
FIGURE 2.
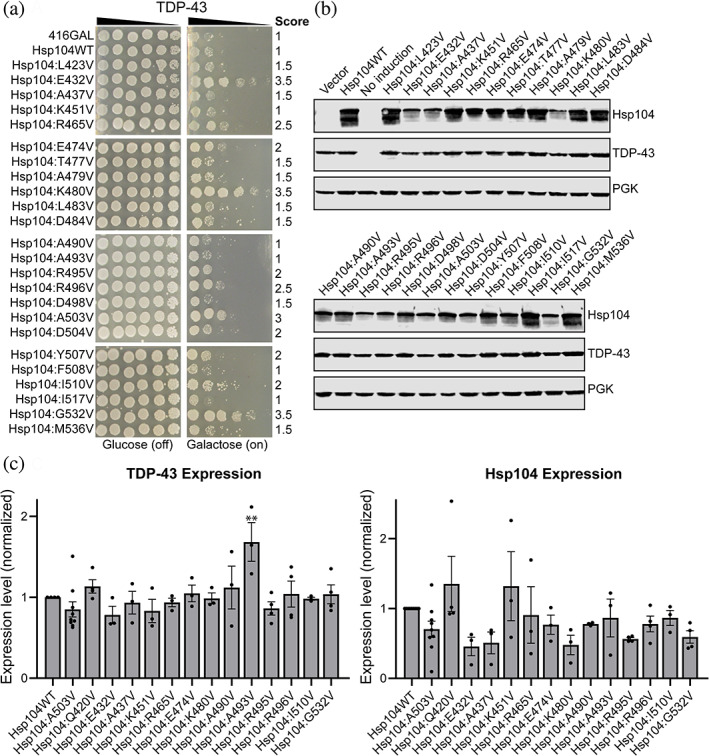
Scanning mutagenesis of the Hsp104 middle domain reveals Hsp104 variants that suppress TDP‐43 toxicity. (a) w303aΔhsp104‐pAG‐303GAL‐TDP‐43 yeast were transformed with Hsp104 variants or vector. Strains were serially diluted five‐fold and spotted in duplicate onto glucose (non‐inducing, left) or galactose (inducing, right) media. Scores are based on number of spots from an average of three replicates. Only strains that suppress FUS toxicity are shown, see Figure S2 for full set of spotting assays. (b) Strains in (a) were induced for 5 hr, lysed, and immunoblotted for Hsp104, TDP‐43, and 3‐phosphogylcerate kinase (PGK; loading control). (C) Quantification of TDP‐43 (left) and Hsp104 (right) immunoblots for selected strains. Values were normalized to the 3‐PGK loading control and are expressed relative to Hsp104WT. Expression of TDP‐43 and Hsp104 levels were compared to the strain expressing TDP‐43 and Hsp104WT using a one‐way ANOVA with a Dunnett's multiple comparisons test (N ≥ 3, individual data points shown as dots, bars show mean ± SEM, all p values >.05 except where denoted: **p < .01)
2.3. Potentiated MD variants suppress α‐syn toxicity
FUS and TDP‐43 are both RNA‐binding proteins with prion‐like domains, which shuttle between the nucleus and cytoplasm. 39 α‐Syn is a lipid‐binding protein that localizes to the plasma membrane. 3 We therefore tested the MD library against α‐syn to assess trends in substrate‐specificity (Figure 3a, Figure S3, Table 1). We found that 20 variants suppressed α‐syn toxicity with a score of 3.5 or greater. As with TDP‐43, all the variants that rescued α‐syn toxicity also rescued FUS toxicity. Interestingly, nine variants: Hsp104A437V, Hsp104T477V, Hsp104L483V, Hsp104D484V, Hsp104A490V, Hsp104A493V, Hsp104D498V, Hsp104F508V, and Hsp104I517V rescued α‐syn and FUS toxicity but did not rescue TDP‐43 toxicity. These nine variants are distributed throughout the MD, and their suppression of FUS and α‐syn toxicity is relatively modest (Figures 1, 2, 3). We confirmed expression of Hsp104 and α‐syn for each of the strains (Figure 3b). As with TDP‐43, expression of α‐syn does not appear to be impaired by expression of the more robustly potentiated Hsp104 variants, and we observed no significant differences in α‐syn expression in quantitative immunoblotting (Figure 3c). We did observe significant variation in expression level of the Hsp104 variants, with Hsp104E432V, Hsp104A437V, Hsp104R495V, and Hsp104G532V showing the greatest decrease in expression level. Each of these variants, with the exception of Hsp104E432V, also displayed significant impairment of expression when co‐expressed with FUS. As when co‐expressed with FUS, expression of Hsp104A437V is impaired when co‐expressed with α‐syn, but it only modestly rescues α‐syn toxicity. In contrast, Hsp104R465V robustly rescues α‐syn toxicity, yet its expression is not impaired (Figure 3c), further supporting the idea that potentiation is not directly linked to expression levels. To compare the rescue of toxicity against FUS, TDP‐43, and α‐syn, we plotted the rescue scores for each of the missense mutants against each substrate (Figure 3d). Comparison indicates that nearly all of the variants that suppress the toxicity of one substrate, suppress the toxicity of all three. While there are some variants that specifically rescue just one substrate, these are generally restricted to variants that are more weakly potentiated. Surprisingly, the collection of Hsp104 variants that rescue both FUS and α‐syn toxicity appear to be more similar than the collection that rescues both FUS and TDP‐43 toxicity, despite the greater similarities between FUS and TDP‐43.
FIGURE 3.
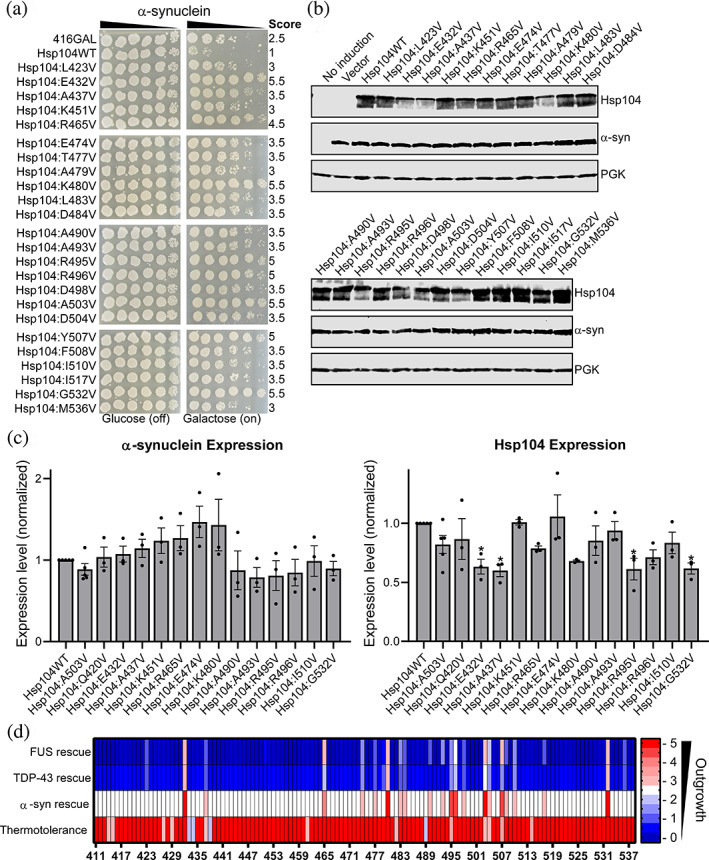
Scanning mutagenesis of the Hsp104 middle domain reveals Hsp104 variants that suppress α‐synuclein toxicity. (a) w303aΔhsp104‐pAG‐303GAL‐α‐syn‐YFP‐304GAL‐α‐syn‐YFP yeast were transformed with Hsp104 variants or vector. Strains were serially diluted five‐fold and spotted in duplicate onto glucose (non‐inducing, left) or galactose (inducing, right) media. Scores are based on number of spots from an average of three replicates. Only strains that suppress FUS toxicity are shown, see Figure S3 for full set of spotting assays. (b) Strains in (a) were induced for 8 hr, lysed, and immunoblotted for Hsp104, YFP (α‐syn), and 3‐phosphogylcerate kinase (PGK; loading control). (c) Quantification of α‐synuclein (left) and Hsp104 (right) immunoblots for selected strains. Values were normalized to the 3‐PGK loading control and are expressed relative to Hsp104WT. Expression of α‐syn and Hsp104 levels were compared to the strain expressing α‐syn and Hsp104WT using a one‐way ANOVA with a Dunnett's multiple comparisons test (N ≥ 3, individual data points shown as dots, bars show mean ± SEM, all p values >.05 except where denoted: *p < .05). (d) Rescue scores of the valine missense variants against FUS, TDP‐43, and α‐syn are plotted. Thermotolerance results are shown in the bottom row, where here a score of 5.5 corresponds to a variant that confers thermotolerance
2.4. Most missense MD mutants confer thermotolerance
We next examined the link between native Hsp104 disaggregase activity and potentiation against human disease substrates. First we aimed to determine which Hsp104 variants in our library, particularly those that do not confer potentiation, retain broad disaggregase activity and have not been inactivated via our mutagenesis strategy. Second, we also sought to investigate the possible links between native disaggregase activity and potentiation, to determine if activity against human disease substrates conflicts with activity against the native substrate repertoire. We therefore screened the 128 variant MD library to identify variants that confer thermotolerance, because thermotolerance activity reflects the primary disaggregase function of Hsp104 in yeast. Normally, upon exposure to environmental stress, Hsp104 expression is upregulated to broadly counter protein aggregation and promote cell survival. 18 , 40 Following extreme heat shock at temperatures 50°C and above, cells expressing Hsp104 are 1,000 times more viable than cells that do not express Hsp104. 40 A brief pre‐treatment at 37°C before heat stress provides further protection via upregulation of the heat shock response (HSR). 40
We screened the full library of MD variants for thermotolerance in high throughput. Here we expressed Hsp104 for several hours, pretreated the cells at 37°C for 30 min to induce the HSR, and then heat stressed the cells at 50°C for 0 or 60 min. Strains were then analyzed after 48 hr outgrowth at 30°C (Figure 3d, Figure S4). We find that 88% of the missense variants confer thermotolerance at levels similar to Hsp104WT. Thus, most of the variants retain their broad disaggregase activity but are incapable of suppressing FUS, TDP‐43, or α‐syn toxicity. Just 16 variants display diminished thermotolerance: Hsp104L414V, Hsp104D415V, Hsp104E427V, Hsp104K429V, Hsp104R433V, Hsp104D434V, Hsp104A437V, Hsp104D438V, Hsp104P461V, Hsp104K482V, Hsp104K489V, Hsp104R495V, Hsp104A503V, Hsp104D504V, Hsp104Y507V, and Hsp104K514V (Table 2, Figure S4). We observed a range of phenotypes, with certain variants such as Hsp104A503V displaying a more subtle growth defect, while other variants such as Hsp104R433V and Hsp104R434V appear to be completely inactive in conferring thermotolerance. Of these 16 variants, 5 are potentiated (Hsp104A437V, Hsp104R495V, Hsp104A503V, Hsp104D504V, and Hsp104Y507V) while the remaining 11 are not potentiated. Additionally, 4 of these 16 variants harbor mutations in the distal loop region (residues 430–446) between helices 1 and 2, and mutations in this region are known to impair collaboration of Hsp104 with Hsp70. 41 For these 4 variants, decreased thermotolerance may not be due to structural perturbation, but instead impaired collaboration with Hsp70. Eleven of these 16 variants with impaired thermotolerance normally harbor a charged side chain at the site of mutagenesis. Therefore, introduction of a hydrophobic valine likely perturbs the protein sufficiently to impair its native function. We also found that Hsp104P461V displays impaired thermotolerance (Table 2, Figure S4). This proline is found in the middle of helix 2, where it would likely break the helix and is hypothesized to serve as a flexible hinge. 42 Hsp104P461A was found to display reduced disaggregase activity in an in vitro luciferase remodeling assay, 42 supporting our findings. Overall, our results suggest that Hsp104 is remarkably tolerant to our mutagenesis strategy and the majority of the variants in the library retain their broad disaggregase activity. To assess the link between potentiation and retention of thermotolerance, we included thermotolerance in the heat map (Figure 3d). We find that there is no clear link between potentiation and retention of thermotolerance, suggesting that FUS, TDP‐43, and α‐syn have distinct properties as compared to the native substrate repertoire of Hsp104. Additionally Hsp104R465V, which rescues all three substrates but has diminished off‐target effects, is active in thermotolerance. Therefore, potentiation against non‐native substrates does not necessarily come at the cost of recognition of the diverse native substrate repertoire. Remarkably, while we hypothesized that a large number of variants in this library would be inactivated via mutation to valine, approximately twice as many variants are potentiated than inactivated.
TABLE 2.
Hsp104 variants conferring diminished thermotolerance
| Variants with diminished thermotolerance | Potentiated (yes/no) |
|---|---|
| L414V | No |
| D415V | No |
| E427V | No |
| K429V | No |
| R433V | No |
| D434V | No |
| A437V | Yes |
| D438V | No |
| P461V | No |
| K482V | No |
| K489V | No |
| R495V | Yes |
| A503V | Yes |
| D504V | Yes |
| Y507V | Yes |
| K514V | No |
Note: All missense variants confer thermotolerance at levels similar to Hsp104WT, with the exception of those shown here. Please see Figure S4 for complete data.
2.5. Certain potentiated MD variants confer temperature sensitivity
Potentiated Hsp104 MD variants isolated in previous studies exhibit a temperature sensitive phenotype, whereby adverse effects are conferred on targets other than the desired target substrate. 8 , 9 , 12 , 24 This temperature sensitive phenotype has been hypothesized to be the result of nonspecific disaggregation and inactivation of metastable proteins, leading to a loss of protein function and impaired cell growth. 9 This link between potentiation and temperature sensitivity is not invariable and certain potentiated Hsp104 variants, specifically those harboring missense mutations in NBD1, have been uncovered that rescue toxicity without conferring a temperature sensitive phenotype. 12 In identifying optimal variants to pursue for potential therapeutic applications, it is important that variants which robustly dissolve and reactivate disease‐associated proteins without off‐target effects be identified. We therefore screened the 24 potentiated MD variants for off‐target effects by comparing growth upon expression at 30 and 37°C (Figure 4). We found that 15 of the 24 variants rescue toxicity of one or more disease substrates without conferring temperature sensitivity, growing at levels similar to a strain expressing Hsp104WT. These variants include: Hsp104L423V, Hsp104K451V, Hsp104R465V, Hsp104E474V, Hsp104T477V, Hsp104A479V, Hsp104L483V, Hsp104D484V, Hsp104A490V, Hsp104R496V, Hsp104D504V, Hsp104F508V, Hsp104I510V, Hsp104I517V, and Hsp104M536V (Figure 4). Most of these variants that confer diminished temperature sensitivity are also the weakest in rescuing FUS toxicity (Figure 1b). However, Hsp104R496V robustly rescues FUS toxicity while conferring no temperature sensitivity (Figures 1b and 4). This finding is especially surprising because the adjacent mutant, Hsp104R495V, confers a slightly weaker rescue of FUS toxicity than Hsp104R496V but is highly temperature sensitive. Thus, potentiation and temperature sensitivity are not invariably linked. Another notable exception to this trend is Hsp104R465V. Hsp104R465V does not confer temperature sensitivity, but it robustly suppresses FUS and α‐syn toxicity, while modestly suppressing TDP‐43 toxicity. Furthermore, expression of Hsp104R465V is not significantly impaired and its expression does not significantly impair FUS, TDP‐43, or α‐syn expression. Therefore, Hsp104 can be potentiated via mutation to the MD without necessarily leading to off‐target effects. These desirable properties are not limited to potentiated Hsp104 NBD1 variants as has been suggested, 12 and Hsp104R465V has emerged as a key variant to pursue for possible therapeutic applications. Furthermore, these results substantiate the importance of including a counterselection against temperature sensitivity as a tool to identify potentiated Hsp104 variants with diminished off‐target effects.
FIGURE 4.
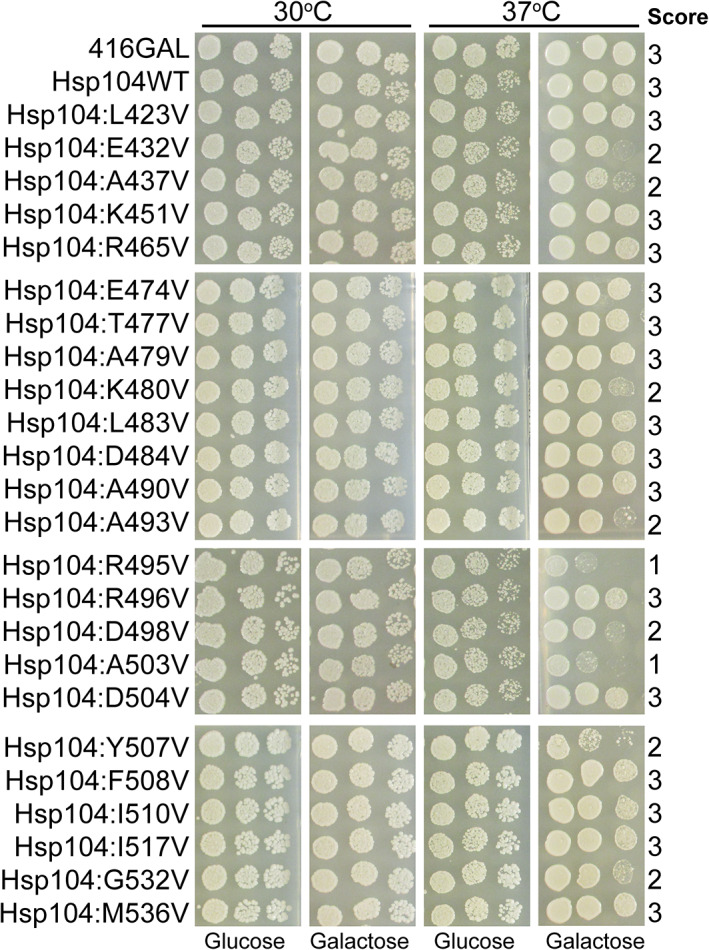
Potentiated Hsp104 MD variants do not uniformly exhibit reduced growth at 37°C. Hsp104 variants were expressed in the 416GAL vector in Δhsp104 yeast in the absence of disease proteins. Strains were serially diluted five‐fold and spotted in duplicate on glucose (non‐inducing) and galactose (inducing) media. One set of plates was incubated at 30°C while a second was incubated at 37°C
2.6. Potentiating mutations broadly disrupt Hsp104 MD secondary structure
In our study, we find that mutation by substitution to valine at 24 of the 128 residues in the MD confers potentiation (Figure 1). Due to the large number and diverse nature of mutations identified that potentiate Hsp104, along with structural data, it has been hypothesized that the MD serves as an autoinhibitory domain whereby potentiation largely stems from destabilization of Hsp104 rather than introduction of specific new interactions. 8 , 12 , 28 , 31 However, understanding the specific structural changes underpinning Hsp104 potentiation has been challenging. In many published structures, the MD has remained a poorly resolved region of the protein. 16 , 33 Recent cryo‐EM studies have revealed new insights into the mechanism of Hsp104 translocation and aggregate dissolution, and HX MS studies have revealed that introduction of the Hsp104A503S mutation destabilizes the outer portion of the MD and makes it more dynamic. 16 , 31 , 33 , 34 , 43 While the HX MS study suggested that Hsp104A503S confers only localized structural destabilization, we hypothesized that because such disparate mutations can confer potentiation (Figures 1, 2, 3), that broad structural perturbation may underpin potentiation. The decreased expression levels we observe for most of the potentiated Hsp104 variants further supports this hypothesis (Figures 1, 2, 3), as less stable proteins are typically expressed at decreased levels. We therefore sought to use pure protein biochemistry to better understand these changes. We purified Hsp104E432V and Hsp104K480V, two of the variants that most potently suppressed FUS, TDP‐43, and α‐syn toxicity (Figures 1, 2, 3), along with Hsp104A503V and previously reported constructs: Hsp104Δ412–459 (Hsp104ΔMotif 1) and Hsp104Δ412–524 (Hsp104ΔMD). 24 Using ATPase and luciferase reactivation assays, 19 we first sought to confirm that the selected variants are potentiated. We find that each of the potentiated variants display ~two to three‐fold higher ATPase activity than Hsp104WT, while deletion of motif 1 or the entire MD inactivates Hsp104 (Figure 5a). While Hsp104 requires Hsp70 and Hsp40 for luciferase reactivation, potentiated MD and NBD1 variants do not (Figure 5b). 8 , 12 , 24 In these assays, we again find that deletion of motif 1 or the entire MD inactivates Hsp104 (Figure 5b), corresponding with previous findings. 24 Similar to Hsp104A503V and other potentiated variants, Hsp104E432V and Hsp104K480V do not require Hsp70 or Hsp40 for luciferase reactivation (Figure 5b). Even in the absence of Hsp70 and Hsp40, the potentiated variants were more active than Hsp104 with Hsp70 and Hsp40. The addition of Hsp70 and Hsp40 increased luciferase reactivation activity substantially, increasing Hsp104K480V activity more than three‐fold. We therefore conclude that Hsp104E432V and Hsp104K480V are potentiated variants.
FIGURE 5.
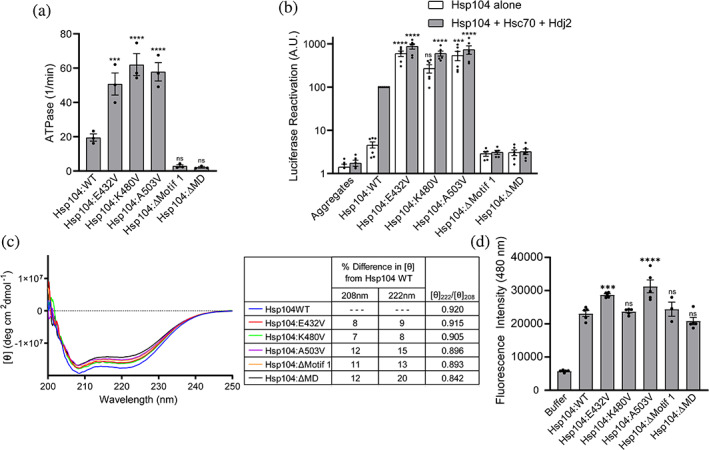
Potentiated Hsp104 MD variants display enhanced disaggregase activity and loss of coiled‐coil secondary structure. (a) MD variants display elevated ATPase activity. Hsp104 variants were compared to Hsp104WT using a one‐way ANOVA with a Dunnett's multiple comparisons test (individual data points are shown as dots, bars represent means ± SEM, N = 3, ***p < .01, ****p < .001). (b) MD variants display elevated disaggregase activity. Luciferase aggregates were incubated with Hsp104 variants (0.167 μM hexamer) plus (gray bars) or minus (white bars) Hsc70 (0.167 μM) and Hdj2 (0.167 μM). Luciferase activity was normalized to Hsp104 plus Hsc70 and Hdj2. Hsp104 variant activities were compared to Hsp104WT plus Hsc70 and Hdj2 using a one‐way ANOVA with a Dunnett's multiple comparisons test (individual data points are shown as dots, bars represent means ± SEM, N = 5, ***p < .01, ****p < .001). (c) Circular dichroism spectroscopy of Hsp104 variants (1–2 μM monomer). Left, average values of molar ellipticity from four independent trials are shown. Right, average [θ] values for the variants at 208 and 222 nm were compared to Hsp104WT. The percent difference in the average [θ] value at 208 and 222 nm between the variants and Hsp104WT are shown, as well as the ratio of [θ]222/[θ]208. (d) ANS fluorescence. Hsp104 (2.5 μM monomer) was incubated with 50 μM ANS at room temperature. Values were compared to Hsp104WT using a one‐way ANOVA with a Dunnett's multiple comparisons test (N ≥ 3, individual data points are shown as dots, bars represent means ± SEM, ***p < .01, ****p < .001)
We next sought to assess whether potentiated variants displayed any differences in secondary structure using circular dichroism (CD) spectroscopy. A single point mutation in a large hexameric protein like Hsp104 would be unlikely to elicit a significant change in CD signal. Indeed, it has been reported that Hsp104A430W and other distal loop mutants have a very similar spectrum to Hsp104WT. 41 However, we hypothesized that, if broader secondary structure perturbations were elicited by the missense mutations, these changes in signal might be detectable. CD spectroscopy is also a useful technique for studying Hsp104 because we can collect spectra in solution and under the same conditions as the luciferase reactivation assays, conditions under which we verified that the variants are active. While CD is highly subject to buffer interference, we wanted to ensure that we were monitoring spectra of the active protein; thus, we used identical buffer conditions as in the luciferase reactivation assays. This precluded monitoring CD signal below 200 nm.
We collected spectra for Hsp104WT, Hsp104A503V, Hsp104E432V, Hsp104K480V, Hsp104ΔMotif 1, and Hsp104ΔMD (Figure 5c). We chose Hsp104E432V and Hsp104K480V because they are robustly potentiated, likely via disruption of electrostatic interactions. The CD spectra reveal troughs at 208 and 222 nm, characteristic of a predominantly α‐helical protein. Strikingly, we found that all of the variants tested displayed decreased molar ellipticity relative to Hsp104WT (Figure 5c). The coiled‐coil MD comprises 14% of the Hsp104 structure, so we would anticipate that complete loss in helicity of the MD would confer a loss of approximately 14% of the α‐helical signal. Matching our predictions, we found that deletion of motif 1 or the entire MD corresponded to an 11% and 12% difference in molar ellipticity relative to Hsp104 at 208 nm, respectively, and a 13% and 20% difference at 222 nm (Figure 5c). The potentiated variants show a similar decrease in molar ellipticity, ranging from 7% to 12% at 208 nm and 8% to 15% at 222 nm (Figure 5c). These changes suggest that these single missense potentiating mutations disrupt the α‐helical character of the MD beyond the small localized changes that would be anticipated for a single point mutation. Instead, these single missense potentiating mutations confer signal changes similar to deletion of the entire MD. Because deletion of the entire MD renders Hsp104 inactive in luciferase reactivation, while the variants display enhanced activity, our results also suggest that the presence of the MD remains important for allosteric control, even when the coiled‐coil is disrupted.
To more specifically assess the coiled‐coil content of the variants, and confirm that the changes in spectra we observed were due to modulation of the MD, we calculated the ratio of molar ellipticities at 222 and 208 nm ([θ]222/[θ]208). This ratio has been used to evaluate the presence of coiled‐coil helices. 44 , 45 Two stranded coiled‐coils typically display a ratio of ~1.03 while non‐interacting α‐helices display a ratio of ~0.83. 44 , 45 Because the MD comprises just 14% of the entire Hsp104 protein, we would anticipate that the [θ]222/[θ]208 would not approach 1.03 even with a fully intact coiled‐coil MD. Correlating with our predictions, the [θ]222/[θ]208 for Hsp104 is 0.920 while the [θ]222/[θ]208 for Hsp104ΔMD is 0.842. The [θ]222/[θ]208 for Hsp104E432V, Hsp104K480V and Hsp104A503V each showed a decreased [θ]222/[θ]208 to 0.915, 0.905, and 0.896, respectively, consistent with loss of coiled‐coil structure (Figure 5c). We suggest that missense potentiating mutations likely confer increased activity not just by breaking interactions between MDs of adjacent protomers or interactions between the MD and NBD1, but that they also confer broad disruption of coiled‐coil structure of the MD, thereby relieving autoinhibition.
To further support our observations in the CD experiments, we also assessed the variants using the hydrophobic dye ANS (8‐anilino‐1‐naphthalene sulfonic acid). ANS can be used to study conformational changes in proteins, as ANS binds hydrophobic regions of proteins thereby increasing fluorescence. We anticipated that if the potentiated Hsp104 variants displayed loss of secondary structure, we would observe increased binding to ANS and therefore increased fluorescence (Figure 5d). We first studied Hsp104WT, Hsp104ΔMotif 1, and Hsp104ΔMD in this assay and observed no significant change in ANS binding as compared to Hsp104WT. This is likely because removal of the entire MD does not confer a substantial change in structure beyond complete removal of the coiled‐coil domain. Importantly, these results support the premise that in these biochemical experiments we are primarily observing changes restricted to the coiled‐coil region, and these mutations do not broadly disrupt the structure of the protein outside the MD. Supporting our conclusions from the CD experiments, we observed increased binding to ANS for both Hsp104E432V and Hsp104A503V. Hsp104K480V displayed a small, but not statistically significant, increase in ANS binding. In sum, our results suggest that missense mutations that potentiate Hsp104 confer these effects via destabilization of the coiled‐coil MD.
2.7. Potentiating mutations disrupt interactions that stabilize the Hsp104 structure
Our CD and ANS binding data suggest a model whereby potentiation is conferred by destabilization of the coiled‐coil. To further investigate this idea, we more closely analyzed the pool of potentiated variants. Many potentiating mutations identified in the Hsp104 MD likely disrupt intra‐ and inter‐protomer MD contacts. 16 , 31 Further supporting this idea of potentiation via destabilization, our scanning mutagenesis studies reveal that mutation of 24 of the 128 residues in the MD, or nearly 19% of the MD, confer potentiation when mutated to valine (Figure 6a, Table 1). We pooled our data with previous studies reporting potentiated Hsp104 MD variants. 8 , 12 , 13 , 24 To minimize confounding factors in selecting variants from the literature, we chose to only consider variants demonstrated to rescue disease substrate toxicity in yeast in our analysis. Remarkably, missense mutations at 32 of the 128 residues in the MD, or 25% of the residues, potentiate Hsp104 via mutation to valine or another residue (Figure 6a, Table 1). We noted that 12 of these 32 positions harbor a charged residue in the native sequence: E432, K451, R465, E469, K470, E474, K480, D484, R495, R496, D498, and D504, suggesting that ablation of electrostatic interactions is a key mechanism that enhances Hsp104 activity (Figure 6b, orange sticks). An additional 15 of these 32 positions harbor a hydrophobic residue in the native sequence: L423, V426, A437, Y466, A479, L483, A490, A493, A503, Y507, F508, I510, I513, I517, and M536, whereby modulation of these sites might disrupt hydrophobic interactions (Figure 6b, green sticks). The final five positions whereby potentiation can be conferred are more diverse: T477, T499, P511, G532, S535 (Figure 6b, red sticks). Electrostatic and hydrophobic interactions are known to be key drivers of coiled‐coil formation. 46 , 47 Therefore, our results support the idea that disruption of electrostatic and hydrophobic interactions that maintain the coiled‐coil structure of the MD could be a key driver of Hsp104 potentiation.
FIGURE 6.
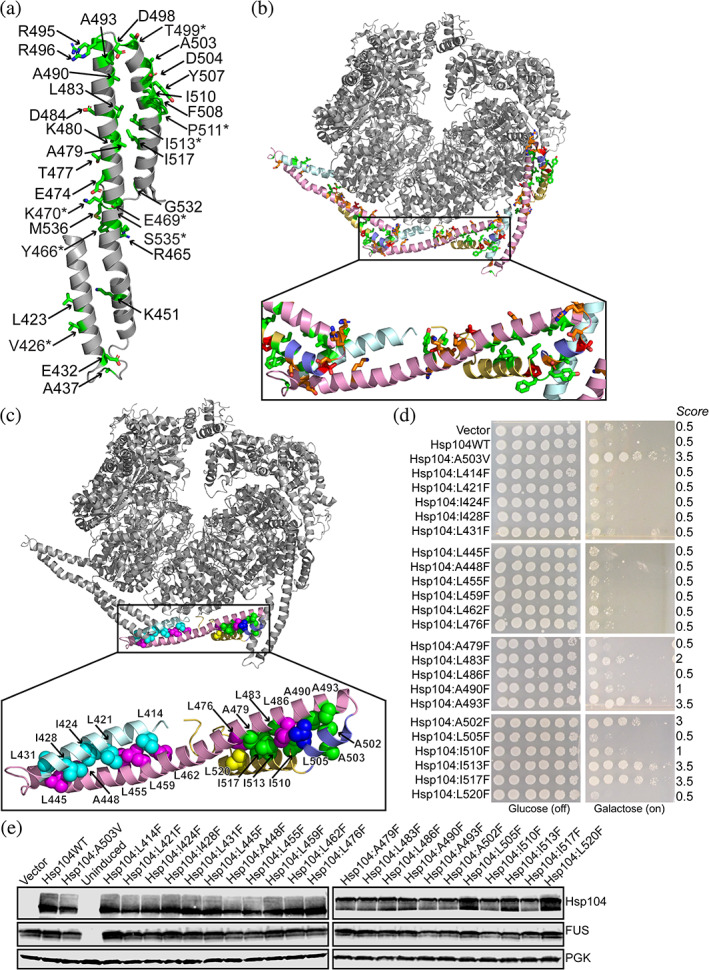
Diverse missense mutations throughout the Hsp104 MD confer potentiation. (a) Sites whereby mutations that potentiate Hsp104, conferring rescue of FUS toxicity, are shown as green sticks. Additional sites where Hsp104 is not potentiated via mutation to valine but potentiation has been reported via mutation to residues other than valine 8 , 24 are denoted with an asterisk. (b) Sites where introduction of missesene mutations confer Hsp104 potentiation are shown. Sites where mutation to valine could ablate electrostatic interactions are shown in orange, sites where mutation to valine could perturb hydrophobic interactions are shown in green, and other sites whereby mutation to valine confers potentiation are shown in red (PDB: 5VY8). (c) Potentiating mutations cluster in the helix 2 – helix 3/4 interface of the MD. Sites that mediate hydrophobic packing and coiled‐coil formation are shown as spheres, Hsp104A503 shown for reference. Sites where missense mutations confer potentiation are shown in green. Sites where missense mutations do not confer potentiation are colored by helix: helix 1: cyan, helix 2: pink, helix 3: blue, helix 4: yellow. (d) w303aΔhsp104‐pAG‐303GAL‐FUS yeast were transformed with Hsp104 variants or vector. Strains were serially diluted five‐fold and spotted in duplicate onto glucose (non‐inducing, left) or galactose (inducing, right) media. (e) Strains in (d) were induced for 5 hr, lysed, and immunoblotted for Hsp104, FUS, and 3‐phosphogylcerate kinase (PGK; loading control)
Hydrophobic residues were the most common type of amino acid whereby mutation conferred potentiation. While hydrophobic residues where valine substitutions conferred potentiation are found in all four helices of the MD, as well as in regions contacting NBD1, we noted that these sites were particularly enriched in helices 3 and 4 (Figure 6b). We analyzed the positioning of all hydrophobic residues in the Hsp104 structure and noted that the coiled‐coil structure of the MD is presumably maintained by the interactions mediated via a strip of hydrophobic residues lining the interface between helices 1 and 2 as well as a second strip lining the interface between helix 2 and helices 3/4. However, positions whereby Hsp104 can be potentiated are far more abundant in the second interface between helix 2 and helices 3/4 rather than in the interface between helices 1 and 2 (Figure 6b, c). Furthermore, sites whereby potentiation can be conferred in helix 1, such as L423 and V426, may contact helices 3 and 4 of the adjacent MD, thereby still conferring potentiation via modulation of this same interface (Figure 6b).
If structural destabilization does confer potentiation as our biochemical data suggests (Figure 5), we would predict that a more significant structural perturbation would confer more robust potentiation. Additionally, because our scanning mutagenesis experiments employed valine, we were cautious that at many sites introduction of valine might result in too modest of a perturbation to confer potentiation. We therefore sought to validate our findings from the circular dichroism and ANS binding assays in our yeast system by testing more significant structural perturbations. We noted that while Hsp104I513V does not rescue FUS, TDP‐43, or α‐syn toxicity (Figures S1‐S3), Hsp104I513F does suppress FUS, TDP‐43, and α‐syn toxicity. 24 Therefore, we chose to introduce a bulkier residue, phenylalanine, at the sites that appeared crucial to maintaining these two hydrophobic interfaces and assayed for suppression of FUS toxicity (Figure 6c,d). We also confirmed expression of each of the Hsp104 variants and that FUS expression levels remained fairly consistent across all strains (Figure 6e). Even with the bulkier phenylalanine mutation, none of the 10 variants with mutations in the interface between helices 1 and 2 (Hsp104L414F, Hsp104L421F, Hsp104I424F, Hsp104I428F, Hsp104L431F, Hsp104L445F, Hsp104A448F, Hsp104L455F, Hsp104L459F, and Hsp104L462F) displayed potentiation (Figure 6c,d). In contrast, in the interface between helix 2 and helices 3/4, we found that mutation to valine, phenylalanine, or other residues yielded potentiated variants at 9 of 13 sites tested: Hsp104A479V, Hsp104L483V/F, Hsp104A490V/F, Hsp104A493F, Hsp104A502F, Hsp104A503X (where x = any amino acid other than alanine), Hsp104I510V/F, Hsp104I513F, and Hsp104I517V/F (Figure 6c,d). 8 , 24 Mutations to valine or phenylalanine do not confer potentiation at positions L476, L486, L505, or L520. However, L476 and L520 are positioned at the end of this interface while L486 and L505 may be positioned suboptimally to mediate hydrophobic contacts (Figure 6c,d). With the identification of Hsp104A502F as a potentiated variant, in total we find that mutation at 33 of 128 sites, or 26% of residues in the MD, can be potentiated via single missense mutations. Furthermore, we find that FUS is more robustly rescued by variants harboring substitutions to phenylalanine instead of valine at positions A493 and I517, with increases in growth score of 1.5 to 2 spots. Additionally, at positions A502 and I513, substitution of phenylalanine, but not valine, confers potentiation (Figures 1 and 6d, Figure S1). These results support the hypotheses from our biochemical assays, indicating that a larger perturbation due to introduction of a bulkier side chain does lead to increased potentiation. In sum, our findings suggest that perturbation specifically in the hydrophobic interface between helix 2 and helices 3/4, but not in the analogous interface between helices 1 and 2, disrupts the coiled‐coil structure of the MD thereby eliciting potentiation.
3. DISCUSSION
Hsp104 disaggregase activity is controlled by the coiled‐coil MD, which wraps around the hexamer and maintains Hsp104 in an autoinhibited state. 16 , 28 , 31 Binding of Hsp70 to the MD is thought to release this autoinhibition, boosting disaggregase activity. Hsp104 can be potentiated via single missense mutations that enhance Hsp104 activity and also eliminate the requirement for Hsp70. 8 , 24 Potentiated variants are more active than Hsp104 WT in combination with Hsp70, and such variants can remodel a more diverse repertoire of substrates. While Hsp104 is inactive against TDP‐43 and FUS, both of which underpin ALS, potentiated Hsp104 variants robustly suppress the toxicity and mislocalization of these proteins. 8 , 27 Specifically, how single point mutations can replace the requirement for Hsp70 binding and confer potentiation has remained elusive. Additionally, understanding how to precisely manipulate the properties of Hsp104 is a key goal for furthering the development of therapeutic disaggregases. 7
Here, we have comprehensively explored the scope of missense mutations in the Hsp104 MD that elicit Hsp104 potentiation. Using scanning mutagenesis, we have constructed a library of 128 missense mutants of Hsp104, wherein each position is mutated to valine. From this library of valine mutants, we have found that 24 variants (~19% of the variants in the library) suppress FUS toxicity, 20 suppress α‐syn toxicity, and 11 suppress TDP‐43 toxicity. Each of the variants that suppresses α‐syn and/or TDP‐43 toxicity also suppresses FUS toxicity. While we anticipated that the pool of potentiated variants that rescue FUS and TDP‐43 toxicity would be the most similar, we find that the requirements for rescue of FUS and α‐syn are actually more similar than those for rescue of FUS and TDP‐43. Mutations to diverse amino acids have been found to confer a therapeutic gain‐of‐function, and when compiling the results of our scanning mutagenesis with earlier studies, 8 , 12 , 24 we find that single missense mutations at 26% of the sites in the Hsp104 MD confer potentiation, suppressing FUS, TDP‐43, and/or α‐syn toxicity. Of these 128 variants, 88% retain the capacity to confer thermotolerance at levels similar to Hsp104 WT. It is surprising that the introduction of a single point mutation more often potentiates Hsp104 than disrupts its native function.
Typically, potentiated Hsp104 variants harboring missense mutations in the MD display impaired expression of the Hsp104 variant and disease substrate, along with impaired growth at elevated temperatures. 8 , 9 , 24 In contrast, several potentiated Hsp104 NBD1 variants have been uncovered that do not display these off‐target effects. 12 We find that Hsp104R465V, which is also robustly potentiated, is expressed at similar levels to Hsp104 WT and yeast expressing Hsp104R465V do not exhibit significantly impaired expression of the disease substrate. Also Hsp104R465V does not confer temperature sensitivity and is active in thermotolerance. Thus we conclude that while off‐target effects may be generally characteristic of Hsp104 variants potentiated via MD mutations, Hsp104 MD variants do not uniformly exhibit off‐target toxicity. Therefore, potentiated Hsp104 variants can be generated to safely counter TDP‐43, FUS, and α‐syn toxicity via missense mutations to the MD as well as NBD1. Additionally, engineering Hsp104 activity against human disease substrates does not necessarily come at the cost of activity against the native yeast repertoire. Further, it appears that off‐target effects can be diminished while still retaining activity against the diverse range of native yeast substrates. Future work will likely uncover the determinants of these off‐target effects, as well as how to further delineate the drivers of temperature sensitivity and thermotolerance, which will be important to precisely control in the further engineering of therapeutic Hsp104 variants.
We have uncovered several new insights regarding mechanistic drivers of Hsp104 potentiation. Hydrophobic residues line the interfaces between helix 1 and helix 2, as well as between helix 2 and helices 3/4, and these strips are likely the key residues that drive the MD to form its characteristic coiled‐coil structure. We have now used an iterative approach to comprehensively characterize the scope of potentiating mutations. We find that disruption of hydrophobic packing in the helix 2—helix 3/4 interface, but not the helix 1—helix 2 interface, confers potentiation. A perhaps equally important mechanism leading to this gain‐of‐function is via disruption of intra‐ and inter‐protomer electrostatic interactions. Potentiating mutations that likely disrupt electrostatic interactions are found throughout the MD, oriented to disrupt both inter‐ and intra‐protomer MD interactions as well as MD—NBD1 interactions.
To comprehensively elucidate the mechanism of Hsp104 potentiation, it is important to understand the structural changes that accompany functional potentiation. Previous work has suggested two primary mechanisms that might confer this therapeutic gain‐of‐function. Disruption of inter‐protomer MD contacts between adjacent protomers can confer Hsp104 potentiation, as can disruption of intra‐protomer MD‐NBD1 contacts. 12 , 16 , 31 Breaking these contacts has been found to enhance activity by speeding ADP dissociation from NBD1. 34 Potentiating mutations may also mimic the effects of Hsp70 activation of Hsp104. 42 , 48 Here, we propose an additional mechanism: broadscale disruption of the coiled‐coil structure itself can also confer potentiation. While certain mutations may break interactions and loosen the dynamics of the coiled‐coil MD, we propose that some missense mutations lead to broad unfolding of most or all of the coiled‐coil region. Unfolding of the MD presumably relieves its autoinhibitory pressure, thereby enhancing substrate disaggregation. Based on this model, we designed a series of variants to be more substantially modulated and confirmed that greater modulation of this interface leads to greater enhancements in potentiation. Our findings correlate with earlier work employing HX MS to study a series of potentiated Hsp104 variants, with some important differences. 34 In the HX MS studies Hsp104A503S, another variant potentiated via mutation to the MD, was found to destabilize helix 3 thereby increasing dynamics of the outer region of the MD. 34 Here, increased dynamics between helices 1 and 3 was identified as a key change accompanying potentiation. While the authors conclude that the A503S mutation destabilizes the MD, they do not detect unfolding of the entire MD as we find in our studies. However, it is important to note that Hsp104A503S and Hsp104A503V have different properties, and Hsp104A503S is generally more stable than Hsp104A503V. 8 Additionally, some differences might be due to the slightly different conditions used in the two studies. We suggest that while certain potentiated variants display broad unfolding of the MD, this is not an absolute requirement for potentiation, and some potentiated variants display more localized unfolding of the MD. In future work, it will be important to further examine the MD structure of potentiated Hsp104 variants using a range of high‐resolution approaches.
It remains puzzling why Hsp104 is not naturally potentiated. Many potentiated variants confer temperature sensitivity or impaired thermotolerance, properties which would likely select against such naturally potentiated variants. However, some variants such as Hsp104R465V display enhanced disaggregase activity without these off‐target effects. Considering that mutation to approximately 26% of positions in the Hsp104 MD confers potentiation, and at many sites degenerate mutations enhance Hsp104 activity, 8 , 24 considerable selective pressures likely have been imposed to converge on the natural Hsp104 sequence. However, if the potentiated form of Hsp104 were deleterious, it is further confounding that additional mutations have not emerged to limit the possibility of accessing a naturally potentiated Hsp104 sequence. We therefore postulate that the MD may serve as a functional switch, whereby Hsp104 activity can be unleashed under certain conditions to promote disaggregation. Secondary mutations might be detrimental because they could restrict access to the potentiated state.
The targeted disaggregation and reactivation of misfolded substrates could simultaneously counter a loss‐ or gain‐of‐function mechanism and is therefore an attractive approach for therapeutically targeting neurodegenerative disease. Potentiated Hsp104 variants could be a valuable tool for these purposes. A remarkably broad number of mutations are capable of potentiating Hsp104 likely via multiple mechanisms. Ultimately, it will be crucial to comprehensively understand the scope of mutations in the entire Hsp104 protein that can confer potentiation, as well as the ways in which these mutations drive Hsp104 potentiation. Our findings reveal important new insights into the mechanism of Hsp104 activity and the requirements for its potentiation, and we have demonstrated that these insights can be applied to rationally tune variants with improved properties.
4. MATERIALS AND METHODS
4.1. Yeast strains, media, and plasmids
All yeast were WT W303aΔhsp104 (MATa, can1‐100, his3‐11,15, leu2‐3, 112, trp1‐1, ura3‐1, ade2‐1). 40 Yeast were grown in rich medium (YPD) or in synthetic media lacking the appropriate amino acids. Media was supplemented with 2% glucose, raffinose, or galactose. Strains integrated with TDP‐43, FUS, and α‐synuclein (pAG303GAL‐TDP‐43, pAG303GAL‐FUS, pAG303GAL‐α‐synuclein‐YFP, and pAG304GAL‐α‐synuclein‐YFP) have been previously described. 8 pAG416GAL‐Hsp104 has been previously described. 8 All mutations were constructed using QuikChange site‐directed mutagenesis (Agilent) and confirmed by DNA sequencing.
4.2. Yeast transformation and spotting assays
Yeast were transformed according to standard protocols using polyethylene glycol and lithium acetate. 49 Hsp104 variants in the pAG416GAL‐Hsp104 plasmid were transformed into the indicated strains. For the spotting assays, yeast were grown to saturation overnight in raffinose supplemented dropout media at 30°C, serially diluted, and spotted in duplicate onto synthetic dropout media containing glucose or galactose. Plates were analyzed after growth for 2–3 days at 30°C. Each experiment was repeated with at least three independent transformations.
4.3. Temperature sensitivity
W303Δhsp104 yeast were transformed with the indicated pAG416GAL‐Hsp104 plasmid. Yeast were grown overnight in synthetic raffinose medium to A600nm = 2.0 and spotted in duplicate onto SD‐Ura or SGal‐Ura media and incubated at both 30 and 37°C. Plates were analyzed after 2–3 days of growth. Experiments were repeated with at least three independent transformations.
4.4. Immunoblotting
Yeast were grown and induced in galactose containing medium for 5 h from overnight cultures supplemented with raffinose. Cultures were normalized to A600nm = 0.6, 3 ml cells were harvested, treated in 0.1 M NaOH for 5 min at room temperature, and cell pellets were then resuspended into 1× SDS sample buffer and boiled for 4 min. Lysates were cleared by centrifugation at 14,000 rpm for 2 min and then separated by SDS‐PAGE (4%–20% gradient, BioRad), and transferred to a PVDF membrane. Membranes were blocked in Odyssey Blocking Buffer (LI‐COR). Primary antibody incubations were performed at 4°C overnight. Antibodies used: anti‐GFP monoclonal (Roche Applied Science), anti‐TDP‐43 polyclonal (Proteintech), anti‐FUS polyclonal (Bethyl Laboratories), anti‐Hsp104 polyclonal (Enzo Life Sciences), and anti‐PGK monoclonal (Invitrogen). Membranes were imaged using a LI‐COR Odyssey FC Imaging system.
For quantitation of expression level, a minimum of three immunoblots were obtained per condition. Quantitation was performed using LI‐COR Image Studio. Values were normalized to a 3‐PGK loading control, expressed relative to levels in a strain expressing Hsp104WT, and averaged. Comparisons were made using a one‐way ANOVA with a Dunnett's multiple comparisons test.
4.5. Thermotolerance
W303aΔhsp104 yeast were transformed in 96‐well plate format with the indicated 416GAL Hsp104 plasmid or 416GAL vector control. Cultures were grown in synthetic raffinose medium to saturation and diluted to A600nm = 0.3 in synthetic galactose medium. After 5 h outgrowth at 30°C, cells were treated for 30 min at 37°C, heat shocked for 0 or 60 min at 50°C, cooled for 2 min on ice, serially diluted, and spotted onto synthetic dropout media supplemented with glucose or galactose. Plates were analyzed after growth for 2–3 days at 30°C. Experiments were repeated with at least three independent transformations. To represent the data in the heat map, rescue scores were adjusted to normalize a WT level of growth to a score of 5.5.
4.6. Protein purification
Hsp104 proteins were expressed and purified as untagged proteins from Escherichia coli. Proteins were overexpressed in BL21(DE3) RIL. Cells were harvested, lysed with lysis buffer (50 mM Tris pH 8.0, 10 mM MgCl2, 2.5% glycerol, 2 mM β‐mercaptoethanol) supplemented with protease inhibitors, and the protein was purified using Affi‐Gel Blue Gel (Bio‐Rad). The protein was eluted with elution buffer (50 mM Tris pH 8.0, 1 M KCl, 10 mM MgCl2, 2.5% glycerol, 2 mM β‐mercaptoethanol). The eluate was buffer exchanged into high salt storage buffer (40 mM HEPES‐KOH pH 7.4, 500 mM KCl, 20 mM MgCl2, 10% glycerol, 1 mM DTT). The protein was then further purified by ResourceQ anion exchange chromatography using running buffer Q (20 mM Tris pH 8.0, 0.5 mM EDTA, 5 mM MgCl2, 50 mM NaCl) and eluted with a linear gradient of buffer Q+ (20 mM Tris pH 8.0, 0.5 mM EDTA, 5 mM MgCl2, 1 M NaCl). Immediately before loading the column, the protein was diluted in buffer Q to achieve 150 mM NaCl and loaded onto the column using a sample pump. The eluted protein was then concentrated and exchanged into high salt storage buffer, flash frozen, and stored at −80°C until use. All proteins were used within 2 days of thawing and no proteins were re‐frozen. Hsp104 concentrations refer to the hexamer concentration. Firefly luciferase was from Sigma and creatine kinase was from Roche. Hsc70 and Hdj2 were from Enzo Life Sciences.
4.7. CD spectroscopy
Purified Hsp104 variants were thawed and dialyzed exhaustively using Slide‐A‐Lyzer Dialysis Cassettes (ThermoFisher) into CD buffer (50 mM Na3PO4, 50 mM K3PO4, 10 mM MgOAc, 1 mM DTT, pH 7) at 4°C. For each set of replicates, all samples were dialyzed in the same container to ensure a buffer match. Following dialysis, concentrations were normalized to 1 or 2 μM monomer and measured again for later calculation of molar ellipticity. CD spectra were recorded on a JASCO‐J‐815 CD spectrometer using a quartz cuvette of 0.1 cm optical path length at room temperature. CD spectra were scanned from 300 to 190 nm at 50 nm/min with a bandwidth of 0.1 nm. Final data were converted to molar ellipticity [θ] in deg cm2 dmol−1 using equation [θ]λ = θλ/(10 * d * c) where θ = millidegrees at λ, d is the pathlength in cm, and c is the concentration in M. Experiments were repeated at least four times.
4.8. Luciferase reactivation assay
Luciferase reactivation was performed as described in Reference 8 with some modifications. To assemble aggregates, firefly luciferase (50 μM) in luciferase‐refolding buffer (LRB: 25 mM HEPES‐KOH pH 7.4, 150 mM KAOc, 10 mM MgAOc, 10 mM DTT) plus 8 M urea was incubated at 30°C for 30 min. The sample was then rapidly diluted 100‐fold into LRB. Aliquots were snap frozen and stored at −80°C until use. Aggregated luciferase (50 nM) was incubated with Hsp104 (0.167 μM hexamer) with ATP (5.1 mM) and an ATP regeneration system (1 mM creatine phosphate, 0.25 μM creatine kinase) in the presence or absence of Hsc70 (0.167 μM) and Hdj2 (0.167 μM) for 90 min at 25°C in CD buffer. At the end of the reaction, luciferase activity was assessed with a luciferase assay system (Promega). Recovered luminescence was monitored using a Tecan Spark plate reader.
4.9. ATPase assay
ATPase assays were performed as described. 8 Hsp104 (0.25 μM monomer) was incubated with ATP (1 mM) for 5 min at 25°C. ATPase activity was assessed by the release of inorganic phosphate, which was determined using a malachite green phosphate detection kit (Innova). Background hydrolysis was determined at time zero and subtracted.
4.10. ANS fluorescence assay
Hsp104WT and variants (2.5 μM monomer) were incubated with 50 μM ANS (8‐anilino‐1‐naphthalene sulfonic acid) at room temperature in CD buffer. Fluorescence spectra were recorded from 390 to 650 nm with a 10 nm bandwidth following excitation at 370 nm using a Tecan Spark plate reader. Average fluorescence values at 480 nm are shown.
AUTHOR CONTRIBUTIONS
Jeremy Ryan: Conceptualization; formal analysis; funding acquisition; investigation; writing‐original draft; writing‐review & editing. Aaron Bao: Investigation; writing‐review & editing. Braxton Bell: Investigation; writing‐review & editing. Cendi Ling: Investigation; writing‐review & editing. Meredith Jackrel: Conceptualization; funding acquisition; investigation; methodology; resources; supervision; writing‐original draft; writing‐review & editing.
Supporting information
Appendix S1: Supporting Information
ACKNOWLEDGEMENTS
We thank Jai Rudra for feedback and assistance with the CD spectroscopy experiments. This work was supported by a Center for Science and Engineering of Living Systems (CSELS) Fellowship (to Jeremy J. Ryan), a WUSTL uSTAR Award (to Aaron Bao), a WUSTL HHMI BioSURF Award (to Cendi Ling), NIH grant R35GM128772, a TargetALS Springboard Award, a Frick Foundation for ALS Award, and an ALS Association Award (to Meredith E. Jackrel).
Ryan JJ, Bao A, Bell B, Ling C, Jackrel ME. Drivers of Hsp104 potentiation revealed by scanning mutagenesis of the middle domain. Protein Science. 2021;30:1667–1685. 10.1002/pro.4126
Funding information Amyotrophic Lateral Sclerosis Association; Bruno and Ilse Frick Foundation for Research on ALS; Center for Science and Engineering of Living Systems Fellowship; National Institute of General Medical Sciences, Grant/Award Number: R35GM128772; Target ALS
REFERENCES
- 1. Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci. 2013;14:248–264. [DOI] [PubMed] [Google Scholar]
- 2. Taylor JP, Brown RH Jr, Cleveland DW. Decoding ALS: From genes to mechanism. Nature. 2016;539:197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Forman MS, Trojanowski JQ, Lee VM. Neurodegenerative diseases: A decade of discoveries paves the way for therapeutic breakthroughs. Nat Med. 2004;10:1055–1063. [DOI] [PubMed] [Google Scholar]
- 4. Lagier‐Tourenne C, Polymenidou M, Cleveland DW. TDP‐43 and FUS/TLS: Emerging roles in RNA processing and neurodegeneration. Hum Mol Genet. 2010;19:R46–R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jackrel ME, Shorter J. Engineering enhanced protein disaggregases for neurodegenerative disease. Prion. 2015;9:90–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jackrel ME, Shorter J. Reversing deleterious protein aggregation with re‐engineered protein disaggregases. Cell Cycle. 2014;13:1379–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jackrel ME, Shorter J. Protein‐remodeling factors as potential therapeutics for neurodegenerative disease. Front Neurosci. 2017;11:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jackrel ME, DeSantis ME, Martinez BA, et al. Potentiated Hsp104 variants antagonize diverse proteotoxic misfolding events. Cell. 2014;156:170–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jackrel ME, Shorter J. Potentiated Hsp104 variants suppress toxicity of diverse neurodegenerative disease‐linked proteins. Dis Model Mech. 2014;7:1175–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guo L, Kim HJ, Wang H, et al. Nuclear‐import receptors reverse aberrant phase transitions of RNA‐binding proteins with prion‐like domains. Cell. 2018;173:677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ryan JJ, Sprunger ML, Holthaus K, Shorter J, Jackrel ME. Engineered protein disaggregases mitigate toxicity of aberrant prion‐like fusion proteins underlying sarcoma. J Biol Chem. 2019;294:11286–11296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tariq A, Lin J, Jackrel ME, et al. Mining disaggregase sequence space to safely counter TDP‐43, FUS, and α‐synuclein proteotoxicity. Cell Rep. 2019;28:2080–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tariq A, Lin J, Noll MM, et al. Potentiating Hsp104 activity via phosphomimetic mutations in the middle domain. FEMS yeast res. 2018;18:foy042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Newby GA, Lindquist S. Blessings in disguise: Biological benefits of prion‐like mechanisms. Trends Cell Biol. 2013;23:251–259. [DOI] [PubMed] [Google Scholar]
- 15. Park YN, Zhao X, Yim YI, et al. Hsp104 overexpression cures Saccharomyces cerevisiae [PSI+] by causing dissolution of the prion seeds. Eukaryot Cell. 2014;13:635–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gates SN, Yokom AL, Lin J, et al. Ratchet‐like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science. 2017;357:273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee S, Sowa ME, Y‐h W, et al. The structure of ClpB: A molecular chaperone that rescues proteins from an aggregated state. Cell. 2003;115:229–240. [DOI] [PubMed] [Google Scholar]
- 18. Parsell DA, Kowal AS, Singer MA, Lindquist S. Protein disaggregation mediated by heat‐shock protein Hsp104. Nature. 1994;372:475–478. [DOI] [PubMed] [Google Scholar]
- 19. Glover JR, Lindquist S. Hsp104, Hsp70, and Hsp40: A novel chaperone system that rescues previously aggregated proteins. Cell. 1998;94:73–82. [DOI] [PubMed] [Google Scholar]
- 20. Lo Bianco C, Shorter J, Regulier E, et al. Hsp104 antagonizes alpha‐synuclein aggregation and reduces dopaminergic degeneration in a rat model of Parkinson disease. J Clin Invest. 2008;118:3087–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. DeSantis ME, Leung EH, Sweeny EA, et al. Operational plasticity enables hsp104 to disaggregate diverse amyloid and nonamyloid clients. Cell. 2012;151:778–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cushman‐Nick M, Bonini NM, Shorter J. Hsp104 suppresses polyglutamine‐induced degeneration post onset in a Drosophila MJD/SCA3 model. PLoS Genet. 2013;9:e1003781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vacher C, Garcia‐Oroz L, Rubinsztein DC. Overexpression of yeast hsp104 reduces polyglutamine aggregation and prolongs survival of a transgenic mouse model of Huntington's disease. Hum Mol Genet. 2005;14:3425–3433. [DOI] [PubMed] [Google Scholar]
- 24. Jackrel ME, Yee K, Tariq A, Chen AI, Shorter J. Disparate mutations confer therapeutic gain of Hsp104 function. ACS Chem Biol. 2015;10:2672–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jackrel ME, Tariq A, Yee K, Weitzman R, Shorter J. Isolating potentiated Hsp104 variants using yeast proteinopathy models. J Vis Exp. 2014;93:e52089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Michalska K, Zhang K, March ZM, et al. Structure of Calcarisporiella thermophila Hsp104 disaggregase that antagonizes diverse proteotoxic misfolding events. Structure. 2019;27:449–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yasuda K, Clatterbuck‐Soper SF, Jackrel ME, Shorter J, Mili S. FUS inclusions disrupt RNA localization by sequestering kinesin‐1 and inhibiting microtubule detyrosination. J Cell Biol. 2017;216:1015–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Oguchi Y, Kummer E, Seyffer F, et al. A tightly regulated molecular toggle controls AAA+ disaggregase. Nat Struct Mol Biol. 2012;19:1338–1346. [DOI] [PubMed] [Google Scholar]
- 29. Schirmer EC, Homann OR, Kowal AS, Lindquist S. Dominant gain‐of‐function mutations in Hsp104p reveal crucial roles for the middle region. Mol Biol Cell. 2004;15:2061–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Howard MK, Sohn BS, von Borcke J, Xu A, Jackrel ME. Functional analysis of proposed substrate‐binding residues of Hsp104. PLoS One. 2020;15:e0230198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Heuck A, Schitter‐Sollner S, Suskiewicz MJ, et al. Structural basis for the disaggregase activity and regulation of Hsp104. Elife. 2016;5:e21516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. DeSantis ME, Shorter J. The elusive middle domain of Hsp104 and ClpB: Location and function. Biochim Biophys Acta. 2012;1823:29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yokom AL, Gates SN, Jackrel ME, et al. Spiral architecture of the Hsp104 disaggregase reveals the basis for polypeptide translocation. Nat Struct Mol Biol. 2016;23:830–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ye X, Lin J, Mayne L, Shorter J, Englander SW. Structural and kinetic basis for the regulation and potentiation of Hsp104 function. Proc Natl Acad Sci U S A. 2020;117:9384–9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Johnson BS, McCaffery JM, Lindquist S, Gitler AD. A yeast TDP‐43 proteinopathy model: Exploring the molecular determinants of TDP‐43 aggregation and cellular toxicity. Proc Natl Acad Sci U S A. 2008;105:6439–6444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sun Z, Diaz Z, Fang X, et al. Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol. 2011;9:e1000614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ju S, Tardiff DF, Han H, et al. A yeast model of FUS/TLS‐dependent cytotoxicity. PLoS Biol. 2011;9:e1001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Outeiro TF, Lindquist S. Yeast cells provide insight into alpha‐synuclein biology and pathobiology. Science. 2003;302:1772–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. King OD, Gitler AD, Shorter J. The tip of the iceberg: RNA‐binding proteins with prion‐like domains in neurodegenerative disease. Brain Res. 2012;1462:61–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sanchez Y, Lindquist SL. HSP104 required for induced thermotolerance. Science. 1990;248:1112–1115. [DOI] [PubMed] [Google Scholar]
- 41. DeSantis ME, Sweeny EA, Snead D, et al. Conserved distal loop residues in the Hsp104 and ClpB middle domain contact nucleotide‐binding domain 2 and enable Hsp70‐dependent protein disaggregation. J Biol Chem. 2014;289:848–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sweeny EA, Tariq A, Gurpinar E, et al. Structural and mechanistic insights into Hsp104 function revealed by synchrotron X‐ray footprinting. J Biol Chem. 2020;295:1517–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Durie CL, Lin J, Scull NW, et al. Hsp104 and potentiated variants can operate as distinct nonprocessive translocases. Biophys J. 2019;116:1856–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Choy N, Raussens V, Narayanaswami V. Inter‐molecular coiled‐coil formation in human apolipoprotein E C‐terminal domain. J Mol Biol. 2003;334:527–539. [DOI] [PubMed] [Google Scholar]
- 45. Fiumara F, Fioriti L, Kandel ER, Hendrickson WA. Essential role of coiled coils for aggregation and activity of Q/N‐rich prions and PolyQ proteins. Cell. 2010;143:1121–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lupas A. Coiled coils: New structures and new functions. Trends Biochem Sci. 1996;21:375–382. [PubMed] [Google Scholar]
- 47. Adamson JG, Zhou NE, Hodges RS. Structure, function and application of the coiled‐coil protein folding motif. Curr Opin Biotechnol. 1993;4:428–437. [DOI] [PubMed] [Google Scholar]
- 48. Torrente MP, Chuang E, Noll MM, Jackrel ME, Go MS, Shorter J. Mechanistic insights into Hsp104 potentiation. J Biol Chem. 2016;291:5101–5115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gietz RD, Schiestl RH. High‐efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protocols. 2007;2:31–34. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information