

Abstract
Precision oncology is premised on identifying and drugging proteins and pathways that drive tumorigenesis or are required for survival of tumor cells. Across diverse cancer types, the signaling pathway emanating from receptor tyrosine kinases on the cell surface to RAS and the MAP kinase pathway is the most frequent target of oncogenic mutations, and key proteins in this signaling axis including EGFR, SHP2, RAS, BRAF, and MEK have long been a focus in cancer drug discovery. In this review, we provide an overview of historical and recent efforts to develop inhibitors targeting these nodes with an emphasis on the role that an understanding of protein structure and regulation has played in inhibitor discovery and characterization. Beyond its well‐established role in structure‐based drug design, structural biology has revealed mechanisms of allosteric regulation, distinct effects of activating oncogenic mutations, and other vulnerabilities that have opened new avenues in precision cancer drug discovery.
Keywords: cancer biology, drug discovery, kinase, structural biology
Short abstract
PDB Code(s): 2GS7, 2GS6, 2EB3, 4LRM, 2ITZ, 4G5J, 1XKK, 3IKA, 6JX0, 6V5N, 6DUK, 6P1L, 6P1D, 4LYF, 6JG7, 6NYB, 6Q0J, 1S9J, 6PP9, 7JUX, 2SHP, 5EHR, 1UWH, 4MNF and 3OG7.
1. INTRODUCTION
To a large extent, cellular proliferation is controlled by signaling from receptor tyrosine kinases (RTKs) positioned on the cell surface through a cascade of intracellular signaling via the RAS/mitogen‐activated protein (MAP) kinase pathway. 1 The central role of this pathway in controlling cellular physiology requires that it is tightly regulated, and is axiomatic that activating somatic mutations in key components of this pathway are frequent causes of cancer. Ras proteins (HRAS, KRAS, and NRAS) are a central switch in this pathway, 2 and the discovery of the oncogenic G12V substitution in the early 1980's showed that even a single base substitution, leading to alteration of one amino acid in the encoded protein, was sufficient to transform cells. 3 , 4 , 5 In the ensuing decades, activating alterations in additional components of the pathway were identified as cancer drivers, perhaps most prominently EGFR 6 , 7 , 8 and BRAF. 9
The recent explosion in genomic analysis enabled by massively parallel sequencing has led to near‐comprehensive characterization of genetic alterations in diverse tumor types. 10 , 11 Analysis of whole cancer exomes and even genomes across diverse cancer types has allowed measurement of the prevalence of previously known driver mutations and identification of still more. The RTK/Ras/MAP kinase pathway is the most frequent target of these oncogenic mutations. 10 Recognition of the role of this pathway in cancer has led to intense efforts to develop drugs targeting pathway components. Multiple approved drugs that inhibit components of this pathway are now available, and sequencing of biopsy specimens is widely used to deploy these agents in a targeted fashion. 12
Development of cancer therapeutics against diverse molecular targets has long been guided and enabled by structural analysis. 13 , 14 Indeed, it is rare for a drug discovery effort to proceed in the absence of structural understanding of the intended receptor. In addition to traditional structure‐guided inhibitor optimization, structural tools are increasingly important for initial discovery of chemical matter that binds a target of interest through fragment‐based approaches. 15 Historically, X‐ray crystallography and solution NMR have been the dominant tools in this area, but cryo‐electron microscopy (cryo‐EM) promises to play an increasingly important role, in particular for large macromolecular complexes that are not amenable to X‐ray crystallography. 16 , 17 Beyond traditional structure‐guided inhibitor optimization, structural insights advance drug discovery via mechanistic information; for example, understanding and exploiting allosteric regulation, overcoming mechanisms of inhibitor resistance, or identifying approaches to obtaining mutant selectivity.
In this review, we highlight these diverse ways in which structural biology has advanced cancer drug discovery, taking examples from inhibitors targeting the RTK/Ras/MAP kinase pathway. This signaling cascade is summarized graphically in Box 1. Each of the nodes in this pathway has been the subject of recent in‐depth reviews focused on inhibitor discovery and application in relevant cancers. 12 , 18 , 19 , 20 , 21 , 22 , 23 Here, through a structural lens, we discuss multiple generations of inhibitors targeting the epidermal growth factor receptor (EGFR) as a representative RTK, as well as relatively recent advances in targeting K‐Ras, which had long been considered “undruggable.” We also consider allosteric inhibitors of SHP2, a tyrosine phosphatase that plays a crucial but still poorly understood role in regulation of this pathway. Finally, we discuss development of inhibitors targeting the BRAF and MEK kinases, the first two of three kinase links in the MAP kinase cascade. Owing to space considerations, we omit consideration of inhibitors of Erk1/2, the terminal kinase in the cascade and also an important target for anticancer drug discovery. 21
BOX 1.
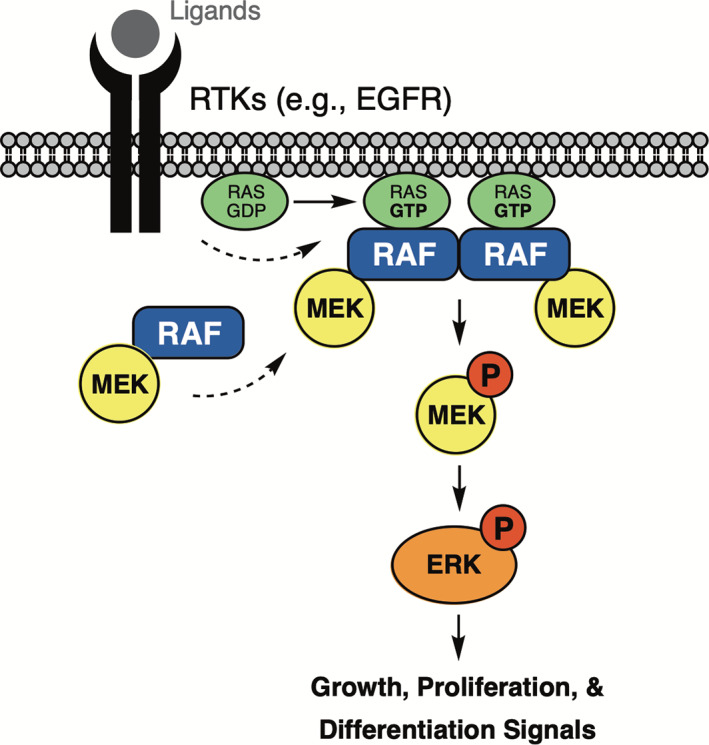
The RTK/Ras/MAP kinase signaling cascade. Activation of the epidermal growth factor receptor (EGFR) and other RTKs on the cell surface promotes recruitment of guanine‐nucleotide exchange factors that induce loading of RAS with GTP. GTP‐bound RAS recruits RAF‐family kinases to the membrane (there are three Raf isoforms, ARAF, BRAF, and CRAF/RAF‐1), where they are activated by dimerization. Once activated, they phosphorylate their only known substrates, MEK1 and MEK2. Active, phosphorylated MEK1/2 in turn phosphorylates ERK1 and ERK2, which have multiple targets for phosphorylation, including other kinases and transcription factors that control gene expression. Somatic mutations in each of the nodes in this pathway can lead to cancer; oncogenic activation of EGFR, K‐Ras, and BRAF is common, while activating mutations in MEK1/2 and ERK1/2 are relatively rare. The SHP2 tyrosine phosphatase (not illustrated) is also an important contributor to this pathway
2. EGFR—FIRST‐GENERATION TO THIRD‐GENERATION INHIBITORS AND BEYOND
The EGFR is one of four members of the ErbB‐family of RTKs that also includes ErbB2, ErbB3, and ErbB4. 24 , 25 These receptors homo‐ or heterodimerize upon binding their cognate growth factor ligands, which induces receptor autophosphorylation and activation. Oncogenic mutations in the receptor can bypass this normal ligand‐driven activation, resulting in constitutive signaling activity.
Somatic activating mutations within the EGFR kinase domain were first recognized in non‐small cell lung cancer (NSCLC), where their presence predicts for response to treatment with EGFR tyrosine kinase inhibitors (TKIs). 6 , 7 , 26 A number of activating alterations have been identified, including in‐frame deletions in exon 19, insertions in exon 20, and point mutations in exons 18 (e.g., G719S) and 21 (e.g., L858R). Despite their diversity, all effectively alter the structure of the kinase domain to override standard ligand‐dependent regulation. 27
The dual‐lobed structure of the EGFR kinase is representative of many protein kinases and contains two key regulatory components: the αC‐helix in the N‐lobe and the activation loop (A‐loop), which emanates from the C‐lobe (Figure 1a). In the inactive state, the αC‐helix is rotated outward and reinforced in this position by an α‐helical segment in the A‐loop. Dimerization of the intracellular kinase domains promoted by ligand binding to the ectodomains produces an asymmetric dimer (Figure 1b) involving kinase‐kinase domain interaction between the C‐lobe of one kinase (activator, yellow) and the N‐lobe of the other kinase. 31 This EGFR kinase dimer promotes activation of the receiver kinase through direct interaction with the activating kinase C‐lobe with the N‐lobe of the receiving kinase that cause inward rotation of the αC‐helix along with concomitant rearrangement of the A‐loop. 28 In general, oncogenic activating mutations override this regulatory mechanism by destabilizing the inactive state of the receptor, promoting a shift in equilibrium of the EGFR kinase to the active state. A well‐characterized example is the L858R point‐mutation, which lies in the helical segment of the A‐loop. 29 Substitution to an arginine is not structurally compatible with the hydrophobic environment at this position in the inactive state, and so it leads reorganization of the activation segment where the positively charged arginine residue is solvent exposed (Figure 1c). Although no crystal structures are available for an Exon19 deletion variant, these alterations shorten the loop that leads into the αC‐helix and thus are expected to enforce its inward, active position. In exon20 insertion mutants, the most common variants add 2–3 amino acid residues at the C‐terminus of the αC‐helix. 30 The inserted residues form a “wedge” that sterically stabilizes the αC‐helix in the inward, active position (Figure 1d).
FIGURE 1.
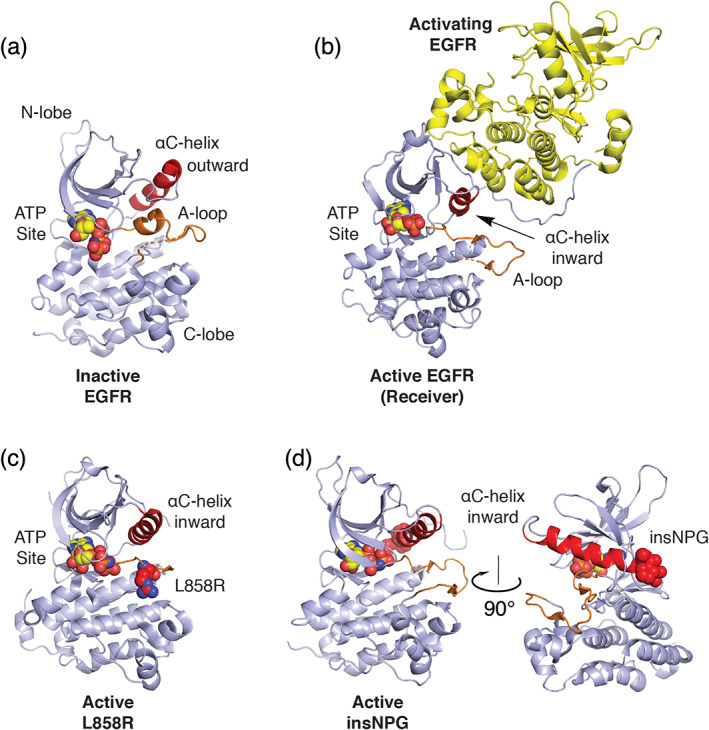
Structural basis for normal and oncogenic EGFR activation. (a) Structure of the EGFR kinase in its inactive state (PDB ID 2GS7). 28 A helical segment in the activation loop (A‐loop, orange) locks the αC‐helix (red) in an outward conformation. (b) Structure of the active kinase (receiver) promoted through asymmetric dimerization with an activating kinase domain. Based on PDB ID 2GS6. 28 (c) Structure of the EGFR kinase containing the oncogenic L858R activating mutation. The mutant Arg858 side chain is shown in space fill with carbon atoms in red (PDB ID 2ITV). 29 Residue L858 lies in the helical portion of the activation segment in the inactive state and the resulting Arginine (shown as red spheres) induces activation by destabilizing this element to allow the inward active position of the αC‐helix. (d) exon20 insNPG adds three amino acids (shown in red spheres) to the base of the αC‐helix sterically reinforcing the kinase in the active conformation (PDB ID 4LRM). 30 In all structures, ATP analogues that define the substrate site are shown in spacefill
First‐generation EGFR TKIs gefitinib and erlotinib are approved for treatment of NSCLC driven by the EGFR L858R point mutation and exon19 deletions. 6 , 7 These 4‐anilinoquinazoline‐based inhibitors (Scheme 1) are ATP‐competitive; they bind in the adenosine binding site of the kinase (Figure 2a). Importantly, these compounds turned out to be mutant‐selective, despite the fact that they were developed to inhibit the WT kinase. They owe their serendipitous mutant‐selectivity to an Achilles heel of the mutants; the mutants have lower affinity for substrate ATP. 26 , 29 Selectivity for mutant EGFR (or put another way, relative sparing of WT EGFR) is crucial for an effective therapeutic because inhibition of the normal receptor in the skin and gastrointestinal tract is the source of dose‐limiting toxicities for EGFR TKIs.
SCHEME 1.
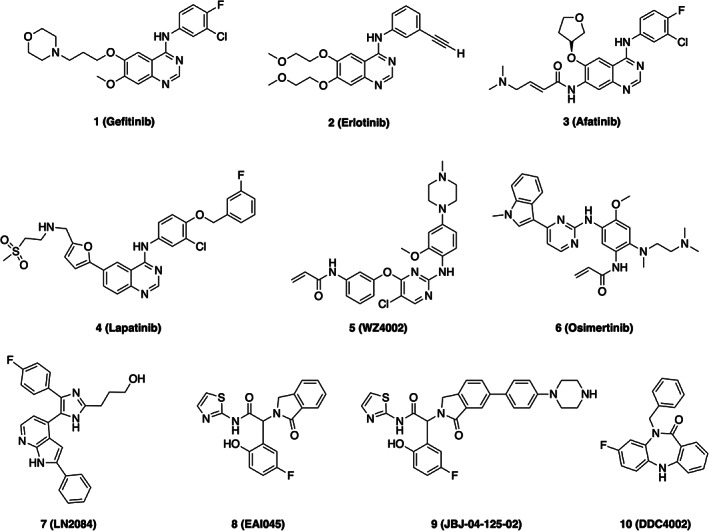
Chemical structures of selected EGFR inhibitors
FIGURE 2.
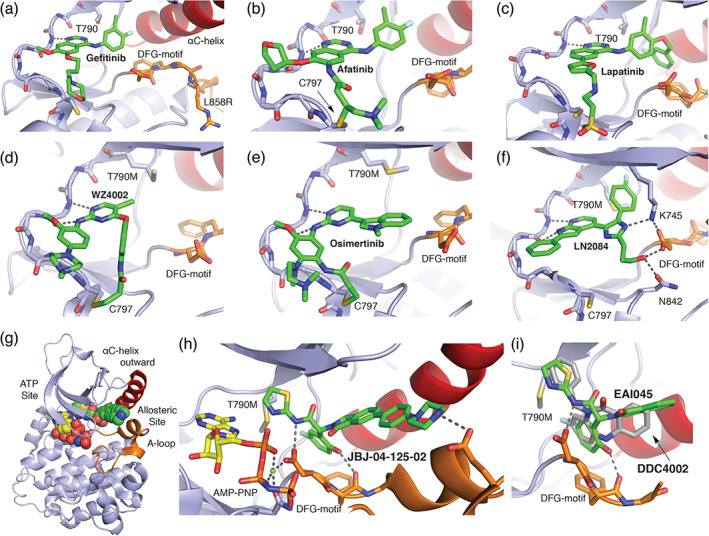
The diverse binding modes of EGFR inhibitors. (a) 1 Gefitinib co‐crystal structure with L858R EGFR (PDB ID 2ITZ). 29 For anilinoquinazoline inhibitors such as gefitinib, afatinib (b), and lapatinib (c), the quinazoline group hydrogen bonds with the “hinge” region of the kinase (dashed lines) and the aniline substituent extends into a hydrophobic pocket at the back of the ATP‐site. (b) 3 Afatinib co‐crystal structure with WT EGFR (PDB ID 4G5J). 32 Afatinib incorporates an acrylamide Michael acceptor that forms a covalent bond with C797 at the edge of the ATP‐site. (c) 4 Lapatinib co‐crystal structure PDB 1XKK. 33 Lapatinib is a “Type 1.5” inhibitor; the fluoro‐benzyl extension on the right‐hand side of the molecule enforces an inactive, C‐helix‐out conformation of the kinase. (d) 5 WZ4002 in complex with T790M EGFR (PDB ID 3IKA). 34 Anilinopyrimidine inhibitors WZ4002 and osimertinib (e) form dual hydrogen bonds with the kinase hinge (dashed lines), and both contain an acrylamide positioned to form a covalent bond with C797. (e) 6 Osimertinib co‐crystal structure with T790M EGFR (PDB ID 6JX0). 35 Note the alternate location of the acrylamide substituent as compared with WZ4002, which nevertheless takes a trajectory that allows formation of a covalent bond with C797. (f) 7 LN2084 in complex with T790M/V948R EGFR PDB ID 6V5N. 36 (g) Co‐crystal structure of T790M/V948R EGFR with allosteric inhibitor 9 JBJ‐04‐125‐02, and (h) zoom in on allosteric inhibitor binding mode PDB ID 6DUK. 37 The allosteric inhibitor occupies a pocket adjacent to, but separate from the ATP‐site. (i) Overlay of binding modes of allosteric inhibitors 8 EAI045 (PDB ID 6P1L) and 10 DDC4002 (PDB ID 6P1D). 38 Note that despite their dissimilar chemical structures, the compounds exhibit a related binding mode
Lapatinib (Figure 2c) is also an anilinoquinazoline‐based first generation inhibitor, but contains an additional fluoro‐benzyl substituent that extends into an allosteric pocket that requires the outward, inactive position of the αC‐helix. 33 , 39 Interestingly, a lapatinib co‐crystal structure with the EGFR kinase provided a first view of the inactive state of the EGFR kinase, including a stereotypical “αC‐helix out” inactive configuration previously seen in Src and cyclin‐dependent kinases. The authenticity of this inactive conformation was subsequently confirmed by drug‐free structures. 28 Though a potent inhibitor of EGFR, it was developed primarily as an inhibitor of the closely related ErbB2 (HER2) kinase, and is approved for treatment of certain breast cancers that overexpress HER2. Importantly, it has not proven effective in EGFR‐mutant lung cancers, in part because these mutations bias against the inactive conformation that this compound requires, and also in part due to its potency on the WT receptor, which leads to a lack of a therapeutic window.
Second‐generation EGFR TKIs build upon first‐generation inhibitors by adding a reactive “warhead” that is positioned to form a covalent bond with C797, which lies at the edge of the ATP site. The first of these irreversible EGFR TKIs were made in the late 1990s by Parke Davis chemists who developed the concept using homology models of the EGFR kinase that revealed the well‐positioned cysteine residue. 40 Introduction of an appropriately placed Michael‐acceptor, typically an acrylamide or derivative thereof, to EGFR‐targeting anilinoquinazoline inhibitors allows highly selective, irreversible inhibition of EGFR and closely related family member ErbB2, which shares the target cysteine residue. Afatinib and dacomitinib are two second generation EGFR TKIs that have received regulatory approval for EGFR‐mutant lung cancer (Figure 2b). 32 , 41 , 42 , 43 Neratinib is a second‐generation inhibitor that is structurally similar to lapatinib and also binds the inactive conformation of the EGFR kinase. 44 Like lapatinib, it has found clinical application in HER2‐amplified breast cancer, but not in EGFR‐mutant lung cancer.
Drug resistance that emerges during the course of treatment is a recurring problem with targeted cancer therapies, and EGFR TKIs are no exception. Resistance invariably develops to the first‐ and second‐generation agents described above, most commonly due to a second mutation in the EGFR kinase domain, T790M. 45 , 46 The T790M mutation lies at the back of the ATP‐site, altering the shape of the inhibitor binding site. An analogous resistance mutation arises in other kinase oncoproteins, most notably the T315I mutation in BCR‐Abl in chronic myelogenous leukemia. 47 In addition to its steric effects on inhibitor binding, the T790M mutation reverts the ATP affinity of the mutant kinase to near‐WT levels, effectively closing the therapeutic window that allowed mutant‐selective inhibition by first and second generation inhibitors. 46 The T790M resistance mutation motivated development of a third‐generation of EGFR TKIs; irreversible inhibitors built on alternate scaffolds with selectivity for the mutant kinase.
The anilinopyrimidine compound WZ4002 provided a proof‐of‐concept breakthrough for mutant‐selective inhibition of T790M‐mutant EGFR (Figure 2d). 34 The chlorine‐substituted anilinopyrimidine core of this compound provides specificity for the T790M mutant, while covalent bond formation with C797 via its acrylamide Michael‐acceptor overcomes the enhanced ATP‐affinity. Thus, unlike second generation EGFR inhibitors, which are also irreversible, the alternate scaffold of WZ4002 provided the crucial mutant selectivity. Work with WZ4002 provided a roadmap for development of multiple third generation EGFR inhibitors, and at least seven such agents advanced to clinical trials. To date, osimertinib is the only third‐generation EGFR TKI to receive regulatory approval, initially for treatment of patients who developed resistance to first‐generation inhibitors due to the T790M mutation). 48 More recently, osimertinib has been approved as a first‐line therapy. 49 Like WZ4002 and all other third‐generation EGFR TKIs, osimertinib relies on formation of a covalent bond with C797 for its efficacy (Figure 2e). 48 Predictably, mutation of this residue (C797S) has emerged as a recurring mechanism of resistance to osimertinib. 50 , 51
Diverse approaches have been undertaken to develop inhibitors that can overcome both the T790M and C797S resistance mutations. 52 Most efforts have focused on development of reversible, ATP‐competitive compounds that are effective against the T790M mutant. Examples include trisubstituted imidazoles (Figure 2f), 53 , 54 , 55 BI‐4020 56 and brigatinib, 57 an inhibitor of the ALK kinase that also has activity against EGFR T790M. All of these compounds appear to make important interactions with conserved residues that are crucial for receptor function, such as K745, the catalytic lysine. 36 While this may restrict potential resistance mutations, it likely also makes it challenging to achieve kinome‐wide selectivity. In effect, targeting C797 with irreversible compounds provided a “shortcut” to selectivity because only a handful of protein kinases have a cysteine in this position (12 of more than 500 across the kinome). 58
Allosteric inhibitors represent an alternative approach to obtaining efficacy against both the T790M and C797S resistance mutations. 37 , 38 , 59 Mutant‐selective allosteric EGFR inhibitors were discovered in a high‐throughput screen designed to identify compounds that were selective inhibitors of L858R/T790M EGFR and exhibited a non‐ATP competitive mechanism of action. 59 This effort led to discovery of EAI045, a potent and selective allosteric inhibitor of L858R/T790M and L858R/T790M/C797S EGFR. Although EAI045 is a 3 nM inhibitor of the mutant kinase in vitro, it was ineffective in vivo in a genetically engineered mouse model unless co‐administered with cetuximab, an antibody that blocks EGFR dimerization. 59 A co‐crystal structure with an analog of EAI045 explained this disconnect; the compound binds in a pocket formed only in the inactive, αC‐helix out conformation of the kinase (the same pocket accessed by the inactive conformation binders lapatinib and neratinib). Thus, receptor dimerization antagonizes binding of these allosteric compounds. Subsequent optimization of the EAI045 scaffold yielded JBJ‐04‐125‐02 (Figure 2g,h), a much more potent allosteric inhibitor that is effective as a single agent in genetically engineered mouse models of EGFR L858R/T790M/C797S cancer. 37 Interestingly, JBJ‐04‐125‐02 was found to co‐bind to the receptor with Osimertinib. 37 Thus, a unique avenue for treatment may involve combination therapy with a structurally compatible third‐generation TKI such as osimertinib. In preclinical studies, this combination treatment has been shown to render EGFR mutant NSCLC cells unable to acquire resistance, likely due to the orthogonal mechanisms of resistance to osimertinib versus the allosteric inhibitor. 60 A second compound class with a dibenzodiazepinone core was discovered in the same screen that led to EAI045. 38 A co‐crystal structure with a representative dibenzodiazepinone, DDC4002, demonstrated that these compounds bind the same pocket as EAI045 and its derivatives, with related ligand‐protein interactions despite their very different chemical structures (Figure 2I). 38
3. DRUGGING K‐RAS, AT LAST
The small GTPase K‐Ras is a key regulator of MAP kinase pathway. Somatic mutations in K‐Ras are the most common activating mutations in cancer and are strongly associated with poor response to therapy. 12 Despite its long‐standing prominence as a target in cancer drug development, only very recently have compounds directly targeting K‐Ras begun to show promise. Its reputation as an “undruggable” target stem from its very high affinity for activating GTP nucleotides, which makes GTP‐competitive agents unworkable given high cellular concentrations of GTP, and from the absence of obvious allosteric regulatory sites. 61
Ras proteins consist of a single structured domain, known as the GTPase or “G domain”, and a more variable C‐terminal tail that terminates with a so‐called CaaX box, a sequence motif that directs prenylation at its C‐terminus. This process involves attachment of a farnesyl or geranylgeranyl group to the cysteine residue of the CaaX motif, as well as subsequent processing, and is required for membrane association of Ras. Ras activity is controlled via well understood structural changes within the GTPase domain. 62 The four key regions of the G domain are the P‐loop, switch I (red), switch II (blue) and the base‐binding loops (Figure 3). Ras signaling is regulated by cycling between the GTP‐bound active and GDP‐bound inactive states. This cycle is regulated by actions of regulatory protein partners. Ras signaling is activated by guanine nucleotide exchange factors (for K‐Ras, this is son of sevenless or SOS), which promote GTP‐loading. Their signaling is terminated by GTPase‐activating proteins, or GAPs, which effectively provide a key catalytic residue that allows rapid hydrolysis of GTP to GDP, restoring the quiescent state. The “on” state of RAS occurs with GTP bound with Thr‐35 and Gly‐60 engaged in H‐bonds with the γ‐phosphate of GTP. Upon hydrolysis, and release of the γ‐phosphate, these two residues relax into the GDP “off” state. 62 The most common oncogenic mutations in K‐Ras occur in positions 12, 13, and 61, with the vast majority occurring at residue 12. 65 Mutations in these positions all generally interfere with GTP hydrolysis resulting in accumulation of the GTP‐bound “on” state. 12 , 61 More recently, it was determined that GDP‐GTP exchange rates vary among Ras mutants, and may be important in determining the biological consequences of distinct K‐Ras mutants. 66
FIGURE 3.
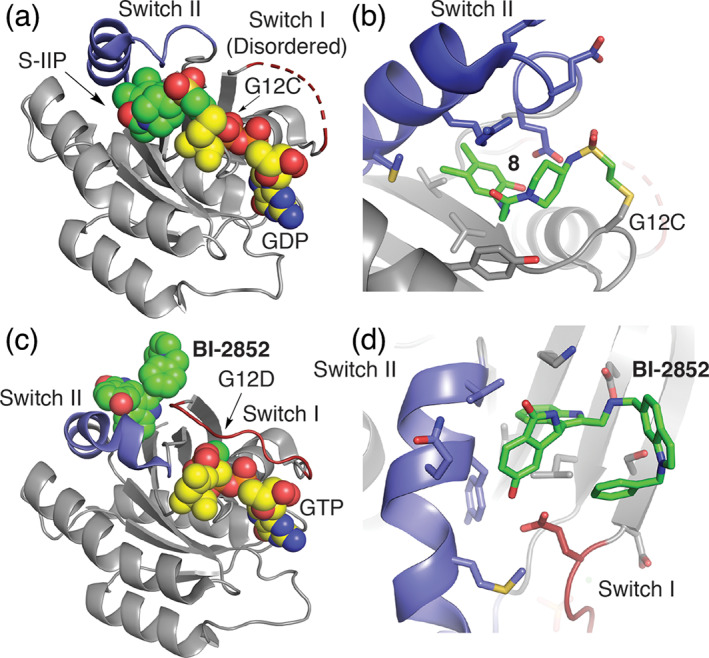
Binding modes of inhibitors in complex with mutant K‐Ras. (a) Co‐crystal structure of a K‐Ras C12‐targeting covalent inhibitor (PDB ID 4LYF 8 ) and (b) zoom in on 8 binding pose. 63 K‐Ras inhibitor forms a covalent bond with the mutant residue G12C cysteine residue and anchors the molecule within a previously unappreciated allosteric binding pocket underneath the Switch II (SII‐P, blue). (c) BI‐2852 based on PDB ID 6JG7 bond to the SI/II pocket of G12D with bound GTP and (d) zoom in on SI/II site. 64 The BI‐2852 covalent drug binds an allosteric site spanning structural elements of Switch I and II that is unchanged in active and inactive states of K‐Ras. GTP or GDP substrate sites are shown in spacefill with yellow carbon atoms
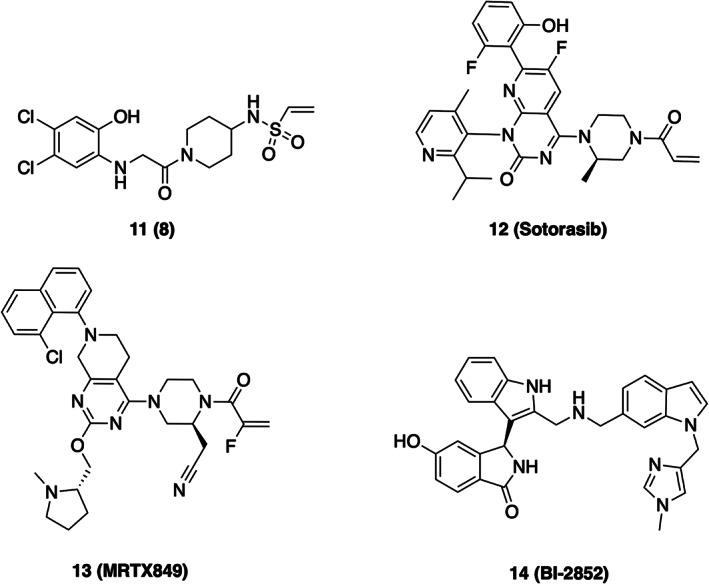
The prevalence of mutations at G12 in K‐Ras presents an opportunities to develop small‐molecule binders that target the mutant alleles in a manner that intrinsically spare WT K‐Ras, in particular for the cysteine mutation at this site. 61 Exploiting the nucleophilicity of the G12C mutant Shokat and co‐workers performed a disulfide‐fragment screen to identify novel molecules that bind the G12C cysteine residue. 63 From this screen, a molecule was discovered that, indeed, formed a covalent bond to G12C and anchored to K‐Ras in a novel allosteric pocket distinct from GTP/GDP binding site directly beneath Switch II (SII‐P, Figure 3a and Scheme 2). 63 Incorporation of an acrylamide warhead, and structural optimization of the inhibitor improved inhibitor potency. Collective structural information from co‐crystal structures indicates that these inhibitors [e.g., 8] bind in the switch II pocket (SII‐P), a previously unappreciated site for drug interaction. Additionally, these inhibitors likely alter the dynamic nature of Switch II by shifting the affinity of K‐Ras for GDP compared to GTP, thus stabilizing K‐Ras in the inactive state, in addition to diminishing interactions with effector and regulatory proteins. 63 , 67 This breakthrough has led to several efforts by pharmaceutical companies resulting in K‐Ras(G12C) inhibitors, such as Sotorasib (AMG510) and MRTX849 (Scheme 2), some with highly promising Phase I results in K‐Ras(G12C) NSCLC. 68 , 69 , 70 , 71 Despite a variety of effective clinical candidates, resistance to G12C inhibitors will likely arise. One mechanism that has been identified in preclinical studies involves new synthesis of K‐Ras(G12C). Cells that are actively making new K‐Ras(G12C), and in which active EGF receptor signaling is promoting GTP‐loading of the newly synthesized mutant Ras, appear to be resistant to these inhibitors. 72 This is because the inhibitors, although irreversible, selectively target the GDP‐loaded form of K‐Ras.
SCHEME 2.
Chemical structures of K‐Ras inhibitors
In another approach, a series of fragment screens and an ensuing intensive structure‐guided drug design campaign generated BI‐2852. 73 This inhibitor differs from the G12C‐targeting inhibitors as it binds to a distinct pocket in between Switch I and Switch II (the SI/II pocket), affording the ability to block interactions with a variety of regulatory partners and effectors. Structurally, the SI/II pocket is present in both active and inactive conformations of K‐Ras. BI‐2852 has shown efficacy in a variety of cell line models. Interestingly, it has also been shown to induce formation of unproductive K‐Ras dimers, although it remains unclear whether induction of these dimers plays a role its cellular efficacy. 64 , 74 Since the SI/II pocket is conserved and not impacted by the oncogenic mutations, BI‐2852 is not especially selective for the mutations motivating further efforts to generate mutant‐selective analogues. 73
4. BRAF, A HOTSPOT FOR MUTATIONS AT THE HEART OF MAPK SIGNALING
Raf‐family kinases ARAF, BRAF and CRAF (also called Raf‐1) sit at the top of the three‐tiered MAP kinase cascade (Box 1). 75 As with Ras, their activity must also be tightly regulated, and mutations that disrupt their normal regulation can lead to cancer. In particular, BRAF is very frequently mutated in cancer. 9 The V600E mutation within its kinase domain drives more than half of all malignant melanoma, and is also found in lung adenocarcinoma, papillary thyroid carcinoma, and other cancers. 76
In the absence of Ras signals, BRAF is maintained in an autoinhibited state as a complex with its substrate, MEK, and a 14‐3‐3 dimer. 77 14‐3‐3 proteins bind to specific phosphoserine or phosphothreonine sites to regulate many signaling proteins, and they are crucial for Raf regulation in both active and inactive states. 78 In the autoinhibited state, the 14‐3‐3 dimer spans between phosphoserine motifs flanking the BRAF kinase domain, forming a “cradle” that sequesters the membrane‐targeting cysteine‐rich domain of BRAF, and also blocks dimerization of the BRAF kinase domain (Figure 4a). In this configuration, the Ras‐binding domain is exposed and accessible to binding by GTP‐bound Ras. The detailed mechanism of activation by Ras has not been fully elucidated, but it is clear that it requires prenylation of Ras, and thus recruitment of RAF by Ras to the plasma membrane. 82 , 83 Upon activation the N‐terminal RBD/CRD regions mediate binding to Ras and the plasma membrane, and the 14‐3‐3 dimer rearranges to bind to the C‐terminal (pSer729) binding motifs in two RAFs, thereby driving formation of an active RAF dimer (Figure 4b). 77 , 84 , 85 , 86
FIGURE 4.
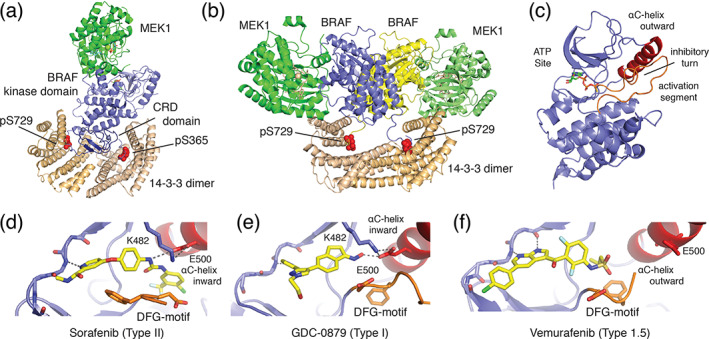
Structural basis for BRAF autoregulation and small‐molecule inhibition. (a) Autoinhibited BRAF:MEK1:14‐3‐3 complex (PDB ID 6NYB) and B) active BRAF:MEK1:14‐3‐3 complex, in which the 14‐3‐3 dimer rearranges to bind the C‐terminal pSer729 site of two BRAF protomers to form the active back‐to‐back BRAF kinase dimer (PDB ID 6Q0J). 77 The Ras‐binding domain is not visualized in these structures. (c) Zoom in on BRAF kinase domain (PDB ID 6NYB) from autoinhibited complex. Key structural features of the autoinhibited state are labeled. 77 (d–f) Binding modes of representative classes of BRAF inhibitor, including (d) Sorafenib (Type II, PDB ID 1UWH), 79 (e) GCD‐0879 (Type I, PDB ID 4MNF), 80 and vemurafenib (Type 1.5, PBD ID 3OG7) 81 showing defining orientations of the DFG‐motif (orange) and αC‐helix (red)
In the monomeric state, the BRAF kinase domain adopts an inactive conformation with its regulatory αC‐helix pivoted in an outward position (Figure 4c). 77 The displaced orientation of the αC‐helix is stabilized by an “inhibitory turn” in the kinase activation loop. In the active state, the BRAF kinase domain forms a “back‐to‐back” dimer that impinges on the αC‐helix, promoting rearrangement to its inward, active position (Figure 4b). 80 , 87 Importantly, V600 lies in the middle of the inhibitory turn, and it packs with several other hydrophobic residues in the kinase N‐lobe. The oncogenic V600E mutation disrupts the inhibitory turn, and thereby allows rearrangement of the αC‐helix to its inward, active position without a requirement for Ras‐driven dimerization. 77 This mechanism of activation is closely analogous to that of the L858R mutation in EGFR (Figure 1a,c). In both kinases, these point mutations destabilize the inhibitory portion of the activation segment to allow the C‐helix to rearrange to the active state irrespective of dimerization. 77
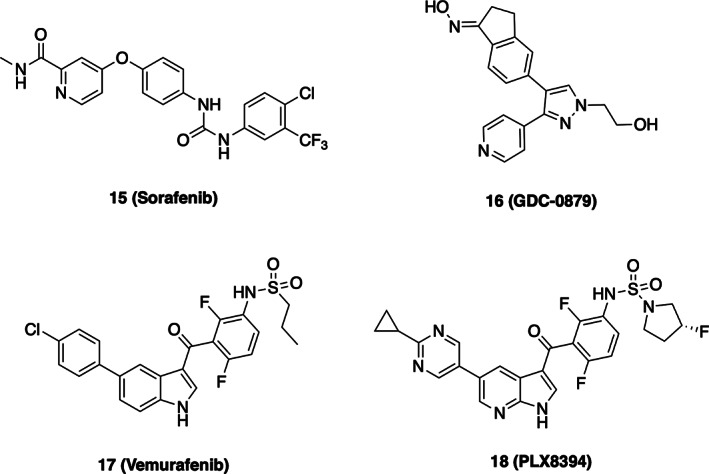
The prevalence of BRAF mutations in cancer and the key role of RAFs in regulating signaling through the MAP kinase cascade have made it a prominent target of inhibitor development. 12 , 76 Structural information has been crucial in this respect, more than 50 structures of the BRAF kinase domain with various inhibitors have been reported to date. The first such structure was determined in complex with the first approved Raf inhibitor, sorafenib (also known a BAY 43‐9006) (Figure 4d and Scheme 3). 79 Though developed initially as a Raf inhibitor, Sorafenib is now recognized as a “multi‐targeted” kinase inhibitor that is potent against a number of kinases, including PDGFR and VEGFR. 88 Indeed, it is approved for certain liver, kidney, and thyroid cancers, but has not found clinical application in melanoma or other cancers driven by BRAF V600E. 89
SCHEME 3.
Chemical structures of BRAF inhibitors
Appreciation of the binding mode and pharmacologic properties of sorafenib and other Raf inhibitors requires a brief discursion into the arcana of kinase inhibitors. ATP‐competitive kinases inhibitors can be divided into two classes, Type I or Type II, based on their effect on the conformation of the “DFG” motif. 90 This three residue motif lies at the N‐terminus of the kinase activation segment and is absolutely conserved; the aspartic acid side chain extends into the ATP‐site, where it coordinates Mg2+ ions together with the triphosphate moiety of ATP, while the phenylalanine is oriented in the opposite direction, toward the αC‐helix. Type I inhibitors bind with this native DFG conformation intact (Figure 4e), while Type II inhibitors induce a crankshaft‐like “flip” of the DFG‐motif, such that the phenylalanine extends into the ATP‐site (Figure 4d). Typically, an aromatic group on the Type II inhibitor assumes the approximate position of the displaced phenylalanine side chain, and an amide or urea group in the Type II inhibitor hydrogen bonds with the flipped DFG segment. Sorafenib is a “Type II” inhibitor, and exhibits both of these salient features. 80 Its urea moiety hydrogen bonds with E500 in the αC‐helix, and its chloro‐trifluoromethyl phenyl group packs in the hydrophobic pocket opened by the DFG rearrangement (Figure 4d). By contrast, Type I Raf inhibitor GDC‐0879 occupies the ATP‐site but does not induce the DFG flip and does not extend beyond the DFG segment (Figure 4e). 80
A second distinction made with certain kinase inhibitors is whether they recognize or induce an inactive kinase conformation in which the αC‐helix is rotated into an outward, inactive conformation. As noted above, RAFs (and many other kinases) adopt inactive conformations in which the αC‐helix rotates and pivots outward so as to remove a key glutamic acid residue from the kinase active site. Certain BRAF inhibitors exploit this inactive conformation by occupying the pocket created by outward displacement of the αC‐helix (Figure 4f). 91 , 92 These have been referred to as Type 1.5 inhibitors, or perhaps more informatively as “αC‐out inhibitors.” To date, three BRAF inhibitors have received FDA approval for treatment of BRAF V600E‐driven melanoma (vemurafenib, dabrafenib, and encorafenib), and all are Type 1.5.
The αC‐out binding mode of these agents is both a key strength and a disabling weakness. The advantage of this binding mode is that it affords mutant‐selective inhibition of the BRAF V600E. Mutant‐selectivity is important because it contributes to a crucial therapeutic window that allows inhibition of the oncogenic variant in the tumor, with relative sparing of WT RAF signaling in normal cells. Unlike WT BRAF, which requires Ras‐driven dimerization for activation, the V600E mutant is active as a monomer. 93 In the monomeric state, binding of vemurafenib and other αC‐out inhibitors can readily induce the inactive, αC‐out conformation of the kinase. 91 By contrast, the αC‐helix is constrained in active BRAF dimers, which enforce its inward, active position. Thus, RAF dimerization antagonizes binding of αC‐out inhibitors is an important mechanism of acquired resistance to these drugs. 92 , 93 BRAF V600E can be driven to dimerize by acquired mutations upstream that promote Ras‐driven dimerization (e.g., in NRas) or by expression of BRAF V600E splicing variants that lack the N‐terminal autoregulatory regions that are required to maintain it in its monomeric state. 93
While the αC‐out inhibitors are effective against the monomeric BRAF(V600E), Type I or II inhibitors with an αC‐helix inward binding mode more potently inhibit BRAF dimers. 91 , 94 In addition to point mutations such as V600E, BRAF can also be activated by chromosomal translocations that result in constitutive dimerization of the kinase. As one example, a large percentage of pediatric low‐grade gliomas are driven by a truncation‐fusion form of BRAF in which a portion of the membrane‐associated KIAA1549 protein is fused to the BRAF kinase domain, replacing its regulatory N‐terminal region. 95 The KIAA1549:BRAF truncation fusion is constitutively dimeric and therefore active. Type II inhibitor TAK‐580 (formerly MLN2480, now called DAY101) potently inhibits this dimer, 96 and is now in clinical trials in children with this tumor type.
No discussion of Raf inhibitors would be complete without mention of “paradoxical activation,” a phenomenon in which Raf inhibitors activate the MAP kinase pathway via activation of wild‐type Rafs. 20 , 76 , 92 The phenomenon is particularly pronounced with αC‐out inhibitors vemurafenib and dabrafenib, and can lead to formation of drug‐induced secondary lesions (squamous cell carcinomas or keratoacanthomas) in melanoma patients treated with these agents. Suppression of this effect is in part the rationale underlying combination treatment with a MEK inhibitor; RAF/MEK inhibitor combinations are now standard of care for melanomas caused by BRAF(V600E). 97 The paradoxical activation is incompletely understood, but is known to stem from action of the inhibitor at the BRAF ATP‐site. 98 , 99 , 100 Raf inhibitors are known to drive formation of Raf dimers, and a prevailing model posits that binding of inhibitor in one side of a Raf dimer induces a catalytically active but inhibitor‐resistant conformation in the contralateral subunit. 94 Although seductively elegant, this negative cooperativity model at present lacks biophysical validation.
Recent structural studies reveal that coordination of ATP in the kinase active site is crucial for maintaining BRAF in its autoinhibited state. 77 , 86 This finding provides an obvious explanation for the genesis of inhibitor‐induced activation—any ATP‐competitive inhibitor could potentially promote conformational activation of RAF via displacement of ATP. More recently developed compounds that are “paradox breakers” may appear to overcome the effect because they are more potent inhibitors of dimeric BRAF. 101 Indeed, PLX8394 is a dimer‐breaker—sufficiently potent in forcing the αC‐helix outward conformation of the kinase that it can induce dissociation of the dimer (Scheme 3). 102 Although PLX8394 and related compounds do not exhibit paradoxical pathway activation at a cellular level, one must worry that their increased potency against Raf dimers comes with the cost of a narrowed therapeutic window due to inhibition of WT Raf dimers. Similarly, certain type II Raf inhibitors are reported to avoid paradoxical activation, 103 but this may also stem from their potent inhibition of Raf dimers. In effect, paradox breaking inhibitors may simply be more potent inhibitors of the WT Raf dimers that they have induced.
5. MEK1/2 INHIBITORS—ALLOSTERIC BY CHANCE
Unlike BRAF, its substrate MEK is very infrequently mutated in cancer. 104 Thus the prominence of MEK (there are two MEK proteins in mammalian cells, MEK1 and MEK2) as a therapeutic target stems largely from its role in propagating signaling through the MAP kinase cascade, downstream of oncogenic Ras and BRAF. Nevertheless, rare oncogenic mutations in MEK provide strong support for development of MEK inhibitors as anticancer agents. More than a dozen MEK inhibitors have advanced to clinical trials and to date four have received FDA approval; three as combination agents with BRAF inhibitors in melanoma (trametinib, cobimetinib, and binimetinib) 97 and a fourth (selumetinib) that has recently been approved due to its efficacy in children with neurofibromatosis Type 1 who have inoperable plexiform neurofibromas. 105 Here, we highlight a few representative examples of MEK inhibitors with a focus on the structural aspects of their mechanism of action.
The first MEK inhibitor, PD098059, was reported more than 25 years ago. 106 Unlike the vast majority of kinase inhibitors known at that time or developed since, this compound exhibited an allosteric mechanism of action and was observed to inhibit activation of MEK by Raf. 106 Studies of this early MEK inhibitors proved prophetic; virtually all clinical‐stage MEK inhibitors described to date bind in the same allosteric site, despite very different chemical structures and their independent origins.
Structural studies with analogs of MEK inhibitors CI‐1040 and PD0325901 provided the first three‐dimensional views of MEK1 and MEK2 (Figure 5a,b and Scheme 4). 107 In these structures, MEK adopts an inactive conformation in which its αC‐helix is displaced outward, not unlike the αC‐helix outward inactive conformations of certain other kinases including EGFR and BRAF, as described above. The displacement of the αC‐helix creates a pocket adjacent to, but distinct from the ATP‐site in which the allosteric inhibitor binds (Figure 5). The bound inhibitors (PD318088 in the MEK1 structure) also interact extensively with the MEK activation segment, which forms a short α‐helix that encloses the allosteric site. Interestingly, hydroxyl groups of these inhibitors form hydrogen bonds with the phosphate oxygens of the bound nucleotide. This structure‐based design feature contributes to the potency and cooperative binding of this compound class with ATP. 107
FIGURE 5.
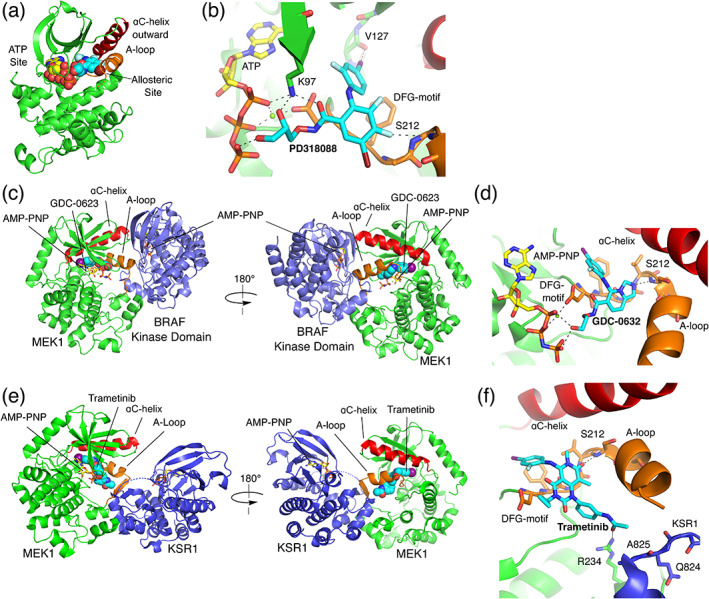
Allosteric MEK inhibitors. (a) Structure of the MEK1 kinase in its inactive state in complex with early MEK inhibitor PD318088 and Mg2+ ATP. The inhibitor and ATP are shown in spacefill to indicate the respective locations of the ATP and allosteric inhibitor binding sites (PDB ID 1S9J). 107 (b) Detailed view of inhibitor interactions in the structure shown in Panel A. (c) Crystal structure of a MEK1: BRAF dimer, with inhibitor GDC‐0623 bound in the MEK allosteric site (PDB ID 6PP9). 77 (d) Detail of the interactions of GDC‐0623 in the MEK allosteric site. (e) Crystal structure of a KSR:MEK1 dimer, with inhibitor trametinib bound in the MEK allosteric site (PDB ID 7JUX). 108 (f) Detailed view of the interactions of trametinib in the MEK1 allosteric site
SCHEME 4.
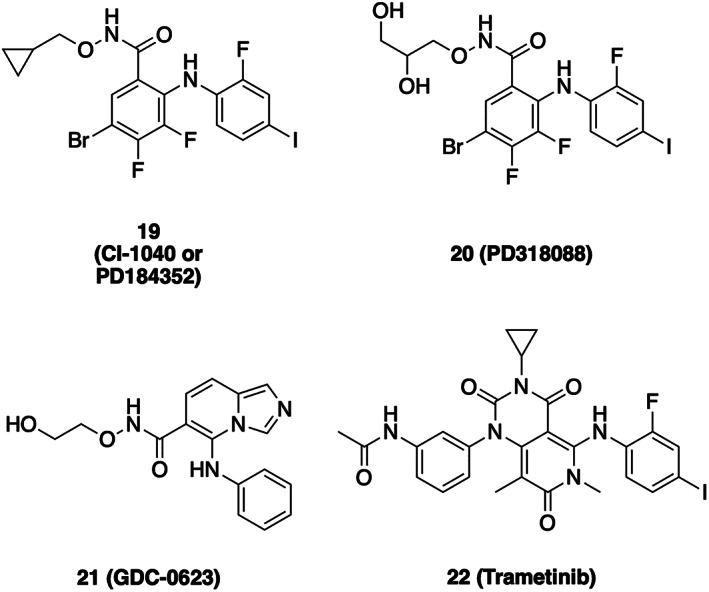
Chemical structures of MEK inhibitors
Subsequent structural studies using a longer MEK construct showed that the general features of this inactive conformation were not induced by binding of the allosteric inhibitor; MEK adopts a closely similar autoinhibited conformation in the absence of an inhibitor. 109 Importantly, this later study also showed that a key region at the N‐terminus of the MEK kinase domain that was known to serve an autoinhibitory role forms an α‐helix that packs across the N‐lobe and helps to stabilize the outward, inactive position of the αC‐helix. 109
The compound class that led to development of trametinib was discovered in a cell‐based screen for induction of p15INK4b, an inhibitor of cyclin D‐bound cyclin‐dependent kinases CDK4 and CDK6. 110 Compound JTP‐70902 induced G1 cell cycle arrest, and MEK1/2 was identified as its molecular target using compound‐immobilized affinity chromatography. Trametinib is a close structural analog of JTP‐70902, and like PD098059, it was found to inhibit phosphorylation of MEK by Raf. 81 , 111 Interestingly, a mass spectrometry study revealed that it selectively inhibited phosphorylation of Ser218, but not Ser222 in the MEK activation segment. 81 Dual phosphorylation of MEK on both Ser218/Ser222 is required for its full activation. Furthermore, trametinib is a much more potent inhibitor of Ser218‐phosphorylated MEK than of the doubly phosphorylated pS218/pS222 species. Thus, the efficacy of trametinib stems at least in part from its ability to block MEK activation by Raf, rather than solely its ability to inhibit fully activated MEK1/2. Although not as thoroughly studied, this mechanism of action is likely relevant to other allosteric MEK inhibitors.
As with trametinib, MEK inhibitor CH5126766 has its origins in a cell‐based screen for compounds that could induce an inhibitor of cyclin‐dependent kinases, in this case p27Kip1. 112 CH5126766 was shown to potently inhibit phosphorylation of MEK by Raf proteins, and to stabilize complexes of inhibitor‐bound MEK with Raf. 112 Thus CH5126766 inhibits signaling through the pathway by inducing a phosphorylation‐resistant conformation of MEK that can also act in a “dominant‐negative” manner to inhibit Raf. 113 A co‐crystal structure confirms that this compound binds in the same allosteric site as PD318088 and trametinib. 113
Despite their common binding site, allosteric MEK inhibitors vary considerably in their efficacy the context of differing mechanisms of pathway activation. 18 While most or all MEK inhibitors potently inhibit proliferation of cell lines bearing BRAF V600E, they differ in their efficacy in the context of oncogenic K‐Ras. For example, trametinib, CH5126766 and GDC‐0623 are relatively effective in inhibiting proliferation of K‐Ras mutant cell lines, whereas PD0325901 and cobimetinib are not. 113 , 114 Although the underlying structural basis for these differences is not understood in detail, it likely stems in large part from their allosteric mechanism and the fact that they are acting primarily on unphosphorylated MEK or Raf/MEK complexes to block phosphorylation of MEK by Raf, rather than via inhibition of active, phosphorylated MEK. Structural and biophysical studies of MEK inhibitors bound to different Raf/MEK complexes may inform development of more selective agents in the future. A co‐crystal structure of GDC‐0623 bound to the MEK1/BRAF complex shows how it stabilizes the MEK activation loop in a phosphorylation resistant conformation (Figure 5c,d) that is essentially the same as that seen in the absence of the MEK inhibitor. 77 , 86 A recent study of trametinib and other MEK inhibitors bound to MEK in complex with Kinase Suppressor of Ras (KSR) is also of interest in this respect. 108 KSR is a RAF‐like scaffolding protein important for MAPK pathway signaling. In this work, trametinib was found to stabilize MEK/KSR complexes (Figure 5e,f), and a sulfamide‐containing derivative termed “trametiglue” was found to stabilize MEK/BRAF complexes whereas trametinib did not. 108
6. SHP2, ALLOSTERIC BY DESIGN
The non‐receptor protein tyrosine phosphatase SHP2, encoded by PTPN11, plays important roles in signal transduction through growth factor receptors and in oncogenesis. 115 Although perhaps counterintuitive because it is a phosphatase, numerous studies indicate that SHP2 is important in cancer cell survival in RTK driven tumors. 116 , 117 Although the specific role of SHP2 in MAPK pathway activation remains unclear, present understanding suggests that SHP2 acts as a scaffolding protein that binds GAB1 and SOS1, 118 , 119 , 120 and more recently has been shown that SHP2 is important for oncogenesis in K‐Ras mutant tumors. 121
Structurally, SHP2 comprises two tandem SH2 phosphotyrosine recognition domains (Figure 6a,N‐terminal in yellow, and C‐terminal in green) followed by the catalytic protein tyrosine phosphatase (PTP, blue) domain. 123 The activity of SHP2 is controlled via an autoregulatory mechanism dependent on intramolecular domain‐domain interactions. 123 In the inactive “closed” arrangement, both SH2 domains (denoted N and C based on their order in the protein sequence) are engaged in intramolecular interactions with the PTP domain (Figure 6a). Importantly, in the autoinhibited state, the N‐SH2 domain structurally obstructs the active site of the phosphatase domain, thereby gating catalytic activity. Phosphatase activity is switched on by simultaneous binding of appropriate tyrosine‐phosphorylated protein segments to the tandem SH2 domains, which releases them from the PTP domain and to allow substrate access to the phosphatase active site. Note that the C‐SH2 domain does not directly block access to the PTP domain, instead it contributes to the specificity of SHP2 activation contributing binding energy and specificity.
FIGURE 6.
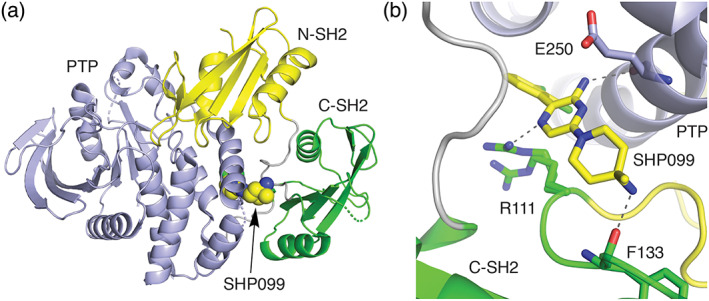
(a) Structure of autoinhibited SHP2 and (b) detail of the binding mode of SHP099 (PDB ID 5EHR). 122 In the inactive state, the tandem SH2 domains of SHP2 bind to the tyrosine phosphatase domain (blue) and the N‐terminal SH2 domain (yellow) directly blocks the phosphatase active site. Allosteric inhibitor SHP099 binds in pocket between the C‐terminal SH2 domain (green) and the phosphatase domain, acting as a molecular glue to stabilize the inhibitory interactions of the SH2 domains
Development of inhibitors directed against the catalytic site of SHP2 and other PTPases has been challenging due to the highly solvated and polar nature of its active site. 23 This challenge motivated efforts to discover SHP2 inhibitors that operate through allosteric mechanisms. 122 Taking advantage of the natural mode of SHP2 autoinhibition revealed in the crystal structure described above, a screen was devised to discover small molecules that work by “locking up” SHP2 in its autoinhibited state. 122 The key was to search for compounds that inhibited SHP2 catalytic activity with a partially activated SHP2 through the inclusion of a biphosphorylated peptide that competes for binding at the SH2 domains. Hits from this screen were subjected to counter screening with the isolated phosphatase domain where productive inhibition would allow for the filtering out compounds that bind the PTP active site from those that operate through allosteric inhibitors. A compound identified from this campaign, with subsequent optimization, yielded in SHP099 (Scheme 5), an allosteric inhibitor of SHP2 that binds a central pocket between the C‐terminal SH2 domain and the PTP domain (Figure 6b). In effect, SHP099 acts as a “molecular glue” to cement the phosphatase in its closed, autoinhibited state. The discovery and characterization of SHP099 has motivated development of additional SHP2 allosteric inhibitors that are now in various stages of clinical assessment for safety and efficacy in settings of advanced or metastatic solid tumors, including K‐Ras G12X mutants (Scheme 5). 23 , 124 , 125 Clinical trials are also underway testing combinations of SHP2 allosteric inhibitors with other drugs targeting the MAPK pathway, such as TNO155 in combination with the K‐Ras drug MRTX849 126 and additionally RMC4630 with MEK‐targeting cobimetinib in contexts of K‐Ras G12C tumors 127 and other cancers. 23 , 125
SCHEME 5.
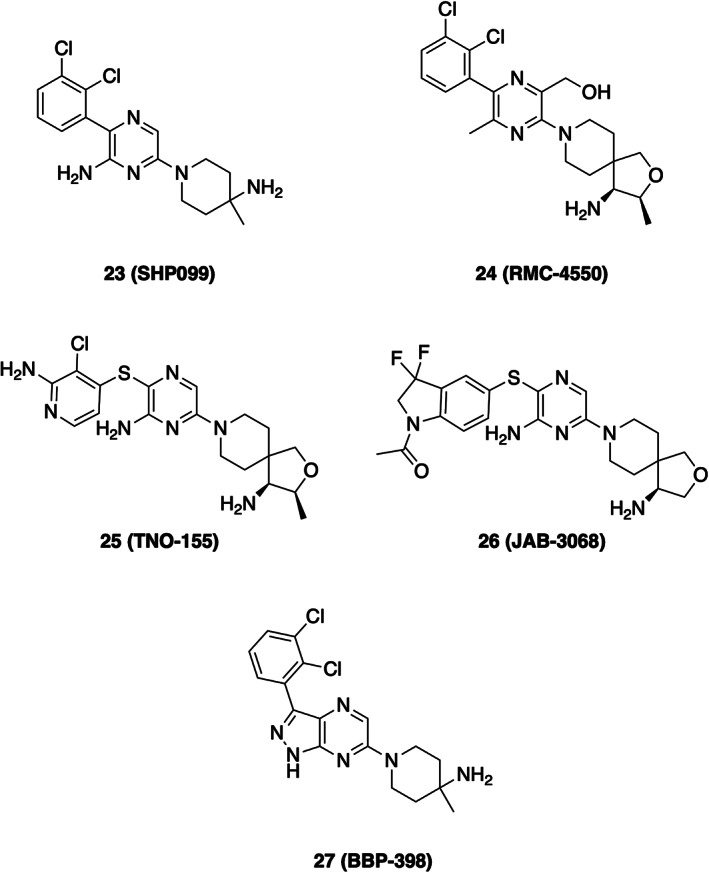
Chemical structures of SHP2 allosteric inhibitors
7. CONCLUDING REMARKS
Drug hunters, like hunters of all stripes, are well‐served by an intimate understanding of their target prey. The tools of structural biology allow target visualization that can guide molecular design, but they also reveal regulatory “habits” and other vulnerabilities that can inspire and inform screens for inhibitors with novel mechanisms of action. Recognition of a cysteine residue conveniently positioned adjacent to the inhibitor binding site in EGFR‐guided evolutionary development of irreversible second‐generation EGFR inhibitors via addition of a covalent “warhead” to an already‐potent anilinoquinazoline scaffold (Figure 2b). Similarly, third‐generation EGFR inhibitors WZ4002 and osimertinib exploit covalent binding to this cysteine to enhance the potency of their weak, but mutant‐selective anilinopyrimidine core (Figure 2d,e). By contrast, appreciation that the G12C mutation in Ras conferred a vulnerability—a surface exposed cysteine residue unique to the mutant protein—inspired a screen for chemical matter targeting that residue in order to gain a toehold on the Switch II pocket, and led to the revolutionary development of mutant‐selective inhibitors of this oncogenic Ras variant. 63 More generally, deep structural and mechanistic understanding of the allosteric regulation of signaling proteins has guided design of screens for inhibitors with new mechanisms of action; for example, the “trap” that captured the first allosteric SHP2 inhibitors. 122
With protein kinase targets in this pathway, elucidation of the structural basis for their normal regulation and oncogenic activation has been valuable for inhibitor development and for understanding drug‐sensitivity and resistance. The inactive conformation of these kinases is exploited by Type 1.5 inhibitors (of EGFR and BRAF) and by allosteric inhibitors (for EGFR and MEK) that extend into or occupy the pocket opened by the outward displacement of the regulatory αC‐helix. These kinases share similarities and differences in the way in which their normal regulation induces the inward, active conformation of the αC‐helix; formation of an asymmetric dimer in EGFR, a symmetric back‐to‐back dimer in BRAF, or by activation‐loop phosphorylation in MEK. Interestingly, oncogenic deletion mutations that shorten the loop leading into the αC‐helix have been identified in BRAF and MEK in addition to EGFR. As in EGFR, these BRAF and MEK mutations lock the kinase in an active state, and also confer resistance to drugs that require the C‐helix‐out inactive conformation. 128 , 129 Similarly, BRAF dimerization induced by pathway activation, or by truncation of its N‐terminal regulatory domains, confers resistance to Type 1.5 inhibitors such as vemurafenib and dabrafenib. 93 , 94
Due in part to the challenges and idiosyncrasies of protein crystallization, actionable structural information for drug targets of interest has often lagged inhibitor discovery. Indeed, the first views of both the active and inactive states of the EGFR kinase domain were enabled by a co‐crystal structures with inhibitors, and the first BRAF inhibitors were developed in the absence of a BRAF structure. Going forward, advances in our understanding of the structure and regulation of the key nodes in the RTK/Ras/MAP kinase pathway should increasingly allow structural biology to lead the way in development of new therapeutic agents targeting this pathway.
AUTHOR CONTRIBUTIONS
David Heppner: Conceptualization; writing‐original draft; writing‐review & editing. Michael Eck: Conceptualization; writing‐original draft; writing‐review & editing.
ACKNOWLEDGMENTS
We acknowledge financial support from the National Institutes of Health R01CA201049, R01CA116020, and R35CA242461 (to M.J.E.) and start‐up funds from the University at Buffalo, State University of New York (to D.E.H.).
Heppner DE, Eck MJ. A structural perspective on targeting the RTK/Ras/MAP kinase pathway in cancer. Protein Science. 2021;30:1535–1553. 10.1002/pro.4125
Funding information National Cancer Institute, Grant/Award Numbers: R01CA116020, R01CA201049, R35CA242461; University at Buffalo, Grant/Award Number: Start‐up Funds
Contributor Information
David E. Heppner, Email: davidhep@buffalo.edu.
Michael J. Eck, Email: eck@crystal.harvard.edu.
REFERENCES
- 1. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Simanshu DK, Nissley DV, McCormick F. RAS proteins and their regulators in human disease. Cell. 2017;170:17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Reddy EP, Reynolds RK, Santos E, Barbacid M. A point mutation is responsible for the acquisition of transforming properties by the T24 human bladder carcinoma oncogene. Nature. 1982;300:149–152. [DOI] [PubMed] [Google Scholar]
- 4. Tabin CJ, Bradley SM, Bargmann CI, et al. Mechanism of activation of a human oncogene. Nature. 1982;300:143–149. [DOI] [PubMed] [Google Scholar]
- 5. Taparowsky E, Suard Y, Fasano O, Shimizu K, Goldfarb M, Wigler M. Activation of the T24 bladder carcinoma transforming gene is linked to a single amino acid change. Nature. 1982;300:762–765. [DOI] [PubMed] [Google Scholar]
- 6. Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non–small‐cell lung cancer to gefitinib. New Engl J Med. 2004;350:2129–2139. [DOI] [PubMed] [Google Scholar]
- 7. Paez JG, Jänne PA, Lee JC, et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. [DOI] [PubMed] [Google Scholar]
- 8. Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. [DOI] [PubMed] [Google Scholar]
- 10. Sanchez‐Vega F, Mina M, Armenia J, et al. Oncogenic signaling pathways in the cancer genome atlas. Cell. 2018;173:321–337. e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. The ICGC/TCGA Pan‐Cancer Analysis of Whole Genomes Consortium . Pan‐cancer analysis of whole genomes. Nature. 2020;578:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moore AR, Rosenberg SC, McCormick F, Malek S. RAS‐targeted therapies: Is the undruggable drugged? Nat Rev Drug Discov. 2020;19:533–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Noble MEM, Endicott JA, Johnson LN. Protein kinase inhibitors: Insights into drug design from structure. Science. 2004;303:1800–1805. [DOI] [PubMed] [Google Scholar]
- 14. van Montfort RL, Workman P. Structure‐based design of molecular cancer therapeutics. Trends Biotechnol. 2009;27:315–328. [DOI] [PubMed] [Google Scholar]
- 15. Erlanson DA, Fesik SW, Hubbard RE, Jahnke W, Jhoti H. Twenty years on: The impact of fragments on drug discovery. Nat Rev Drug Discov. 2016;15:605–619. [DOI] [PubMed] [Google Scholar]
- 16. Renaud J‐P, Chari A, Ciferri C, et al. Cryo‐EM in drug discovery: Achievements, limitations and prospects. Nat Rev Drug Discov. 2018;17:471–492. [DOI] [PubMed] [Google Scholar]
- 17. Van Drie JH, Tong L. Cryo‐EM as a powerful tool for drug discovery. Bioorgan Med Chem Lett. 2020;30:127524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Caunt CJ, Sale MJ, Smith PD, Cook SJ. MEK1 and MEK2 inhibitors and cancer therapy: The long and winding road. Nat Rev Cancer. 2015;15:577–592. [DOI] [PubMed] [Google Scholar]
- 19. Fiskus W, Mitsiades N. B‐Raf inhibition in the clinic: Present and future. Ann Rev Med. 2016;67:29–43. [DOI] [PubMed] [Google Scholar]
- 20. Hymowitz SG, Malek S. Targeting the MAPK pathway in RAS mutant cancers. Cold Spring Harb Perspect Med. 2018;8:a031492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kidger AM, Sipthorp J, Cook SJ. ERK1/2 inhibitors: New weapons to inhibit the RAS‐regulated RAF‐MEK1/2‐ERK1/2 pathway. Pharmacol Therapeut. 2018;187:45–60. [DOI] [PubMed] [Google Scholar]
- 22. Huang L, Jiang S, Shi Y. Tyrosine kinase inhibitors for solid tumors in the past 20 years (2001–2020). J Hematol Oncol. 2020;13:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yuan X, Bu H, Zhou J, Yang C‐Y, Zhang H. Recent advances of SHP2 inhibitors in cancer therapy: Current development and clinical application. J Med Chem. 2020;63:11368–11396. [DOI] [PubMed] [Google Scholar]
- 24. Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol. 2009;21:177–184. [DOI] [PubMed] [Google Scholar]
- 25. Yarden Y, Pines G. The ERBB network: At last, cancer therapy meets systems biology. Nat Rev Cancer. 2012;12:553–563. [DOI] [PubMed] [Google Scholar]
- 26. Carey KD, Garton AJ, Romero MS, et al. Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Res. 2006;66:8163–8171. [DOI] [PubMed] [Google Scholar]
- 27. Eck MJ, Yun C‐H. Structural and mechanistic underpinnings of the differential drug sensitivity of EGFR mutations in non‐small cell lung cancer. Biochim Biophys Acta Proteins Proteom. 2010;1804:559–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–1149. [DOI] [PubMed] [Google Scholar]
- 29. Yun C‐H, Boggon TJ, Li Y, et al. Structures of lung cancer‐derived EGFR mutants and inhibitor complexes: Mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 2007;11:217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yasuda H, Park E, Yun C‐H, et al. Structural, Biochemical, and Clinical Characterization of Epidermal Growth Factor Receptor (EGFR) Exon 20 Insertion Mutations in Lung Cancer. Sci Transl Med. 2013;5:216ra177–216ra177. 10.1126/scitranslmed.3007205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lemmon MA. Ligand‐induced ErbB receptor dimerization. Exper Cell Res. 2009;315:638–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Solca F, Dahl G, Zoephel A, et al. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J Pharmacol Exper Therap. 2012;343:342–350. [DOI] [PubMed] [Google Scholar]
- 33. Wood ER, Truesdale AT, McDonald OB, et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib). Relationships among protein conformation, inhibitor off‐rate, and receptor activity in tumor cells. Cancer Res. 2004;64:6652–6659. [DOI] [PubMed] [Google Scholar]
- 34. Zhou W, Ercan D, Chen L, et al. Novel mutant‐selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462:1070–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yan X‐E, Ayaz P, Zhu S‐J, et al. Structural basis of AZD9291 selectivity for EGFR T790M. J Med Chem. 2020;63:8502–8511. [DOI] [PubMed] [Google Scholar]
- 36. Heppner DE, Günther M, Wittlinger F, Laufer SA, Eck MJ. Structural basis for EGFR mutant inhibition by trisubstituted imidazole inhibitors. J Med Chem. 2020;63:4293–4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. To C , Jang J, Chen T, et al. Single and dual targeting of mutant EGFR with an allosteric inhibitor. Cancer Discov. 2019;9:926–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. De Clercq DJH, Heppner DE, To C , et al. Discovery and optimization of dibenzodiazepinones as allosteric mutant‐selective EGFR inhibitors. ACS Med Chem Lett. 2019;10:1549–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Moy B, Kirkpatrick P, Kar S, Goss P. Lapatinib. Nat Rev Drug Discov. 2007;6:431–432. [DOI] [PubMed] [Google Scholar]
- 40. Fry DW, Bridges AJ, Denny WA, et al. Specific, irreversible inactivation of the epidermal growth factor receptor and erbB2, by a new class of tyrosine kinase inhibitor. Proc Natl Acad Sci U S A. 1998;95:12022–12027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dungo RT, Keating GM. Afatinib: First global approval. Drugs. 2013;73:1503–1515. [DOI] [PubMed] [Google Scholar]
- 42. Ellis P, Coakley N, Feld R, Kuruvilla S, Ung Y. Use of the epidermal growth factor receptor inhibitors gefitinib, erlotinib, afatinib, dacomitinib, and icotinib in the treatment of non‐small‐cell lung cancer: A systematic review. Curr Oncol. 2015;22:e183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sun H, Wu Y‐L. Dacomitinib in non‐small‐cell lung cancer: A comprehensive review for clinical application. Future Oncol. 2019;15:2769–2777. [DOI] [PubMed] [Google Scholar]
- 44. Bose P, Ozer H. Neratinib: An oral, irreversible dual EGFR/HER2 inhibitor for breast and non‐small cell lung cancer. Expert Opin Investig Drugs. 2009;18:1735–1751. [DOI] [PubMed] [Google Scholar]
- 45. Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non–small‐cell lung cancer to gefitinib. New Engl J Med. 2005;352:786–792. [DOI] [PubMed] [Google Scholar]
- 46. Yun C‐H, Mengwasser KE, Toms AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008;105:2070–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nardi V, Azam M, Daley GQ. Mechanisms and implications of imatinib resistance mutations in BCR‐ABL. Curr Opin Hematol. 2004;11:35–43. [DOI] [PubMed] [Google Scholar]
- 48. Oxnard GR, Thress KS, Alden RS, et al. Association between plasma genotyping and outcomes of treatment with osimertinib (AZD9291) in advanced non–small‐cell lung cancer. J Clin Oncol. 2016;34:3375–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Soria J‐C, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR‐mutated advanced non‐small‐cell lung cancer. New Engl J Med. 2018;378:113–125. [DOI] [PubMed] [Google Scholar]
- 50. Niederst MJ, Hu H, Mulvey HE, et al. The allelic context of the C797S mutation acquired upon treatment with third‐generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin Cancer Res. 2015;21:3924–3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Thress KS, Paweletz CP, Felip E, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non–small cell lung cancer harboring EGFR T790M. Nat Med. 2015;21:560–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen L, Fu W, Zheng L, Liu Z, Liang G. Recent progress of small‐molecule epidermal growth factor receptor (EGFR) inhibitors against C797S resistance in non‐small‐cell lung cancer: Miniperspective. J Med Chem. 2017;61:4290–4300. [DOI] [PubMed] [Google Scholar]
- 53. Günther M, Juchum M, Kelter G, Fiebig H, Laufer S. Lung cancer: EGFR inhibitors with low nanomolar activity against a therapy‐resistant L858R/T790M/C797S mutant. Angew Chem Intl Ed. 2016;55:10890–10894. [DOI] [PubMed] [Google Scholar]
- 54. Günther M, Lategahn J, Juchum M, et al. Trisubstituted pyridinylimidazoles as potent inhibitors of the clinically resistant L858R/T790M/C797S EGFR mutant: Targeting of both hydrophobic regions and the phosphate binding site. J Med Chem. 2017;60:5613–5637. [DOI] [PubMed] [Google Scholar]
- 55. Juchum M, Günther M, Döring E, Sievers‐Engler A, Lämmerhofer M, Laufer S. Trisubstituted imidazoles with a rigidized hinge binding motif act as single digit nM inhibitors of clinically relevant EGFR L858R/T790M and L858R/T790M/C797S mutants: An example of target hopping. J Med Chem. 2017;60:4636–4656. [DOI] [PubMed] [Google Scholar]
- 56. Engelhardt H, Böse D, Petronczki M, et al. Start selective and rigidify: The discovery path toward a next generation of EGFR tyrosine kinase inhibitors. J Med Chem. 2019;62:10272–10293. [DOI] [PubMed] [Google Scholar]
- 57. Uchibori K, Inase N, Araki M, et al. Brigatinib combined with anti‐EGFR antibody overcomes osimertinib resistance in EGFR‐mutated non‐small‐cell lung cancer. Nat Commun. 2017;8:14768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liu Q, Sabnis Y, Zhao Z, et al. Developing irreversible inhibitors of the protein kinase cysteinome. Chem Biol. 2013;20:146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jia Y, Yun C‐H, Park E, et al. Overcoming EGFR (T790M) and EGFR (C797S) resistance with mutant‐selective allosteric inhibitors. Nature. 2016;534:129–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Niggenaber J, Heyden L, Grabe T, Müller MP, Lategahn J, Rauh D. Complex crystal structures of EGFR with third‐generation kinase inhibitors and simultaneously bound allosteric ligands. ACS Med Chem Lett. 2020;11:2484–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ostrem JM, Shokat KM. Direct small‐molecule inhibitors of KRAS: From structural insights to mechanism‐based design. Nat Rev Drug Discov. 2016;15:771–785. [DOI] [PubMed] [Google Scholar]
- 62. Scheffzek K, Ahmadian MR, Kabsch W, et al. The Ras‐RasGAP complex: Structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science. 1997;277:333–339. [DOI] [PubMed] [Google Scholar]
- 63. Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K‐Ras (G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kessler D, Gollner A, Gmachl M, et al. Reply to Tran et al.: Dimeric KRAS protein–protein interaction stabilizers. Proc Natl Acad Sci U S A. 2020;117:3365–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72:2457–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, Westover KD. Biochemical and structural analysis of common cancer‐associated KRAS mutations. Mol Cancer Res. 2015;13:1325–1335. [DOI] [PubMed] [Google Scholar]
- 67. Janes MR, Zhang J, Li L‐S, et al. Targeting KRAS mutant cancers with a covalent G12C‐specific inhibitor. Cell. 2018;172:578–589. [DOI] [PubMed] [Google Scholar]
- 68. Canon J, Rex K, Saiki AY, et al. The clinical KRAS (G12C) inhibitor AMG 510 drives anti‐tumour immunity. Nature. 2019;575:217–223. [DOI] [PubMed] [Google Scholar]
- 69. Papadopoulos KP, Ou S‐HI, Johnson ML, et al. A phase I/II multiple expansion cohort trial of MRTX849 in patients with advanced solid tumors with KRAS G12C mutation. J Clin Oncol. 2019;37:TPS3161. [Google Scholar]
- 70. Hallin J, Engstrom LD, Hargis L, et al. The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS‐mutant cancers in mouse models and patients. Cancer Discov. 2020;10:54–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kim D, Xue JY, Lito P. Targeting KRAS(G12C): From inhibitory mechanism to modulation of antitumor effects in patients. Cell. 2020;183:850–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Xue JY, Zhao Y, Aronowitz J, et al. Rapid non‐uniform adaptation to conformation‐specific KRAS(G12C) inhibition. Nature. 2020;577:421–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kessler D, Gmachl M, Mantoulidis A, et al. Drugging an undruggable pocket on KRAS. Proc Natl Acad Sci U S A. 2019;116:15823–15829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tran TH, Alexander P, Dharmaiah S, et al. The small molecule BI‐2852 induces a nonfunctional dimer of KRAS. Proc Natl Acad Sci U S A. 2020;117:3363–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lavoie H, Therrien M. Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol. 2015;16:281–298. [DOI] [PubMed] [Google Scholar]
- 76. Holderfield M, Deuker MM, McCormick F, McMahon M. Targeting RAF kinases for cancer therapy: BRAF‐mutated melanoma and beyond. Nat Rev Cancer. 2014;14:455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Park E, Rawson S, Li K, et al. Architecture of autoinhibited and active BRAF–MEK1–14‐3‐3 complexes. Nature. 2019;575:545–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gardino AK, Smerdon SJ, Yaffe MB, et al. Structural determinants of 14‐3‐3 binding specificities and regulation of subcellular localization of 14‐3‐3‐ligand complexes: A comparison of the X‐ray crystal structures of all human 14‐3‐3 isoforms. Seminars in Cancer Biology, 2006;16(3):173–182. 10.1016/j.semcancer.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 79. Wan PT, Garnett MJ, Roe SM, et al. Mechanism of activation of the RAF‐ERK signaling pathway by oncogenic mutations of B‐RAF. Cell. 2004;116:855–867. [DOI] [PubMed] [Google Scholar]
- 80. Haling JR, Sudhamsu J, Yen I, et al. Structure of the BRAF‐MEK complex reveals a kinase activity independent role for BRAF in MAPK signaling. Cancer Cell. 2014;26:402–413. [DOI] [PubMed] [Google Scholar]
- 81. Gilmartin AG, Bleam MR, Groy A, et al. GSK1120212 (JTP‐74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2011;17:989–1000. [DOI] [PubMed] [Google Scholar]
- 82. Leevers SJ, Paterson HF, Marshall CJ. Requirement for Ras in Raf activation is overcome by targeting Raf to the plasma membrane. Nature. 1994;369:411–414. [DOI] [PubMed] [Google Scholar]
- 83. Stokoe D, Macdonald SG, Cadwallader K, Symons M, Hancock JF. Activation of Raf as a result of recruitment to the plasma membrane. Science. 1994;264:1463–1467. [DOI] [PubMed] [Google Scholar]
- 84. Kondo Y, Ognjenović J, Banerjee S, et al. Cryo‐EM structure of a dimeric B‐Raf:14‐3‐3 complex reveals asymmetry in the active sites of B‐Raf kinases. Science. 2019;366:109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Liau NPD, Venkatanarayan A, Quinn JG, et al. Dimerization induced by C‐terminal 14‐3‐3 binding is sufficient for BRAF kinase activation. Biochemistry. 2020;59:3982–3992. [DOI] [PubMed] [Google Scholar]
- 86. Liau NPD, Wendorff TJ, Quinn JG, et al. Negative regulation of RAF kinase activity by ATP is overcome by 14‐3‐3‐induced dimerization. Nat Struct Mol Biol. 2020;27:134–141. [DOI] [PubMed] [Google Scholar]
- 87. Rajakulendran T, Sahmi M, Lefrançois M, Sicheri F, Therrien M. A dimerization‐dependent mechanism drives RAF catalytic activation. Nature. 2009;461:542–545. [DOI] [PubMed] [Google Scholar]
- 88. Wilhelm S, Carter C, Lynch M, et al. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006;5:835–844. [DOI] [PubMed] [Google Scholar]
- 89. Escudier B, Worden F, Kudo M. Sorafenib: Key lessons from over 10 years of experience. Expert Rev Anticancer Therapy. 2019;19:177–189. [DOI] [PubMed] [Google Scholar]
- 90. Zhao Z, Wu H, Wang L, et al. Exploration of type II binding mode: A privileged approach for kinase inhibitor focused drug discovery? ACS Chem Biol. 2014;9:1230–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Karoulia Z, Wu Y, Ahmed Tamer A, et al. An integrated model of RAF inhibitor action predicts inhibitor activity against oncogenic BRAF signaling. Cancer Cell. 2016;30:485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Karoulia Z, Gavathiotis E, Poulikakos PI. New perspectives for targeting RAF kinase in human cancer. Nat Rev Cancer. 2017;17:676–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Poulikakos PI, Persaud Y, Janakiraman M, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature. 2011;480:387–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Yao Z, Torres Neilawattie M, Tao A, et al. BRAF mutants evade ERK‐dependent feedback by different mechanisms that determine their sensitivity to pharmacologic inhibition. Cancer Cell. 2015;28:370–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Jones DTW, Kocialkowski S, Liu L, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008;68:8673–8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Sun Y, Alberta JA, Pilarz C, et al. A brain‐penetrant RAF dimer antagonist for the noncanonical BRAF oncoprotein of pediatric low‐grade astrocytomas. Neuro Oncol. 2017;19:774–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Subbiah V, Baik C, Kirkwood JM. Clinical development of BRAF plus MEK inhibitor combinations. Trends Cancer. 2020;6:797–810. [DOI] [PubMed] [Google Scholar]
- 98. Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild‐type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–435. [DOI] [PubMed] [Google Scholar]
- 99. Heidorn SJ, Milagre C, Whittaker S, et al. Kinase‐dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild‐type BRAF. Nature. 2010;464:427–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zhang C, Spevak W, Zhang Y, et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature. 2015;526:583–586. [DOI] [PubMed] [Google Scholar]
- 102. Yao Z, Gao Y, Su W, et al. RAF inhibitor PLX8394 selectively disrupts BRAF dimers and RAS‐independent BRAF‐mutant‐driven signaling. Nat Med. 2019;25:284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Noeparast A, Giron P, Brakeleer SD, et al. Type II RAF inhibitor causes superior ERK pathway suppression compared to type I RAF inhibitor in cells expressing different BRAF mutant types recurrently found in lung cancer. Oncotarget. 2018;9:16110–16123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Yaeger R, Corcoran RB. Targeting alterations in the RAF–MEK pathway. Cancer Discov. 2019;9:329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Gross AM, Wolters PL, Dombi E, et al. Selumetinib in children with inoperable plexiform neurofibromas. New Engl J Med. 2020;382:1430–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen‐activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. [DOI] [PubMed] [Google Scholar]
- 107. Ohren JF, Chen H, Pavlovsky A, et al. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat Struct Mol Biol. 2004;11:1192–1197. [DOI] [PubMed] [Google Scholar]
- 108. Khan ZM, Real AM, Marsiglia WM, et al. Structural basis for the action of the drug trametinib at KSR‐bound MEK. Nature. 2020;588:509–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Fischmann TO, Smith CK, Mayhood TW, et al. Crystal structures of MEK1 binary and ternary complexes with nucleotides and inhibitors. Biochemistry. 2009;48:2661–2674. [DOI] [PubMed] [Google Scholar]
- 110. Yamaguchi T, Yoshida T, Kurachi R, et al. Identification of JTP‐70902, a p15INK4b‐inductive compound, as a novel MEK1/2 inhibitor. Cancer Sci. 2007;98:1809–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Abe H, Kikuchi S, Hayakawa K, et al. Discovery of a highly potent and selective MEK inhibitor: GSK1120212 (JTP‐74057 DMSO solvate). ACS Med Chem Lett. 2011;2:320–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Ishii N, Harada N, Joseph EW, et al. Enhanced inhibition of ERK signaling by a novel allosteric MEK inhibitor, CH5126766, that suppresses feedback reactivation of RAF activity. Cancer Res. 2013;73:4050–4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Lito P, Saborowski A, Yue J, et al. Disruption of CRAF‐mediated MEK activation is required for effective MEK inhibition in KRAS mutant tumors. Cancer Cell. 2014;25:697–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Hatzivassiliou G, Haling JR, Chen H, et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS‐ versus BRAF‐driven cancers. Nature. 2013;501:232–236. [DOI] [PubMed] [Google Scholar]
- 115. Grossmann KS, Rosário M, Birchmeier C, Birchmeier W. The tyrosine phosphatase Shp2 in development and cancer. Adv Cancer Res. 2010;106:53–89. [DOI] [PubMed] [Google Scholar]
- 116. Prahallad A, Heynen GJ, Germano G, et al. PTPN11 is a central node in intrinsic and acquired resistance to targeted cancer drugs. Cell Rep. 2015;12:1978–1985. [DOI] [PubMed] [Google Scholar]
- 117. Schneeberger VE, Ren Y, Luetteke N, et al. Inhibition of Shp2 suppresses mutant EGFR‐induced lung tumors in transgenic mouse model of lung adenocarcinoma. Oncotarget. 2015;6:6191–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Bennett AM, Tang TL, Sugimoto S, Walsh CT, Neel BG. Protein‐tyrosine‐phosphatase SHPTP2 couples platelet‐derived growth factor receptor beta to Ras. Proc Natl Acad Sci U S A. 1994;91:7335–7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Li W, Nishimura R, Kashishian A, et al. A new function for a phosphotyrosine phosphatase: Linking GRB2‐Sos to a receptor tyrosine kinase. Mol Cell Biol. 1994;14:509–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Dance M, Montagner A, Salles J‐P, Yart A, Raynal P. The molecular functions of Shp2 in the Ras/mitogen‐activated protein kinase (ERK1/2) pathway. Cell Signal. 2008;20:453–459. [DOI] [PubMed] [Google Scholar]
- 121. Ruess DA, Heynen GJ, Ciecielski KJ, et al. Mutant KRAS‐driven cancers depend on PTPN11/SHP2 phosphatase. Nat Med. 2018;24:954–960. [DOI] [PubMed] [Google Scholar]
- 122. Chen Y‐NP, LaMarche MJ, Chan HM, et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature. 2016;535:148–152. [DOI] [PubMed] [Google Scholar]
- 123. Hof P, Pluskey S, Dhe‐Paganon S, Eck MJ, Shoelson SE. Crystal structure of the tyrosine phosphatase SHP‐2. Cell. 1998;92:441–450. [DOI] [PubMed] [Google Scholar]
- 124. Jingwei W, Huan Z, Guilong Z, Runling W. Allosteric inhibitors of SHP2: An updated patent review (2015‐2020). Curr Med Chem. 2020;27:1–17. [DOI] [PubMed] [Google Scholar]
- 125. Kerr DL, Haderk F, Bivona TG. Allosteric SHP2 inhibitors in cancer: Targeting the intersection of RAS, resistance, and the immune microenvironment. Curr Opin Chem Biol. 2021;62:1–12. [DOI] [PubMed] [Google Scholar]
- 126. Ryan MB, Fece de la Cruz F, Phat S, et al. Vertical pathway inhibition overcomes adaptive feedback resistance to KRASG12C inhibition. Clin Cancer Res. 2020;26:1633–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Nichols RJ, Haderk F, Stahlhut C, et al. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF‐, NF1‐ and RAS‐driven cancers. Nat Cell Biol. 2018;20:1064–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Foster SA, Whalen DM, Özen A, et al. Activation mechanism of oncogenic deletion mutations in BRAF, EGFR, and HER2. Cancer Cell. 2016;29:477–493. [DOI] [PubMed] [Google Scholar]
- 129. Gao Y, Chang MT, McKay D, et al. Allele‐specific mechanisms of activation of MEK1 mutants determine their properties. Cancer Discov. 2018;8:648–661. [DOI] [PMC free article] [PubMed] [Google Scholar]