

Abstract
Astrocytes are the most abundant cell type in the central nervous system (CNS), performing complex functions in health and disease. It is now clear that multiple astrocyte subsets or activation states (plastic phenotypes driven by intrinsic and extrinsic cues) can be identified, associated to specific genomic programs and functions. The characterization of these subsets and the mechanisms that control them may provide unique insights into the pathogenesis of neurologic diseases, and identify potential targets for therapeutic intervention. In this article, we provide an overview of the role of astrocytes in CNS inflammation, highlighting recent discoveries on astrocyte subsets and the mechanisms that control them.
Astrocytes: Once Overlooked, Now Stepping into the Spotlight
Cells in the adult CNS (see Glossary) can be classified in two major groups: neurons and glia. Glial cells (oligodendrocytes, microglia, and astrocytes) share a common feature: they are unable to produce an electrical impulse. Oligodendrocytes are specialized in myelin production, microglia are CNS-resident macrophages, and astrocytes were traditionally viewed as cells involved in supporting neurons. However, it is now clear that astrocytes perform a broad array of functions, including ion and water homeostasis, neurotransmitter recycling, blood–brain barrier (BBB) formation and maintenance, and immune signaling, as well as the regulation of neuronal synaptogenesis [1–5]. The list of astrocyte functions continues to grow, reflecting the identification of novel astrocyte subpopulations with defined phenotypic and functional features. Bulk RNA-seq and single-cell RNA-seq (scRNA-seq) have been key to advancing our understanding of astrocyte subpopulations and activation states. The characterization of their role in disease, as well as the mechanisms that control them, could pave the way for the development of new therapies to treat neurologic conditions. In this review we discuss recent advances regarding the roles of astrocytes in CNS inflammation and the specific astrocyte subpopulations involved.
Astrocyte Heterogeneity
More than 100 years ago, the understanding of the structure and function of the nervous system was revolutionized by the contributions of Camillo Golgi and Santiago Ramon y Cajal (1906 Nobel Prize winners). Golgi developed ‘the black reaction’, a silver staining technique that revealed entire neurons including their processes. Ramon y Cajal would later use this method to provide evidence on the individual nature of nerve cells, a foundational idea of modern neuroscience.
Among many contributions, Ramon y Cajal provided some of the first evidence suggesting that astrocytes are not a uniform population. Ramon y Cajal’s drawings highlighted the morphological heterogeneity of gray matter protoplasmic astrocytes and white matter fibrous astrocytes. Novel technologies have shed new light on astrocyte heterogeneity, including – but not limited to – cell morphology, function, and gene expression and the signaling pathways that control it.
In a recent study, a combination of tools – including a reporter mouse strain expressing GFP under the control of the promoter of the astrocyte marker aldehyde dehydrogenase 1 family member L1 (Aldh1l1–GFP), flow cytometry, and transcriptional profiling – were used to characterize astrocyte heterogeneity [6]. Five astrocyte populations were defined based on the expression of Aldh1l1, CD51, CD63, and CD71. These astrocyte populations showed different frequencies in the cortex, brainstem, thalamus, olfactory bulb, cerebellum, and spinal cord of mice. RNA-seq analysis identified transcriptional modules associated with these astrocyte subsets, providing insights into their potential functional specialization [6].
Astrocyte heterogeneity is also detected within the same CNS region. For example, the cerebral cortex is the outer layer of the cerebrum of humans and other mammals, divided into six layers. Astrocytes in different cortical layers display morphological heterogeneity, as revealed by differences in volume, orientation, and arborization, which are associated with specific transcriptional programs as evidenced by RNA-seq analyses [7,8]. A recent study also analyzed astrocyte heterogeneity in the mouse cerebral cortex [9]. Using an in situ transcriptomic approach focused on the expression of 46 candidate genes, specific transcriptional characteristics were defined in different layers of the cerebral cortex (superficial, mid-, and deep), and in distinct mouse cortical areas (dorsoventral versus rostrocaudal). Taken together, these findings highlight the broad molecular and functional diversity of astrocytes, while they also raise questions about the mechanisms that drive astrocyte diversity. Neurons, for example, have been suggested to play a role in the control of astrocyte diversity in the homeostatic CNS [10], but additional cell types participate in the control of astrocyte phenotype in the context of inflammation, as discussed below.
Reactive Astrocytes
Based on the heterogeneity of astrocytes detected under homeostatic conditions, it is unsurprising that astrocytes mount diverse responses that can contribute to tissue repair and promote CNS pathology in the context of trauma, infection, and neurodegenerative diseases. Thus, astrocyte reactivity is not an all-or-none process; rather, it constitutes a highly heterogeneous process.
One common feature of reactive astrocytes across different species is the upregulation of glial fibrillary acidic protein (GFAP) detectable both at the mRNA and the protein level [11]. GFAP is upregulated in astrocytes in response to most types of CNS injury and is widely used as a marker of astrocyte reactivity [11]. However, GFAP expression in reactive astrocytes varies significantly depending on their location in the CNS, their proximity to the injury site, and the type of injury [11]. Moreover, GFAP upregulation is a feature of many but not all reactive astrocytes, which does not inform on their function or underlying heterogeneity.
Based on the expression of a selected set of genes, one study defined two activation states of mouse reactive astrocytes: ‘A1 and A2’ [12], by analogy with the ‘M1 and M2’ macrophage polarization states. Of relevance, this study identified tumor necrosis factor (TNF), interleukin (IL)-1α, and complement component 1q (C1q) produced by lipopolysaccharide (LPS)-stimulated microglia as differentiation factors that promote an A1 astrocyte phenotype [12]. Although it is now clear that additional astrocyte activation states exist in vivo besides A1 and A2, resembling the diversity of microglia/macrophage activation states [13], these studies shed new light on astrocyte heterogeneity and its potential implications for CNS pathology. A1 astrocytes display neurotoxic activity in vitro and were proposed to participate in the pathogenesis of multiple neurologic diseases. astrocytes expressing complement component 3, a candidate marker of A1 astrocytes, were detected in CNS samples from Alzheimer’s disease (AD), Parkinson’s disease, and multiple sclerosis (MS) patients. Conversely, A2 astrocytes, identified during the analysis of ischemia samples, have been assigned neuroprotective activity. For example, in a rat model of transient middle cerebral artery occlusion/reperfusion, lentivirus-mediated knockdown of protein deglycase DJ-1 decreased glutathione concentrations and reduced neuronal viability, suggesting that DJ-1 could mediate astrocyte neuroprotection by increasing the release of glutathione, used by neurons to resist oxidative stress [15]. Ongoing single-cell genomic studies of clinical samples and experimental models are likely to identify new subsets of reactive astrocytes and the mechanisms controlling them. An important issue related to the identification of disease-associated subsets of reactive astrocytes is whether they are stable or whether they show some degree of plasticity, a major question in determining the best approach to target these cell populations therapeutically.
Signaling Pathways Controlling Reactive Astrocytes
Multiple signaling pathways initiate and modulate astrocyte reactivity, including the Janus kinase/signal transducer and activator of transcription 3 (JAK/STAT3) pathway [16,17], the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway [18], the calcineurin (CN) pathway [19], and the mitogen-activated protein kinase (MAPK) pathway [20] (Table 1). Although NF-κB, CN, and MAPK constitute important pathways in the control of reactive astrocytes, they seem to modulate rather than initiate this process. For instance, in a mouse model of amyotrophic lateral sclerosis (ALS), the inhibition of NF-κB signaling in astrocytes had only a minor impact on the number of GFAP-positive cells during the onset of the disease and none at later stages [21]. Additionally, according to the experimental model used, CN triggered astrocyte reactivity (demonstrated by adenoviral transfer of activated CN to primary rat hippocampal cultures [22]) but also prevented it [demonstrated by a study that used insulin-like growth factor 1 to favor anti-inflammatory interactions between CN, NF-κB, and peroxisome proliferator-activated receptor-γ (PPARγ) in AD mice] [23]. Current evidence supports the idea that a central pathway in the initiation of astrocyte reactivity is the JAK/STAT3 pathway [16,17,24], a concept supported by the detection of JAK/STAT3 activation as a common feature of reactive astrocytes in many neurodegenerative disease animal models [25] as well as in brain metastases associated with lung cancer, breast cancer, and melanoma in humans [26]. However, it is important to point out that multiple pathways crosstalk to fine-tune the phenotype of reactive astrocytes. For example, STAT3 and NF-κB can physically interact to synergistically control target genes, while the aryl hydrocarbon receptor (AHR) can suppress NF-κB signaling in mouse astrocytes via suppressor of cytokine signaling 2 (SOCS2) [27,28]. In addition, deubiquitinating enzymes are emerging as important regulators of astrocyte reactivity. For example, A20 deletion in Gfap-Cre A20fl/fl mice led to increased NF-κB activation and worsened experimental autoimmune encephalomyelitis (EAE) (a model for MS); in addition, otubain-1 (OTUB1) ablation in Gfap-Cre Otub1fl/fl mice led to SOCS1 destabilization, increased proinflammatory gene expression, and worsened EAE relative to controls [29,30]. Thus, the integration of multiple cues in the local microenvironment, and the subsequent activation of interacting downstream signaling pathways, determines the final phenotype and function of reactive astrocytes, affecting their production of effector molecules (Table 2).
Table 1.
Pathways Associated with the Modulation of Murine and Human Reactive Astrocytesa
| Functional category | Molecule | Signaling pathway | Effect on reactive astrocytes | Refs |
|---|---|---|---|---|
| Cytokines | IL-6 | GP130–JAK/STAT3 | Regulates expression of GFAP, vimentin, STAT3 and other genes with STAT responsive elements | [24] |
| CNTF | GP130–JAK/STAT3 | Regulates expression of GFAP, vimentin, STAT3, and other genes with STAT responsive elements | [24] | |
| TGF-β | SMAD | Increases vimentin, actin, and GFAP expression; contributes to scar formation | [115] | |
| IL-1β | NF-κB | Proinflammatory | [24] | |
| TNF | NF-κB/CN–NFAT | Activates expression of target genes | [24] | |
| IFN-α/β | STAT1/STAT2 | Antiviral | [37] | |
| IFN-γ | IFNGR–JAK/STAT1 | Potentiates astrocyte reactivity | [116] | |
| DAMPS | ATP | P2XR/P2YR–Ca2+ | Proinflammatory | [117] |
| ROS | NF-κB, PI3K, MAPK | Proinflammatory | [118] | |
| NO | Soluble guanylyl cyclase | Proinflammatory (mitochondrial impairment, neuronal injury and death) | [119] | |
| Growth factors | EGF | MAPK | Expression of inducible NO synthase, GFAP, modification of the extracellular matrix, proliferation | [120] |
| FGF | MAPK | FGF family members show differentially regulatory roles | [24] | |
| TGF-α | MAPK | Induces expression of GFAP and vimentin and morphological features of reactive astrocytes | [121] | |
| Hormones | Progesterone | Progesterone receptors | Anti-inflammatory | [122] |
| Androgens | Androgen receptors | Anti-inflammatory | [123] | |
| Estradiol | Estrogen receptors | Anti-inflammatory | [124] | |
| Neurodegeneration | Amyloid beta | RAGE/NF-κB | Proinflammatory (inflammatory cytokines and NO) | [125] |
| Neurotransmitters | Glutamate | CN–NFAT | Context dependent (triggers or inhibits astrocyte reactivity) | [24] |
| PAMPs | LPS | TLR4/NF-κB | Proinflammatory | [126] |
| dsRNA (poly I:C) | TLR3 | Antiviral and proinflammatory | [127] | |
| ssRNA | TLR7–Myd88/NF-κB | Antiviral and proinflammatory | [128] | |
| CpG DNA | TLR9–Myd88/NF-κB | Antiviral and proinflammatory | [128] | |
| Cytosolic DNA | cGAS–STING/IFN | Antiviral and proinflammatory | [129] |
Abbreviations: cGAS–STING, cGMP-AMP synthase–stimulator of interferon genes; CN–NFAT, calcineurin-nuclear factor of activated T cells; CNTF, ciliary neurotrophic factor; DAMPs, damage-associated molecular patterns; EGF, epidermal growth factor; FGF, fibroblast growth factor; GP130, glycoprotein 130; IFNGR, IFN-γ receptor; Myd88, myeloid differentiation primary response 88; NO, nitric oxide; P2XR, P2X receptor; P2YR, P2Y receptor; PI3K, phosphatidylinositol-3-kinase; RAGE, receptor for advanced glycation end products; ROS, reactive oxygen species; ssRNA, single-stranded RNA.
Table 2.
Effector Molecules Produced by Murine and Human Reactive Astrocytesa
| Functional category | Effector molecule (e.g., upregulated, secreted) | Refs |
|---|---|---|
| Antimicrobial | ISGs, β-defensins | [37,39] |
| Cytokines | IL-1β, IL-6, TNF, IFN-γ, TGF-β | [71] |
| Chemokines | CCL2, CCL3, CCL5, CCL20, CXCL1, CXCL10 | [71] |
| Complement cascade components | C3, CFB | [12] |
| Cell proliferation/growth factors | VEGFA, VEGFB, FGF, BMP1, NGF, BDNF, PDGFD | [130] |
| Extracellular matrix | CCN family, MMPs, Tenascin-C, ADAMTS-4, TIMP1 | [131] |
| Intermediate filaments | GFAP, nestin, vimentin, synemin | [132] |
| Neurotransmitters | Glutamate, ROS, ATP | [133,134] |
Abbreviations: ADAMTS-4, a disintegrin and metalloproteinase with thrombospondin motifs 4; BDNF, brain-derived neurotrophic factor; BMP1, bone morphogenetic protein 1; C3, complement component 3; CCN, connective tissue growth factor, cysteine-rich protein, nephroblastoma overexpressed gene; CFB, complement factor B; MMPs, matrix metalloproteinases; NGF, nerve growth factor; PDGFD, platelet-derived growth factor D; TIMP1, TIMP metallopeptidase inhibitor 1.
Astrocytes in the Response against Microbial Infections Targeting the CNS
Many viruses target the human CNS, including the flaviviruses Zika virus (ZIKV), West Nile virus (WNV) and Japanese encephalitis virus (JEV) [31]. Astrocytes in mice and humans express a wide variety of pattern recognition receptors (PRRs) responsive to viral pathogen-associated molecular patterns (PAMPS). In flavivirus-infected cells, double-stranded RNA (dsRNA) – an intermediate product of viral genome replication – is a major PAMP recognized by Toll-like receptor (TLR) 3, retinoic acid-inducible gene I (RIG-I), and melanoma differentiation-associated gene 5 (MDA5) [32–34]. TLR signaling, together with mitochondrial antiviral signaling (MAVS) [33] triggered by RIG-I-like receptors and MDA5, controls the activation of transcription factors such as NF-κB, interferon (IFN) regulatory factor 1 (IRF1), IRF3, and, eventually, antiviral responses driven by type I IFNs (IFN-Is) [35]. Ablation of the IFN-I receptor (IFNAR) using Gfap-Cre Ifnarfl/fl mice reduced survival and increased neuronal death after WNV infection [36] and promoted mouse hepatitis virus A59 replication, severe encephalomyelitis, and mortality [37].
IFN-Is drive the expression of hundreds of IFN-stimulated genes (ISGs), which constitute the core of the antiviral innate immune response [35]. Many ISGs exert direct antiviral effects. For instance, RNase L cleaves RNA in virally infected cells [38]. Using RNA-seq analysis, mouse primary astrocytes were shown to express high basal amounts of ISGs, which could play an important role in limiting virus replication in the CNS [39]. IFN-Is also induce the expression of the transcription factor AHR in astrocytes, suppressing IFN-I production and other mechanisms of intrinsic immunity as part of a negative feedback loop that limits immunopathology but is exploited by ZIKV and other pathogens to evade antiviral immunity [40,41] (Figure 1). Although interesting, the significance of AHR upregulation in astrocytes in response to neurotropic viruses remains to be further investigated. Finally, it is important to mention that astrocytes also display heterogeneity in the expression of PRRs and ISGs, both under homeostatic conditions and in response to viral infections – as shown for human astrocytes in vitro and for mice infected with WNV [36]. Hence, future studies should identify the roles of specific astrocyte subsets in the control of viral infections in the CNS.
Figure 1. Reactive Astrocytes in Mouse and Humans in the Context of Central Nervous System (CNS) Viral Infection.
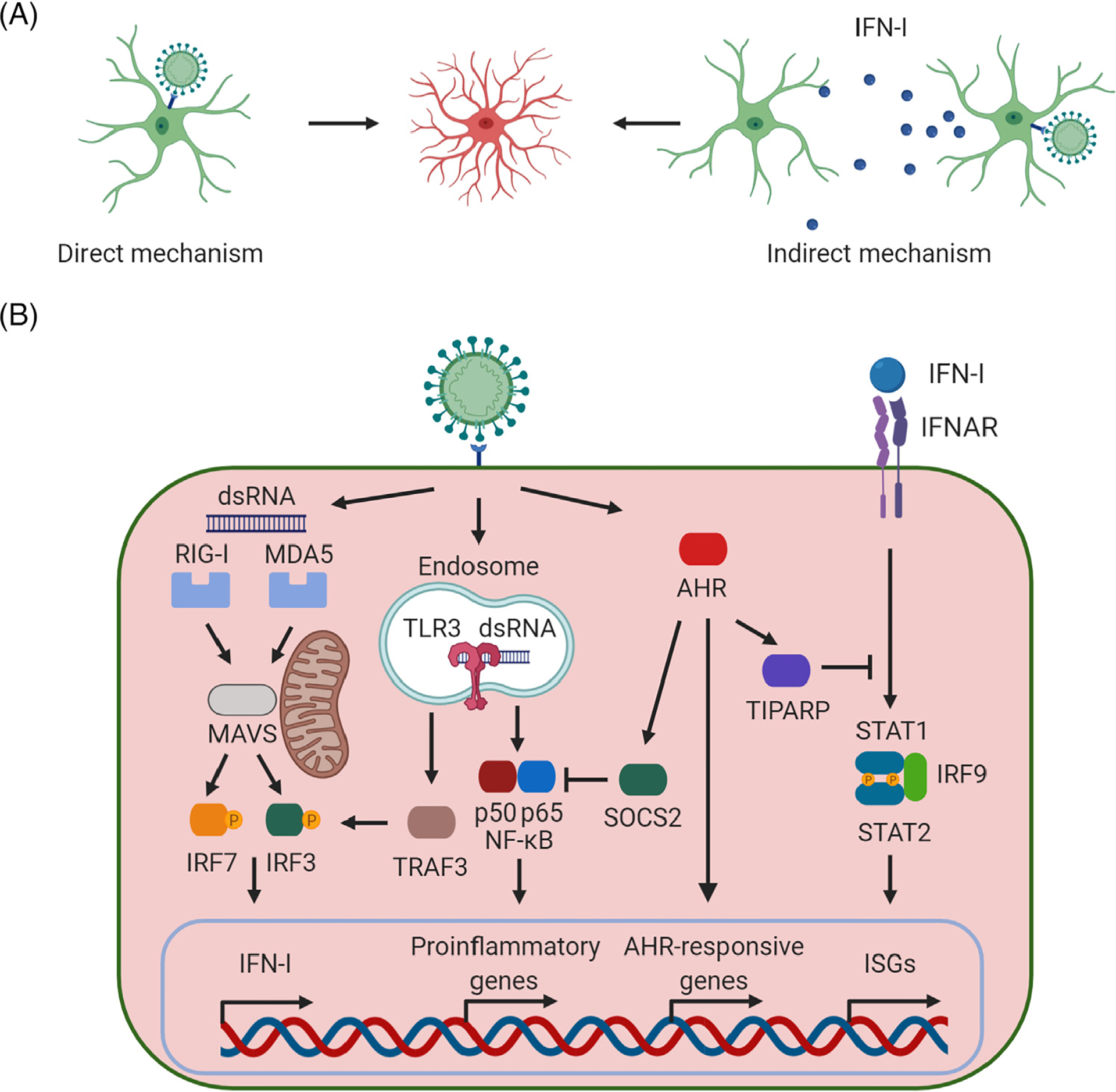
(A) Reactive astrocytes are induced in the context of viral infections through two major mechanisms: direct and indirect. The direct mechanism involves infection of astrocytes, which then become reactive astrocytes. Conversely, the indirect mechanism involves signaling molecules [e.g., interferon type I (IFN-I)] produced by neighboring infected cells (including other astrocytes as well as other cell types in the CNS). (B) Major signaling pathways involved in the generation of reactive astrocytes on viral infection. Double-stranded RNA (dsRNA), one of the major viral pathogen-associated molecular patterns (PAMPs), is recognized by the endosomal Toll-like receptor 3 (TLR3) as well as the cytoplasmic retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated protein 5 (MDA-5), leading to the synthesis and release of IFN-I. Then, secreted IFN-Is activate autocrine and paracrine signaling cascades through the IFN-I receptor inducing the expression of IFN-stimulated genes (ISGs), mounting an effective antiviral response [32–35]. AHR, aryl hydrocarbon receptor; IFNAR, interferon-α/β receptor; IRF3, interferon regulatory factor 3; IRF7, interferon regulatory factor 7; IRF9, interferon regulatory factor 9; MAVS, mitochondrial antiviral signaling protein; NF-κB, nuclear factor kappalight-chain-enhancer of activated B cells; SOCS2, suppressor of cytokine signaling 2; STAT, signal transducer and activator of transcription; TIPARP, TCDD inducible poly(ADP-ribose) polymerase; TRAF3, tumor necrosis factor receptorassociated factor 3. This figure was created using BioRender (https://biorender.com/).
CNS-targeting viruses and bacteria (Box 1) can cause neurological sequelae such as hearing loss, cognitive impairment, and epilepsy, usually detected weeks to months after pathogen clearance [31]. These neurological deficits are thought to involve both cell death caused by the pathogen and the immunopathological response (Box 2) [31]. The continued emergence of novel neurotropic viruses worldwide demands further studies to understand the role of astrocytes in microbially induced neurological sequelae, with the goal of developing clinical treatments.
Box 1. Astrocytes Respond to Bacterial Challenges
Similar to the situation described in response to viral infections, astrocytes recognize bacterial PAMPS through several TLRs, NOD-like receptors (NOD-1, NOD-2), and scavenger receptors (SRs), triggering signaling cascades that promote pathogen clearance. For instance, astrocytes stimulated with LPS induce various cytokines, including IL-1β, IL-6, and TNF as well as several chemokines such as CCL2, CCL20, and CXCL1 [103]. However, whether LPS triggers astrocyte reactivity directly or indirectly via the activation of microglia remains under debate [12]. Regardless, the induction of these molecules was demonstrated in mouse astrocytes in vitro after infection with Brucella spp. [104] and other bacterial infections of the CNS [103].
Box 2. Virus-Induced Neurological Sequelae: The Case of WNV and ZIKV
WNV is the causative agent of West Nile fever and West Nile virus neuroinvasive disease (WNND). About half of WNND patients experience neurological sequelae [105]. IL-1β produced by astrocytes plays a key role in long-term cognitive sequelae by interfering with the homeostasis of neuronal progenitor cells [106]. While WT mice exhibit neurocognitive defects, Il1r1−/− mice recover from WNND and exhibit normal neurogenesis and resistance to spatial learning defects [106]. Although microglia are thought to contribute to the neurological sequelae of infection via their direct effects on synapses on neurons [107,108], the microglial activation of astrocytes is also likely to contribute to disease pathogenesis [12,86,107,108].
The recent 2015–2016 ZIKV outbreak has been linked to a congenital Zika syndrome characterized by fetal brain abnormalities [109] and to Guillain–Barré syndrome [110]. ZIKV can replicate in the adult human brain and impair synapsis and memory in animal models [107,111]. ZIKV infection in adult mice induces the expression of the proinflammatory cytokines IL-1β and TNF [111]. However, as opposed to the effect observed in WNV infection, Il1r1−/− mice are also susceptible to ZIKV-induced memory impairment [111]. Thus, even for related viruses, distinct mechanisms seem to underlie neurological impairments. The mechanisms responsible for ZIKV-induced neurological complications remain unclear. However, the tropism of ZIKV for astrocytes, the proposed role of astrocytes as ZIKV reservoirs, and the induction of an antiviral and proinflammatory immune response, also proposed to mediate impaired cognition in adoptive transfer EAE [112], warrant further investigation into the role of astrocytes in ZIKV-induced neuropathology.
Astrocytes in MS
MS is an autoimmune disorder targeting the CNS. In most patients, MS initially presents as a relapsing–remitting disease (RRMS) in which episodes of neurological dysfunction (relapses) are followed by a period of partial or full recovery (remission). Adaptive immunity plays a central role in MS pathogenesis, as indicated by the therapeutic success of drugs such as natalizumab, alemtuzumab, rituximab, and ocrelizumab, which target B cells and T cells in RRMS [42]. A significant fraction of RRMS patients eventually develop a progressive form of the disease [secondary progressive MS (SPMS)] characterized by the progressive and irreversible accumulation of neurological disability unresponsive to most available therapeutic interventions [42]. Moreover, astrocytes and other CNS-resident cells are thought to play a major role in SPMS pathogenesis (see below) [43].
EAE is a mouse model that recapitulates many features of MS, including inflammation, demyelination, and the generation of reactive astrocytes (Box 3). The depletion of reactive astrocytes around the time of EAE induction has resulted in increased disease severity [44], concomitant with greater infiltration of peripheral leukocytes into the CNS, relative to wild-type (WT) mice [45]. Similarly, the loss of reactive astrocytes in two mouse models (Gfap-TK and Gfap-Cre Stat3fl/fl) after spinal cord injury also increased the severity [46]. Furthermore, in the context of inflammation, astrocytes also display a decreased ability to uptake and degrade extracellular glutamate to limit N-methyl-d-aspartate receptor (NMDAR)-mediated excitotoxicity, as shown by the reduced expression of glutamine synthetase and glutamate dehydrogenase in EAE mice (detected by western blot and RT-PCR) as well as by the decrease in glutamate transporters observed in samples from MS patients (revealed via immunohistochemistry) [47,48]. These findings suggest that at least a subset of reactive astrocytes limits both the recruitment of immune cells to the CNS and the development of neuropathology following acute inflammation or injury. By sharp contrast, reactive astrocyte depletion, during chronic progressive EAE in F1 non-obese diabetic (NOD)/C57BL/6 (Gfap-TK) mice decreased the recruitment of neurotoxic monocytes to the CNS, reduced demyelination and axonal loss, and ameliorated disease severity relative to WT controls [49], suggesting that a subset of reactive astrocytes at this disease stage promotes CNS pathogenesis. The disease-promoting activities of astrocytes in MS seem to involve the recruitment of inflammatory monocytes to the CNS, the activation of CNS-resident microglia, and, potentially, the intrinsic neurotoxic activity of the latter [43]. Taken together, this dual role of astrocytes in EAE highlights their heterogeneity and ability to function as regulators of CNS inflammation.
Box 3. EAE: A Widely Accepted Mouse Model of MS
Multiple EAE models have been developed to study specific aspects of MS [113]. For example, EAE induced by immunization of B6 mice with myelin oligodendrocyte glycoprotein derived peptide (MOG35–55) mostly mimics an acute demyelinating event, while EAE induced in NOD mice on immunization with MOG35–55 models certain aspects of SPMS, including partial recovery after an initial acute phase of the disease followed by a chronic progressive disease course [114]. These EAE models have helped to define the contribution of astrocytes to MS pathogenesis by both the acquisition of disease-promoting activities and their reduced participation in homeostatic processes such as the metabolic support of neurons and the reduced production of neurotrophic factors [43,81].
The central role of T cells in MS pathology has triggered multiple investigations on the ability of CNS-resident cells to act as antigen presenting cells (APCs) involved in the local reactivation of self-reactive T cells. These studies identified important roles for CNS-resident myeloid cells [50–53]. For example, mass cytometric analyses combined with cell-specific deletion of the major histocompatibility complex (MHC)-II identified a population of conventional dendritic cells (CD11b+ CD172a+ cDC2s) as essential for the reactivation of encephalitogenic T cells during EAE [51]. Great attention has also been given to the potential role of astrocytes as APCs, although this remains a controversial subject. Nevertheless, MHC-II – needed for antigen presentation – is upregulated in IFN-γ-stimulated astrocytes in vitro [54], chronic active MS plaques [55], and Parkinson’s disease [56]. Astrocytes were recently suggested to act as APCs in Parkinson’s disease, a hypothesis supported by histological analyses of patient postmortem brain tissue showing CD4+ T cells surrounding MHC-II-positive astrocytes [56]. MHC-I is usually expressed in low amounts, although it has been induced in mouse primary astrocytes in vitro on infection with St. Louis encephalitis virus and JEV [57]. Similar findings were reported for astrocytes in vivo, using a mouse model of infection with the JHM strain of mouse hepatitis virus [58].
Besides interacting with peptide-MHC molecules, T cells require co-stimulatory signals to become fully activated. Accordingly, the expression of co-stimulatory molecules by astrocytes is also controversial. Some studies detected the expression of the co-stimulatory molecule B7 in murine astrocytes in vitro after IFN-γ stimulation [59] and in chronic active lesions of MS [60]. However, other studies using human fetal astrocytes in vitro failed to detect B7 even after stimulation with IFN-γ and granulocyte-macrophage colony-stimulating factor (GM-CSF) [61]. These inconsistencies may reflect species-specific differences in addition to artifacts associated with neonatal astrocytes in culture. Therefore, further studies are required to conclude whether astrocytes play a role in antigen presentation and T cell activation.
Perhaps more important than their contested role in priming naïve T cells, astrocytes control the adaptive immune response in the CNS via the production of cytokines and chemokines (Table 2). Mouse astrocytes in vitro have been shown to produce IL-12 and IL-23, which amplify T helper 1 (Th1)/Th17 cell responses [62]. Conversely, mouse astrocytes treated in vitro with Th17 cell-conditioned medium produce IL-6, which in turn promotes Th17 cell differentiation [63], suggesting a positive feedback loop that might boost effector T cell responses in the CNS. However, in agreement with the existence of diverse astrocyte activation states during CNS inflammation, stimulated mouse astrocytes have also been shown to produce IL-27, as demonstrated by ELISA [64]. IL-27 expression has also been detected in primary cultures of human astrocytes after treatment with proinflammatory cytokines as well as in MS lesions [65]. IL-27, in turn, limits Th17 cell differentiation, modulates dendritic cell functions [66], and promotes the differentiation of IL-10-producing anti-inflammatory type 1 regulatory (Tr1) cells [64,66–69]. IL-10, produced by Tr1 cells, suppresses inflammatory transcriptional programs in astrocytes [70]. Thus, astrocytes are engaged in a bidirectional dialog with T cells that results in the modulation of CNS inflammation. Future studies should investigate the specific T cell and astrocyte subsets involved.
Astrocytes are also a key source of chemokines (Table 2) [71]. Some of the key chemokines produced by astrocytes include (C-C motif) ligand 2 (CCL2), CCL20, C-X-C chemokine ligand 8 (CXCL8), and CXCL10. CCL2, for example, was detected in astrocytes from MS patients and plays a key role in recruiting monocytes, macrophages, basophils, T cells, and microglia to the site of inflammation [49], while CCL20 expressed in astrocytes from post-ischemic brains of mice is a strong chemoattractant for dendritic cells [72] and lymphocytes. CXCL8 recruits neutrophils and monocytes when acting synergistically with CCL2. Moreover, CXCL8 contributes to the Th1 proinflammatory response in MS [73]. In situ studies should determine how astrocyte regional heterogeneity establishes local chemokine gradients that control the recruitment of immune cells to specific CNS regions.
Control of Astrocyte Responses in MS
From a therapeutic perspective, the identification of these astrocyte activation states and the molecular mechanisms that control them may offer novel targets for the therapeutic control of astrocytes in MS. Below we discuss recent advances on mechanisms associated with the control of astrocytes in MS.
Metabolic Control of Astrocyte Responses
Cellular metabolism controls peripheral immune cells [67,74–77] and microglial activity in Alzheimer’s disease [78], but the metabolic mechanisms controlling astrocyte subsets are unknown. In a model of high-fat diet (HFD), mice knocked out for c-Jun N-terminal Kinase 1 (Jnk1−/−) were protected from HFD-induced cognitive impairment and showed reduced astrocyte reactivity compared with WT mice. Thus, JNK-1 was suggested to link metabolic dysregulation with cognitive impairment [79]. In another study, the transcriptional and metabolic analysis of astrocytes from MS lesions and in the NOD mouse model of chronic progressive EAE revealed dysregulated sphingolipid metabolism leading to the accumulation of lactosylceramide (LacCer) [49,80]. LacCer activates a newly described signaling pathway involving cytosolic phospholipase A2 (cPLA2) and MAVS [81]. Although MAVS is known to control antiviral responses [33], it had not been previously linked to virus-free CNS pathology. In this study, a combination of proteomic, metabolomic, transcriptomic, and perturbation studies in astrocytes in vitro and the NOD EAE mouse model showed that LacCer-triggered cPLA2–MAVS signaling drives the expression of NF-κB-dependent transcriptional programs in astrocytes that promote inflammation and neurodegeneration (Figure 2). In addition, MAVS activation decreases lactate production in mouse astrocytes [81], interfering with the ability of astrocytes to provide lactate to support neuron metabolic needs [82]. This novel mechanism connects astrocyte sphingolipid metabolism with the control of neurodegeneration and astrocyte–neuron metabolic coupling. In addition, these studies identified miglustat – a drug that targets sphingolipid metabolism and is used for the treatment of Gaucher disease – as a candidate drug that might be repurposed to modulate astrocyte pathogenic activities in MS. Further studies are needed to characterize the relationship between cell metabolism and the control of astrocyte activation states.
Figure 2. Metabolic Control of Mouse and Human Astrocytes.
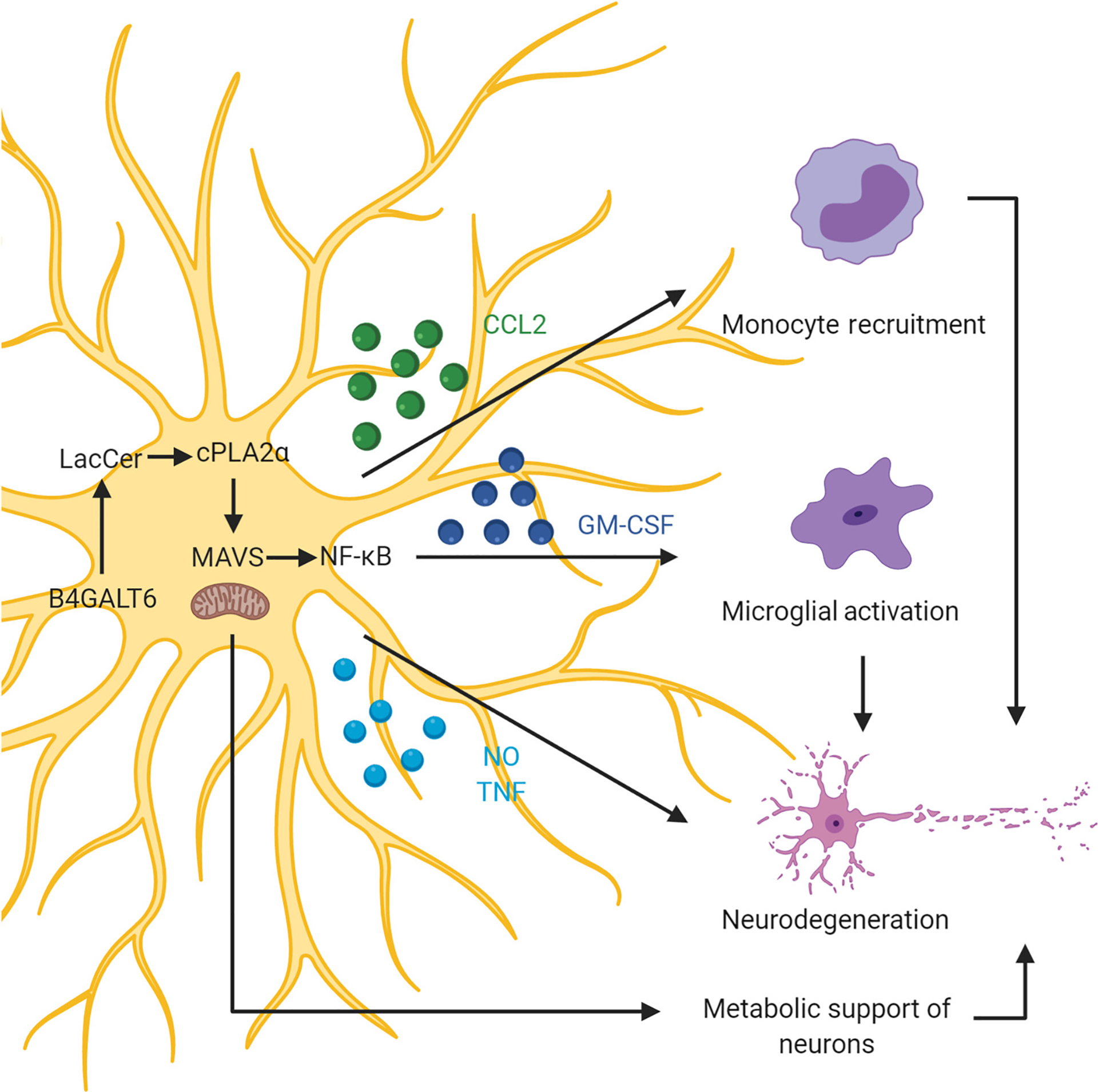
Sphingolipid metabolism in astrocytes activates a novel signaling pathway involving cytosolic phospholipase A2 (cPLA2) and mitochondrial antiviral signaling protein (MAVS). Lactosylceramide (LacCer) produced by sphingolipid metabolism binds to and activates cPLA2, which then activates MAVS inducing nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)-driven proinflammatory transcriptional programs. MAVS activation also triggers metabolic remodeling that reduces lactate production and limits the metabolic support of neurons by astrocytes, boosting neurodegeneration [81]. B4GALT6, beta-1,4-galactosyltransferase 6; CCL2, chemokine (C-C motif) ligand 2; GM-CSF, granulocyte-macrophage colony-stimulating factor; NO, nitric oxide; TNF, tumor necrosis factor. This figure was created using BioRender (https://biorender.com/).
Microbial Metabolites Control Astrocytes Directly and Indirectly via Microglia
The commensal flora is an important source of metabolites for the host [83–85]. Microbial metabolites have multiple roles in host physiology, but their effects on CNS-resident cells are poorly understood. AHR was recently reported to be activated in astrocytes by microbial metabolites derived from dietary tryptophan crossing the BBB [27]. AHR activation was found to suppress proinflammatory transcriptional modules in astrocytes by interfering with NF-κB signaling through a mechanism mediated by SOCS2 [27,28,86–88]. Additional studies using Cx3cr1creERT2Ahrfl/fl (CX3CR1-AHR) mice and a Trp-depleted diet (TDD) demonstrated that microbial metabolites also activate AHR in microglia, limiting NF-κB activation [86]. TDD-fed mice showed limited recovery from EAE in both control and CX3CR1-AHR mice. However, the administration of Trp-derived AHR agonists ameliorated EAE in control but not in CX3CR1-AHR mice, suggesting that microglial AHR activation by microbial Trp metabolites limits EAE. Moreover, RNA-seq analysis showed that AHR also regulates the production of microglial transforming growth factor alpha (TGF-α) and vascular endothelial growth factor (VEGF)-B, which modulate astrocyte proinflammatory responses [86]. Collectively, these findings suggest that the tonic activation of this and potentially additional pathways might contribute to the control of inflammation and other physiological processes [89,90] under homeostatic conditions. Thus, future investigations should focus on the modulation of specific astrocyte activation states by defined members of the commensal flora.
Control of Astrocyte Responses by Environmental Factors
Microbial metabolites reach the CNS to act on resident cells [27,86], raising the possibility that some environmental chemicals may also reach the CNS and modulate the behavior of astrocytes. Both genetic [91] and environmental factors [92] have been linked to MS and other neurological disorders in which astrocytes are implicated.
To systematically evaluate the effects of environmental chemicals on astrocytes, a novel platform that combines bioinformatics, chemical screens in a neuroinflammation zebrafish model, and genetic and small-molecule perturbation studies in mouse and human systems was developed [93]. This platform identified the herbicide linuron as an inducer of astrocyte proinflammatory responses via the activation of sigma non-opioid intracellular receptor 1 (SIGMAR1) and the regulators of the unfolded protein response (UPR) inositol-requiring enzyme 1 α (IRE1α) and X-box binding protein 1 (XBP1) [93]. Importantly, this newly identified Sigmar1–IRE1α–XBP1 pathway was found to control the response of astrocytes during EAE and MS in the absence of linuron. Lentivirus-mediated knockdown of Xbp1 or Ern1 (encoding IRE1α) in astrocytes ameliorated EAE and RNA-seq showed that Xbp1 knockdown limited the activation of inflammatory pathways, linking SIGMAR1–UPR signaling to astrocyte-driven inflammation and neurotoxicity (Figure 3). Of note, herbicides have been reported to increase the risk of development of MS [94] and the UPR was recently shown to drive astrocyte pathogenic activities in prion-induced neurodegeneration [95], validating the role of the UPR in the control of astrocyte pathogenic responses. These studies describe a novel approach to the study of gene–environment interactions modulating astrocyte function and identify SIGMAR1–UPR signaling as a driver of astrocyte neurotoxic activities. This work also highlights the need for further research aimed at characterizing the effects of environmental chemicals on reactive astrocytes.
Figure 3. Control of Mouse and Human Astrocytes by the Unfolded Protein Response (UPR) and Environmental Chemicals.
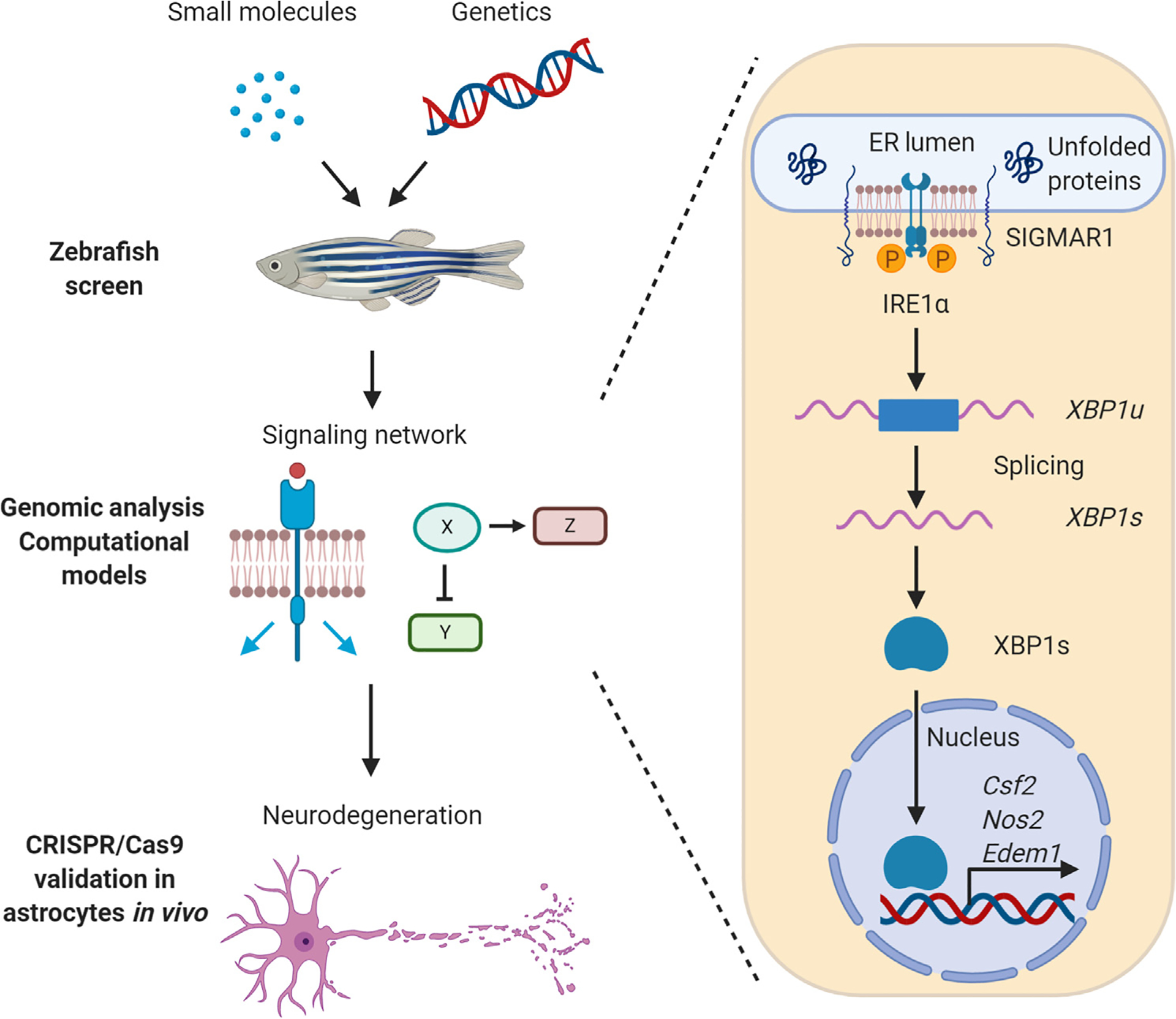
Platform for the unbiased identification of environmental factors that affect astrocyte responses. A small-molecule screen on a collection of environmental chemicals provided by the US Environmental Protection Agency (EPA) identified the herbicide linuron as an environmental chemical that increases inducible nitric oxide synthase (iNOS) expression in astrocyte-like cells in a novel zebrafish model of neurodegeneration; these findings were later validated in mouse and human astrocyte models. Computational modeling, genetic, and small-molecule perturbation studies on murine and human systems identified the mechanism of action of linuron, which activates the UPR via sigma non-opioid intracellular receptor 1 (SIGMAR1)–inositol-requiring enzyme 1 α (IRE1α)–X-box binding protein 1 (XBP1) signaling. UPR signaling is also activated and drives astrocyte proinflammatory programs even in the absence of linuron during experimental autoimmune encephalomyelitis (EAE) and multiple sclerosis (MS) [93]. Csf2, colony-stimulating factor 2; ER, endoplasmic reticulum; Edem1, ER degradation-enhancing alpha-mannosidase-like protein 1; Nos2, nitric oxide synthase 2; XBP1s, X-box binding protein 1 spliced form; XBP1u, X-box binding protein 1 unspliced form. This figure was created using BioRender (https://biorender.com/).
Control of Astrocyte Activation States by T Cells
On a finishing note, it is important to revisit the concept of astrocyte heterogeneity, but this time in the framework of MS. Similar to what has been reported in other neurological diseases [96–98], reactive astrocytes in MS are expected to adopt multiple activation states. Through the use of scRNA-seq, epigenetic analyses, and in vivo cell-specific CRISPR-Cas9-based genetic perturbations, seven astrocyte subpopulations were identified in EAE and MS [99]. Functional studies established that one of these astrocyte subsets was expanded in EAE and MS, promoting CNS pathology [99]. scRNA-seq analysis established that this population is characterized by a decrease in the expression of NRF2-driven transcriptional modules that limit oxidative stress and inflammation. Knockdown of Nfe2l2 (encoding NRF2) in astrocytes worsened EAE and upregulated proinflammatory pathways, as shown by RNA-seq. Of note, this astrocyte subset was also controlled by an epigenetic program driven by MAFG and MAT2A, known to cooperate to promote DNA methylation. These findings suggest that epigenetic modifications stabilize this subset, resembling recent findings in murine microglia [100,101] and identifying epigenetic modifiers as candidate targets for astrocyte modulation in MS.
Finally, T cells and other peripheral cells are recruited to the CNS during MS, EAE, and other neurological disorders [42]. As already mentioned, T cells and astrocytes are engaged in poorly characterized interactions. The analysis of the transcriptional signature of MAFG+MAT2A+ astrocytes revealed increased signaling in response to GM-CSF, a cytokine produced by effector encephalitogenic T cells and known to drive CNS inflammation in EAE [102] but previously unknown to act on astrocytes. Using mice with conditional knockout of the GM-CSF receptor (Aldh1l1-creERT2 Csf2rb), MAFG+MAT2A+ astrocytes were stabilized in EAE by GM-CSF produced by inflammatory T cells, as deletion of Csf2rb decreased the expression of Mafg and Mat2a and ameliorated EAE [102]. Furthermore, scRNA-seq in MS samples revealed an expanded astrocyte population characterized by decreased NRF2 activation and increased MAFG activation, DNA methylation, GM-CSF signaling, and proinflammatory pathways [102]. Thus, these studies identify a novel mechanism by which T cells could control astrocyte activation states, adding to the growing literature on the effects of T cells on astrocytes during CNS inflammation [49,72].
Concluding Remarks
Our understanding of astrocyte heterogeneity remains limited and should be expanded not only to define the breadth of astrocyte activation states but also to identify commonalities among different neurological disorders and processes. In addition, gradients of polarizing factors and cell interactions are likely to be established in different CNS locations to adjust astrocyte activation states to their microenvironments. Hence, it is important to leverage the power of in situ transcriptomics with other methods for single-cell analysis to establish the spatial distribution of astrocyte subsets in the CNS, defining the cell interactions that control and stabilize them (see Outstanding Questions). Addressing these points may guide the development of novel strategies for the targeting of specific astrocyte activation states as novel therapeutic approaches for neurological diseases.
Outstanding Questions.
How are astrocyte activation states controlled by the local microenvironment?
What is the physiological function of neurotoxic astrocyte activities and which mechanisms mediate them?
What are the similarities of diseasespecific features of the astrocyte activation states identified in multiple neurological diseases?
How stable are astrocyte activation states?
What astrocyte cell interactions control astrocyte activation states and which are the mechanisms involved?
Can specific astrocyte subsets be therapeutically targeted in neurological diseases?
Highlights.
Astrocytes display functional and phenotypic heterogeneity across and within CNS regions under homeostatic conditions.
CNS insults (trauma, infection, autoimmune inflammation, protein aggregates) induce a broad array of astrocyte activation states, poorly defined in terms of phenotype, function, and role in human pathology.
Astrocyte activation during acute microbial infection is essential for pathogen clearance but can contribute to long-term neurological impairments.
The plasticity of astrocytes and their ability to adopt either a proinflammatory or an anti-inflammatory phenotype could be targeted for therapeutic intervention.
Acknowledgments
This work was supported by grants NS102807, NS087867, ES02530, AI126880, and AI093903 from the NIH, RSG-14-198-01-LIB from the American Cancer Society, and RG4111A1 and JF2161-A-5 from the National Multiple Sclerosis Society (to F.J.Q.). F.J.Q. received support from the International Progressive MS Alliance (PA-1604-08459). We thank all members of the Quintana laboratory for helpful advice and discussions.
Glossary
- A20
ubiquitin-editing enzyme encoded by the TNFAIP3 gene, which is a key negative regulator of NF-κB signaling.
- Arborization
fine, tree-like branching of astrocyte processes.
- Blood–brain barrier (BBB)
semipermeable barrier in blood vessels vascularizing the CNS; essential for regulation of the exchange of molecules between the CNS and peripheral blood.
- Central nervous system (CNS)
part of the nervous system comprising the brain and spinal cord.
- Complement
component of the immune system mediated by plasma proteins acting as a cascade to perform a broad range of functions, including opsonization of pathogens. Complement participates in CNS development and in the pathology of neurological disorders.
- Damage-associated molecular patterns (DAMPs)
host biomolecules released into the extracellular space as a result of cell damage, promoting a proinflammatory response. Classic examples include ATP and DNA.
- Experimental autoimmune encephalomyelitis (EAE)
the most commonly used mouse in studies of MS; induced by immunization with myelin antigens.
- Glial fibrillary acidic protein (GFAP)
an intermediate filament protein expressed in astrocytes and one of the most commonly used astrocyte markers.
- Interferons (IFNs)
group of cytokines playing key roles in fighting viral infections and in regulating the immune response; divided into three classes – IFN-I, IFN-II, and IFN-III.
- Interferon-stimulated genes (ISGs)
genes whose expression is induced by IFN.
- Janus kinase/signal transducer and activator of transcription (JAK/STAT)
signaling pathway involved in immunity, cell division, and cell death. In the CNS, JAK/STAT is a central player in the induction of astrocyte reactivity.
- Multiple sclerosis (MS)
autoimmune inflammatory disease of the CNS characterized by inflammation, demyelination, axonal damage, and gliosis, leading to neurologic disability.
- Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)
transcription factor regulating the expression of key genes involved in inflammation and the immune response.
- Pathogen-associated molecular patterns (PAMPs)
structural motifs highly conserved in pathogens; recognized by specific receptors in the host, triggering an innate immune response. Examples include LPSs, typically found in Gram-negative bacteria and dsRNA, usually generated by viral RNA polymerases as an intermediate in genome replication.
- Pattern recognition receptors (PRRs)
germline-encoded protein receptors in the host that recognize PAMPs. There are four major families of PRRs.
- Prion-induced neurodegeneration
prions are misfolded proteins that can trigger other proteins to fold abnormally, causing fatal progressive neurodegeneration.
- T helper 1 (Th1)/Th17 cells
proinflammatory CD4+ Th cells linked to the pathogenesis of many autoimmune disorders.
- Toll-like receptors (TLRs)
one of the four major families of proteins that constitute the broader group of PRRs. TLRs comprise ten members in humans (TLR1–10) and 13 in mice (TLR1–13).
- Type 1 regulatory (Tr1) cells
FoxP3− subset of CD4+ T cells producing IL-10 and limiting inflammation.
References
- 1.Allen NJ et al. (2012) Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 486, 410–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alvarez JI et al. (2011) The Hedgehog pathway promotes blood–brain barrier integrity and CNS immune quiescence. Science 334, 1727–1731 [DOI] [PubMed] [Google Scholar]
- 3.Chung WS et al. (2013) Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504, 394–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Molofsky AV et al. (2014) Astrocyte-encoded positional cues maintain sensorimotor circuit integrity. Nature 509, 189–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsai HH et al. (2012) Regional astrocyte allocation regulates CNS synaptogenesis and repair. Science 337, 358–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.John Lin CC et al. (2017) Identification of diverse astrocyte populations and their malignant analogs. Nat. Neurosci 20, 396–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lanjakornsiripan D et al. (2018) Layer-specific morphological and molecular differences in neocortical astrocytes and their dependence on neuronal layers. Nat. Commun 9, 1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zeisel A et al. (2015) Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142 [DOI] [PubMed] [Google Scholar]
- 9.Bayraktar OA et al. (2020) Astrocyte layers in the mammalian cerebral cortex revealed by a single-cell in situ transcriptomic map. Nat. Neurosci 23, 500–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farmer WT et al. (2016) Neurons diversify astrocytes in the adult brain through sonic hedgehog signaling. Science 351, 849–854 [DOI] [PubMed] [Google Scholar]
- 11.Sofroniew MV (2014) Astrogliosis. Cold Spring Harb. Perspect. Biol 7, a020420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liddelow SA et al. (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Colonna M and Brioschi S (2020) Neuroinflammation and neurodegeneration in human brain at single-cell resolution. Nat. Rev. Immunol 20, 81–82 [DOI] [PubMed] [Google Scholar]
- 15.Peng L et al. (2019) Effect of DJ-1 on the neuroprotection of astrocytes subjected to cerebral ischemia/reperfusion injury. J. Mol. Med. (Berl.) 97, 189–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herrmann JE et al. (2008) STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J. Neurosci 28, 7231–7243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ceyzeriat K et al. (2016) The complex STATes of astrocyte reactivity: how are they controlled by the JAK–STAT3 pathway? Neuroscience 330, 205–218 [DOI] [PubMed] [Google Scholar]
- 18.Brambilla R et al. (2005) Inhibition of astroglial nuclear factor κB reduces inflammation and improves functional recovery after spinal cord injury. J. Exp. Med 202, 145–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Furman JL and Norris CM (2014) Calcineurin and glial signaling: neuroinflammation and beyond. J. Neuroinflammation 11, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roy Choudhury G et al. (2014) Involvement of p38 MAPK in reactive astrogliosis induced by ischemic stroke. Brain Res 1551, 45–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crosio C et al. (2011) Astroglial inhibition of NF-κB does not ameliorate disease onset and progression in a mouse model for amyotrophic lateral sclerosis (ALS). PLoS One 6, e17187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Norris CM et al. (2005) Calcineurin triggers reactive/inflammatory processes in astrocytes and is upregulated in aging and Alzheimer’s models. J. Neurosci 25, 4649–4658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fernandez AM et al. (2012) Regulation of the phosphatase calcineurin by insulin-like growth factor I unveils a key role of astrocytes in Alzheimer’s pathology. Mol. Psychiatry 17, 705–718 [DOI] [PubMed] [Google Scholar]
- 24.Ben Haim L et al. (2015) Elusive roles for reactive astrocytes in neurodegenerative diseases. Front. Cell. Neurosci 9, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ben Haim L et al. (2015) The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in Alzheimer’s and Huntington’s diseases. J. Neurosci 35, 2817–2829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Priego N et al. (2018) STAT3 labels a subpopulation of reactive astrocytes required for brain metastasis. Nat. Med 24, 1024–1035 [DOI] [PubMed] [Google Scholar]
- 27.Rothhammer V et al. (2016) Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med 22, 586–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yeste A et al. (2016) Tolerogenic nanoparticles inhibit T cellmediated autoimmunity through SOCS2. Sci. Signal 9, ra61. [DOI] [PubMed] [Google Scholar]
- 29.Wang X et al. (2013) Astrocytic A20 ameliorates experimental autoimmune encephalomyelitis by inhibiting NF-κB-and STAT1-dependent chemokine production in astrocytes. Acta Neuropathol 126, 711–724 [DOI] [PubMed] [Google Scholar]
- 30.Wang X et al. (2019) OTUB1 inhibits CNS autoimmunity by preventing IFN-γ-induced hyperactivation of astrocytes. EMBOJ 38, e100947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klein RS et al. (2017) Infectious immunity in the central nervous system and brain function. Nat. Immunol 18, 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meylan E et al. (2005) Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437, 1167–1172 [DOI] [PubMed] [Google Scholar]
- 33.Seth RB et al. (2005) Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell 122, 669–682 [DOI] [PubMed] [Google Scholar]
- 34.Kato H et al. (2006) Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441, 101–105 [DOI] [PubMed] [Google Scholar]
- 35.McNab F et al. (2015) Type I interferons in infectious disease. Nat. Rev. Immunol 15, 87–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Daniels BP et al. (2017) Regional astrocyte IFN signaling restricts pathogenesis during neurotropic viral infection. J. Clin. Invest 127, 843–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hwang M and Bergmann CC (2018) Alpha/beta interferon (IFN-α/β) signaling in astrocytes mediates protection against viral encephalomyelitis and regulates IFN-γ-dependent responses. J. Virol 92, e01901–e01917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chakrabarti A et al. (2015) RNase L activates the NLRP3 inflammasome during viral infections. Cell Host Microbe 17, 466–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lindqvist R et al. (2016) Fast type I interferon response protects astrocytes from flavivirus infection and virus-induced cytopathic effects. J. Neuroinflammation 13, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamada T et al. (2016) Constitutive aryl hydrocarbon receptor signaling constrains type I interferon-mediated antiviral innate defense. Nat. Immunol 17, 687–694 [DOI] [PubMed] [Google Scholar]
- 41.Giovannoni F et al. (2020) AHR is a Zika virus host factor and a candidate target for antiviral therapy. Nat. Neurosci 23, 939–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baecher-Allan C et al. (2018) Multiple sclerosis: mechanisms and immunotherapy. Neuron 97, 742–768 [DOI] [PubMed] [Google Scholar]
- 43.Wheeler MA and Quintana FJ (2019) Regulation of astrocyte functions in multiple sclerosis. Cold Spring Harb. Perspect. Med 9, a020009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Toft-Hansen H et al. (2011) Inhibition of reactive astrocytosis in established experimental autoimmune encephalomyelitis favors infiltration by myeloid cells over T cells and enhances severity of disease. Glia 59, 166–176 [DOI] [PubMed] [Google Scholar]
- 45.Voskuhl RR et al. (2009) Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. J. Neurosci 29, 11511–11522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anderson MA et al. (2016) Astrocyte scar formation aids central nervous system axon regeneration. Nature 532, 195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hardin-Pouzet H et al. (1997) Glutamate metabolism is downregulated in astrocytes during experimental allergic encephalomyelitis. Glia 20, 79–85 [DOI] [PubMed] [Google Scholar]
- 48.Werner P et al. (2001) Multiple sclerosis: altered glutamate homeostasis in lesions correlates with oligodendrocyte and axonal damage. Ann. Neurol 50, 169–180 [DOI] [PubMed] [Google Scholar]
- 49.Mayo L et al. (2014) Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat. Med 20, 1147–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jordao MJC et al. (2019) Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 363, eaat7554. [DOI] [PubMed] [Google Scholar]
- 51.Mundt S et al. (2019) Conventional DCs sample and present myelin antigens in the healthy CNS and allow parenchymal T cell entry to initiate neuroinflammation. Sci. Immunol 4, eaau8380. [DOI] [PubMed] [Google Scholar]
- 52.Quintana FJ (2019) Myeloid cells in the central nervous system: so similar, yet so different. Sci. Immunol 4, eaaw2841. [DOI] [PubMed] [Google Scholar]
- 53.Waisman A and Johann L (2018) Antigen-presenting cell diversity for T cell reactivation in central nervous system autoimmunity. J. Mol. Med. (Berl.) 96, 1279–1292 [DOI] [PubMed] [Google Scholar]
- 54.Vagaska B et al. (2016) MHC-class-II are expressed in a subpopulation of human neural stem cells in vitro in an IFNγ-independent fashion and during development. Sci. Rep 6, 24251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zeinstra E et al. (2000) Astrocytes in chronic active multiple sclerosis plaques express MHC class II molecules. Neuroreport 11, 89–91 [DOI] [PubMed] [Google Scholar]
- 56.Rostami J et al. (2020) Astrocytes have the capacity to act as antigen-presenting cells in the Parkinson’s disease brain. J. Neuroinflammation 17, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abraham S and Manjunath R (2006) Induction of classical and nonclassical MHC-I on mouse brain astrocytes by Japanese encephalitis virus. Virus Res 119, 216–220 [DOI] [PubMed] [Google Scholar]
- 58.Hamo L et al. (2007) Distinct regulation of MHC molecule expression on astrocytes and microglia during viral encephalomyelitis. Glia 55, 1169–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Magnus T et al. (2005) Microglial expression of the B7 family member B7 homolog 1 confers strong immune inhibition: implications for immune responses and autoimmunity in the CNS. J. Neurosci 25, 2537–2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zeinstra E et al. (2003) Reactive astrocytes in chronic active lesions of multiple sclerosis express co-stimulatory molecules B7–1 and B7–2. J. Neuroimmunol 135, 166–171 [DOI] [PubMed] [Google Scholar]
- 61.Satoh J et al. (1995) T-cell costimulatory molecules B7–1 (CD80) and B7–2 (CD86) are expressed in human microglia but not in astrocytes in culture. Brain Res 704, 92–96 [DOI] [PubMed] [Google Scholar]
- 62.Constantinescu CS et al. (2005) Astrocytes as antigen-presenting cells: expression of IL-12/IL-23. J. Neurochem 95, 331–340 [DOI] [PubMed] [Google Scholar]
- 63.Prajeeth CK et al. (2017) Effectors of Th1 and Th17 cells act on astrocytes and augment their neuroinflammatory properties. J. Neuroinflammation 14, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fitzgerald DC et al. (2007) Suppressive effect of IL-27 on encephalitogenic Th17 cells and the effector phase of experimental autoimmune encephalomyelitis. J. Immunol 179, 3268–3275 [DOI] [PubMed] [Google Scholar]
- 65.Senecal V et al. (2016) Production of IL-27 in multiple sclerosis lesions by astrocytes and myeloid cells: modulation of local immune responses. Glia 64, 553–569 [DOI] [PubMed] [Google Scholar]
- 66.Mascanfroni ID et al. (2013) IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nat. Immunol 14, 1054–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mascanfroni ID et al. (2015) Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-α. Nat. Med 21, 638–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chihara N et al. (2018) Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature 558, 454–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Apetoh L et al. (2010) The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat. Immunol 11, 854–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mayo L et al. (2016) IL-10-dependent Tr1 cells attenuate astrocyte activation and ameliorate chronic central nervous system inflammation. Brain 139, 1939–1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rothhammer V and Quintana FJ (2015) Control of autoimmune CNS inflammation by astrocytes. Semin. Immunopathol 37, 625–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ito M et al. (2019) Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature 565, 246–250 [DOI] [PubMed] [Google Scholar]
- 73.Hyvarinen T et al. (2019) Co-stimulation with IL-1β and TNF-α induces an inflammatory reactive astrocyte phenotype with neurosupportive characteristics in a human pluripotent stem cell model system. Sci. Rep 9, 16944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Du X et al. (2018) Hippo/Mst signalling couples metabolic state and immune function of CD8α+ dendritic cells. Nature 558, 141–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Geiger R et al. (2016) L-Arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell 167, 829–842 e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kornberg MD et al. (2018) Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science 360, 449–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mills EL et al. (2018) Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 556, 113–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ulland TK et al. (2017) TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell 170, 649–663 e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Busquets O et al. (2019) c-Jun N-terminal Kinase 1 ablation protects against metabolic-induced hippocampal cognitive impairments. J. Mol. Med. (Berl.) 97, 1723–1733 [DOI] [PubMed] [Google Scholar]
- 80.Rothhammer V et al. (2017) Sphingosine 1-phosphate receptor modulation suppresses pathogenic astrocyte activation and chronic progressive CNS inflammation. Proc. Natl. Acad. Sci.U. S. A 114, 2012–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chao CC et al. (2019) Metabolic control of astrocyte pathogenic activity via cPLA2-MAVS. Cell 179, 1483–1498.e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Magistretti PJ and Allaman I (2018) Lactate in the brain: from metabolic end-product to signalling molecule. Nat. Rev. Neurosci 19, 235–249 [DOI] [PubMed] [Google Scholar]
- 83.Dodd D et al. (2017) A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature 551, 648–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen H et al. (2019) A forward chemical genetic screen reveals gut microbiota metabolites that modulate host physiology. Cell 177, 1217–1231 e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chu C et al. (2019) The microbiota regulate neuronal function and fear extinction learning. Nature 574, 543–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rothhammer V et al. (2018) Microglial control of astrocytes in response to microbial metabolites. Nature 557, 724–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rothhammer V et al. (2017) Dynamic regulation of serum aryl hydrocarbon receptor agonists in MS. Neurol. Neuroimmunol. Neuroinflamm 4, e359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rothhammer V et al. (2018) Detection of aryl hydrocarbon receptor agonists in human samples. Sci. Rep 8, 4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Juricek L et al. (2017) AhR-deficiency as a cause of demyelinating disease and inflammation. Sci. Rep 7, 9794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ramos-Garcia NA et al. (2020) Aryl hydrocarbon receptor in post-mortem hippocampus and in serum from young, elder, and Alzheimer’s patients. Int. J. Mol. Sci 21, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.International Multiple Sclerosis Genetics Consortium (2019) Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 365, eaav7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rothhammer V and Quintana FJ (2016) Environmental control of autoimmune inflammation in the central nervous system. Curr. Opin. Immunol 43, 46–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wheeler MA et al. (2019) Environmental control of astrocyte pathogenic activities in CNS inflammation. Cell 176, 581–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Graves JS et al. (2017) Maternal and perinatal exposures are associated with risk for pediatric-onset multiple sclerosis. Pediatrics 139, e20162838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Smith HL et al. (2020) Astrocyte unfolded protein response induces a specific reactivity state that causes non-cellautonomous neuronal degeneration. Neuron 105, 855–866.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Diaz-Castro B et al. (2019) Astrocyte molecular signatures in Huntington’s disease. Sci. Transl. Med 11, eaaw8546. [DOI] [PubMed] [Google Scholar]
- 97.Mathys H et al. (2019) Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhou Y et al. (2020) Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med 26, 131–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wheeler MA et al. (2020) MAFG-driven astrocytes promote CNS inflammation. Nature 578, 593–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ayata P et al. (2018) Epigenetic regulation of brain region-specific microglia clearance activity. Nat. Neurosci 21, 1049–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wendeln AC et al. (2018) Innate immune memory in the brain shapes neurological disease hallmarks. Nature 556, 332–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Codarri L et al. (2011) RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat. Immunol 12, 560–567 [DOI] [PubMed] [Google Scholar]
- 103.Geyer S et al. (2019) Immunity against bacterial infection of the central nervous system: an astrocyte perspective. Front. Mol. Neurosci 12, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Garcia Samartino C et al. (2010) Brucella abortus induces the secretion of proinflammatory mediators from glial cells leading to astrocyte apoptosis. Am. J. Pathol 176, 1323–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Patel H et al. (2015) Long-term sequelae of West Nile virus-related illness: a systematic review. Lancet Infect. Dis 15, 951–959 [DOI] [PubMed] [Google Scholar]
- 106.Garber C et al. (2018) Astrocytes decrease adult neurogenesis during virus-induced memory dysfunction via IL-1. Nat. Immunol 19, 151–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Garber C et al. (2019) T cells promote microglia-mediated synaptic elimination and cognitive dysfunction during recovery from neuropathogenic flaviviruses. Nat. Neurosci 22, 1276–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vasek MJ et al. (2016) A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 534, 538–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.França GVA et al. (2016) Congenital Zika virus syndrome in Brazil: a case series of the first 1501 livebirths with complete investigation. Lancet 388, 891–897 [DOI] [PubMed] [Google Scholar]
- 110.Cao-Lormeau VM et al. (2016) Guillain–Barré syndrome outbreak associated with Zika virus infection in French Polynesia: a case-control study. Lancet 387, 1531–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Figueiredo CP et al. (2019) Zika virus replicates in adult human brain tissue and impairs synapses and memory in mice. Nat. Commun 10, 3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Osso LA and Chan JR (2015) Astrocytes underlie neuroinflammatory memory impairment. Cell 163, 1574–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Simmons SB et al. (2013) Modeling the heterogeneity of multiple sclerosis in animals. Trends Immunol 34, 410–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Basso AS et al. (2008) Reversal of axonal loss and disability in a mouse model of progressive multiple sclerosis. J. Clin. Invest 118, 1532–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Schachtrup C et al. (2010) Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-β after vascular damage. J. Neurosci 30, 5843–5854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.John GR et al. (2003) Cytokines: powerful regulators of glial cell activation. Neuroscientist 9, 10–22 [DOI] [PubMed] [Google Scholar]
- 117.Franke H et al. (2012) Pathophysiology of astroglial purinergic signalling. Purinergic Signal 8, 629–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nahirnyj A et al. (2013) ROS detoxification and proinflammatory cytokines are linked by p38 MAPK signaling in a model of mature astrocyte activation. PLoS One 8, e83049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Brahmachari S et al. (2006) Induction of glial fibrillary acidic protein expression in astrocytes by nitric oxide. J. Neurosci 26, 4930–4939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Liu B et al. (2006) Epidermal growth factor receptor activation: an upstream signal for transition of quiescent astrocytes into reactive astrocytes after neural injury. J. Neurosci 26, 7532–7540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rabchevsky AG et al. (1998) A role for transforming growth factor alpha as an inducer of astrogliosis. J. Neurosci 18, 10541–10552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Labombarda F et al. (2011) Progesterone attenuates astro-and microgliosis and enhances oligodendrocyte differentiation following spinal cord injury. Exp. Neurol 231, 135–146 [DOI] [PubMed] [Google Scholar]
- 123.Barreto G et al. (2007) Testosterone decreases reactive astroglia and reactive microglia after brain injury in male rats: role of its metabolites, oestradiol and dihydrotestosterone. Eur. J. Neurosci 25, 3039–3046 [DOI] [PubMed] [Google Scholar]
- 124.Giraud SN et al. (2010) Estradiol inhibits ongoing autoimmune neuroinflammation and NFκB-dependent CCL2 expression in reactive astrocytes. Proc. Natl. Acad. Sci. U. S. A 107, 8416–8421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Akama KT et al. (1998) Amyloid beta-peptide stimulates nitric oxide production in astrocytes through an NFκB-dependent mechanism. Proc. Natl. Acad. Sci. U. S. A 95, 5795–5800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gorina R et al. (2011) Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFκB signaling, MAPK, and Jak1/Stat1 pathways. Glia 59, 242–255 [DOI] [PubMed] [Google Scholar]
- 127.De Miranda J et al. (2009) Astrocytes recognize intracellular polyinosinic-polycytidylic acid via MDA-5. FASEB J 23, 1064–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Butchi NB et al. (2010) Interactions between TLR7 and TLR9 agonists and receptors regulate innate immune responses by astrocytes and microglia. Glia 58, 650–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jeffries AM and Marriott I (2017) Human microglia and astrocytes express cGAS–STING viral sensing components. Neurosci. Lett 658, 53–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Burbach GJ et al. (2004) Induction of brain-derived neurotrophic factor in plaque-associated glial cells of aged APP23 transgenic mice. J. Neurosci 24, 2421–2430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Song I and Dityatev A (2018) Crosstalk between glia, extracellular matrix and neurons. Brain Res. Bull 136, 101–108 [DOI] [PubMed] [Google Scholar]
- 132.Jing R et al. (2007) Synemin is expressed in reactive astrocytes in neurotrauma and interacts differentially with vimentin and GFAP intermediate filament networks. J. Cell Sci 120, 1267–1277 [DOI] [PubMed] [Google Scholar]
- 133.Orellana JA et al. (2011) ATP and glutamate released via astroglial connexin 43 hemichannels mediate neuronal death through activation of pannexin 1 hemichannels. J. Neurochem 118, 826–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sheng WS et al. (2013) Reactive oxygen species from human astrocytes induced functional impairment and oxidative damage. Neurochem. Res 38, 2148–2159 [DOI] [PMC free article] [PubMed] [Google Scholar]