

Abstract
Identifying and purchasing new small molecules to test in biological assays is enabling for ligand discovery, but as purchasable chemical space continues to grow into the tens of billions based on inexpensive make-on-demand compounds, simply searching this space becomes a major challenge. We have therefore developed ZINC20, a new version of ZINC with two major new features: billions of new molecules and new methods to search them. As a fully enumerated database, ZINC can be searched precisely using explicit atomic-level graph-based methods, such as SmallWorld for similarity, Arthor for pattern and substructure search, as well as 3D methods such as docking. Analysis of the new make-on-demand compound sets by these and related tools reveals startling features. For instance, over 97% of the core Bemis-Murcko scaffolds in make-on-demand libraries are unavailable from “in stock” collections. Correspondingly, the number of new Bemis-Murcko scaffolds is rising almost as a linear fraction of the elaborated molecules. Thus, an 88-fold increase in the number of molecules in the make-on-demand vs the in-stock sets is built upon a 16-fold increase in the number of Bemis-Murcko scaffolds. The make-on-demand library is also more structurally diverse than physical libraries, with a massive increase in disc- and sphere-like shaped molecules. The new system is freely available at zinc20.docking.org.
Graphical Abstract
Introduction
Commercially accessible libraries enable new research because they allow rapid and inexpensive exploration of chemical space. Unfortunately, as the libraries have grown into the billions of molecules, traditional search and representation methods have become unwieldy. For instance, a similarity search at ECFP4 Tanimoto 40% to find analogs using pre-calculated and indexed ECFP4 512-bit fingerprints stored in a Postgres database took generally under 20 seconds to search 100 million molecules on our 64-core computer. The same calculation often took over 3 minutes for 1 billion molecules. A key problem of similarity search is that to find the most similar molecules, the entire database must be consulted to be sure one has found the most similar. An incomplete scan could provide misleading results if the most similar molecules were near the end of the database. Whereas the number of in-stock compounds worldwide – now around 14 million – grows only a few percent each year, make-on-demand libraries are growing almost exponentially, and may be bounded only by our ability to construct and represent them computationally; even now, over 1010 are notionally available, with some private commercial collections larger still. It is perhaps only a few years until the number of readily available commercial compounds will reach 1011 to 1012 molecules. New approaches are urgently needed to search this space.
A key benefit of make-on-demand libraries is that compounds contained in them are both new to the planet and can be readily accessed. These never-been-made compounds can usually be reliably synthesized because the building blocks are on the shelf, the parallel synthetic methods are robust, and the viability of synthesis has been predicted in advance based on data from analogous prior reactions. As a result, synthesis-on-demand compounds now make up over 99.9% of the world’s catalog molecules. We have experienced synthesis fulfillment rates of over 85% from these libraries in over a dozen projects, which is about the same success rate as for supposedly in stock compounds.1, 2
ZINC is a publicly available database that aggregates commercially available and annotated compounds3–5. ZINC provides downloadable 2D and 3D versions as well as a website that enables rapid molecule lookup and analog search. ZINC has grown from fewer than 1 million compounds in 2005 to nearly 2 billion now. Its design has changed over time to respond to the needs of the field, the growth of purchasable chemical space, and developments in software and hardware. The ZINC website is used by thousands of investigators each month and many terabytes of data are downloaded each week as can be seen from the usage statistics accessible via the “About” menu followed by “usage” in the top right corner of every ZINC page.
In addition to its original focus on molecular docking3, an important application of ZINC is Analog-by-catalog (ABC), a pragmatic approach based on the molecular similarity principle6, 7 to identify similar compounds with which to explore structure-activity relationships (SAR). Many investigators find it helpful to search for analogs in real time, where “what if” questions about compound availability may be explored.
Real-time and non-real-time search are qualitatively different, although there is no clear single line that separates them. Search in real time provides results rapidly enough that the investigator remains focused on the question at hand, and thus a kind of conversation between the scientists and the database can be said to take place. In non-real-time search, the user moves on to other things, and must return and remember what she was thinking when she asked the query. While individuals vary, our own experience is that searches that reliably return results in under one minute are real time and engaging while searches that take over 3 minutes tend to lose the immediate attention of the investigator and are not real time. Calculations that take between one and three minutes are borderline and will depend on the concentration of the scientist, so we call them near-real-time here. To be most useful, search should be in real time or occasionally near-real time. As the libraries have grown, methods that could search 100 million molecules reliably for similarity in real-time, generally under 20 seconds, became near-real-time taking from one to three minutes as ZINC grew towards a billion molecules, and then became non-real-time, taking over three minutes, at the current size heading towards two billion molecules. For substructure and pattern searches, the problem was even worse. Even at 100 million molecules, substructure and pattern searches often failed to complete in under 3 minutes. The problem became steadily worse as the database grew to and beyond one billion molecules.
One approach to this problem avoids full database enumeration entirely by instead searching in building-block space8 using feature trees8. This approach scales well, searching only the building blocks and not the enumerated compounds, and approximating pharmacophores (“feature trees”) rather than searching at the atomic level. While the searches are only near real time, reportedly taking several minutes to search 1010 implied molecules on a desktop computer, this approach searches 10 times as many molecules as we could search using pre-computed fingerprints in the same time on our server. Moreover, because it scales with building block size and not with the size of the enumerated database, this approach is likely to remain competitive as the implied size of purchasable chemical space grows to 1011, 1012 and beyond.8 Notwithstanding these advantages of speed, fully enumerated chemical databases retain important advantages. The molecules can be interrogated more precisely at the atomic level versus the more approximate pharmacophore represented by feature trees. (Figure 1) For instance, searching building blocks at the atomic level for protein kinase hinge binders can match a specific pattern of hydrogen bond acceptors and donors, whereas approximate feature-based methods often cannot. Searching fully enumerated structures also allows interrogation of the entire molecule. Approaches like feature trees do not allow substructure searches or pattern matching, as widely implemented in SMARTS9, for instance. Explicitly enumerated databases also allow each molecule to be interrogated for synthetic accessibility because only accessible molecules are enumerated, while in building block space there remains some doubt that all the implied molecules are accessible. Finally, for atomic resolution screens, as in molecular docking, the molecules must be fully enumerated in three-dimensions.
Figure 1. Feature trees capture pharmacophores in a “fuzzy” fashion that cannot distinguish atomic-level detail.
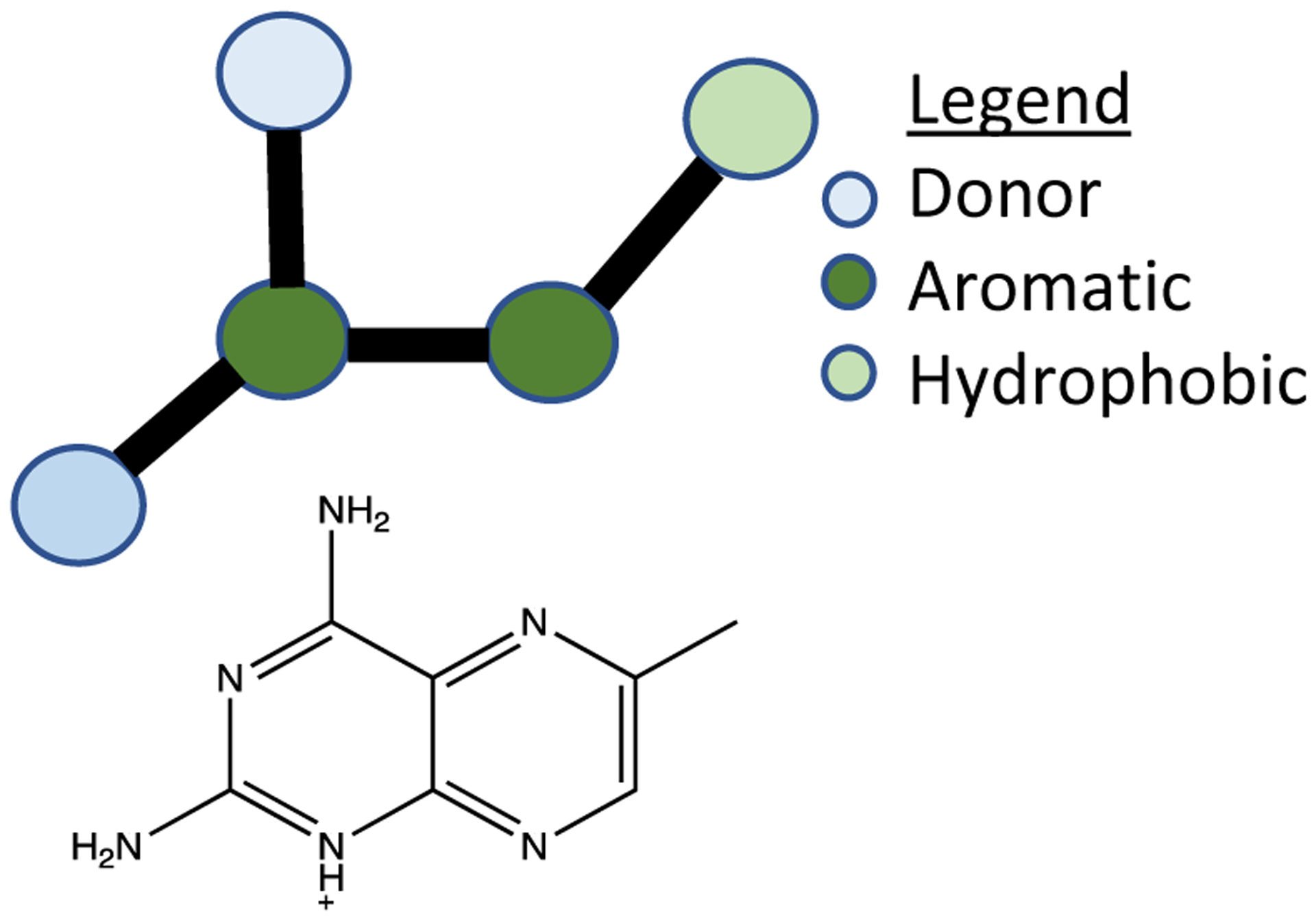
By contrast, searches in SmallWorld and Arthor are atomic and can match precise atom patterns, allowing for finer discrimination at scale.
In this work we focus on three distinct kinds of searches: A) whole molecule search, in which molecules that most resemble the entire query are prioritized, B) substructure search, in which the molecules that contain the entire query molecule are identified and C) pattern search, in which molecules containing specified molecular pattern(s) are selected. These represent the three most common molecular-level searches we are aware of. Substructure and pattern searches have one big advantage over similarity searches: they do not need to complete to give correct and useful results. For similarity, the question is almost always, what are the most similar molecules, and thus the entire database must be consulted. For substructure and pattern searches this is not the case. Molecules either have the substructure or match the pattern, or they do not, so any subset of the database that can be screened provides useful and correct, if incomplete information. Pattern searches can be particularly useful on building block databases to identify functional group combinations for library enumeration.
Fingerprint-based search methods vary in detail but scale roughly linearly in the number of molecules. In our hands all fingerprint-based methods are challenging to use for providing real-time public search of over one billion molecules. Radical new non-fingerprint-based approaches offer the hope that real-time search may remain viable for several more orders of magnitude of library growth.
Here we investigate how the growth of the ZINC library, largely driven by the virtual make-on-demand libraries has affected the compounds, chemotypes, and Bemis-Murcko scaffolds (hereafter “scaffolds”) readily available to the community. We then turn to investigating how this large new chemical space may be efficiently searched, overcoming the liabilities that have made traditional atom-resolution search methods outdated for the growing libraries. Three distinct types of searches are investigated: the whole molecule similarity search, substructure searches, and chemotype pattern searches. We will argue that the advent of the “make-on-demand” libraries has dramatically expanded not only the number of new molecules readily accessible to the community, but also the diversity of accessible compounds. The new tools described here are made freely available via https://zinc20.docking.org, https://sw.docking.org, https://arthor.docking.org and https://cartblanche.docking.org and their http cognates.
Results
New content of the ZINC library
ZINC continues to grow in size with ZINC20 now including 1.4 billion compounds, 1.3 billion of which are purchasable, sourced from 310 catalogs from 150 companies. Over 90% of catalogs are refreshed every 90 days and over 90% of compounds have been verified as purchasable within the last three months (our 90/90/90 rule). Additional databases of molecules not yet added to ZINC, totalling more than 1010 molecules, may also be searched, as described below.
As a resource for lead discovery, ZINC has in the past prioritized screening compounds that follow the rule-of-4 (Ro4): thus, molecular weight less than 400 g/mol and calculated logP less than 4. Recently we have seen an increasing number of targets for which Ro4 molecules are too small or insufficiently lipophilic, and correspondingly, we have begun to load more Ro5 molecules. Of the 736 million lead-like (Ro4) molecules in ZINC, 509 million are available for download in 3D ready for docking.
We wondered how the diversity of the make-on-demand library compares to that of molecules in physical screening decks. To investigate this, we compared the ZINC make-on-demand library, most of which is from the Enamine REAL collection10, with several publicly available physical libraries, including the Molecular Libraries Small Molecule Repository of the NIH (MLSMR)11, the Small Molecule Discovery Center library UCSF (SMDC)12, an academic high throughput screening center, and the “in-stock” Ro4 compounds in ZINC5 (Table 1). To quantify the topological diversity of the molecules within each library, we calculated Bemis-Murcko scaffolds13 for all molecules and plotted the number of compounds within each scaffold (Figure 2). For instance, The MLSMR contains just over 405,000, of which almost 53,000 contain a unique scaffold, with no other analogs containing the same scaffold (leftmost yellow dot in figure Figure 2C). By the time there are ten molecules per scaffold, the number of scaffolds has fallen to 3504, a 15-fold drop. In the six million molecule ZINC “in stock” collection, meanwhile, there are over 534,000 molecules that are found in unique scaffolds, and by the time there are ten molecules per scaffold the number of scaffolds has dropped to 33,542, about a 16-fold drop (light green squares in Figure 2A). Indeed, the two libraries are roughly proportional in the ratio of compounds to scaffolds throughout their distributions with the ZINC “in stock” simply being 15-fold larger. This increase in size largely explains the longer-tail of the “in-stock” library, where a relatively small number of scaffolds have several thousands of compounds within them. For all three physical collections (“in-stock”, SMDC, and MLSMR), the rate of change of cluster sizes going from smallest to largest scaffolds is about the same. Intriguingly, the distribution appears broader in the make-on-demand library. Despite being 100-fold larger than the ZINC “in-stock” collection, for instance, there are only 10-fold more scaffolds that contain only 1 molecule, and by the time one reaches ten molecules per scaffold, the number of scaffolds has only fallen by 3.75-fold, not 19-fold. The slower rate of decline in scaffolds per number of molecules within them holds throughout the distribution, further extending the long tail. While it is certainly true that the rise of the number of scaffolds has not kept up with the rise of compounds, and that the ratio of scaffolds to compounds in the physical libraries is higher than that in the “make-on-demand” library, the number of scaffolds in the “make-on-demand” library is surprisingly high. For instance, while the number of molecules is 88-fold higher in the “make-on-demand” than in the “in-stock” library (Table 1), the scaffolds in the former have also risen by 16-fold versus the latter. These numbers reflect on the frequent ability to find analogs around a Bemis-Murcko scaffold within the “make-on-demand” library. This observation is consistent with the parallel chemistry used to generate the libraries. It is also consistent with the high relative diversity of the collection and the lack of dominance of most scaffolds. The most overrepresented scaffolds are generally also the small and simple ones, such phenyl (3.5 M compounds), pyridine (2.8 M compounds), and cyclohexyl (1.7 M compounds), reflecting a weakness in using Bemis-Murcko scaffolds as a classifier for these ultra-small scaffolds.
Table 1.
In-stock and make-on-demand catalogs
| Catalog name | Physical? | Number | Website |
|---|---|---|---|
| ZINC Ro4 in-stock | Yes | 6,060,000 | https://zinc15.docking.org |
| SMDC | Yes | 690,125 | https://smdc.ucsf.edu |
| MLSMR | Yes | 406,098 | https://pubchem.ncbi.nlm.nih.gov/source/MLSMR |
| ZINC Ro4 make-on-demand | No | 515,000,000 | https://zinc15.docking.org |
Figure 2. Scaffold diversity of “in-stock” and “on-demand” catalogs.
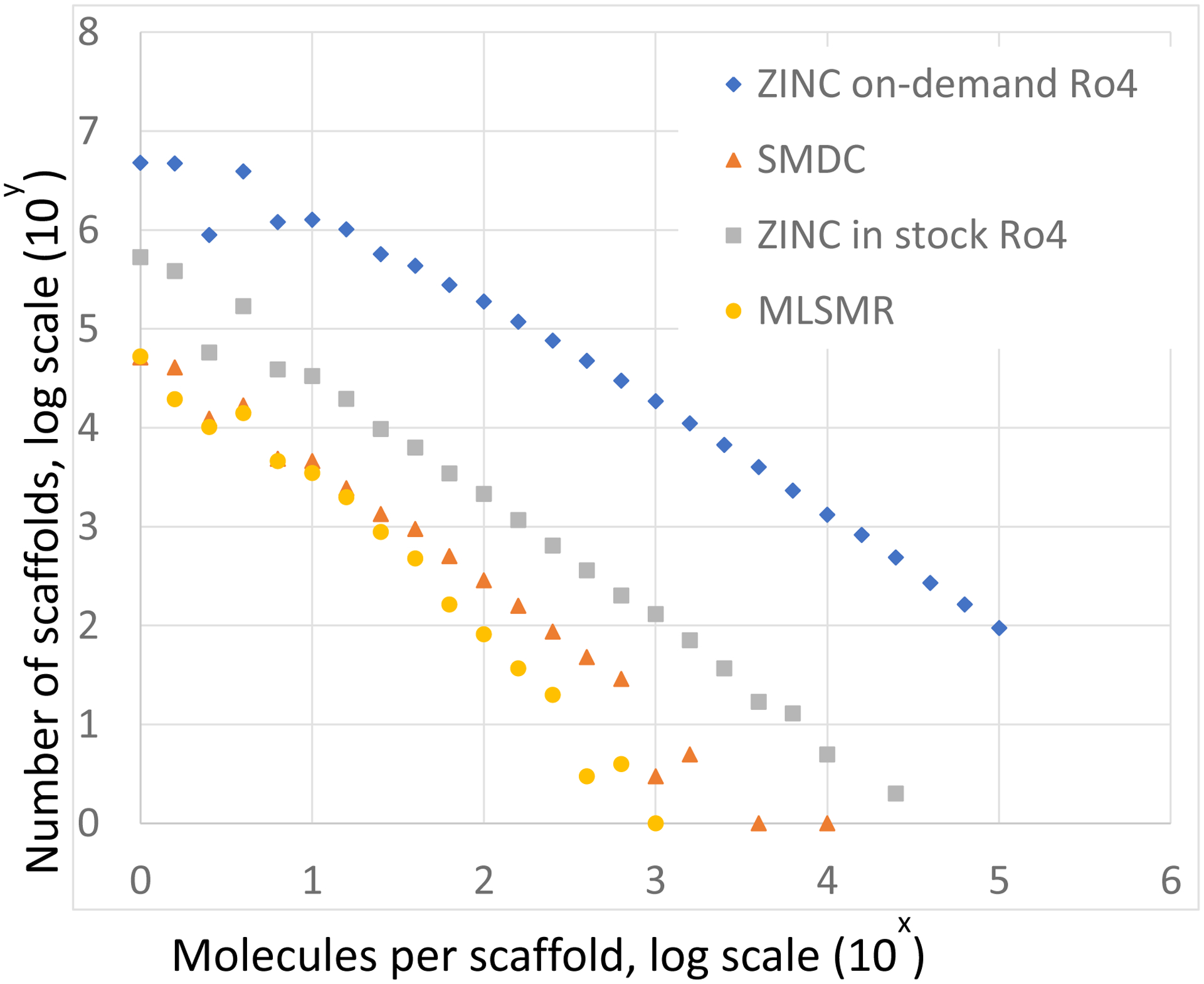
Comparison of the number of scaffolds in bins of molecules per scaffold. Histogram tops shown. Log/log scale. Thus there are 95 on-demand scaffolds having 10,000 examples in the database (bottom right), and about 4.7 million that have only a single representative (top left).
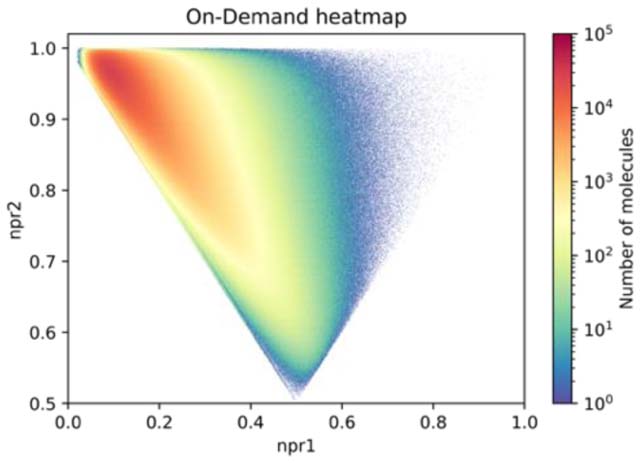
Another approach to assess library diversity is to compare the shapes of molecules.14. This method uses normalized ratios of principle moments of inertia to classify shape with extreme values characterized as rod (1D, top left) , disc (2D, bottom middle) and sphere (3D, top right). Our calculated results show that the make-on-demand molecules cover important areas of this space that are historically underrepresented in physical decks, such as the space in the direction of sphere-like (top right corner). (Figure 3). For reference, benzene is disc shaped (npr1=0.5, npr2=0.5), adamantane is sphere-like (npr1=1, npr2=1) and Gleevec is rod like (npr1=0,npr2=1)
Figure 3. Molecular Shape Distribution.

Normalized ratios of principal moments of inertia analysis. Two principle component magnitude ratios are plotted, npr1 (x-axis) and npr2 (y-axis) (see Methods). Rod-shaped molecules appear in top left of graph, disc-shaped in the bottom middle, and sphere in the top right The heatmaps are colored by the number of molecules in each of 500,000 pixels. A) ZINC in-stock Ro4 B) MLSMR C) SMDC. D) ZINC Ro4 make-on-demand.
We can also ask how much of the make-on-demand library is not represented in the three different physical libraries, by determining the number of scaffolds of the make-on-demand libraries missing in physical decks. Almost all of the make-on-demand molecules have no representation in physical libraries at the scaffold level demonstrating the novelty of the make-on-demand chemical space (Table 2). Overall, the number of molecules in the make-on-demand libraries is massive – about a thousand times bigger than the physical libraries MLSMR and SMDC and about one hundred times bigger than all the molecules in stock anywhere in the world. The physical libraries are expensive to create and can only grow slowly while the make-on-demand libraries are growing rapidly, further re-enforcing these trends.
Table 2.
Scaffolds in physical and make-on-demand libraries.
| Library | Number of molecules | Number of Bemis-Murcko Scaffolds | Number of Scaffolds in Library AND On-Demand | Number of Scaffolds in Library NOT IN On-Demand (%) | Percent of Scaffolds in On-Demand NOT IN Library |
|---|---|---|---|---|---|
| MLSMR | 406,098 | 108,178 | 25,822 | 82,356 | 99.9% |
| SMDC | 690,125 | 136,862 | 34,234 | 102,628 | 99.8% |
| In-stock-Ro4.0 | 6,136,700 | 1,263,063 | 495,474 | 767,589 | 97.5% |
| In-stock-Ro3.5 | 3,546,040 | 744,796 | 400,574 | 344,222 | 97.9% |
| On-Demand | 531,645,834 | 19,590,914 | - | - | 0% |
New tools
ZINC is not only a repository for accessible molecules, and their physical representations (as are widely used in docking, for instance), it is also a suite of tools for searching those molecules. An important use of ZINC tools is to find analogs of a biologically active molecule, perhaps found from a library screen. This is often called analog-by-catalog (ABC) and has the advantage of rapid synthesis and testing at relatively low cost. Previous methods for analog searching, based on stored fingerprints in Postgres/RDKit5 began to suffer in speed-of-search as ZINC grew, owing to the roughly linear scaling of enumerated fingerprints. The speed declines further still when the fingerprint indexes can no longer fit in the computer memory and often get swapped out to disk, which we estimated started becoming frequent after ZINC reached 1.2 billion SMILES. In order to identify a method that could better scale to the growing on-demand libraries, we implemented two fast-search methods with different search algorithms, SmallWorld and Arthor.
SmallWorld is a graph-edit distance and maximum common subgraph (MCS) method. One of its key innovations is to pre-index anonymous graphs of all possible molecules that are specifically enumerated. Searching a database indexed in this way involves simply looking up the anonymized graph of the target molecule, followed by taking small steps in graph-edit-distance space to traverse the pre-indexed map (Figure 4). As a result, SmallWorld search time grows in sub-linear time, almost independent of the number of molecules searched, given a sufficiently large and fast disk to hold the index. In a database of 166 billion molecules, for instance, the most similar molecules were identified consistently within one or two seconds on our computers.
Figure 4.
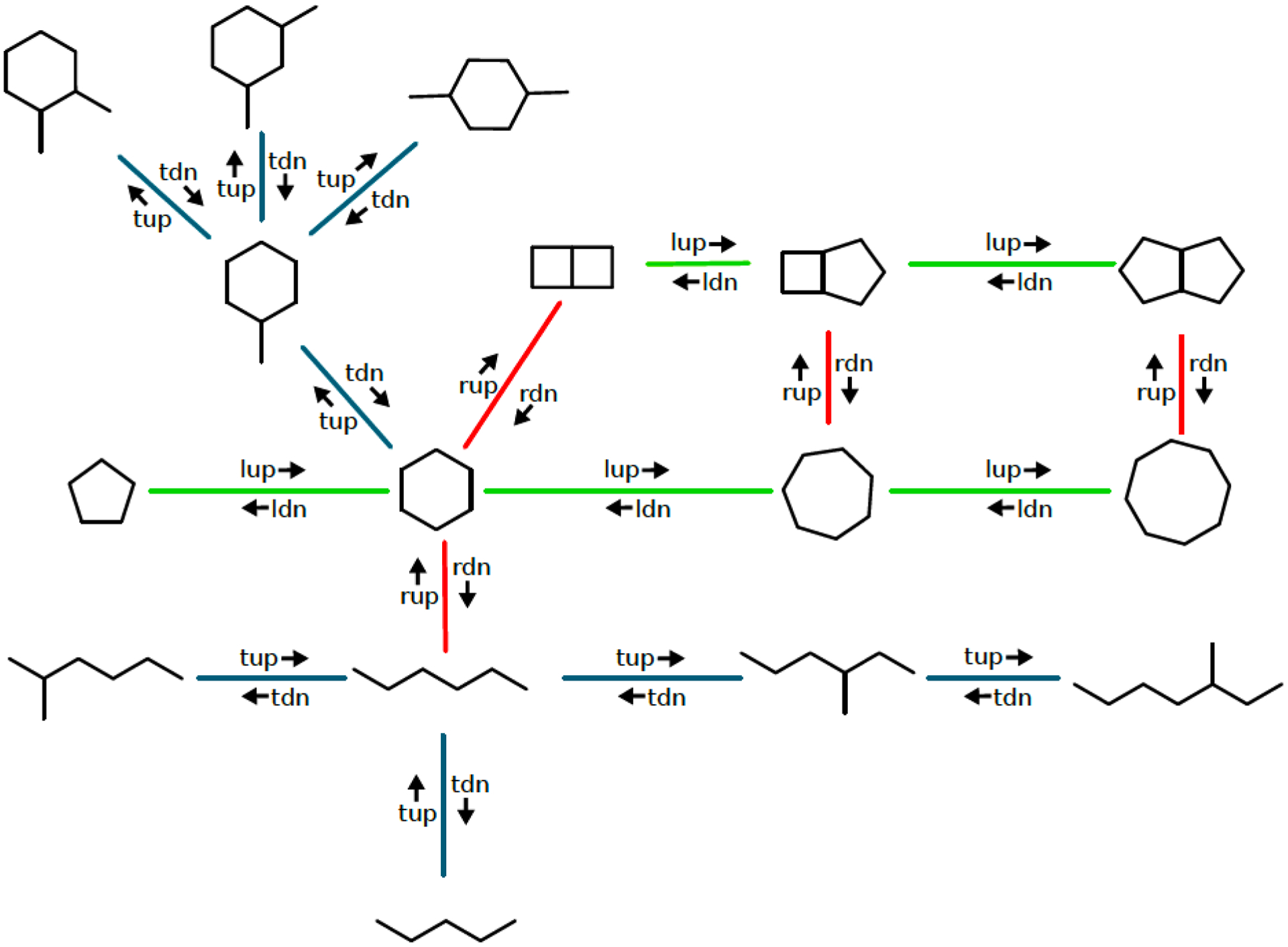
SmallWorld indexes the topological space of organic molecules into anonymous graphs.
Extended figure legend. In this ideographic representation (map) of chemical space, vertices of this graph are labelled with molecular graphs. Each graph is connected to its neighbors by elementary steps in graph-edit-distance space, such as add a terminal atom (tup), delete a terminal atom (tdn), open a ring (rup), close a ring (rdn), insert a linker atom (lup), delete a linker atom (ldn).
A second approach, Arthor15, uses a compact persistent binary representation of molecules and a customized pattern matcher based on the SMARTS language to operate on it. Given a minimium of 128 GB of computer memory, Arthor can search over 1 billion molecules for substructure or chemotype patterns in around one or two seconds on modern commodity computers (see Supporting information). Unlike SmallWorld, Arthor remains roughly linear in performance with database growth, but it is the fastest method we have yet found for atom-level substructure and pattern searches. Arthor can be scaled over an array of several computers using the RoundTable algorithm to allow rapid search of billions and even tens of billions of molecules. Whereas Arthor does support whole-molecule similarity search, we have chosen to use SmallWorld for whole molecule similarity due to its speed, relying on Arthor for substructure and pattern searches where it excels. To make the public search pragmatic, we cap Arthor searches at 10,000 molecules. For common patterns, such as phenyl and cyclohexyl for instance, the first 10,000 molecules are often found in under a second. For rarer, more specific patterns that occur up to a few thousand times, Arthor easily searches 1.4 billion molecules in a second or two. It is molecules or patterns of intermediate frequency that present a challenge to a real-time service. In our hands, Arthor generally completes searching 1.4 billion, or hits the 10,000-molecule cap within at most 10–20 seconds in the worst cases. This has allowed us to support a freely accessible public interface to Arthor, both standalone and via ZINC. We intend to offer a comprehensive asynchronous search feature of larger databases and allowing more results in the near future. For now, those who are interested in how many of the 1.4 billion molecules of ZINC contain a phenyl ring should download the database to their home computer and search it there.
To investigate SmallWorld analog searches of the make-on-demand library, we exported molecules as SMILES for both building blocks and screening compounds, organized by purchasability (see Box 1). We found that analogs of molecules having thousands of analogs among the 515 million (and also bigger multi-billion-size datasets) were fast, with first results typically appearing within seconds. Thousands of analogs - essentially all analogs we would usually be interested in – would usually appear within 15 seconds and often much faster. This is a radical and profound improvement over any fingerprint-based methods we are aware of. Conversely, using the older ECFP4-fingerprint method, where the fingerprint is stored and indexed inside Postgres, searching for the most similar 100 analogs almost always takes more than 3 minutes to search 1.4 billion molecules. In analog searches for over 1000 molecules, each with over 100 analogs, SmallWorld searches on average took two seconds to find the first 100 analogs.
Box 1. Purchasability in ZINC.
ZINC organizes catalogs and the compounds they contain into six purchasability levels, three in stock, one make-on-demand, one boutique, and “annotated” i.e. not for sale.
Premier – these are compounds from the most responsive and most reliable vendors. Most of these compounds are also well priced at around $100 or less.
In-stock – These are compounds where prices are often higher than Premier, or are unknown.
Agent – These are compounds available via resellers that do not make the compounds.
On-demand. These are compounds that are often well priced often near $100, generally less than $200 each. Compounds are not in stock but are synthesized on request, generally requiring around 6 weeks from order to delivery and with – in our experience – an 85% delivery success rate.
Boutique – These are compounds that are advertised for sale. Absent further information, we either have little experience with the vendor, or suspect the compounds are expensive, or both. We generally do not dock boutique compounds, but we will consider them during analog searches.
Annotated (Not-for-sale) – These are compounds such as in ChEMBL, DrugBank or other annotated collections, where the compounds are reported to have biological activity but do not exist is any current commercial catalog according to information available to us.
We assign purchasability based on our own purchasing experience, prices when available, and based on tips from helpful colleagues. Compounds are assigned the highest purchasability based on their current catalog membership. We revise purchasability continually based on information we receive. Please contact us if you have information to share that will help us improve ZINC.
Accordingly, we built a software interface to perform similarity searches using SmallWorld from ZINC (Figure 5). From https://zinc20.docking.org the user selects “Substances” from the menu bar at the top of the page (Figure 5, left panel). On the left-hand side of the page, the bait molecules may be drawn or SMILES pasted into the “Search using one” field. The results are displayed as previously in ZINC155. Alternatively, the user can go directly to https://sw.docking.org and search from there (Figure 5, right panel). Here, the subsets of ZINC to search are selected from the DataSet popup, which includes some datasets such as WuXi GalaXi and Mcule Ultimate that are not yet fully loaded into ZINC. Dataset names contain the date they were prepared and the approximate number of molecules. Other parameters for controlling the search are available (left in the panel). Two Tanimoto similarities based on legacy fingerprints, ECFP4, and Daylight, are calculated and displayed for historical comparison to graph-edit distances and may be used to sort the results.
Figure 5. SmallWorld for whole-molecule similarity in ZINC.
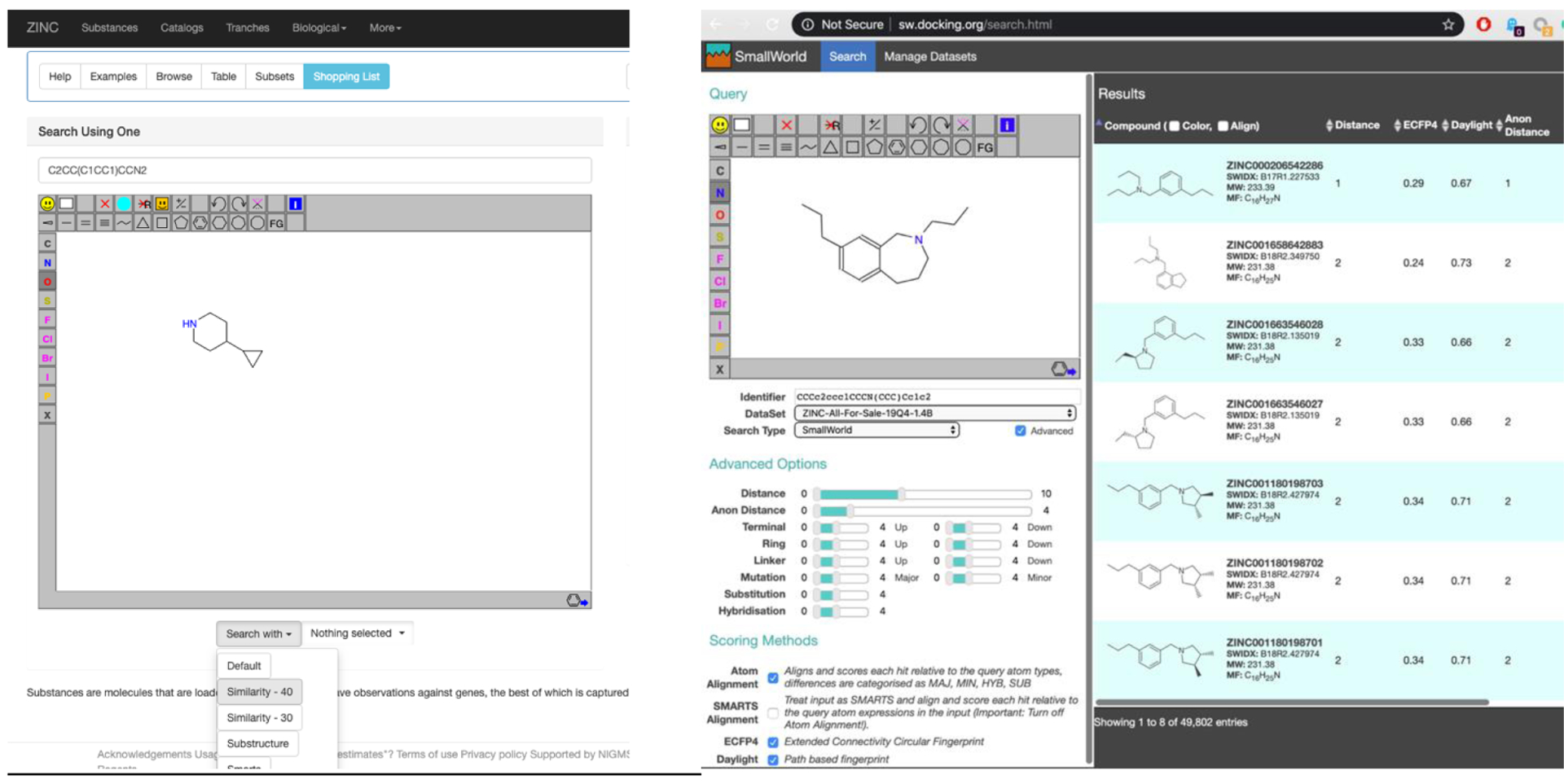
Left, the ZINC interface, that now routes similarity searches to SmallWorld. A new popup selector now also allows the selection by purchasability subset and/or building blocks. Right, the SmallWorld interface at sw.docking.org. The subset of ZINC to search is selected from the DataSet popup (middle left).
Two widely used variations of analog searching are substructure searches and chemotype pattern matching, which allow investigators to specify “wildcard” atom types, functional group combinations, and much more. As with fingerprint-based analog searches, substructure, and chemotype pattern searches in ZINC15/RDKit became increasingly challenging with the advent of the ultra-large libraries.
Substructure and pattern searches do not need to finish to produce useful and correct, if incomplete, results, because both search types are answering yes/no questions to whether molecules contain the substructure or pattern. Accordingly, we have limited Arthor results to the first 10,000 molecules. Our public version of Arthor is capable of finding molecules containing a given substructure or pattern, but if there are more than 10,000 matches in the database the results will be incomplete. When we have more computers online dedicated for Arthor we may revise this limit upwards. Our goal is provide a useful public service in a pragmatic fashion.
To integrate Arthor natively into ZINC searches, indexes were created on statically exported subsets of ZINC as well as databases of Enamine, Mcule, and WuXi, that have not yet been incorporated into ZINC. New software was written to combine the results of an Arthor search with other search constraints in ZINC. A new interface was written to allow investigators to access the Arthor tool from ZINC, selecting the substructure or the SMARTS patterns options from the popup (Figure 6, left panel). To search by substructure or SMARTS patterns, investigators browse to https://zinc20.docking.org and click on the “Substance” in the menu bar at the top. To search by substructure, a substructure is entered in the drawing tool (Figure 6, left). To search by a chemotype pattern, a SMARTS pattern is typed in the input field. Optionally, investigators can select the subsets to search from the second popup (Figure 6, left, bottom panel). Some results should appear within a minute. ZINC may also be searched using the Arthor tool directly at https://arthor.docking.org (Figure 6). To search using substructure (left panel) the user selects Substructure, selects the database subset to use, and then draws the molecule or pastes in the SMILES. To search using chemotype patterns, the user selects SMARTS, and enters the SMARTS pattern. In each case, results appear on the right. Clicking on the ZINC number in the right panel opens the ZINC database for that molecule is a separate browser window. Clicking on the download button (top right of the page) downloads results in a spreadsheet-compatible format. Both the SmallWorld and the Arthor standalone interfaces were developed by NextMove Software. Arthor is also capable of running similarity searches, but we do not currently use this feature because SmallWorld is always faster and complete for the closest analogs.
Figure 6. Arthor for substructure and SMARTS chemotype pattern matching in ZINC.
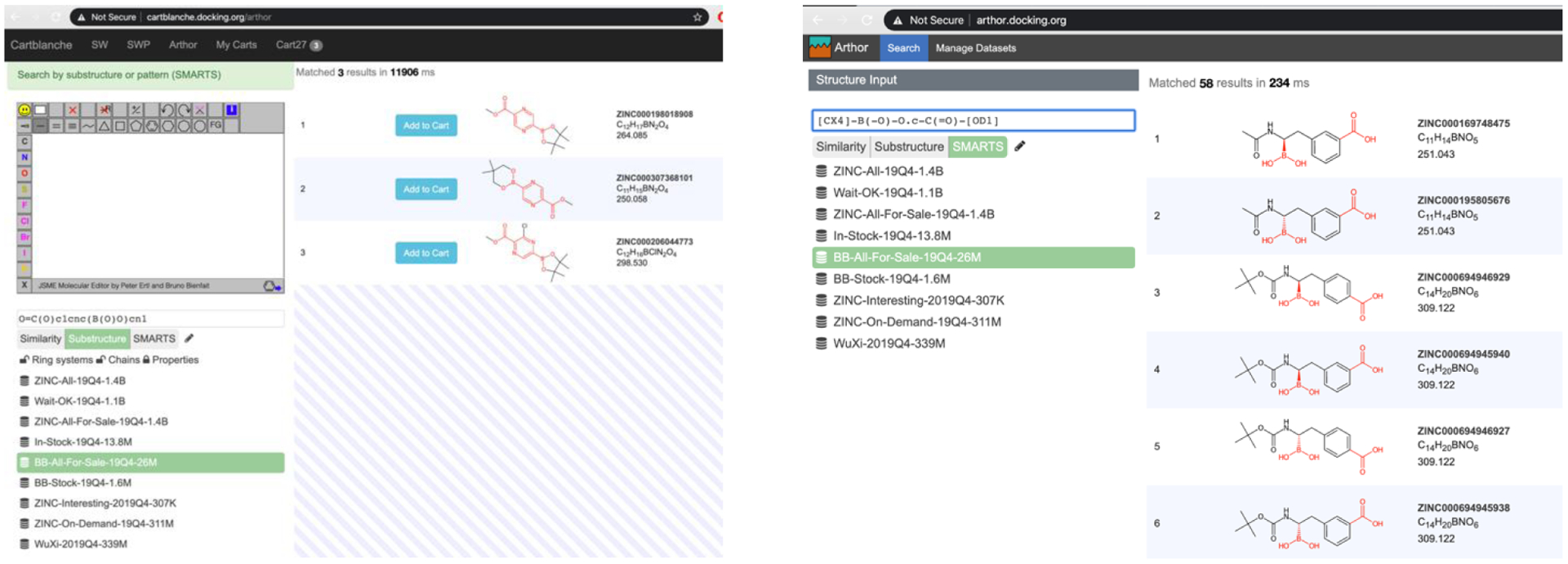
Left, substructure search. Right, SMARTS search for aliphatic boronic acid or ester plus an aromatic carboxylate in the same molecule.
Looking to the future, we have also created a new interface, Cartblanche.docking.org, that combines SmallWorld and Arthor searches into a single interface. Cartblanche is designed to make finding and purchasing molecules easier - a lightweight yet powerful chemical search tool. Cartblanche uses a shopping cart metaphor to allow the user to save molecules, curate lists (“carts”), assign estimated prices, and prepare an order for sending to vendors. Cartblanche may be used anonymously while registration enables additional benefits such as multiple and persistent shopping carts. Additionally, there are databases that cannot be made public because of fears of contaminating chemical space for patenting. Access to these additional resources is available on request to chemistry4biology@gmail.com, including the Enamine REAL Space On-Demand 13.5 Billion molecule set. Cartblanche and these bigger libraries are continuing to evolve and are being made available now as is in the hope that it they will be useful.
Discussion
Four themes emerge from this work. First, purchasable chemical space and the ZINC database that organizes it has become so big that new technologies are needed for real-time search. As purchasable chemical space continues to grow in the coming years, this engineering challenge will continue. Second, ZINC contains many new scaffolds and many molecules that are new to the planet, far exceeding what is available in physical public libraries. The scale and diversity of what can simply be searched and purchased is an opportunity for research. Third, because it is fully enumerated, ZINC can be searched precisely using explicit atomic-level graph-based methods, such as SmallWorld, pattern and substructure methods such as Arthor as well as 3D methods such as docking. The new tools presented here allow ZINC to be searched rapidly, often in real-time. The whole molecule search in SmallWorld should continue to work in real time for several further orders of magnitude of library growth, albeit with considerable effort to curate and support the service. Finally, ZINC and the other tools described here are freely available to everyone via zinc20.docking.org and other docking.org websites.
Purchasable chemical space has grown so much and so rapidly that public tools that could easily handle all of the purchasable chemical space five years ago are no longer fast enough to support real-time search. This growth is an exciting opportunity for investigators seeking chemical novelty, but a big challenge for those implementing tools that are easy to use. Fortunately, radically new technology in the form of SmallWorld and Arthor with the Roundtable algorithm has provided us with a mechanism to continue to offer free public search of public chemical libraries using fully enumerated databases.
As chemical space has grown so has its scaffold and shape diversity. For 88 times as many compounds as those in the make-on-demand library as those molecules that are in stock, the number of Bemis-Murcko scaffolds in the libraries have grown 16-fold. The novelty of the screening compounds is largely due to the novelty of the building blocks that have simply not been previously accessible for enumerating libraries. The structural diversity is also broadly-based, as quantified by the inertial ratios shown in the rod/disk/sphere plots. The coverage of this space by make-on-demand compounds is far denser and broader. There is far more chemical matter in the region tending towards sphere-like than is available in any physical decks, which are notoriously biased towards first rod like and secondarily disc-like. Together, the chemical diversity, scaffold diversity, and shape diversity of the make-on-demand libraries has a diversity that far exceeds public physical libraries and likely private ones as well as 97% of scaffolds in make-on-demand libraries have no representative in physical libraries.
Purchasable chemical space can be searched quickly and precisely at the atomic level. Of the three distinct types of search, whole-molecular similarity has the greatest ability to scale, owing to the SmallWorld algorithm. Chemotype pattern searches are particularly useful for building blocks, where the presence of distinct functional groups and scaffolds can be used to find reagents for library building or synthesis, including for DNA encoded libraries. At the scale of current building block collections, even the most complex Arthor searches take only a few seconds.
Several caveats merit mention. All of these databases are moving targets. Every month, over 20,000 make-on-demand compounds become in stock, i.e. are synthesized, and perhaps a quarter as many in stock become depleted – sold out. Make-on-demand libraries grow and shrink far more rapidly. We have seen 109 new molecules appear in a single month, and it is common to see hundreds of millions of make-on-demand compounds reclassified as unavailable at modest parallel synthesis prices as vendors gain experience and learn they are not as easy to make as once believed. The new tools, inevitably, also have limitations. For all of its advantages, the search time with Arthor scales roughly linearly with database size for substructure and chemotype pattern searches. Eventually the almost unbounded growth in accessible chemical space will pose a problem, as an order of magnitude more molecules will require an order of magnitude more memory and/or cpus, and that will rapidly become limiting. A SmallWorld-like algorithm for substructure and pattern matching with sub-linear scaling would be hugely enabling for the field. The hardware needed to run the services described in this work cost hundreds of thousands of dollars and require hundreds of person-hours monthly simply to maintain with current information.
Notwithstanding these caveats, new free tools are available now. ZINC supports whole molecule similarity search with SmallWorld using graph-edit-distance search, annotated with traditional ECFP4 and Daylight Tanimoto similarity values calculated. Substructure search is supported by Arthor, as is full chemotype pattern search using SMARTS. These new tools may be sufficient to allow molecular searches to scale with the explosion of compounds in ZINC, at least for the next several years. As ZINC grows beyond 10 billion molecules towards 100 billion and beyond, newer approaches may be considered.
Methods
We used RDKit version 2018_03, OpenBabel version 2.3.2 and Molinspiration version 2015. We use Postgres 12.0 for the database to host ZINC and Centos7 operating systems.
Our public SmallWorld (version 4.0.1) is hosted on a computer with 16 Intel Xeon E5-2623 v4 running at 2.60GHz and 64 GB of RAM. The anonymous index is stored on a 114 TB SSD disk array formatted with ZFS z2. Indexes are prepared and stored on a second 114 TB disk array formatted in the same way. The private SmallWorld instance is currently hosted on the same computer.
Smallworld uses a map of anonymous graphs for indexing, which is currently 42 TB, dated March 2020. It should be stored on as fast disk as can be afforded (we currently use SSD). The database indexes are 13–14 million molecules per GB. Thus 13.5 TB takes about 1 TB. The database can be prepared in chunks of 500 M molecules each and then merged and sorted to build the final index.
Our public Arthor server (Version 3.3) is a hosted on a heterogeneous set of 8 computers, each with a minimum of 128 GB of memory, a minimum of 7.6 TB of SSD disk and between 80 and 128 cores. Arthor works best with lots of memory, because the indexes are best accessed when memory cached.
Arthor uses two kinds of index, one for similarity search, which we do not support here, and the other for both substructure and pattern (SMARTS) searches. The substructure/pattern indexes index 7–9 million molecule per GB. Thus 1.7 TB of disk is required to index 13.5 billion molecules. For larger databases (> 2 billion molecules) we create Arthor indexes of 500 million molecules each and split them across as many computers as possible, each with as much memory as possible but never less than 128 GB.
ZINC20 is hosted on an array of computers. The database itself is on a computer with 32 Intel Xeon Gold 5222 CPUs running at 3.80GHz. The Postgres database resides on a 114 TB SSD disk array formatted with ZFS z2. This machine has 1.5 TB of RAM expandable to 3.0 TB. The web server runs on two computers each with 80 cores of Intel Xeon Gold 6138 CPUs at 2.00GHz. One of these has 400 GB of RAM, the other 128 GB of RAM. Database curation and loading is performed on our private cluster of approximately 1000 cores.
The public version of the Small Molecule Discovery Center collection was sourced as SMILES by request. The MLSMR was downloaded from PubChem on September 1, 2019. The Enamine collection and the WuXi GalaXi and Mcule Ultimate collections were made available by agreement with the suppliers.
The normalized ratios of principle moments of inertia (NPMI) for the rod/disc/sphere plots were calculated using RDKit using the following commands.
mol = C.AddHs(mol)
AllChem.EmbedMolecule(mol, useExpTorsionAnglePrefs=True, useBasicKnowledge=True)
npr1 = round(CD.CalcNPR1(mol), 4)
npr2 = round(CD.CalcNPR2(mol), 4)
Bemis Murcko Scaffolds were calculated using the mib toolkit by Molinspiration (molinspiration.com). Histograms were prepared using python scripts, Jupyter notebooks and Microsoft Excel.
Supplementary Material
Acknowledgements.
Supported by NIH grant GM71896 (To JJI). We thank OpenEye Scientific Software for an academic license for Omega, OEChem, and other tools, and thank ChemAxon for an academic license for JChem, Marvin, and other software. We thank Molinspiration (molinspiration.com) for a license. We thank the providers of public databases and free software that ZINC has benefitted from: RDKit, DrugBank, HMDB, ChEBI, ChEMBL as well as other databases and resources cited in the text. We are grateful to the lab of J-L Reymond for access to GDB17 which helped us test SmallWorld at the 166 billion molecule scale. We thank members of the Shoichet Lab for testing the software and for timely feedback. We thank Tia Tummino and Elissa Fink for reading the manuscript. We thank the thoughtful reviewers at JCIM for kind feedback that helped improve the manuscript in many ways.
Footnotes
Supporting Information. The Performance section from the Arthor 3.2 User Manual benchmarking Arthor performance on a variety of hardware. These are all available in the supporting information at ACS.org free of charge in a tar.gz file and on our wiki, wiki.docking.org/index.php/ZINC20.
Declared conflicts of interest. RAS owns NextMove software and JM is an employee. JJI is a scientific advisor to Life Science Data Systems, a company that provides chemical catalog related services in the pharmaceutical sector. YSM is an employee of ChemSpace, a company that markets fine chemicals.
Literature Cited
- 1.Lyu J; Wang S; Balius TE; Singh I; Levit A; Moroz YS; O’Meara MJ; Che T; Algaa E; Tolmachova K; Tolmachev AA; Shoichet BK; Roth BL; Irwin JJ, Ultra-Large Library Docking for Discovering New Chemotypes. Nature 2019, 566, 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stein RM; Kang HJ; McCorvy JD; Glatfelter GC; Jones AJ; Che T; Slocum S; Huang XP; Savych O; Moroz YS; Stauch B; Johansson LC; Cherezov V; Kenakin T; Irwin JJ; Shoichet BK; Roth BL; Dubocovich ML, Virtual Discovery of Melatonin Receptor Ligands to Modulate Circadian Rhythms. Nature 2020, 579, 609–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Irwin JJ; Shoichet BK, Zinc--a Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model 2005, 45, 177–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Irwin JJ; Sterling T; Mysinger MM; Bolstad ES; Coleman RG, Zinc: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model 2012, 52, 1757–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sterling T; Irwin JJ, Zinc 15 - Ligand Discovery for Everyone. J. Chem. Inf. Model 2015, 55, 2324–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muchmore SW; Debe DA; Metz JT; Brown SP; Martin YC; Hajduk PJ, Application of Belief Theory to Similarity Data Fusion for Use in Analog Searching and Lead Hopping. J. Chem. Inf. Model 2008, 48, 941–8. [DOI] [PubMed] [Google Scholar]
- 7.Martin YC; Kofron JL; Traphagen LM, Do Structurally Similar Molecules Have Similar Biological Activity? J. Med. Chem 2002, 45, 4350–8. [DOI] [PubMed] [Google Scholar]
- 8.Klingler FM; Gastreich M; Grygorenko OO; Savych O; Borysko P; Griniukova A; Gubina KE; Lemmen C; Moroz YS, Sar by Space: Enriching Hit Sets from the Chemical Space. Molecules 2019, 24, 3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daylight Smarts - a Language for Describing Molecular Patterns. (February 1, 2020),
- 10.Enamine Real Database. https://enamine.net/library-synthesis/real-compound
- 11.PubChem Molecular Libraries Small Molecule Repository. http://pubchem.ncbi.nlm.nih.gov (January 1, 2020),
- 12.Arkin MR; Ang KK; Chen S; Davies J; Merron C; Tang Y; Wilson CG; Renslo AR, Ucsf Small Molecule Discovery Center: Innovation, Collaboration and Chemical Biology in the Bay Area. Comb Chem High Throughput Screen 2014, 17, 333–42. [DOI] [PubMed] [Google Scholar]
- 13.Bemis GW; Murcko MA, The Properties of Known Drugs. 1. Molecular Frameworks. J . Med. Chem 1996, 39, 2887–2893. [DOI] [PubMed] [Google Scholar]
- 14.Sauer WH; Schwarz MK, Molecular Shape Diversity of Combinatorial Libraries: A Prerequisite for Broad Bioactivity. J. Chem. Inf. Comput. Sci 2003, 43, 987–1003. [DOI] [PubMed] [Google Scholar]
- 15.NextMove Software I Arthor 3.0, NextMove Software, Inc.: Cambridge, England, 2020. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.