

Abstract
We are entering into an exciting era of genomics where truly complete, high-quality assemblies of human chromosomes are available end-to-end, or from ‘telomere-to-telomere’ (T2T). This technological advance offers a new opportunity to include endogenous human centromeric regions in high-resolution, sequence-based studies. These emerging reference maps are expected to reveal a new functional landscape in the human genome, where centromere proteins, transcriptional regulation, and spatial organization can be examined with base-level resolution across different stages of development and disease. Such studies will depend on innovative assembly methods of extremely long tandem repeats (ETRs), or satellite DNAs, paired with the development of new, orthogonal validation methods to ensure accuracy and completeness. This review reflects the progress in centromere genomics, credited by recent advancements in long-read sequencing and assembly methods. In doing so, I will discuss the challenges that remain and the promise for a new period of scientific discovery for satellite DNA biology and centromere function.
Keywords: Centromere, Satellite DNA, Genomics, Long-read assembly, Telomere-to-telomere
1. Introduction
Large, multi-megabase sized arrays of tandem repeats, or satellite DNAs, are a common, yet poorly understood feature of eukaryotic genomes. Genomic regions enriched in satellite DNAs are generally known to be associated with pericentromeric constitutive heterochromatin and span sites involved in kinetochore assembly, or sequences epigenetically marked as the centromere [1–3]. The underlying genetic and genomic contribution to centromere identity has been elusive in part due to the phenomenon known as the “centromere paradox”, where centromere function is conserved over vast evolutionary time yet the underlying centromeric repeat sequences are rapidly evolving [4,5]. Even in the absence of an evolutionary conserved centromeric sequence, it is clear that understanding of the interactions between inner kinetochore proteins and the underlying genome, within and between species, could shape our understanding of centromere biology [6–8]. Unfortunately, centromeric regions have been extremely difficult to study at the base-level due to the challenge of correctly assembling long arrays of near-identical tandem repeats. As a result, most centromere-associated satellite arrays and non-satellite DNAs embedded within these repeat-rich regions remain largely unassembled in the vast majority of sequenced genomes [5] and detached from genome-wide functional studies that rely on short-read mapping [9,10]. This disconnect from contemporary genomics has severely limited our ability to explore and test sequence features (DNA structural conformation or specialized regulatory mechanisms (e.g. transcriptional, replication, and/or DNA repair-based mechanism)) that contribute to centromere function. The use of an incomplete genomic map, where only a small subset of collapsed centromeric sequences are present in the reference assembly, issues an observational bias. This is equivalent to the streetlight effect [11]; we are restricting our studies to the small number of sequences we can currently ‘see’ and in doing so, we are likely ignoring hidden genomic information. Undoubtedly, future access to high-quality and complete reference maps of centromeric regions will issue a new opportunity to comprehensively test the role of genome organization and epigenomic regulation, and will lead to new innovative computational and experimental strategies to dramatically expand studies of satellite DNA biology.
Our current model of centromere sequence organization has been largely shaped by human genomic studies [12–15]. Normal human centromeres are defined at the sequence level by alpha satellite DNA, an AT-rich tandem repeat, or a ~171 bp monomer [16]. Alpha satellite offers a broad range of sequence diversity at the monomer level [17–19]. Ordered arrangements of a small number of divergent monomers form a larger repeating unit, known as a ‘higher-order repeat’ (HOR) [20]. HORs are organized into large, often multi-megabase sized satellite arrays, with limited nucleotide differences between repeat copies [12,21,22]. One or more HOR arrays, each offering different combinations of ordered divergent monomers, are found on each chromosome [17,20,23]. These distinct arrays can often be labeled in a chromosome-specific manner, which has allowed researchers to assign both short and long reads to a single centromeric region [19,24–26]. Interestingly, not all alpha satellite sequences associate with kinetochore proteins or are thought to be competent for centromere function [19]. For example, genomic studies on chromosome 17 (D17Z1 and D17Z1b) have shown that array length, HOR variant composition, and transcriptional regulation can contribute to our understanding of why certain centromere HOR arrays are active while others are defined by pericentromeric heterochromatin [8,27,28]. Cloning of select HOR array sequences into human artificial chromosomes (HACs) has been useful to systematically test alpha satellite sequence competency in centromere establishment and maintenance [19,29,30]. HAC experimental systems, however, suffer from being extremely labor-intensive, thus making it impractical to test a comprehensive collection of centromeric satellite variants. Further, the variability of HAC construct amplification or multimerization in cells and the potential influx of unexpected, non-satellite DNAs may introduce additional unknown variables into a study aimed to understand the genomic mechanisms present at the true endogenous satellite-DNA locus [29,30]. Ultimately, the best way to explore the genomic contribution would be to perform focused, high-resolution base-level studies within an endogenous centromere.
The human haploid sex chromosomes (DXZ1 and DYZ3 on the X and Y chromosome, respectively) have offered the most high-resolution studies of a human centromeric satellite array to date [12,13,15,25,31]. Previous studies have reported that arrays can vary in length by a factor of 10 due to the expansion and contraction repeats (e.g. the DYZ3 array is observed in the range ~200 kb to ~2 Mb) [25,31]. Although the HORs within a single array are observed to be highly similar to one another, single nucleotide variants and larger structural variants due to monomer rearrangement are observed [25,26,32,33]. These ‘repeat imperfections’, either alone or in combination, are incredibly valuable to assembly efforts as they offer unique markers to guide the true linear ordering [13,15,34]. Recent linear assemblies of both DXZ1 [13] and DYZ3 [15] have been released, which credit both dramatic improvements in long-read technologies and repeat assembly strategies.
Technological gains in single molecule sequencing, both in terms of read length [35] and base quality [36], matched with improved assembly strategies are releasing assembly ‘predictions’ of entire centromeric regions without targeted effort [13,37] (Table 1). Evaluations of these emerging assemblies will undoubtedly benefit from decades of previous research, which have laid the groundwork for sequence structure in each human centromeric region. For example, the assembled centromeric region of chromosome 7 from the recent HiCanu assembly provides evidence for two previously characterized HOR arrays, D7Z1 and D7Z2 [37,38] (Fig. 1a and b). The results from previous physical maps, clone-based studies, and array sizing by pulsed-field gel electrophoresis (PFGE) Southerns are largely concordant with the observed array sizes, spacing between the arrays, and ordering relative to the p- and q-arms (Fig. 1b) [38–40]. Further, the assembly supports the presence of pericentromeric satellites, HSat2B [41], monomeric satellites [40,42] with interspersed transposable elements [12], and flanking segmental duplications between the HOR arrays and extending into the centric transitions [43,44] (Fig. 1c). Concordance with these broad genomic expectations offer the first level of critical evaluation. However, the genomic community is now charged with the development of new, high-resolution evaluation strategies to fully assess the assembly completeness and base-level accuracy, as these details will be critical to guide future functional studies.
Table 1.
Existing approaches applied to tandem repeat and centromeric satellite DNA assembly.
| Method | Application | Details | Advantages | Challenges | Pub. |
|---|---|---|---|---|---|
| centro Flye | ETR assembly | Reconstruction using rare k-mers in error-prone long reads. | Use of ultra-long nanopore data. Automated assembly. | Error-prone reads are difficult to resolve near-identical arrays (phasing). Requires polishing. | [34] |
| SV-mediated assembly | ETR assembly | Reconstruction using rare changes in repeat structure in error-prone long reads | Use of ultra-long nanopore data. Manual curation | Slow, requires expertise, and prone to human error. Requires polishing. | [13,15] |
| HiCanu | ETR assembly | Leverages the full potential of mid-length, high-quality reads via ho mo polymer compression, overlap-based error correction, and aggressive false overlap filtering | Use of PacBio HiFi sequence assembly in regions of sufficient repeat heterogeneity. Does not require polishing | Limitations in assembly based on variant spacing and read length. | [37] |
| Deep repeat resolution | ETR assembly | Machine learning algorithms to distinguish patterns of single nucleotide variants of repeats from noisy data | Automated assembly of complex repeat structures (histone cluster in Drosophila) can be used in ultra-long nanopore data | Not yet demonstrated on a wide range of eukaryotic genomes and may require additional polishing | [50] |
Fig. 1. Genomic organization of human centromeric regions.
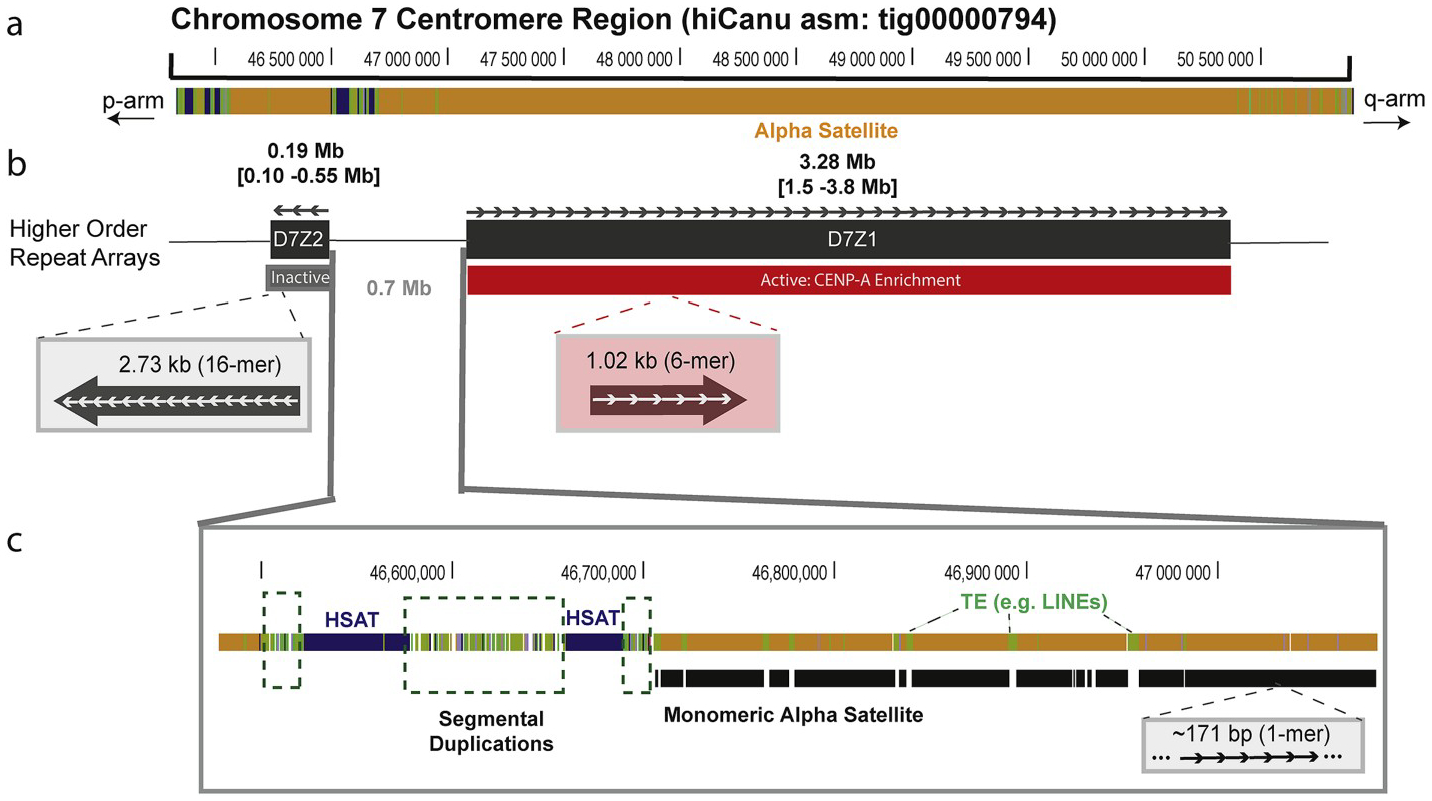
(a) The assembled human centromeric regions on chromosome 7 (HiCanu contig: tig00000794:44788681–50796794) is characterized by RepeatMasker [112] to be defined by long tracts of composed of alpha satellite DNA (ALR/Alpha; shown in orange) with interspersed repeats (green) and pericentromeric satellites (blue). (b) Alpha satellite ~171 bp monomers (shown as white arrows) are organized into a multi-monomeric repeat unit, or higher order repeat (HOR), with the orientation of the repeats indicated. Chromosome 7 has two HOR arrays: D7Z2 (16-mer HOR) and D7Z1 (6-mer HOR) [38]. The D7Z1 and D7Z2 array sizes are within the expected range as determined by PFGE Southerns. The D7Z1 has been previously determined to have CENP-A enrichment (shown in red, live or active array) and D7Z2 array, shown in grey is typically inactive, or not expected to be bound to CENP-A [42] The genomic region between the D7Z1 and D7Z2 array (~700 kb) is concordant with previous physical mapping data for this region [39]. (c) Sequences found flanking HOR arrays are typically pericentromeric satellites, shown in blue (HSat), segmental duplications, and monomeric (divergent 171 bp alpha satellite) with interspersed transposable elements.
Overall, these milestones in centromere assembly mark the beginning of a new genomic revolution, where the ‘goal post’ for a finished genome is now placed on completing gap-free, telomere-to-telomere (T2T) chromosome assemblies and release of high-resolution maps at centromeres. In light of this advance, I aim to review the current progress and challenges in human centromere repeat assembly. Further, I will provide a brief perspective on how these new reference maps and emerging genomic technologies are expected to invigorate the sequence-based studies of centromere biology.
2. Satellite array assembly
Linear assembly of ‘extremely long tandem repeats’, or ETRs [45], relies on having comprehensive read coverage for a single array, confident variant detection, and adequate read lengths to ensure that the maximum distance between two informative variants within the array can be spanned. The necessity for sequence lengths in repeat assembly is supported by a quote by Rodger Staden (1979), “If the overlap is of sufficient length to distinguish it from being a repeat in the sequence the two sequences must be contiguous” [46]. Indeed, the initial human centromere satellite array assemblies benefitted from high-coverage, ultra-long (UL) sequence data (i.e. reads that are at least 100 kb in length) [35], which were capable of spanning large, informative repeat variants (as shown in Fig. 2a) and predicting the linear ordering of positionally unique array markers. From this initial study of an assembled, multi-megabase sized centromeric array (DXZ1) [13], small differences that distinguish one repeat copy from another were identified and the spacing of these events throughout the array. These unique markers were reported on average every 2.3 kbp in the DXZ1 (with max spacing of 42 kb) [13], revealing a new collection of ‘mile markers’ to inform assembly of high-quality mid- and short-read data (Fig. 2a).
Fig. 2. Assembly of extra-long tandem repeats (ETRs).
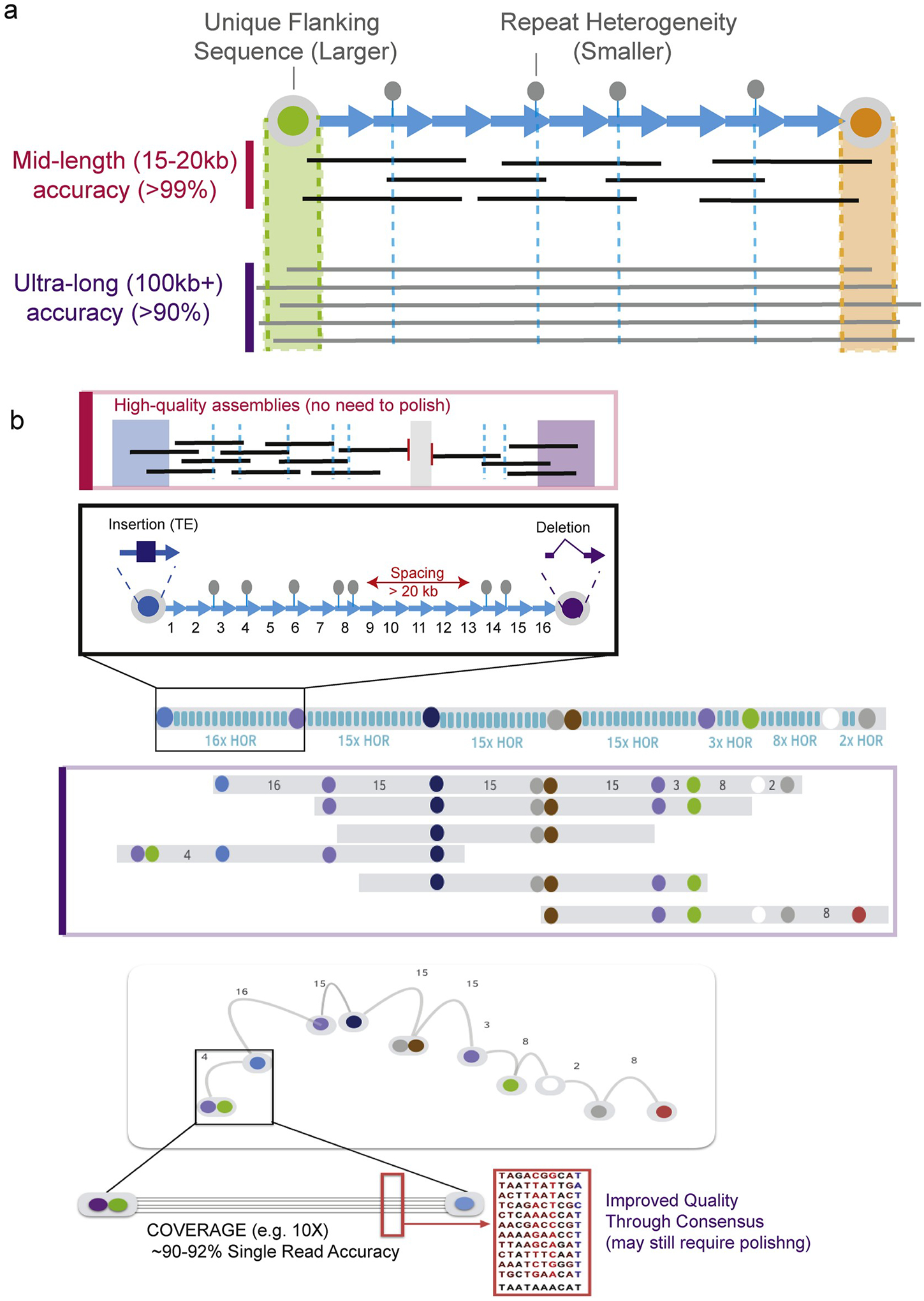
(a) Long tandem repeats (shown as blue arrows) are flanked by unique sequences (indicated in green and orange to mark regions upstream and downstream of the repeat array). These transitions are confidently detected in long-error prone ultra-long reads, and anchoring reads that can fully traverse the repeat region bypasses the need for assembly. Rather, one can derive a consensus of the underlying repeat region. Alternatively, short (often single nucleotide) unique markers within the repeat units are sufficient to distinguish copies of the repeat and lead to assembly of the array using mid-length reads (here defined as a read that is less than the length of the array and incapable of spanning these regions completely). Assembly using these rare, single nucleotide markers requires extraordinary quality, like CCS data from PacBio, to minimize overlaps due to sequencing errors. (b) A structural variant (SV) based assembly strategy shown labels each HOR as canonical (blue), with the number of uninterrupted canonical repeats (e.g. 16x HOR) indicated. Rearranged HOR structures, or structural variants (SV) are indicated as colored circles. Focusing on the first 18 repeats to illustrate the repeat heterogeneity in the array that can be used to guide assemblies of extremely high quality mid-length reads. The challenge is traversing arrays where the spacing between unique markers is longer than the length of the read (e.g. shown in read a spacing greater than 20 kb, and indicated as a break in the assembly above in grey shading). The SV-based assembly method uses the spacing and organization of HOR rearrangements (colored circles) in array-assigned ultra-long reads. Overlap between SV-maps in ultra-long read data results in a repeat contigs with improved sequence quality by consensus.
Single-molecule technologies offer incredible read lengths (i.e. nanopore sequencing reads are commonly reported reaching at least 1 Mbp of alignable sequence) [47], however, it comes at the cost of an increased error rate. Improvement is currently achieved through increasing read coverage and deriving a high-quality consensus. The success of this strategy depends on correctly overlapping, or aligning error-prone read sequences containing long-tracts of repeats. In contrast, high fidelity (HiFi), data from Pacific Biosciences (PacBio) generated from a circular consensus strategy (CCS), in which DNA is topologically circularized and sequenced multiple times to create a high-quality consensus for each read [36]. This new technology is extremely promising for repeat characterization and centromeric satellite assembly [37,48], yet it may have an upper limit in HiFi consensus length that may be insufficient to traverse the maximum variant spacing of all ETRs (Fig. 2b). Early estimates suggest that satellite arrays, at least in the effectively haploid complete hydatidiform mole (CHM13), vary considerably in the spacing of these unique markers. For example, the single HOR array (D8Z2) on chromosome 8 has maximum spacing between variants is ~4 kb, when other arrays, like the DXZ1 array on the X chromosome has been reported to have maximum spacing greater than 40 kb [13]. Therefore, if the spacing of unique markers (or collection of low-frequency markers) within a given satellite array are shorter than the length of extremely high-quality reads (or reads where the sequencing error is so low that it does not confound efforts to identify rare repeat heterogeneity) complete linear assembly is expected. HiCanu [37], the recent modification of the Canu assembler [49], performed a series of filtering steps to further improve the quality of Sanger-like reads (e.g. performing homopolymer compression, overlap-based error correction, and aggressive false overlap filtering). In doing so, HiCanu generated assemblies that fully spanned nine centromeric regions (chromosomes 2, 3, 7, 8, 10, 12, 16, 19, 20). Notably, several centromeric regions remain in fragmented assemblies likely marking sites that lack sufficient marker density or repeat heterogeneity.
High-quality, or high-fidelity reads that span hundreds of kilobases would offer a clear advantage for comprehensive satellite assembly genome-wide. Similar to the CCS strategy, the centromere on the Y chromosome was assembled from high-quality consensus sequence data using a nanopore ultra-long (100 kb+) reads [15]. In this study, bacterial artificial chromosomes (BACs) known to span the DYZ3 locus [49] (with insert lengths ~100–300 kb) were sequenced in their entirety many times [15]. Consensus derived from the global alignments of the full-length BAC inserts results in a high-quality 100 kb + sequence. This was a useful strategy for the DYZ3 array since it is known to be usually small (~300 kb) [31] and could be spanned using a small number of previously characterized BACs [49]. However, dependence on BACs for satellite assembly genome-wide is not ideal since it relies on the labor-intensive construction of the library and satellite DNA may be prone to inherent cloning biases.
High-coverage UL data from the CHM13 genome offered a new consensus-based method broadly similar to the BAC-strategy for DYZ3. Rather than using the BAC vector sequence to anchor and align the satellite insert sequences, this method identified large perturbations in the repeat structure (or structural variants (SVs)), which served as unique flanking sequences to guide the overlap of reads and generate a high-quality consensus [13] (Fig. 2b). Although this SV-based approach has been useful in guiding the T2T-X DXZ1 array assembly, the limited number of SVs in the array and the spacing (the maximum distance between SVs was 493 kbp) severely lowered the coverage and consensus-derived quality in SV-depleted regions within the array. Emerging ETR automated assembly methods bypass this limitation by using distinguishing patterns of single nucleotide variants, or the spacing and organization of low-copy k-mers, from noisy data to resolve repeat structures. For example, efforts to assemble the tandemly repeated histone array in the Drosophila genome used a correction heuristic based on artificial neural networks [50]. That is, after reducing the error rate of long-read data, the corrected clusters of variants can be used to traverse the assembly graph, thus presenting an automated strategy to explore assemble complex tandem repeat structures in other eukaryotic genomes. More recently, centroFlye [34], an algorithm for assembling tandem repeats from long error prone reads, was used to generate the first automated assembly of a human centromere on the X chromosome [13]. This work builds from the approach of resolving unbridged repeats (i.e. positions where repeat copies differ from one another) used in the Flye assembler [51]. The centroFlye pipeline identifies a set of rare k-mers (by default, short sequences 19 bp) that appear in a database error-prone nanopore reads data from a single haploid array. Spacing between rare kmers are modeled using a distance graph, and sets of k-mers with shared distances between reads are used to distinguish the rare markers, useful for assembly, versus those that likely represent sequencing errors.
Assemblies from error-prone long-read sequence data require rigorous consensus polishing to achieve maximum base call accuracy [52]. Critical to the success of this process is the correct placement of reads to the assembly, which is challenging to do correctly within the context of large repeat regions with multiple high-scoring mapping locations. Therefore, efforts to reach a finished, high-quality ETR assembly from UL-nanopore reads require the development of new ‘repeat-aware’ mapping and polishing approaches. The T2T-X centromeric array was polished using a marker-assisted mapping strategy. Here, the placement of all short (21 bp), unique (single-copy) sequences were determined across the entire 3.1 Mb T2T-X centromeric array. Long-read alignments were generated and then the top sites were scored and placed in the location maximizing the unique marker matches [13]. Repeat copies in an ETR are nearly identical, and the number of unique markers is expected to be low and the spacing irregular. Therefore, alignment coverage could drop in regions of the array with a lower density of unique markers. Indeed, the T2T-X centromeric array had only 16,163 unique 21-mers across the multi-megabase array. TandemMapper [45] addresses this challenge, in that it uses locally unique markers (i.e. where a given ETR is subdivided into segments and unique markers are defined within each segment), which have the benefit of being more abundant than unique k-mers. More recently, Winnowmap [53] has been shown to improve long-read mapping (PacBio and UL-Nanopore reads), by optimizing the standard minimizer sampling procedure (minimap2 [54]) to improve mapping accuracy within repeats. Notably, current satellite assemblies from PacBio HiFi data, where the read data Sanger-like quality (> 99%), are of extremely high quality (QV~50). These assemblies bypass the need for downstream polishing methods, which is a huge advantage in both computational run time and avoiding errors due to imprecise mapping [37].
3. Assembly evaluation and validation
Each centromeric satellite assembly strategy results in a ‘hypothesis’, or a predicted linear organization with reference to the long-read data and parameters that are defined by each algorithm. Different assemblers may offer different hypotheses. Orthogonal validation and evaluation strategies are essential, especially in the early days of centromere genomics. Sequence-based evaluation methods that are available to detect misassemblies outside of centromeric regions often rely on direct comparisons with a reference genome or concordant read mapping [55–58]. These methods are not easily extended to satellite assemblies. ‘TandemQUAST’ [45] approaches this challenge by offering several reference-free quality assessment modules (e.g. indels, coverage, breakpoints, and HOR-structural variation) to evaluate ETR assemblies at the base-level. Further, ETR assessment using HiFi alignment coverage has been useful in cross-evaluation of polishing error and marking sites of potential collapse [13,45].
In addition to computational evaluation methods, there is a critical need to develop new experimental validation methods to study array structure. Orthogonal experimental validation methods, such as the comparison with optical map assemblies and/or ordered BACs by FISH [59], are difficult to extend to centromere satellite assemblies. Notably, the short nucleotide 6- or 7-bp recognition sites used in generating standard optical maps are vastly underrepresented in alpha satellites, and as a result, centromeric regions are commonly omitted. To date, PFGE Southerns offers the only ‘gold standard’ to evaluate the larger satellite array structure [22,33]. In this method, high molecular weight (HMW) DNA is digested with one or more restriction enzymes (REs) that are common in the genome, yet are absent or infrequent in the satellite array. As a result, the majority of the genome is digested into small fragments, while the satellite array can be retained more or less intact. The undigested HMW fragments are separated on a PFGE, transferred for Southern blotting, and labeled using a probe specific to a satellite array. This information can be combined with other orthogonal methods, like quantitative digital droplet PCR [13] and base coverage in whole genome sequencing data [25,41], to estimate array HOR copy number. Additionally, PFGE-Southern experiments are extremely useful for predicting the structural organization within a given array. That is, one can compare the fragment lengths observed in the PFGE-Southern against the predicted fragments in the assembled array data [13]. This assay, however, is difficult to scale to support genome-wide studies, is low-resolution in that it primarily evaluates larger structural organization and is not effective in studies of arrays from diploid chromosomes. Array sizing rely on imprecise standards (i.e. Schizosaccharomyces pombe and Saccharomyces cerevisiae chromosomes). Therefore, each sample benefits from the use of a panel of different REs observed to cut infrequently within an array to fully evaluate sizing concordance. The 3.1 Mb T2T-X centromere assembly has been cross-evaluated by all existing validation methods [13,34,45] and offers a unique reference, or benchmark, to launch new genome technologies.
4. New insights in centromere genomics
The T2T-X centromeric satellite array offers a high-quality, structurally validated benchmark centromere assembly [13,45]. With the release of this linear assembly we can now ask: what genomic and epigenomic insights have been gained? And how does this work improve from previous efforts that model repeat variants [25]? Ultimately, how can we use positional information in the context of the multi-megabase sized array to advance questions in centromere biology? The T2T centromeric satellite array is defined by 1537 copies of the ~2 kbp higher order repeat (DXZ1) that are ordered in a head-to-tail tandem array in a single direction [60] (Fig. 3a). The array is interrupted once by a single full-length (6055 bp) L1HS transposable element, which represents a Long Interspersed Nuclear Elements (LINEs) subfamily that is known to jump autonomously in modern humans [61] (Fig. 3a). From the perspective of the HOR structure and orientation alone, the DXZ1 array is highly homogenized, or composed almost entirely by the canonical ~2 kbp (12-mer) HOR (97.9% 1504/1537). Of the 33 identified HOR-SVs, five are represented as single events within the array. The remaining SVs are largely involved in localized expansions, with a small number of examples of long-distance shared SV patterns (with distances greater than 2 Mb, as shown in red for the 1.7 kb 10-mer repeat, Fig. 3a). Mapping the sites of rearrangement relative to the canonical repeat reveals deletions of varying lengths that span every monomer within the 12-mer HOR (Fig. 3b). Notably, a subset (~40% of SVs, 13/33; 6-mer, 10-mer, and 17-mer) are found to delete at least one of the four 17-bp CENP-B binding motif (or CENP-B box) [62,63]. However, given sparse SV representation within the DXZ1 array it is possible that such functional deletions of the CENP-B box could have limited overall effect.
Fig. 3. High-resolution genomic study of the CHM13 T2T DXZ1 centromeric array.
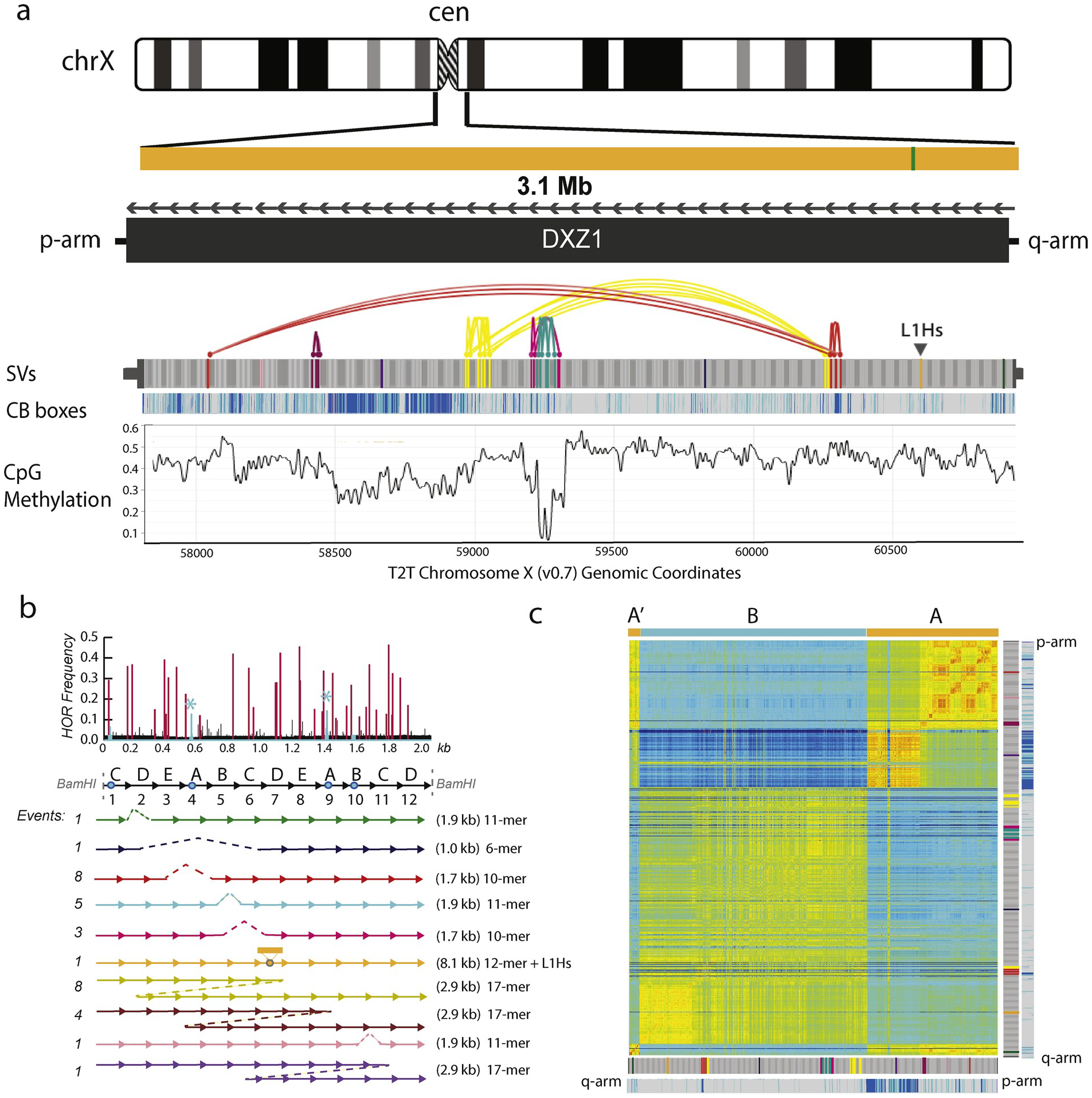
(a) The CHM13 DXZ1 is defined by 3.1 megabases of alpha satellite (shown as orange band, which is interrupted once with an L1Hs LINE insertion (green, closest to q-arm). The DXZ1 repeats are orientation from q-arm to p-arm (relative to the published BamHI DXZ1 repeat (GenBank: X02418)), with no shifts in repeat direction [21]. The ~2 kbp canonical repeat is shown as grey and the position of structural variants, or rearrangements (insertion/deletions) are noted with color. SVs that have shared repeat structure are connected with a line, or an arc. DXZ1 canonical HOR have four active CENP-B boxes. Each HOR in the array was colored based on the number of active CENP-B boxes: light grey (4/4), teal (3/4), blue (2/4), purple (1/4), and dark purple (0/4). HORs with less than two active CENP-B boxes are < 3% of the array and cannot be detected by eye at the resolution of the entire array. The plot of methylation data (obtained from nanopolish [100] from nanopore alignment and signal data) demonstrates a drop in methylation in the middle of the array. (b) Repeat variation patterns relative to the canonical 12-mer HOR, with blue circles mapping the sites of CENP-B boxes. SV HOR structures are colored to match the SV annotation in panel (a). Dashed lines mark sites of deletion. The LIHs/LINE element insertion is indicated as an inserted orange bar. Event numbers reference the occurence of each SV in the CHM13 DXZ1 array. A consensus sequence was derived from the 1537 HORs. Pairwise alignments with each HOR with the derived consensus was used to generate a database of nucleotide differences and positions. The low-frequency variants (< 10% of the array) are shown in black. Light blue is used to show data in regions that span the 17-bp CENP-B box. Stars over the CENP-B boxes in the two A repeats indicate that the variant is high-frequency (greater than 10% of the HORs) and modifies one of the 9 conserved, functional bases in the motif. Red peaks show the remaining 35 high-frequency (37 total with two peaks in the CENP-B boxes) sites, or regions that differ from the consensus sequence in more than 10% of the HOR repeats. (c) Pairwise identity between the ordered 1537 HORs in the 37 high-frequency variant positions is shown (using heatmap function in R), large similarity domains are defined manually into two groups: A/A’ and B. The SV-annotation and CENP-B status data are positioned on either side of the matrix for genomic context. The array similarity matrix data is reverse complemented to the array in (a) to match the orientation of the canonical published repeat.
Perhaps one of the more surprising findings of the T2T-X centromere assembly was the high frequency of repeat heterogeneity (smaller, single nucleotide variants that serve as unique markers within the canonical HOR) [13]. On average, the DXZ1 HOR (12-mers) are near-identical (99.72%, with an observed range of 95.9–100%). However, these small differences, or ‘low-frequency’ variants, either alone or in combination, offer unique markers that span the vast majority of the DXZ1 array. This further supports the finding of unique markers on average every 2.3 kbp [13], and the successful implementation of the centroFlye pipeline in assembling DXZ1 [34]. In contrast to these rare base changes that are useful in distinguishing one HOR copy from all others, there are 37 ‘high-frequency’ base changes within the HOR (defined as any base change observed above the threshold of 10% of all HORs in the DXZ1 array) (Fig. 3b). Notably, such high-frequency base changes in the CENP-B boxes (within the two A-repeats) are observed to interrupt the core recognition sequence at the 10th position: *TTCG****[A]**CGGG* [64]. These changes are observed outside of the CpG sites in the motif, which are expected to contribute to methylation-based regulation of CENP-B binding [65]. Only 53% (827/1537) DXZ1 repeats are observed to contain all four CENP-B boxes, as defined through the identification of the conserved, nine functional bases (Fig. 3a,c) [66]. Over a quarter of the DXZ1 array is missing a single active CENP-B boxes (407/1537), and 17.5% (269/1537) have two out of the four active boxes (Fig. 3a,c). Two DXZ1 HORs lack any active CENP-B boxes, and a small fraction (32 HORs, or 2% of the array) have only one active box. Interestingly, the location of these events appear non-random within the array, where increased CENP-B box loss is observed in the range of 58.5–59.0 Mb of the T2T-X (v07) assembly. Pairwise comparisons between the 1537 HOR repeats at the high-frequency variant sites provide evidence for two larger domains within the DXZ1 array (labeled A and B in Fig. 3c). The similarity block A can be further divided into two subdomains, of which one is associated with increased loss of CENP-B active motifs. Further, we detect evidence of HOR similarity between the p-arm and the q-arm (labeled A’ in Fig. 3c). The larger similarity domain (B) appears to have a smaller subdomain ~60.5 Mb.
Understanding the marker spacing and underlying genomic structure, with attention to CENP-B for its noted enhancement in the fidelity of human centromere function [19,30,67,68], will help inform the genomic interpretation and future epigenetic studies. One surprising finding in the T2T-X centromeric array was the drop of CpG methylation (59.2–59.3 Mb, 93 kb) detected in the nanopore signal files that confidently map to the array (Fig. 3a). No notable decreases in CpGs were detected in this region (with 39 CpGs on average for DXZ1 HORs, STDEV 4.6). The drop in methylation is found in the ‘B’-similarity domain (Fig. 3c) and overlaps a subset of SVs (10-mer, dark pink and 11-mer, teal in Fig. 3a and b). Additional studies are needed to test if this region is correlated with centromeric chromatin [69] and/or sites of satellite transcription [70,71]. Further collections of other T2T-X assemblies and matched epigenetic profiles across diverse human genomes will undoubtedly contribute to our understanding of how these findings differ between individuals, how the X array evolves, and how these genomic features contribute to our understanding of the influence in chromosomal aneuploidies [68].
5. Remaining challenges
Current assembly and evaluation methods are only applicable when read datasets can be confidently assigned to a single, haploid array. Even in effectively haploid genomes, like CHM13 [72,73], there are satellite arrays present on different homologous chromosomes that are known to be similar to one another at the sequence level. Therefore, the error-prone long-read data for these arrays are combined together initially and are difficult to fully phase, or assign specifically to a single chromosomal location. This is a recognized challenge on the acrocentric chromosomes, for example, where arrays on chromosome 13 are very difficult to distinguish from those on chromosome 21 [74,75]. Additionally, a single centromeric region can contain the intermixing of arrays that hybridize to the same HOR sequence, as is the case of the satellite arrays that are shared between chromosomes 4 and 9 [76]. It is also important to consider that satellite DNAs may rearrange or change in copy number over multiple cell divisions (i.e. either in cell culture and/or in somatic cells as we age) [77–79]. Subclonal expansion of such events could potentially introduce unexpected array sequence heterogeneity, which may confound assembly and evaluation efforts that assume a single version of a haploid array. Fortunately, with the public release of both high coverage UL nanopore and HiFi data from the CHM13 genome [13,48], it is now possible to initiate new phasing and assembly methods in regions known to present challenges to haploid, or effectively haploid genomes.
New method development and optimization of phasing of similar arrays in CHM13 will be critical in reaching the ultimate goal of T2T diploid phased assembly. This may become less of a challenge as reads become much longer (routinely reach lengths 500 kb + or even megabases) and/or of higher base accuracy. Also the new development of innovative, long-read sequencing technologies or imaging-based methods that include multi-megabase sized repeat regions could recast and revolutionize diploid ETR assembly. Such methods, matched with the ever-improving gains in read length and base-level quality from single molecule reads, are expected to drive studies closer to the goal of reaching high-quality ETR diploid assemblies in single cells. Until then, it is very likely that any ambiguity in phasing or even assembly efforts, can be represented as a graph. In this case, sequences that cannot be fully phased will be represented as a single cluster, or node, with connecting edges to other sequence clusters or assembled regions that can be properly phased and assembled. This data format for satellite DNAs may align with developing ‘pangenomic’ references and benefit from streamlined alignment and variant detection computational methods and software [80].
6. Future centromere studies
Genetic rearrangement [81,82], epigenetic misregulation [83,84], and aberrant transcription [77,85–87] of centromeric and heterochromatic satellite DNAs have been shown to contribute to chromosome instability, aging, and cancers. These specialized satellite array enriched regions are observed to interact with a wide range of DNA-binding proteins (e.g. transcription factors) [88–91], have highly regulated transcriptional activity [92], and spatially-distinct organization in the nucleus that can alter gene activity in cis and trans [93,94]. Further, anomalies the formation and maintenance of constitutive heterochromatin can affect cell division and differentiation. There is a clear need to study epigenetic regulation and genome biology across ETRs, however, centromeric regions are currently omitted from contemporary functional genomic and epigenomic studies (e.g. Refs. [9,95,96]) due to the paucity of genomic information and inability to confidently map functional short read data. Modeled diploid arrays in the human reference genome that serve as short read mapping targets [10,25], have already launched several compelling studies in centromere biology and function. For example, by establishing centromere protein A (CENPA) enrichment profiles to all GRCh38 reference models over different stages of the cell cycle, Nechemia-Arbely et al. was able to demonstrate that DNA replication acts as an error correction mechanism for the precise reloading of centromeric CENP-A and maintenance of centromere identity [7]. Similar short read CENP-A mapping to these early modeled maps has been useful in evaluating DNA sequence variation and aneuploidy, with special attention to the spacing and regularity of the centromere protein B motif, or CENP-B box [68]. Indeed, even efforts to explore transcriptional based mechanisms in centromere and pericentromeric based studies are expected to benefit from array-specific markers [97,98].
As more centromere assemblies become available it will finally be possible to perform mapping of functional datasets across satellite arrays. This will enable the first high-resolution studies of the epigenetic and transcriptional regulation of human centromeric regions. Doing so will require careful identification and evaluation of the limited ‘mappable’ sites, or marked regions of known single copy variants within repeat-rich regions. Marking these regions with low-frequency k-mers has been shown to be useful in correctly mapping long read data across centromeric arrays [13]. Mapping of long read data also offers a unique opportunity to explore the signatures of methylated bases, which can be predicted from both PacBio [99] and Nanopore [100,101] data. Extended functional characterization of centromeric arrays may require a new toolkit of technologies that benefit from longer reads. Full length RNA sequencing, either direct RNA or cDNA sequencing, is available using long-read sequencing [102–106]. Early strategies have been released for chromatin capture (Hi-C) strategies that involve long-read sequencing on the nanopore platform (Pore-C) [107]. New innovative long-read methods to predict sites of open chromatin [108,109] and/or imaging-based methods (for example [110]) may dramatically improve the detection of protein association to ETRs to improve assessment of cell populations or single cells.
Our understanding of centromere biology will greatly benefit from comparing genomic and epigenomic profiles between individuals that differ at the genomic level. It is well understood that satellite sequences that span centromeres are highly variable in length and repeat structure in the population (reviewed [111]), yet very little is known about the extent of this variation at the genomic level and how this influences centromere identity [27,68]. Therefore, future studies in the T2T era will need to expand from a single, high resolution benchmark reference genome(s) to study how these sequences change over time: that is, between individuals in the population using available both short and long read population datasets, across multi-generational pedigrees, and exploring inter-cellular variation within a single individual by employing single cell sequencing data. Novel streamlined, parallelizable strategies will be needed to expand studies of genomic variation and functional datasets to cohorts of individuals. Such work may change the way we think about satellite variation and the risks with aneuploidies, aging and disease. Further, it will open a new door for discovery with the emergence of synthetic chromosome biology where one might be able to customize array sequence composition in the future.
7. Concluding remarks
The release of the first high-quality, base-level maps of human centromeric regions will undoubtedly set a new standard for how we study centromere biology. With this advance, we can begin to explore trends in satellite sequence organization and array molecular mechanisms that influence centromere activity more comprehensively. Gaining access to centromeric regions is expected to open a new world of scientific discovery, where changes in epigenetic regulation at centromere regions can be studied through development, cellular stress, and disease models. Although the focus of this review has been placed on the recent progress in issuing new benchmarking efforts in the human genome, the emerging genome technology and innovation is expected to extend broadly to other complex genomes where it is possible to phase chromosome-assigned satellite sequence data.
Acknowledgements
This work was supported by NIH/NHGRI R21 (1R21HG010548-01) and NIH/NHGRI U01 (1U01HG010971). I would like to thank Mark Diekhans for the assistance with RepeatMasker annotation data and Ariel Gershman for methylation profile information.
References
- [1].Yunis JJ, Yasmineh WG, Heterochromatin, satellite DNA, and cell function, Science 174 (1971) 1200–1209. [DOI] [PubMed] [Google Scholar]
- [2].Vafa O, Sullivan KF, Chromatin containing CENP-A and α-satellite DNA is a major component of the inner kinetochore plate, Curr. Biol 7 (1997) 897–900. [DOI] [PubMed] [Google Scholar]
- [3].Pardue ML, Gall JG, Chromosomal localization of mouse satellite DNA, Science 168 (1970) 1356–1358. [DOI] [PubMed] [Google Scholar]
- [4].Henikoff S, Ahmad K, Malik HS, The centromere paradox: stable inheritance with rapidly evolving DNA, Science 293 (2001) 1098–1102. [DOI] [PubMed] [Google Scholar]
- [5].Melters DP, Bradnam KR, Young HA, Telis N, May MR, Ruby JG, Sebra R, Peluso P, Eid J, Rank D, Garcia JF, DeRisi JL, Smith T, Tobias C, Ross-Ibarra J, Korf I, Chan SWL, Comparative analysis of tandem repeats from hundreds of species reveals unique insights into centromere evolution, Genome Biol. 14 (2013) R10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Malik HS, Henikoff S, Major evolutionary transitions in centromere complexity, Cell 138 (2009) 1067–1082. [DOI] [PubMed] [Google Scholar]
- [7].Nechemia-Arbely Y, Miga KH, Shoshani O, Aslanian A, McMahon MA, Lee AY, Fachinetti D, Yates JR 3rd, Ren B, Cleveland DW, DNA replication acts as an error correction mechanism to maintain centromere identity by restricting CENP-A to centromeres, Nat. Cell Biol 21 (2019) 743–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kuo ME, Sullivan LL, Chew K, Sullivan BA, Genomic variation within alpha satellite DNA influences centromere location on human chromosomes with metastable epialleles, Genome 26 (2016) 1301–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].ENCODE project consortium, the ENCODE (ENCyclopedia of DNA elements) project, Science 306 (2004) 636–640. [DOI] [PubMed] [Google Scholar]
- [10].Miga KH, Eisenhart C, Kent WJ, Utilizing mapping targets of sequences underrepresented in the reference assembly to reduce false positive alignments, Nucleic Acids Res. 43 (2015) e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Freedman DH, Why Scientific Studies Are So Often Wrong: the Streetlight Effect vol. 26, Discover Magazine, 2010, http://www.sjsu.edu/people/fred.prochaska/courses/ScWk240/s3/Freedman-Week-15.pdf. [Google Scholar]
- [12].Schueler MG, Higgins AW, Rudd MK, Gustashaw K, Willard HF, Genomic and genetic definition of a functional human centromere, Science 294 (2001) 109–115. [DOI] [PubMed] [Google Scholar]
- [13].Miga KH, Koren S, Rhie A, Vollger MR, Gershman A, Bzikadze A, Brooks S, Howe E, Porubsky D, Logsdon GA, Schneider VA, Potapova T, Wood J, Chow W, Armstrong J, Fredrickson J, Pak E, Tigyi K, Kremitzki M, Markovic C, Maduro V, Dutra A, Bouffard GG, Chang AM, Hansen NF, Thibaud-Nissen F, Schmitt AD, Belton J-M, Selvaraj S, Dennis MY, Soto DC, Sahasrabudhe R, Kaya G, Quick J, Loman NJ, Holmes N, Loose M, Surti U, Risques RA, Graves Lindsay TA, Fulton R, Hall I, Paten B, Howe K, Timp W, Young A, Mullikin JC, Pevzner PA, Gerton JL, Sullivan BA, Eichler EE, Phillippy AM, Telomere-to-telomere Assembly of a Complete Human X Chromosome, bioRxiv, 2019735928, 10.1101/735928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Aldrup-Macdonald ME, Sullivan BA, The past, present, and future of human centromere genomics, Genes 5 (2014) 33–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jain M, Olsen HE, Turner D, Stoddart D, Paten B, Haussler D, Willard HF, Akeson M, Miga KH, Linear assembly of a human centromere on the Y chromosome, Nat. Biotechnol 36 (2018) 321–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Manuelidis L, Wu JC, Homology between human and simian repeated DNA, Nature 276 (1978) 92–94. [DOI] [PubMed] [Google Scholar]
- [17].Waye JS, Willard HF, Nucleotide sequence heterogeneity of alpha satellite repetitive DNA: a survey of alphoid sequences from different human chromosomes, Nucleic Acids Res. 15 (1987) 7549–7569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Rudd MK, Willard HF, Analysis of the centromeric regions of the human genome assembly, Trends Genet. 20 (2004) 529–533. [DOI] [PubMed] [Google Scholar]
- [19].Hayden KE, Strome ED, Merrett SL, Lee H-R, Rudd MK, Willard HF, Sequences associated with centromere competency in the human genome, Mol. Cell Biol 33 (2013) 763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Willard HF, Waye JS, Hierarchical order in chromosome-specific human alpha satellite DNA, Trends Genet. 3 (1987) 192–198. [Google Scholar]
- [21].Willard HF, Waye JS, Chromosome-specific subsets of human alpha satellite DNA: analysis of sequence divergence within and between chromosomal subsets and evidence for an ancestral pentameric repeat, J. Mol. Evol 25 (1987) 207–214. [DOI] [PubMed] [Google Scholar]
- [22].Wevrick R, Willard HF, Long-range organization of tandem arrays of alpha satellite DNA at the centromeres of human chromosomes: high-frequency array-length polymorphism and meiotic stability, Proc. Natl. Acad. Sci. Unit. States Am 86 (1989) 9394–9398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Alexandrov I, Kazakov A, Tumeneva I, Shepelev V, Yurov Y, Alpha-satellite DNA of primates: old and new families, Chromosoma 110 (2001) 253–266. [DOI] [PubMed] [Google Scholar]
- [24].Mravinac B, Sullivan LL, Reeves JW, Yan CM, Kopf KS, Farr CJ, Schueler MG, Sullivan BA, Histone modifications within the human X centromere region, PloS One 4 (2009) e6602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Miga KH, Newton Y, Jain M, Altemose N, Willard HF, Kent WJ, Centromere reference models for human chromosomes X and Y satellite arrays, Genome Res. 24 (2014) 697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Suzuki Y, Myers G, Morishita S, Long-read Data Revealed Structural Diversity in Human Centromere Sequences, bioRxiv, (2019) https://www.biorxiv.org/content/10.1101/784785v1.abstract. [Google Scholar]
- [27].Maloney KA, Sullivan LL, Matheny JE, Strome ED, Merrett SL, Ferris A, Sullivan BA, Functional epialleles at an endogenous human centromere, Proc. Natl. Acad. Sci. U.S.A 109 (2012) 13704–13709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Aldrup-MacDonald ME, Kuo ME, Sullivan LL, Chew K, Sullivan BA, Genomic variation within alpha satellite DNA influences centromere location on human chromosomes with metastable epialleles, Genome Res. 26 (2016) 1301–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Logsdon GA, Gambogi CW, Liskovykh MA, Barrey EJ, Larionov V, Miga KH, Heun P, Black BE, Human artificial chromosomes that bypass centromeric DNA, Cell 178 (2019) 624–639.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Grimes BR, Rhoades AA, Willard HF, α-Satellite DNA and vector composition influence rates of human artificial chromosome formation, Mol. Ther 5 (2002) 798–805. [DOI] [PubMed] [Google Scholar]
- [31].Oakey R, Tyler-Smith C, Y chromosome DNA haplotyping suggests that most European and Asian men are descended from one of two males, Genomics 7 (1990) 325–330. [DOI] [PubMed] [Google Scholar]
- [32].Schindelhauer D, Schwarz T, Evidence for a fast, intrachromosomal conversion mechanism from mapping of nucleotide variants within a homogeneous alpha-satellite DNA array, Genome Res. 12 (2002) 1815–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mahtani MM, Willard HF, Pulsed-field gel analysis of alpha-satellite DNA at the human X chromosome centromere: high-frequency polymorphisms and array size estimate, Genomics 7 (1990) 607–613. [DOI] [PubMed] [Google Scholar]
- [34].Bzikadze AV, Pevzner PA, centroFlye: Assembling Centromeres with Long Error-Prone Reads, bioRxiv, (2019), p. 772103, 10.1101/772103. [DOI] [Google Scholar]
- [35].Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, Tyson JR, Beggs AD, Dilthey AT, Fiddes IT, Malla S, Marriott H, Nieto T, O’Grady J, Olsen HE, Pedersen BS, Rhie A, Richardson H, Quinlan AR, Snutch TP, Tee L, Paten B, Phillippy AM, Simpson JT, Loman NJ, Loose M, Nanopore sequencing and assembly of a human genome with ultra-long reads, Nat. Biotechnol 36 (2018) 338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wenger AM, Peluso P, Rowell WJ, Chang P-C, Hall RJ, Concepcion GT, Ebler J, Fungtammasan A, Kolesnikov A, Olson ND, Töpfer A, Alonge M, Mahmoud M, Qian Y, Chin C-S, Phillippy AM, Schatz MC, Myers G, DePristo MA, Ruan J, Marschall T, Sedlazeck FJ, Zook JM, Li H, Koren S, Carroll A, Rank DR, Hunkapiller MW, Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome, Nat. Biotechnol 37 (2019) 1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Nurk S, Walenz BP, Rhie A, Vollger MR, Logsdon GA, HiCanu: Accurate Assembly of Segmental Duplications, Satellites, and Allelic Variants from High-Fidelity Long Reads, bioRxiv, 2020, https://www.biorxiv.org/content/10.1101/2020.03.14.992248v2.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Waye JS, England SB, Willard HF, Genomic organization of alpha satellite DNA on human chromosome 7: evidence for two distinct alphoid domains on a single chromosome, Mol. Cell Biol 7 (1987) 349–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wevrick R, Willard HF, Physical map of the centromeric region of human chromosome 7: relationship between two distinct alpha satellite arrays, Nucleic Acids Res. 19 (1991) 2295–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].de la Puente A, Velasco E, Pérez Jurado LA, Hernández-Chico C, van de Rijke FM, Scherer SW, Raap AK, Cruces J, Analysis of the monomeric alphoid sequences in the pericentromeric region of human chromosome 7, Cytogenet, Genome Res. 83 (1998) 176–181. [DOI] [PubMed] [Google Scholar]
- [41].Altemose N, Miga KH, Maggioni M, Willard HF, Genomic characterization of large heterochromatic gaps in the human genome assembly, PLoS Comput. Biol 10 (2014) e1003628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Uralsky L, Shepelev VA, Alexandrov AA, Yurov YB, Rogaev EI, Alexandrov IA, Classification and monomer-by-monomer annotation of supra-chromosomal family 1 alpha satellite higher-order repeats in hg38 human genome assembly, Data in Brief 24 (2019) 103708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].She X, Horvath JE, Jiang Z, Liu G, Furey TS, Christ L, Clark R, Graves T, Gulden CL, Alkan C, Bailey JA, Sahinalp C, Rocchi M, Haussler D, Wilson RK, Miller W, Schwartz S, Eichler EE, The structure and evolution of centromeric transition regions within the human genome, Nature 430 (2004) 857–864. [DOI] [PubMed] [Google Scholar]
- [44].Genovese G, Handsaker RE, Li H, Altemose N, Lindgren AM, Chambert K, Pasaniuc B, Price AL, Reich D, Morton CC, Pollak MR, Wilson JG, McCarroll SA, Using population admixture to help complete maps of the human genome, Nat. Genet 45 (2013) 406–14, 414e1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Mikheenko A, Bzikadze AV, Gurevich A, Miga KH, Pevzner PA, TandemMapper and TandemQUAST: mapping long reads and assessing/improving assembly quality in extra-long tandem repeats, bioRxiv 2019 (2019), 10.1101/2019.12.23.887158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Staden R, A strategy of DNA sequencing employing computer programs, Nucleic Acids Res. 6 (1979) 2601–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Payne A, Holmes N, Rakyan V, Loose M, Whale watching with BulkVis: a graphical viewer for Oxford Nanopore bulk fast5 files, bioRxiv (2018) 312256, 10.1101/312256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Vollger MR, Logsdon GA, Audano PA, Sulovari A, Porubsky D, Peluso P, Wenger AM, Concepcion GT, Kronenberg ZN, Munson KM, Baker C, Sanders AD, Spierings DCJ, Lansdorp PM, Surti U, Hunkapiller MW, Eichler EE, Improved assembly and variant detection of a haploid human genome using single-molecule, high-fidelity long reads, Ann. Hum. Genet 84 (2020) 125–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Tilford CA, Kuroda-Kawaguchi T, Skaletsky H, Rozen S, Brown LG, Rosenberg M, McPherson JD, Wylie K, Sekhon M, Kucaba TA, Waterston RH, Page DC, A physical map of the human Y chromosome, Nature 409 (2001) 943–945. [DOI] [PubMed] [Google Scholar]
- [50].Bongartz P, Schloissnig S, Deep repeat resolution—the assembly of the Drosophila Histone Complex, Nucleic Acids Res. 47 (2018) e18–e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kolmogorov M, Yuan J, Lin Y, Pevzner PA, Assembly of long, error-prone reads using repeat graphs, Nat. Biotechnol 37 (2019) 540–546. [DOI] [PubMed] [Google Scholar]
- [52].Koren S, Phillippy AM, Simpson JT, Loman NJ, Loose M, Reply to “Errors in long-read assemblies can critically affect protein prediction, Nat. Biotechnol 37 (2019) 127–128. [DOI] [PubMed] [Google Scholar]
- [53].Jain C, Rhie A, Zhang H, Chu C, Koren S, Phillippy A, Weighted minimizer sampling improves long read mapping, bioRxiv (2020) 2020, 10.1101/2020.02.11.943241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Li H, Minimap2: pairwise alignment for nucleotide sequences, Bioinformatics 34 (2018) 3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Gurevich A, Saveliev V, Vyahhi N, Tesler G, QUAST: quality assessment tool for genome assemblies, Bioinformatics 29 (2013) 1072–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T, Hansen N, Teague B, Alkan C, Antonacci F, Haugen E, Zerr T, Yamada NA, Tsang P, Newman TL, Tüzün E, Cheng Z, Ebling HM, Tusneem N, David R, Gillett W, Phelps KA, Weaver M, Saranga D, Brand A, Tao W, Gustafson E, McKernan K, Chen L, Malig M, Smith JD, Korn JM, McCarroll SA, Altshuler DA, Peiffer DA, Dorschner M, Stamatoyannopoulos J, Schwartz D, Nickerson DA, Mullikin JC, Wilson RK, Bruhn L, Olson MV, Kaul R, Smith DR, Eichler EE, Mapping and sequencing of structural variation from eight human genomes, Nature 453 (2008) 56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Jiang Y, Wang Y, Brudno M, PRISM: pair-read informed split-read mapping for base-pair level detection of insertion, deletion and structural variants, Bioinformatics 28 (2012) 2576–2583. [DOI] [PubMed] [Google Scholar]
- [58].Salzberg SL, Phillippy AM, Zimin A, Puiu D, Magoc T, Koren S, Treangen TJ, Schatz MC, Delcher AL, Roberts M, Marçais G, Pop M, Yorke JA, GAGE: a critical evaluation of genome assemblies and assembly algorithms, Genome Res. 22 (2012) 557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Udall JA, Dawe RK, Is it ordered correctly? Validating genome assemblies by optical mapping, Plant Cell 30 (2018) 7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Waye JS, Willard HF, Chromosome-specific alpha satellite DNA: nucleotide sequence analysis of the 2.0 kilobasepair repeat from the human X chromosome, Nucleic Acids Res. 13 (1985) 2731–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Brouha B, Schustak J, Badge RM, Lutz-Prigge S, Farley AH, Moran JV, Kazazian HH Jr., Hot L1s account for the bulk of retrotransposition in the human population, Proc. Natl. Acad. Sci. U.S.A 100 (2003) 5280–5285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Masumoto H, Masukata H, Muro Y, Nozaki N, Okazaki T, A human centromere antigen (CENP-B) interacts with a short specific sequence in alphoid DNA, a human centromeric satellite, J. Cell Biol 109 (1989) 1963–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Muro Y, Masumoto H, Yoda K, Nozaki N, Ohashi M, Okazaki T, Centromere protein B assembles human centromeric alpha-satellite DNA at the 17-bp sequence, CENP-B box, J. Cell Biol 116 (1992) 585–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Masumoto H, Nakano M, Ohzeki J-I, The role of CENP-B and α-satellite DNA: de novo assembly and epigenetic maintenance of human centromeres, Chromosome Res. 12 (2004) 543–556. [DOI] [PubMed] [Google Scholar]
- [65].Tanaka Y, Kurumizaka H, Yokoyama S, CpG methylation of the CENP-B box reduces human CENP-B binding, FEBS J. 272 (2005) 282–289. [DOI] [PubMed] [Google Scholar]
- [66].Ohzeki J-I, Nakano M, Okada T, Masumoto H, CENP-B box is required for de novo centromere chromatin assembly on human alphoid DNA, J. Cell Biol 159 (2002) 765–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Fachinetti D, Han JS, McMahon MA, Ly P, Abdullah A, Wong AJ, Cleveland DW, DNA sequence-specific binding of CENP-B enhances the fidelity of human centromere function, Dev. Cell 33 (2015) 314–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Dumont M, Gamba R, Gestraud P, Klaasen S, Worrall JT, De Vries SG, Boudreau V, Salinas-Luypaert C, Maddox PS, Lens SMA, Others, Human chromosome-specific aneuploidy is influenced by DNA-dependent centromeric features, EMBO J. 39 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Sullivan BA, Karpen GH, Centromeric chromatin exhibits a histone modification pattern that is distinct from both euchromatin and heterochromatin, Nat. Struct. Mol. Biol 11 (2004) 1076–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].McNulty SM, Sullivan BA, Alpha satellite DNA biology: finding function in the recesses of the genome, Chromosome Res. 26 (2018) 115–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Biscotti MA, Canapa A, Forconi M, Olmo E, Barucca M, Transcription of tandemly repetitive DNA: functional roles, Chromosome Res. 23 (2015) 463–477. [DOI] [PubMed] [Google Scholar]
- [72].Chin CS, Korlach J, Wilson RK, Eichler EE, Discovery and genotyping of structural variation from long-read haploid genome sequence data, Genome 27 (2017) 677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Durbin R, Wilson RK, Flicek P, Eichler EE, Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly, Genome (2017), http://genome.cshlp.org/content/27/5/849.short. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Ikeno M, Masumoto H, Okazaki T, Distribution of CENP-B boxes reflected in CREST centromere antigenic sites on long-range alpha-satellite DNA arrays of human chromosome 21, Hum. Mol. Genet 3 (1994) 1245–1257. [DOI] [PubMed] [Google Scholar]
- [75].Greig GM, Warburton PE, Willard HF, Organization and evolution of an alpha satellite DNA subset shared by human chromosomes 13 and 21, J. Mol. Evol 37 (1993) 464–475. [DOI] [PubMed] [Google Scholar]
- [76].Finelli P, Antonacci R, Marzella R, Lonoce A, Archidiacono N, Rocchi M, Structural organization of multiple alphoid subsets coexisting on human chromosomes 1, 4, 5, 7, 9, 15, 18, and 19, Genomics 38 (1996) 325–330. [DOI] [PubMed] [Google Scholar]
- [77].Bersani F, Lee E, Kharchenko PV, Xu AW, Liu M, Xega K, MacKenzie OC, Brannigan BW, Wittner BS, Jung H, Ramaswamy S, Park PJ, Maheswaran S, Ting DT, Haber DA, Pericentromeric satellite repeat expansions through RNA-derived DNA intermediates in cancer, Proc. Natl. Acad. Sci. U.S.A 112 (2015) 15148–15153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Pluhar SA, Erickson L, Pauls KP, Effects of tissue culture on a highly repetitive DNA sequence (E180 satellite) in Medicago sativa, Plant Cell Tissue Organ Cult. 67 (2001) 195–199. [Google Scholar]
- [79].Ershova ES, Malinovskaya EM, Konkova MS, Veiko RV, Umriukhin PE, Martynov AV, Kutsev SI, Veiko NN, Kostyuk SV, Copy number variation of human satellite III (1q12) with aging, Front. Genet 10 (2019) 704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Paten B, Novak AM, Eizenga JM, Garrison E, Genome graphs and the evolution of genome inference, Genome Res. 27 (2017) 665–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Tyler-Smith C, Oakey RJ, Larin Z, Fisher RB, Crocker M, Affara NA, Ferguson-Smith MA, Muenke M, Zuffardi O, Jobling MA, Localization of DNA sequences required for human centromere function through an analysis of rearranged Y chromosomes, Nat. Genet 5 (1993) 368–375. [DOI] [PubMed] [Google Scholar]
- [82].Black EM, Giunta S, Repetitive fragile sites: centromere satellite DNA as a source of genome instability in human diseases, Genes 9 (2018) 615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Qu G, Dubeau L, Narayan A, Yu MC, Ehrlich M, Satellite DNA hypomethylation vs. overall genomic hypomethylation in ovarian epithelial tumors of different malignant potential, Mutat. Res 423 (1999) 91–101. [DOI] [PubMed] [Google Scholar]
- [84].Peng JC, Karpen GH, Epigenetic regulation of heterochromatic DNA stability, Curr. Opin. Genet. Dev 18 (2008) 204–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Ting DT, Lipson D, Paul S, Brannigan BW, Akhavanfard S, Coffman EJ, Contino G, Deshpande V, Iafrate AJ, Letovsky S, Rivera MN, Bardeesy N, Maheswaran S, Haber DA, Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers, Science 331 (2011) 593–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Perea-Resa C, Blower MD, Centromere biology: transcription goes on stage, Mol. Cell Biol 38 (2018) e00263–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Zhu Q, Pao GM, Huynh AM, Suh H, Tonnu N, Nederlof PM, Gage FH, Verma IM, BRCA1 tumour suppression occurs via heterochromatin-mediated silencing, Nature 477 (2011) 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Cobb BS, Morales-Alcelay S, Kleiger G, Brown KE, Fisher AG, Smale ST, Targeting of Ikaros to pericentromeric heterochromatin by direct DNA binding, Genes Dev. 14 (2000) 2146–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Pilipuk GP, Galigniana MD, Schwartz J, Subnuclear localization of C/EBPβ is regulated by growth hormone and dependent on MAPK, J. Biol. Chem 278 (2003) 35668–35677. [DOI] [PubMed] [Google Scholar]
- [90].Steensma DP, Higgs DR, Fisher CA, Gibbons RJ, Acquired somatic ATRX mutations in myelodysplastic syndrome associated with alpha thalassemia (ATMDS) convey a more severe hematologic phenotype than germline ATRX mutations, Blood 103 (2004) 2019–2026. [DOI] [PubMed] [Google Scholar]
- [91].Shestakova EA, Mansuroglu Z, Mokrani H, Ghinea N, Bonnefoy E, Transcription factor YY1 associates with pericentromeric γ-satellite DNA in cycling but not in quiescent (G0) cells, Nucleic Acids Res. 32 (2004) 4390–4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Smurova K, De Wulf P, Centromere and pericentromere transcription: roles and regulation … in sickness and in health, Front. Genet 9 (2018) 674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Misteli T, Spatial positioning; a new dimension in genome function, Cell 119 (2004) 153–156. [DOI] [PubMed] [Google Scholar]
- [94].Henikoff S, Dosage-dependent modification of position-effect variegation in Drosophla, Bioessays 18 (1996) 401–409. [DOI] [PubMed] [Google Scholar]
- [95].Dekker J, Belmont AS, Guttman M, Leshyk VO, Lis JT, Lomvardas S, Mirny LA, O’Shea CC, Park PJ, Ren B, Politz JCR, Shendure J, Zhong S, 4D Nucleome Network, the 4D nucleome project, Nature 549 (2017) 219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, Farnham PJ, Hirst M, Lander ES, Mikkelsen TS, Thomson JA, The NIH roadmap epigenomics mapping consortium, Nat. Biotechnol 28 (2010) 1045–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].McNulty SM, Sullivan LL, Sullivan BA, Human centromeres produce chromosome-specific and array-specific alpha satellite transcripts that are complexed with CENP-A and CENP-C, Dev. Cell 42 (e6) (2017) 226–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Johnson WL, Yewdell WT, Bell JC, McNulty SM, Duda Z, O’Neill RJ, Sullivan BA, Straight AF, RNA-dependent stabilization of SUV39H1 at constitutive heterochromatin, Elife 6 (2017) e25299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, Korlach J, Turner SW, Direct detection of DNA methylation during single-molecule, real-time sequencing, Nat. Methods 7 (2010) 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Simpson JT, Workman RE, Zuzarte PC, David M, Dursi LJ, Timp W, Detecting DNA cytosine methylation using nanopore sequencing, Nat. Methods 14 (2017) 407–410. [DOI] [PubMed] [Google Scholar]
- [101].Rand AC, Jain M, Eizenga JM, Musselman-Brown A, Olsen HE, Akeson M, Paten B, Mapping DNA methylation with high-throughput nanopore sequencing, Nat. Methods 14 (2017) 411–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Workman RE, Tang AD, Tang PS, Jain M, Tyson JR, Razaghi R, Zuzarte PC, Gilpatrick T, Payne A, Quick J, Sadowski N, Holmes N, de Jesus JG, Jones KL, Soulette CM, Snutch TP, Loman N, Paten B, Loose M, Simpson JT, Olsen HE, Brooks AN, Akeson M, Timp W, Nanopore native RNA sequencing of a human poly(A) transcriptome, Nat. Methods 16 (2019) 1297–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Gonzalez-Garay ML, Introduction to isoform sequencing using pacific Biosciences technology (Iso-Seq), in: Wu J (Ed.), Transcriptomics and Gene Regulation, Springer, Netherlands, Dordrecht, 2016, pp. 141–160. [Google Scholar]
- [104].Garalde DR, Snell EA, Jachimowicz D, Sipos B, Lloyd JH, Bruce M, Pantic N, Admassu T, James P, Warland A, Jordan M, Ciccone J, Serra S, Keenan J, Martin S, McNeill L, Wallace EJ, Jayasinghe L, Wright C, Blasco J, Young S, Brocklebank D, Juul S, Clarke J, Heron AJ, Turner DJ, Highly parallel direct RNA sequencing on an array of nanopores, Nat. Methods 15 (2018) 201–206. [DOI] [PubMed] [Google Scholar]
- [105].Cole C, Byrne A, Adams M, Volden R, Vollmers C, Complete characterization of the human immune cell transcriptome using accurate full-length cDNA sequencing, bioRxiv (2019) 761437, 10.1101/761437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Byrne A, Cole C, Volden R, Vollmers C, Realizing the potential of full-length transcriptome sequencing, Philos. Trans. R. Soc. Lond. B Biol. Sci 374 (2019) 20190097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Ulahannan N, Pendleton M, Deshpande A, Schwenk S, Behr JM, Dai X, Tyer C, Rughani P, Kudman S, Adney E, Tian H, Wilkes D, Mosquera JM, Stoddart D, Turner DJ, Juul S, Harrington E, Imielinski M, Nanopore sequencing of DNA concatemers reveals higher-order features of chromatin structure, bioRxiv (2019) 833590, 10.1101/833590. [DOI] [Google Scholar]
- [108].Lee I, Razaghi R, Gilpatrick T, Sadowski N, Sedlazeck FJ, Timp W, Simultaneous profiling of chromatin accessibility and methylation on human cell lines with nanopore sequencing, bioRxiv (2018) 504993, 10.1101/504993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Shipony Z, Marinov GK, Swaffer MP, Sinnott-Armstrong NA, Skotheim JM, Kundaje A, Greenleaf WJ, Long-range single-molecule mapping of chromatin accessibility in eukaryotes, Nat. Methods 17 (2020) 319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Altemose N, Maslan A, Lai A, White JA, Streets AM, μDamID: a microfluidic approach for imaging and sequencing protein-DNA interactions in single cells, bioRxiv (2019) 706903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Miga KH, Centromeric satellite DNAs: hidden sequence variation in the human population, Genes 10 (2019) 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Smit AFA, Hubley R, Green P, RepeatMasker Open-4.0. 2013–2015, (2015), pp. 289–300. [Google Scholar]