

Abstract
Human mesenchymal stromal cells (MSCs) are promising candidates for cell therapy due to their ease of isolation and expansion and their ability to secrete antiapoptotic, pro‐angiogenic, and immunomodulatory factors. Three‐dimensional (3D) aggregation “self‐activates” MSCs to augment their pro‐angiogenic and immunomodulatory potential, but the microenvironmental features and culture parameters that promote optimal MSC immunomodulatory function in 3D aggregates are poorly understood. Here, we generated MSC aggregates via three distinct methods and compared them with regard to their (a) aggregate structure and (b) immunomodulatory phenotype under resting conditions and in response to inflammatory stimulus. Methods associated with fast aggregation kinetics formed aggregates with higher cell packing density and reduced extracellular matrix (ECM) synthesis compared to those with slow aggregation kinetics. While all three methods of 3D aggregation enhanced MSC expression of immunomodulatory factors compared to two‐dimensional culture, different aggregation methods modulated cells' temporal expression of these factors. A Design of Experiments approach, in which aggregate size and aggregation kinetics were systematically covaried, identified a significant effect of both parameters on MSCs' ability to regulate immune cells. Compared to small aggregates formed with fast kinetics, large aggregates with slow assembly kinetics were more effective at T‐cell suppression and macrophage polarization toward anti‐inflammatory phenotypes. Thus, culture parameters including aggregation method, kinetics, and aggregate size influence both the structural properties of aggregates and their paracrine immunomodulatory function. These findings underscore the utility of engineering strategies to control properties of 3D MSC aggregates, which may identify new avenues for optimizing the immunomodulatory function of MSC‐based cell therapies.
Keywords: bone marrow stromal cells, cell therapy, cytokines, mesenchymal stromal cells
Mesenchymal stromal cell (MSC) aggregates display augmented immunomodulatory potential that is dependent on aggregate size and formation kinetics. Large, slow‐forming MSC aggregates outperform small, fast‐forming aggregates in their capacity to induce M2 macrophage polarization and suppress T‐cell proliferation. Aggregate size and aggregation kinetics also influence ECM synthesis and packing density, thereby linking aggregation‐dependent structural features to immunomodulatory function in MSC aggregates.
Significance statement.
Mesenchymal stromal cells (MSCs) have potential as cell therapies to treat immune and inflammatory diseases, based on their capacity to secrete trophic and immunomodulatory factors. A bioengineered platform enabling systematic control over aggregate size and aggregation kinetics was developed in this study. This platform was used, in combination with Design of Experiments (DOE) methodology, to evaluate the effects of aggregation parameters on MSC immunomodulatory function. This is the first study to identify aggregation kinetics as a critical factor affecting MSC‐mediated immunomodulation, and the strategy and findings described here may pave the way for more efficacious MSC therapies to treat immune.
1. INTRODUCTION
Human mesenchymal stromal cells (hMSCs) are of significant interest in regenerative medicine, as evidenced by their use in >600 clinical trials for indications, including stroke, osteoarthritis, diabetes, graft‐vs‐host disease (GvHD), and myocardial infarction. 1 , 2 A subset of therapeutic indications uses MSCs' multilineage differentiation potential for tissue engineering applications, where MSC differentiation and physical contribution to tissue repair are thought to be the primary mechanism of action. However, following in vitro and in vivo studies showing their unique capacity to suppress inflammation, 3 , 4 MSCs have been widely explored for their ability to enhance functional outcomes in immune‐ and inflammatory‐related diseases by regulating excessive responses from innate and adaptive immune cells, and by stimulating resident cells involved in tissue repair. 5 , 6 , 7 , 8 , 9 In these contexts, the mechanism‐of‐action of MSCs has been largely attributed to their secretion of trophic and immunomodulatory factors, such as vascular endothelial growth factor (VEGF), prostaglandin E2 (PGE2), tumor necrosis factor‐stimulated gene 6 (TSG‐6), and indoleamine dioxygenase (IDO). 10 , 11 For example, MSC‐secreted PGE2 inhibits natural killer cell cytotoxicity and cytokine production, 12 pushes resident pro‐inflammatory (M1‐like phenotype) macrophages toward an anti‐inflammatory (M2‐like) phenotype, 8 , 13 and inhibits T‐cell activation and proliferation. 14 Similarly, TSG‐6 inhibits macrophage intracellular signaling to limit their secretion of inflammatory mediators, 15 and IDO mediates MSC suppression of T‐cell proliferation. 16
While a multitude of animal studies have established the benefits of MSC paracrine activity in inflammation‐ and immune‐related diseases, clinical outcomes of MSC administration have been less clear. The safety of MSC administration in humans is well established, yet clear demonstrations of clinical efficacy in well‐designed trials have been limited. These discrepancies in treatment efficacy between animal and clinical trials may be ascribed to differences in dosing, as MSC dosing per weight is more than an order of magnitude higher in mouse models (typically 50 × 106 cells/kg) compared to what has been delivered intravenously in human trials (typically 2 × 106 cells/kg). 17 Irrespective of the dose dependency for a specific application, clinical experience to date indicates that the required MSC doses for beneficial effect in humans are considerable. In recent phase III clinical trials that met their primary endpoints, Remestemcel‐L (GvHD, Mesoblast) dosed MSCs at 2 million cells/kg twice a week for 4 weeks (NCT02336230), while Alofisel (Crohn's disease‐associated treatment‐refractory complex perianal fistulas, TiGenix) delivered 120 million cells in a single treatment (NCT01541579). However, the cost of a cell therapy product is proportional to the number of cells per dose. Therefore, from the standpoint of biomanufacturing feasibility and accessibility of the therapy to patients, there is a strong case for minimizing the number of cells per dose by increasing the effective potency of the cells delivered. 18
The goal of providing functionally potent MSCs for clinical use has led to the investigation of in vitro conditioning or “activation” regimens to enhance MSC paracrine function immediately before delivery. For instance, stimulation of MSCs with interferon‐gamma (IFN‐γ), an inflammatory cytokine that is produced by neutrophils and T cells and is abundant in injury sites, increases MSC immunosuppressive function in vitro 19 , 20 , 21 , 22 and appears to be required for maximal MSC effect in some animal models. 23 Interestingly, three‐dimensional (3D) aggregation of MSCs has been proposed to independently “self‐activate” MSCs' paracrine function, leading to enhanced secretion of molecules associated with immune cell regulation and tissue regeneration, compared to two‐dimensional (2D) monolayer MSCs 24 , 25 , 26 —even in the absence of inflammatory cytokine activation. Complementary advantages of MSC aggregates have been proposed as well. These include the ability of 3D aggregates to survive and persist longer in vivo compared to dissociated cells 27 , 28 , 29 , 30 and the potential for aggregation to promote more sustained enhancements to MSC function compared to cytokine activation, 31 for which the effects of activation are transient. While studies to date have consistently shown that assembly into 3D aggregates primes MSCs to upregulate their secretion of specific paracrine factors, a limited understanding of how the aggregate microenvironment influences the MSC secretome has hindered efforts to optimize MSC aggregates for enhanced immunomodulatory functions. In particular, hanging drops (HDs) and forced aggregation via centrifugation—two commonly published methods for generating size‐controlled cell aggregates—promote 3D aggregation of MSCs with relatively rapid kinetics (<12 hours) based on gravity‐driven sedimentation and centrifugation force, which are not readily tunable. Novel approaches that enable MSC aggregation to be systematically controlled may broaden the experimental space for identifying aggregation parameters that maximize MSC immunomodulatory function.
We previously described the development of a bioengineered platform encompassing patterned labile cell culture substrates that enable control over the size and aggregation kinetics of self‐assembled (SA) pluripotent stem cell aggregates. Using this platform, we showed that aggregation method and kinetics impact 3D aggregate structure and lineage bias during differentiation of human embryoid bodies. 32 In the current study, we used a similar approach to evaluate the effects of aggregation method on MSC immunomodulatory phenotype and function. We demonstrated that different kinetics of aggregation induced distinct temporal profiles of MSC paracrine factor secretion that correlated with changes in aggregate structural features including cell packing density, extracellular matrix (ECM) synthesis, and differential expression of mechanotransduction‐related genes. We then used a multifactorial Design of Experiments (DOE) approach to identify a combination of culture conditions that significantly enhanced the anti‐inflammatory and immunomodulatory potential of MSC aggregates. By tuning aggregate size and aggregation kinetics, we co‐optimized the paracrine immunomodulatory function of MSC aggregates to suppress T‐cell proliferation and polarize macrophages toward an M2 anti‐inflammatory phenotype.
2. MATERIALS AND METHODS
2.1. Cell culture
2.1.1. Mesenchymal stromal cells
hMSCs were purchased from Lonza and maintained in adherent culture in MSC growth medium (alpha‐MEM with 10% fetal bovine serum [FBS; Gibco] and 1x penicillin/streptomycin [Hyclone]). hMSCs were passaged at 70% confluence and used for all experiments between passages 4 and 7.
2.1.2. Macrophages
THP‐1 monocytes (ATCC) were thawed and maintained in RPMI1640 supplemented with 10% heat‐inactivated FBS and pen/strep. Cells were propagated by centrifugation and resuspension in fresh media every 3 days, maintained at a density of 105 to 106 cells/mL, and used for experiments between passages 6 and 20. A macrophage differentiation protocol was adapted from previously published work. 33 , 34 Briefly, THP‐1 cells were seeded into 24‐well plates at a density of 1.5 × 105 cells/well and differentiated into macrophages with 500 nM phorbol myristate acetate (PMA) for 72 hours followed by a 24 hours resting period in serum‐free RPMI. Macrophages were then polarized toward an M1‐like phenotype by treating with THP‐1 growth medium containing 20 ng/mL IFN‐γ for an additional 24 hours. Following an additional 24 hours rest period in serum‐free RPMI, macrophages were treated with MSC‐conditioned media (MSC‐CM) or control media for 36 hours prior to collecting macrophage‐conditioned media (Mϕ‐CM). Control media treatments included peripheral blood mononuclear cell (PBMC) media alone or containing 2 ng/mL PGE2 (Cayman Chemical).
2.1.3. PBMCs
Human peripheral blood mononuclear cells were purchased frozen (ZenBio) or isolated fresh by density gradient centrifugation from whole blood (Interstate Blood Bank) using SepMate‐50 (IVD) tubes with Lymphoprep (STEMCELL Technologies) following manufacturer's instructions. PBMCs were thawed and maintained in PBMC medium (RPMI1640 supplemented with 10% heat‐inactivated FBS, pen/strep, nonessential amino acids [Hyclone], and sodium pyruvate [Gibco]), and used within 96 hours following thaw.
2.2. Generation of SA MSC aggregates
Engineered labile substrates were fabricated by patterning self‐assembled monolayer (SAM) arrays presenting cycRGDfC peptide, following methods previously published by our group. 32 Briefly, alkanethiol solutions were prepared by combining 1 mM ethanolic solutions of carboxylic acid‐terminated hexa(ethylene glycol) undecanethiol (EG6COOH) and 11‐tri(ethylene glycol)‐undecane‐1‐thiol (EG3OH) at varying molar ratios based on the desired cycRGDfC peptide concentration. For example, for a desired 1% cycRGDfC substrate, 1 mol equivalent EG6COOH was combined with 99 mol equivalents EG3OH. SAM arrays were patterned onto gold‐coated glass slides (100 Å Au ‹111›, 20 Å Ti adhesion layer; Platypus Technologies) by filling microfabricated polydimethylsiloxane (PDMS) wells with alkanethiol solutions and incubating for 10 minutes for local SAM formation. After rinsing with deionized water, carboxylate groups were converted to active ester groups by incubating PDMS wells in a solution of 100 mM N‐hydroxysuccinimide and 250 mM n‐(3‐dimethylaminopropyl)‐N′‐ethylcarbodiimide hydrochloride for 15 minutes. After an additional rinse with deionized water, cycRGDfC peptide (Peptides International) was covalently coupled to patterned SAMs via incubation of the peptide solution (0.3 mM) in PDMS wells for 1 hour. After peptide coupling, PDMS wells were rinsed with deionized water, and regions surrounding array spots were backfilled with 0.1 mM EG3OH. Following removal of the PDMS wells from the gold slide, substrates were rinsed with 0.1% sodium dodecyl sulfate, deionized water, and EtOH, and dried with N2 gas. Substrates were incubated in 70% EtOH for 20 minutes and rinsed with sterile‐filtered deionized water before placing into cell culture media.
For single‐cell seeding of MSCs onto labile and nonlabile substrates, cells were washed with phosphate buffered saline and incubated with 0.05% trypsin/EDTA at 37°C for 5 minutes to singularize cells. Following singularization, trypsin was quenched and cells pelleted by centrifugation at 200g for 5 minutes. Cell pellets were resuspended in MSC growth medium before seeding at desired densities. After 1 hour incubation in a humidified incubator at 37°C and 5% CO2 to allow cell adhesion, seeded SAM arrays were immersed in basal medium to remove nonspecifically adhered cells. SAM arrays were then placed into new wells containing MSC growth medium and maintained in this medium unless otherwise indicated. High seeding densities were used to ensure confluence shortly after seeding. Unless otherwise stated, MSCs were seeded onto SAMs at a density of 9 × 104 cells/cm2.
As formation of compact and morphologically defined SA aggregates from 5% cycRGDfC substrates occurred within 72 hours after seeding, we denoted this time point “day 0.” This nomenclature is used throughout to describe 2D and 3D MSCs at 72 hours postseed. Unless otherwise stated, “SA MSC aggregates” referred to throughout Figures 1, 2, 3 were generated from 5% cycRGDfC substrates.
FIGURE 1.
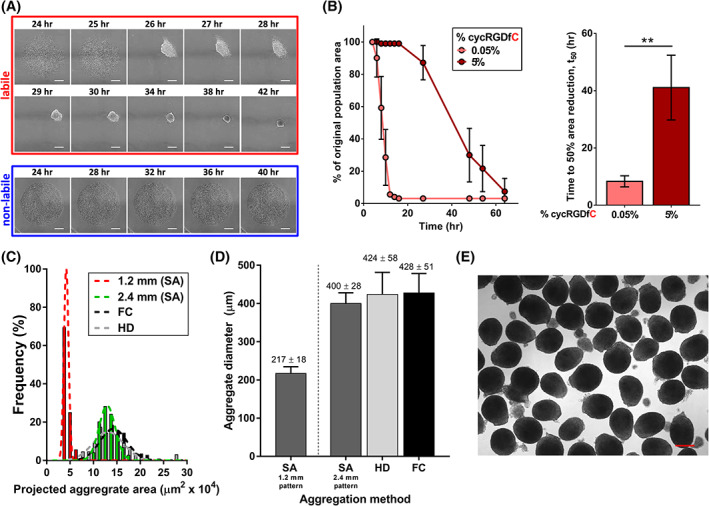
Generation of self‐assembled (SA) MSC aggregates with controllable size and kinetics from labile substrates. A, Time‐lapse images of MSCs on substrates presenting cyclic RGD (cycRGD) via a labile (top) or nonlabile (bottom) linkage. Labile substrates promote MSC self‐assembly into 3D cell aggregates, while nonlabile substrates promote continued MSC culture in 2D. Scale bars = 250 μm. B, (left) MSC aggregate self‐assembly kinetics (measured as percentage of original population area over time) as a function of initial peptide density on labile substrates. Error bars represent 95% CI (right). Mean t 50 values of self‐assembly for MSC aggregates generated from labile substrates of varying cycRGDfC density. “t 50” refers to the time required for the cell population to reach 50% of the original population area, previously established as a metric of aggregation kinetics. 32 Error bars represent 95% CI, **P < .01. C, Histogram of projected aggregate area for SA MSC aggregates generated from labile substrates (1.2 or 2.4 mm diameter circular patterns). Size distributions of HD and FC aggregates are overlaid for comparison (black and gray bars, respectively). D, Measurement of average size for day 0 MSC aggregates generated by SA from 1.2 and 2.4 mm diameter circular patterns, vs size‐matched HD and FC aggregates. E, Representative bright‐field image of day 0 SA MSC aggregates generated from 2.4 mm diameter circular patterns. Scale bar = 250 μm. FC, forced centrifugation; HD, hanging drop; MSC, mesenchymal stromal cell
FIGURE 2.
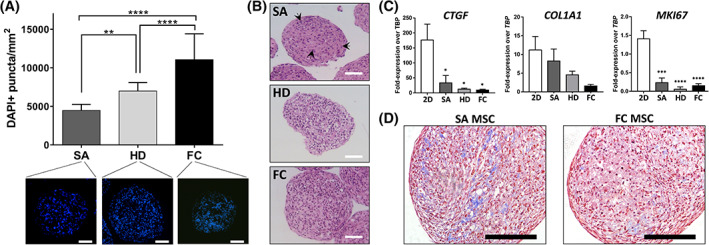
Method of cell aggregation influences cell density and extracellular matrix synthesis in MSC aggregates. A, Quantification of cell density in cryosectioned day 0 MSC aggregates formed by SA, HD, or FC. Size‐matched aggregates between all three methods (see Figure 1D) were used for analysis. Values represent the mean ± SD (**P < .01, ****P < .0001, one‐way analysis of variance with Tukey's post hoc test). Scale bars = 100 μm. B, Representative H&E stains of day 0 SA, HD, and FC aggregates showing differences in aggregate structure and nuclear shape based on aggregation method. Cell nuclei within SA aggregates demonstrated elongated morphology (black arrows), whereas nuclei in HD and FC aggregates were rounded and compact. Scale bars = 100 μm. C, Expression of extracellular matrix‐ and proliferation‐associated genes in day 0 MSC aggregates compared to 2D MSCs. Asterisks denote a significant difference relative to 2D MSCs (*P < .05, ***P < .001, ****P < .0001; one‐way analysis of variance with Tukey's post hoc test). D, Histological sections of day 10 SA and FC MSC aggregates, stained with Masson's Trichrome, showing differences in aggregate structure and collagen deposition between aggregation methods. Scale bars = 100 μm. FC, forced centrifugation; HD, hanging drop; MSC, mesenchymal stromal cell; SA, self‐assembled
FIGURE 3.
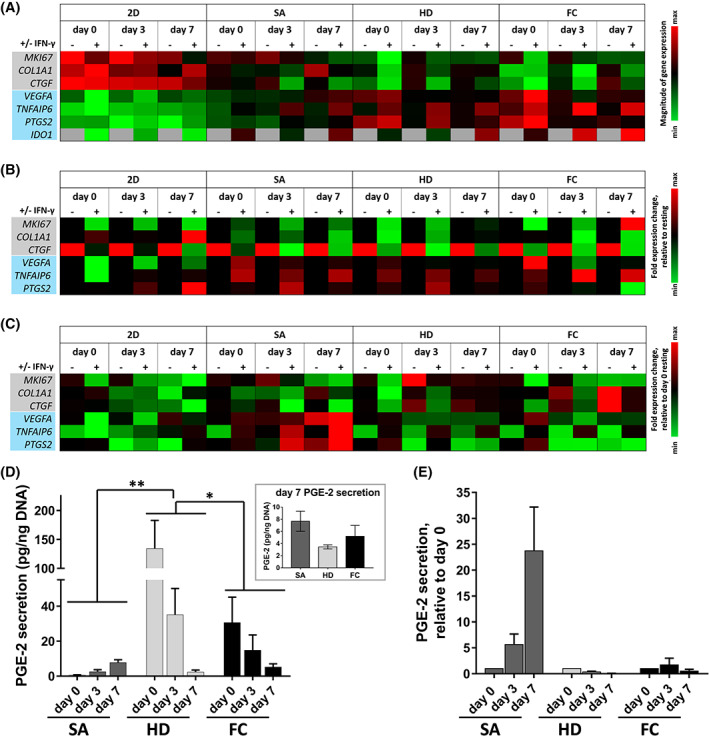
Culture format and aggregation method influences MSC immunomodulatory phenotype and temporal response to pro‐inflammatory environments. Gene expression profiles for MSCs cultured in 2D monolayer (2D) or in 3D aggregates (SA, HD, or FC aggregation methods), in the presence (+) or absence (−) of IFN‐γ. The assessed genes were associated with cell proliferation and extracellular matrix synthesis (gray), or pro‐angiogenic and immunomodulatory paracrine factors (blue). Gene expression heatmaps are displayed as (A) Expression relative to TBP, (B) Fold expression relative to resting (ie, without IFN‐γ), and (C) Fold expression relative to day 0 resting conditions. D, Time course of PGE2 production by IFN‐γ‐primed MSC aggregates formed via SA, HD, and FC approaches. PGE2 was measured from media conditioned for 24 hours following IFN‐γ priming. Asterisks indicate statistically significant difference between denoted aggregation methods (*P < .05, **P < .01; two‐way analysis of variance with Tukey's post hoc test). (inset) Comparison of PGE2 secretion by day 7 aggregates. E, Fold change in PGE2 production by IFN‐γ‐primed MSC aggregates over 7 days, relative to day 0. FC, forced centrifugation; HD, hanging drop; IFN‐γ, interferon‐gamma; MSC, mesenchymal stromal cell; PGE2, prostaglandin E2; SA, self‐assembled
2.3. Generation of HD and forced centrifugation aggregates
2.3.1. Hanging drop
MSCs were trypsinized as described above and resuspended at desired densities in MSC growth medium. Aggregates were formed using the hanging drop technique 35 with 20 000 cells per 25 μL droplet (size‐matched to 2.4 mm SA aggregates) unless otherwise stated.
2.3.2. Forced centrifugation
Agarose microwells were fabricated based on previously published methods. 32 MSCs were trypsinized as described above and seeded into agarose molds for a final seeding density of 25 000 cells per aggregate (size‐matched to 2.4 mm SA aggregates) unless stated otherwise. Plates were centrifuged at 300g for 5 minutes and incubated at 37°C, 5% CO2.
2.4. Quantification of aggregation kinetics and aggregate size distribution
All time‐lapse images were acquired using a Nikon Ti Eclipse inverted microscope (×10 PhL objective) equipped with a TIZ Tokai Hit incubated stage that was humidified and maintained at 37°C and 5% CO2. Image analysis was performed using NIS Elements as previously described. 32
2.5. Maintenance of MSCs for gene expression and immune cell assays
2D MSCs were maintained in tissue culture‐treated polystyrene plates, while 3D MSC aggregates were maintained in ultra‐low adhesion plates (Corning) in MSC growth medium unless otherwise specified. IFN‐γ stimulation of MSCs was performed for 24 hours in alpha‐MEM containing 0.5% FBS and 20 ng/mL human IFN‐γ (R&D Systems, 285‐IF‐100). For MSC‐CM experiments, treatment media were removed and replaced with PBMC media for conditioning for 24 hours. For media conditioning and PBMC coculture experiments, total MSC number was matched across all SA aggregates conditions based on scaling by the pattern area of labile substrates. In comparisons between SA and forced centrifugation (FC) aggregates, total MSC number was matched based on quantification of DNA per SA aggregate that was converted to cell number per aggregate using a MSC DNA standard curve; total cell number in FC conditions was then adjusted to match based on the cell number per aggregate and number of aggregates used in each assay.
2.5.1. Trilineage differentiation
2D MSCs (7.5 × 103 cells/cm2) or 3D MSC aggregates formed by SA, HD, or FC were plated on collagen I‐coated plates (Corning) in MSC growth medium. At 72 hours after plating, growth medium was exchanged with differentiation media. Media were changed every 3 to 4 days, and cells were fixed in 10% neutral buffered formalin for analysis after 21 days of differentiation.
Adipogenic medium: High‐glucose Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% FBS, 1 μM dexamethasone, 1 μg/mL insulin, 500 μM isobutylmethylxanthine, and pen/strep. Chondrogenic medium: High‐glucose DMEM supplemented with 10% FBS, ITS Premix, 0.9 mM sodium pyruvate, 50 μg/mL L‐ascorbic acid 2‐phosphate, 40 μg/mL L‐proline, 0.1 μM dexamethasone, and 10 ng/mL transforming growth factor‐β1, and pen/strep. Osteogenic medium: alpha‐MEM supplemented with 10% FBS, 0.1 μM dexamethasone, 10 mM β‐glycerophosphate, 50 μM L‐ascorbic acid 2‐phosphate, and pen/strep.
2.5.2. Histology
MSC aggregates were fixed in 10% neutral buffered formalin for 1 to 2 hours at room temperature, washed with PBS, and incubated in 10% sucrose solution overnight at 4°C before embedding in either Histogel for processing into paraffin blocks or O.C.T. compound at −80°C for cryosectioning.
Frozen samples were sectioned into 5 to 7 μm slices on SuperFrost slides using a Leica CM1900 cryostat. Slides were equilibrated to room temperature, fixed/permeabilized in ice‐cold acetone for 10 minutes, and blocked with 10% bovine serum albumin for 1 hour at room temperature. For quantification of cell density in aggregates, slides were stained with 4′,6‐diamidino‐2‐phenylindole (DAPI) and mounted in ProLong Gold Antifade Reagent before imaging on a Nikon Ti Eclipse microscope. Counts were analyzed from histological sections of n = 10 distinct aggregates per condition, from two independent experimental replicates.
Paraffin‐embedded aggregates were sectioned at a thickness of 5 μm, deparaffinized in xylene, and stained with hematoxylin and eosin.
2.5.3. Quantitative reverse transcription polymerase chain reaction (RT‐PCR)
2D MSCs or aggregates were washed with PBS and total RNA was isolated using the RNeasy mini kit (Qiagen) according to manufacturer's instructions. RNA was reverse transcribed into cDNA using the QuantiTect Reverse Transcription Kit or RT2 First Strand Synthesis Kit (Qiagen). For 384‐well format RT2 MSC Profiler Array, cDNA samples were run on a Roche LightCycler 4800 system according to the manufacturer's protocol. Results were analyzed using Qiagen's GeneGlobe Data Analysis Center.
2.5.4. Cytokine production analysis
Enzyme‐linked immunosorbent assays (ELISAs) for PGE‐2, IL‐10, and TNF‐α (R&D Systems) and cytokine analysis using Inflammatory Cytokine Arrays (Raybiotech) were performed following the manufacturer's recommendations. Where indicated, cytokine production was normalized to total DNA measured by Quant‐iT PicoGreen dsDNA Assay Kit (Thermo Fisher Scientific).
2.5.5. T‐cell proliferation assay
Unless otherwise stated, T‐cell proliferation assays were carried out in 24‐well tissue culture plates (2D adherent MSCs) or ultra‐low adhesion plates (3D suspension MSC aggregates) with a total media volume of 550 μL PBMC media per well, following protocols adapted from previously published work. 36 , 37 MSCs, PBMCs, and anti‐CD2/CD3/CD28 antibody‐functionalized beads (MSC Suppression Inspector; Miltenyi Biotec) were all prepared in PBMC culture media as described above. Indirect MSC and PBMC cocultures were performed in a Transwell setup (1.0 μm pore size), where PBMCs were cultivated in the top chamber for 96 hours in the presence of anti‐CD2/CD3/CD28 activating beads, separated from MSCs in the bottom chamber.
2.5.6. Flow cytometry
Unless otherwise stated, flow cytometry preparations were carried out in 96‐well V‐bottom polypropylene plates. Briefly, PBMCs were collected, centrifuged at 300g/5 minutes, washed with PBS to remove traces of serum, and incubated in suspension with Ghost Dye Red 780 viability dye (2 μL dye per mL PBS; Tonbo Biosciences) for 30 minutes on ice. Samples were centrifuged to pellet, decanted, and incubated with Human TruStain FcX (BioLegend; 5 μL/100 μL cell suspension in 2% FBS in PBS) for 10 minutes at room temperature to block FcR‐mediated antibody binding. After centrifugation and decanting, cells were stained for CD3 for 30 minutes at room temperature, fixed with 1% paraformaldehyde for 20 minutes at room temperature, permeabilized with ice‐cold 90% methanol for 15 minutes at 4°C, and stored at −20°C. Samples were washed twice with Flow Buffer 1 (PBS containing 0.5% BSA) to remove residual methanol and incubated for 1 hour at room temperature with primary antibody in Flow Buffer 2 (PBS containing 0.5% BSA and 0.1% Triton X‐100). Samples were washed with Flow Buffer 2, resuspended in Flow Buffer 1, and stored on ice prior to data collection. Data were collected on a MACSQUANT flow cytometer (Miltenyi Biotec) and analyzed using FlowJo software.
Primary antibodies and dilutions used: AlexaFluor488 mouse IgG1 anti‐human Ki67 primary conjugate, Clone B56 (BD Biosciences; 1:80), APC mouse IgG2a anti‐human CD3 primary conjugate, Clone HIT3a (BD Biosciences; 1:10).
2.5.7. Western blotting
Day 0 MSC aggregates were collected, washed with PBS, and resuspended in ice‐cold RIPA buffer containing 1X Halt Protease/Phosphatase Inhibitor Cocktail. Aggregates were homogenized in buffer using a polypropylene pestle, agitated for 15 minutes at 4°C and centrifugation at 12 000g for 15 minutes at 4°C. The supernatants from samples were collected and total protein was quantified by microBCA assay. Protein samples were combined with Laemmli buffer containing beta‐mercaptoethanol, denatured for 5 minutes at 100°C, loaded in 10% polyacrylamide gels and separated by sodium dodecyl‐sulfate polyacrylamide gel electrophoresis (SDS‐PAGE). Proteins were transferred to polyvinylidene difluoride (PVDF) membranes and incubated in 5% nonfat dry milk in TBST for 1 hour at room temp. Membranes were incubated in rabbit anti‐human phosphoYAP Ser127 primary antibody (Cell Signaling Technology, 4911S, 1:1000) in Antibody Diluent (Thermo Fisher, 00‐3118) overnight at 4°C, washed with Tris‐buffered saline with 0.1% Tween® 20 (TBST), and incubated in horseradish‐peroxidase (HRP)‐conjugated goat anti‐rabbit IgG secondary antibodies in blocking buffer (Abcam, 1:10 000) for 1 hour at RT. Membranes were washed with TBST and incubated with ECL™ Western Blotting substrate (Pierce) for 1 minute. Chemiluminescence was detected using a LAS4000 Mini imager and analyzed by densitometry using ImageJ's gel plug‐in. β‐actin was used as a load control.
2.5.8. DOE design and modeling
JMP software (SAS) was used to create a full‐factorial DOE design encompassing two factors (aggregate size and %cycRGD) at three levels. The DOE was carried out in triplicate and quadruplicate for T cell and macrophage assays, respectively. Based on the considerable variability in Mϕ IL‐10 and TNF‐α secretion that we observed between experimental replicates, normalized “scores” for immune cell assays were used as the response variables in the DOE analysis. Within a given experiment, scores from 0 to 1 were assigned to each experimental condition, corresponding to its performance in each immune cell assay. The lowest and highest conditions within each assay were assigned scores of 0 and 1, respectively, and the scores of the remaining conditions were calculated based on their distribution within this range. The DOE model was generated with least squares fitting of linear dependences for aggregate size and %cycRGD and included a first‐order interaction term (aggregate size*%cycRGD). The Factor Profiler in JMP was used to maximize desirability, which was defined as minimization of T‐cell proliferation (%Ki67+ CD3+ PBMCs) and maximization of macrophage polarization toward an M2‐like phenotype (maximize IL‐10, minimize TNF‐α secretion; equally weighted).
2.5.9. Figure generation and statistical analysis
With the exception of statistics related to the DOE model (ie, Figure 5), all statistical analyses were performed using GraphPad Prism. Unless otherwise indicated, gene expression data were analyzed by one‐way analysis of variance with multiple comparisons and Tukey's post hoc test. The method of statistical analysis and degree of significance for all other statistically significant comparisons is denoted in each figure/caption. The DOE models, graphs, and statistics were generated in JMP while all other graphs were generated in GraphPad Prism.
3. RESULTS
3.1. Labile substrates supported self‐assembly of MSC aggregates with tunable size and aggregation kinetics
We previously demonstrated that engineered substrates presenting patterns of the cyclized cell adhesion peptide RGD (cycRGDfC) via a labile bond could control pluripotent stem cell aggregation kinetics based on initial substrate cycRGDfC density. 32 Controllable aggregation from these substrates occurred via a mechanism whereby chemical lability of a thioester bond between the substrate and RGD moieties allowed for short‐term two‐dimensional cell adhesion, followed by subsequent self‐assembly of patterned cell collectives into three‐dimensional aggregates. In the current study, we verified that MSC self‐assembly (SA) into aggregates on these substrates was controllable in a similar manner, dependent on both lability of the substrate and cycRGDfC density. MSCs seeded on 0.05% cycRGDfC labile substrates formed aggregates within <16 hours (t 50 of self‐assembly = 8.3 ± 1.9 hours) while those on 5% cycRGDfC aggregated significantly more slowly (t 50 = 41.1 ± 11.3 hours), and cells failed to aggregate at all on nonlabile substrates, as expected (Figure 1A,B). The size of SA MSC aggregates depended on 2D pattern geometry, with SA aggregates from 1.2 and 2.4 mm diameter patterns generating aggregates of significantly different diameters (217 ± 18 μm and 400 ± 28 μm, respectively) (Figure 1C,D).
We directly compared MSC aggregates generated by our SA approach to those formed by two conventional methodologies—HD and forced aggregation (FC)—to determine characteristic aggregation kinetics and the degree of control over aggregate size distribution offered by each method. Consistent with prior studies, HD and FC MSCs aggregated on rapid time scales, with loose, clearly three‐dimensional structures formed by 6 hours after seeding, and compact spheroids arising by 24 hours (Figure S1). The size of aggregates generated by HD and FC could be varied by controlling cell number per droplet or per microwell, respectively (Figure S1). HD and FC aggregation methods demonstrated slightly broader distributions of MSC aggregate diameter (424 ± 58 μm and 428 ± 51 μm, respectively) compared to SA aggregates of similar size (Figure 1C‐E). We chose SA MSCs formed on 5% cycRGDfC substrates for further characterization, as this slowly aggregating condition provided the largest disparity in aggregation kinetics, compared to HD and FC methods.
3.2. Aggregation method affected structural properties and trilineage differentiation of MSC aggregates
The method used to generate MSC aggregates had a significant effect on 3D cell packing density. Analysis of DAPI‐stained sections of size‐matched aggregates revealed that FC and HD aggregates had higher numbers of nuclei per cross‐sectional area (11.06 ± 3.35 and 6.99 ± 1.11 × 103 nuclei/mm−2, respectively) compared to SA aggregates (4.48 ± 0.78 × 103 per mm2) (Figure 2A). Interestingly, aggregation method also appeared to affect MSC nuclear shape. Cell nuclei in HD and FC aggregates were smaller and more rounded in morphology, compared to nuclei within SA aggregates which were more elongated and associated with a spindle‐like cell morphology (Figure 2B). 3D culture and aggregation method also influenced MSC synthesis of ECM‐ and proliferation‐related genes. Day 0 aggregates, particularly those formed by HD and FC methods, exhibited significantly lower expression of CTGF, COL1A1, and MKI67 compared to 2D MSCs (Figure 2C). SA aggregates expressed these genes at higher levels than HD or FC aggregates, although these differences were not statistically significant. In accordance with the gene expression results, we also observed more collagen deposition in SA aggregates compared to FC aggregates (Figure 2D).
To investigate whether 3D aggregation affected trilineage differentiation potential, we plated aggregates or 2D MSCs on collagen‐coated dishes in the presence of appropriate induction media. Aggregates attached within 24 hours of plating and individual cells migrated out to varying degrees depending on the type of outgrowth and the media environment. All three methods of aggregation produced MSCs capable of differentiating toward adipogenic and chondrogenic fates. We observed high densities of Oil Red O‐positive cells in HD and FC aggregates, whereas intermediate and low densities were observed in SA aggregates and 2D MSCs, respectively (Figure S2A). 3D aggregates in chondrogenic medium stained positively for Alcian Blue, with the strongest staining in regions within or in close proximity to the original aggregate (Figure S2B). In contrast, all three aggregation methods exhibited poor osteogenic differentiation. Following 3 weeks of osteogenic induction, weak Alizarin Red staining was observed in plated SA aggregates, while no areas of positive staining were seen in HD or FC aggregates. 2D MSCs retained osteogenic differentiation potential as evidenced by strong Alizarin Red staining after the 3‐week differentiation (Figure S2C).
3.3. Culture format and aggregation method influenced MSC immunomodulatory phenotype and temporal response to pro‐inflammatory environments
Previous work indicated that exposure to pro‐inflammatory stimuli 20 , 38 , 39 , 40 (eg, IFN‐γ) and three‐dimensional culture of MSCs 24 , 25 , 41 , 42 can independently enhance aspects of MSC immunomodulatory phenotype. To better understand the influence of 3D culture geometry and aggregation method on the sustained immunomodulatory properties of MSCs, we performed gene expression analysis for IFN‐γ‐stimulated vs unstimulated (resting) MSCs cultured in 2D monolayers or 3D cell aggregates, over a 7‐day time course. Irrespective of the method used to aggregate the cells and the presence of IFN‐γ, 3D MSC aggregates demonstrated higher expression of VEGFA, a gene associated with angiogenesis and wound healing, as well as increased expression of genes associated with paracrine induction of tolerogenic immune cell phenotypes (TNFAIP6, PTGS2, and IDO1), relative to the corresponding 2D MSC conditions across all time points assessed (Figure 3A). On the other hand, 2D MSCs exhibited elevated expression of genes associated with cell proliferation (MKI67) and ECM synthesis (COL1A1, CTGF), relative to 3D MSC aggregates.
Treatment with IFN‐γ had a significant influence on MSC expression of a subset of immunomodulatory genes. Specifically, IDO1 expression was undetectable in resting MSCs in 2D and 3D culture but was induced at high levels upon IFN‐γ stimulation (Figure 3A). Expression of TNFAIP6 was also significantly affected by IFN‐γ treatment; at day 0, IFN‐γ induced a 2.4‐fold downregulation of TNFAIP6 in 2D MSCs but upregulated TNFAIP6 expression >3.1‐fold in all three 3D aggregate conditions (Figures 3B and S3). This IFN‐γ‐dependent inducibility of TNFAIP6 expression persisted at days 3 and 7 in all 3D MSC aggregates irrespective of aggregation method (3.0‐ to 7.1‐fold upregulation relative to resting MSC), while IFN‐γ had only a marginal effect on expression in 2D MSCs (<1.6‐fold upregulation; Figure S3). Additionally, IFN‐γ treatment decreased the expression of MKI67, COL1A1, and CTGF in MSCs across all time points and culture formats, with the exception of COL1A1 which was marginally upregulated in 2D MSCs in response to IFN‐γ (Figure 3B).
Immunomodulatory gene expression profiles were dynamic over the course of 7 days in culture, and MSC secretion of paracrine factors depended on the method of 3D aggregation. HD and FC aggregates expressed high levels of VEGFA, TNFAIP6, and PTGS2 at day 0 but significantly downregulated their expression at days 3 and 7, whereas expression of these genes gradually increased over time in SA aggregates (Figures 3C and S3). In agreement with trends observed in the gene expression profiles, production of PGE2 by SA aggregates steadily increased over time (0.50 ± 0.49 pg PGE2/ng DNA at day 0, vs 7.68 ± 2.85 pg PGE2/ng at day 7). This trend of increasing PGE2 secretion over time was in stark contrast to PGE2 production by HD and FC aggregates, which dramatically decreased between day 0 (134.30 ± 83.81 pg PGE2/ng DNA for HD, 30.56 ± 25.26 pg PGE2/ng DNA for FC) and day 7 (3.46 ± 2.00 and 5.20 ± 3.11 pg PGE2/ng DNA, respectively) (Figure 3D). Strikingly, the amount of PGE2 secreted by MSCs in 2D monolayer was 40‐fold lower than that of the lowest 3D aggregate condition (Figure S4). Taken together, culturing MSCs in 3D aggregates enhanced their secretion of paracrine factors involved in immune cell modulation, under both resting conditions and following inflammatory stimulus. Furthermore, 3D aggregation not only influenced MSC response to pro‐inflammatory environmental cues but also affected temporal regulation of MSC immunomodulatory phenotype depending on the method of aggregation used.
3.4. Multifactorial DOE identified the size and aggregation kinetics of MSC aggregates as parameters that influence immunomodulatory function
Prior studies have implicated aggregate size 26 , 43 , 44 , 45 , 46 , 47 and kinetics of aggregation 32 , 48 as factors that independently instructed cell phenotype in 3D. We hypothesized that each of these factors may have both distinct and interacting effects on MSC capacity to regulate specific immune cell types, and that a combination of these factors could be identified to promote the overall anti‐inflammatory and immune‐suppressive functions of MSCs. To address this hypothesis, we used a DOE approach to systematically investigate the influence of aggregate size and aggregation kinetics on the immunomodulatory function of IFN‐γ‐treated SA MSC aggregates. As before, size of SA aggregates was controlled by varying pattern size of labile substrates (Figure 1C,D), while aggregation kinetics were controlled by varying %cycRGD, where higher %cycRGD corresponded to slower aggregation rates (Figure 1B).
We constructed a two‐factor, three‐level full factorial design that resulted in nine unique conditions that we tested for their capacity to suppress T‐cell proliferation (ie, minimize %Ki67+ CD3+ PBMCs) and polarize macrophages toward an M2‐like phenotype (ie, maximize IL‐10 and minimize TNF‐α secretion by Mϕ) (Figures 4, S5, and S6). Multivariate analysis of immune cell responses as a function of MSC aggregation parameters identified independent and interacting effects of aggregate size and aggregation kinetics on MSC immunomodulatory function (Figure 5). Specifically, the model identified a negative linear relationship between %cycRGD and T‐cell proliferation (P < .0001, Figure 5A) and a positive linear relationship between %cycRGD and Mϕ secretion of IL‐10 (P < .05, Figure 5B), suggesting that slower aggregation kinetics promote both T‐cell suppression and M2 Mϕ polarization by SA MSC aggregates. Aggregate size alone had no significant effect on T‐cell suppression (P = .14) or Mϕ IL‐10 secretion (P = .23) (Figure 5A,B). In contrast, the model identified a negative linear relationship between aggregate size and Mϕ TNF‐α secretion (P < .01), but no significant effect of %cycRGD on TNF‐α secretion (Figure 5C).
FIGURE 4.
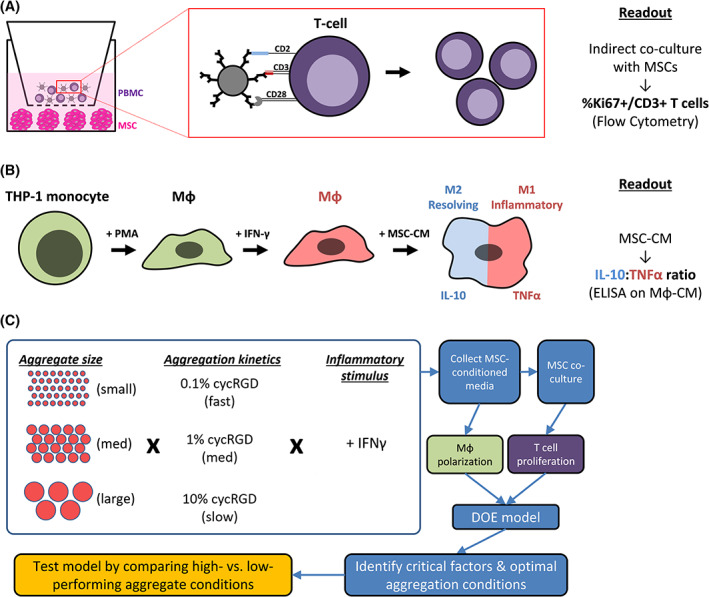
Experimental approach to assessing the role of aggregate size and aggregation kinetics on MSC immunomodulatory function. A, Schematic representation of T‐cell proliferation assay. MSC aggregates are cultured on a low‐adhesion surface in the bottom chamber of a Transwell setup. Peripheral blood mononuclear cells (PBMCs) are added to the top chamber, separated by a porous membrane (1.0 μm pore size), and cocultured with MSC aggregates for 96 hours in the presence of anti‐CD2/CD3/CD28 activating beads. MSCs are cultured in the lower chamber and produce paracrine factors that suppress T‐cell proliferation. B, Schematic representation of macrophage polarization. THP‐1 monocytes are differentiated into macrophages (Mϕ) by PMA treatment and further polarized toward M1 Mϕ phenotype with IFN‐γ treatment. MSC immunomodulatory function in this assay is determined by the ability of MSC‐CM to polarize Mϕ toward M2 (high IL‐10‐, low TNF‐α‐secreting) phenotype. C, Schematic of Design of Experiments (DOE) approach and tested conditions. Varying SA aggregate sizes (1.2, 2.4, and 5.0 mm patterns) and aggregation kinetics (0.1%, 1%, and 10% cycRGDfC) were tested in a two‐factor, three‐level full factorial design in which all conditions included IFN‐γ stimulation of MSCs. A subset of additional experiments included a comparison of resting and IFN‐γ‐treated MSC aggregates. IFN‐γ, interferon‐gamma; Mϕ‐CM, macrophage‐conditioned media; MSC‐CM, mesenchymal stromal cell‐conditioned media; PMA, phorbol myristate acetate; SA, self‐assembled
FIGURE 5.
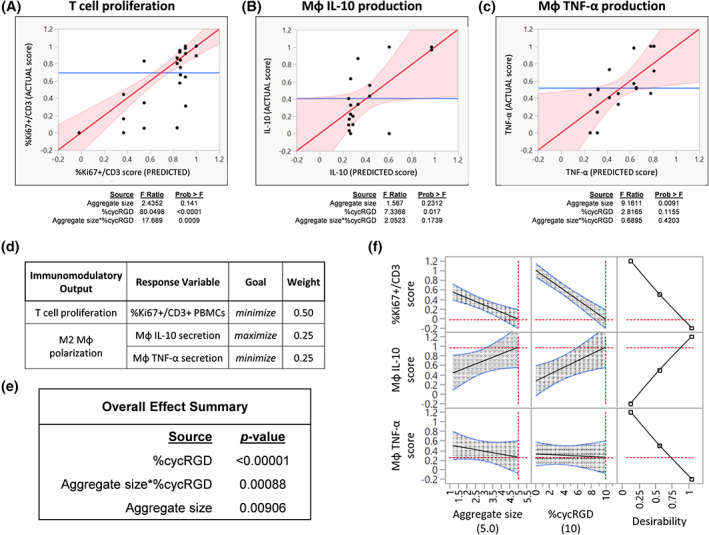
Results of DOE‐generated models to optimize MSC aggregate immunomodulatory function. Model predictions for, A, T‐cell suppression, B, Mϕ IL‐10 production, and, C, Mϕ TNF‐α production. The fits of the models were visualized by plotting predicted vs actual scores in immune cell assays upon quantifying (i) %Ki67+ fraction of CD3+ PBMCs following indirect coculture with MSC aggregates and (ii) Mϕ secretion of IL‐10 and TNF‐α (ELISA) following culture in media conditioned by MSC aggregates. Results of effect tests for each response variable are shown below the model fit. D, Overall desirability in the DOE model was defined as minimization of T‐cell proliferation (%Ki67+ CD3+ peripheral blood mononuclear cells) and maximization of macrophage polarization toward an M2‐like phenotype (maximize IL‐10, minimize TNF‐α secretion). E, Overall effect summary for factors influencing desirability in the model. F, Maximizing desirability within the experimental space identifies optimal aggregate size (5.0 mm) and aggregation kinetics (10% cycRGD) for T‐cell suppression and macrophage polarization. DOE, Design of Experiments; MSC, mesenchymal stromal cell
3.5. Validation of DOE‐identified conditions to optimize T‐cell suppression and macrophage polarization by MSC aggregates
We tested the DOE model by generating SA MSC aggregates from two additional MSC donors (Table 1), specifically comparing aggregates that were predicted by the DOE model to perform well (5.0 mm pattern/10% cycRGD, ie, “large/slow”) vs poorly (1.2 mm pattern/0.1% cycRGD, ie, “small/fast”) in assays of immunomodulatory function. To determine whether the effects identified by the model were specific to MSC aggregates generated by the SA approach, we also compared FC MSC aggregates of different sizes (“small” = ~800 cells per aggregate; “large” = ~13 500 cells per aggregate). In T‐cell suppression assays, DOE‐optimized SA MSC aggregates outperformed all other aggregate conditions tested for both MSC donors. Large/slow‐assembling SA aggregates from donors 2 and 3 reduced the percentage of proliferating T cells to 76.0% and 69.5%, respectively, while small/fast‐assembling SA aggregates resulted in 90.2% and 84.7% proliferating T cells following coculture (Figure 6A[i]). DOE‐optimized SA MSC aggregates also exhibited the highest capacity for M2 macrophage polarization. Secreted IL‐10:TNF‐α ratio was higher for macrophages cultured in conditioned media from large/slow‐assembling SA aggregates (2.73 ± 0.10 and 10.31 ± 5.29) compared to those in media conditioned by small/fast‐assembling SA aggregates (1.05 ± 0.02 and 2.48 ± 0.70) from donors 2 and 3, respectively (Figure 6B,D).
TABLE 1.
Donor information for MSCs used in the study
| ID | Source | Supplier | Sex | Donor age | Passage # at use |
|---|---|---|---|---|---|
| Donor 1 | Bone marrow | Lonza (commercial) | F | 26 | 5‐7 |
| Donor 2 | Bone marrow | Wan‐Ju Li lab (UW‐Madison) | F | 25 | 5‐7 |
| Donor 3 | Bone marrow | Lonza (commercial) | F | 21 | 5‐7 |
Note: Donor 1 MSCs were used for all experiments characterizing differences between 2D monolayer and 3D MSC aggregates, as well as for experiments informing the DOE model. Validation of the DOE model was performed using MSCs from donors 2 and 3.
Abbreviations: DOE, Design of Experiments; MSCs, mesenchymal stromal cells.
FIGURE 6.
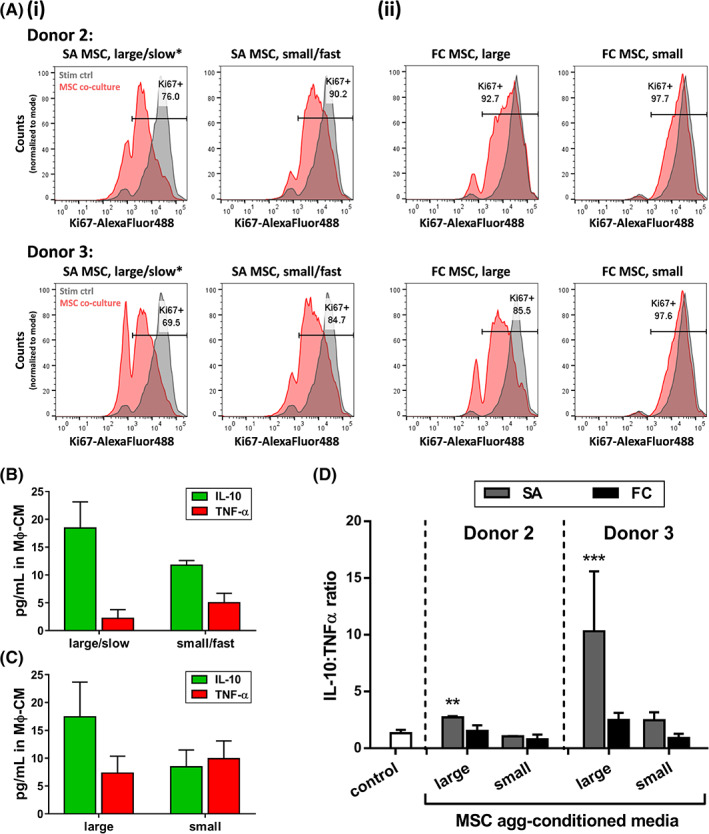
Validation of DOE model in two different MSC donors. Immunomodulatory function of DOE‐optimized SA MSC aggregates was tested in two additional MSC donors. A, T‐cell suppression: Flow cytometry histograms of %Ki67+ CD3 T cells following indirect coculture with (i) SA MSC aggregates predicted by DOE to perform well (large/slow*) or poorly (small/fast), and (ii) large or small FC MSC aggregates, from two additional MSC donors. Gray histogram corresponding to stimulated PBMC control is shown in all plots for comparison. y‐axis displays cell counts, normalized to mode. At least 8000 CD3+ cells were counted per experimental condition. B,C, Macrophage polarization: Representative IL‐10 and TNF‐α concentrations measured in Mϕ‐CM from macrophages treated with MSC‐CM from (B) SA MSC and (C) FC MSC aggregates. Data shown for MSC donor 3. D, IL‐10:TNF‐α ratio in Mϕ‐CM from macrophages treated with media conditioned by MSC aggregates described in (A). Asterisks denote statistically significant differences relative to control (no treatment). **P < .01, ***P < .001; one‐way analysis of variance with multiple comparisons. DOE, Design of Experiments; FC, forced centrifugation; Mϕ‐CM, macrophage‐conditioned media; MSC, mesenchymal stromal cell; MSC‐CM, mesenchymal stromal cell‐conditioned media; SA, self‐assembled
Although FC aggregates generally performed worse than SA aggregates in immune cell assays, we nevertheless still observed a positive effect of increasing aggregate size in the FC conditions. Coculture with large FC aggregates resulted in 92.7% and 85.5% proliferating cells (donors 2 and 3), while small FC aggregates had only marginal effects on T‐cell proliferation (97.7% and 97.6%, vs 95.8% Ki67+ in controls without MSCs) (Figure 6A[ii]). Similarly, media conditioned by large FC aggregates induced a higher macrophage IL‐10:TNF‐α ratio compared to that of small aggregates (1.55 ± 0.49 vs 0.79 ± 0.42, 2.48 ± 0.65 vs 0.91 ± 0.37 for donors 2 and 3, respectively) (Figure 6C,D).
4. DISCUSSION
Recent efforts have demonstrated the utility of biomaterials to “engineer” the MSC aggregate microenvironment by incorporating loaded microparticles that sustain release of MSC‐activating cytokines within the aggregate interior. 49 Such strategies show promise for controlling the soluble environment of MSCs but have yet to be matched with approaches to systematically control the aggregation process (eg, aggregation kinetics), which has been shown to influence aggregate structural characteristics and resulting cell phenotype. 48 , 50 In this study, we sought to characterize the influence of different 3D aggregation methods on the structure and function of MSC aggregates. We previously reported that the method and kinetics of aggregation had striking effects on cell density, porosity, and growth factor signaling in human embryoid bodies, which influenced loss of pluripotency and lineage bias during their spontaneous differentiation. 32 Thus, we hypothesized here that the same parameters could affect the structure and immunomodulatory function of MSC aggregates.
We first characterized the structure of MSC aggregates generated via three approaches associated with distinct aggregation kinetics, where aggregates were matched in approximate diameter across conditions. While the size of cell aggregates is commonly reported as a basis for comparison across studies, recent reports suggest that this metric alone is a poor indicator of phenotypic equivalence—even within aggregates generated from the same cell line/source—without also accounting for processing parameters such as the method of aggregation. 32 , 48 , 50 , 51 Indeed, in comparisons of size‐matched SA, HD, and FC aggregates, we observed differences in cell packing density, nuclear morphology, and ECM synthesis within MSC aggregates based on the aggregation method used. Cells within SA aggregates were present at a significantly lower density (Figure 2A), had more elongated nuclear morphologies (Figure 2B), and deposited greater quantities of collagen (Figure 2D) compared to cells from HD or FC aggregates. These marked differences in cell packing density and cell morphologies in SA vs HD and FC aggregates led us to investigate whether genes associated with known cell mechanosensing pathways, specifically yes‐associated protein (YAP)/transcriptional coactivator with PDZ‐binding motif (TAZ) signaling, were affected by the method of aggregation.
YAP and its homolog, TAZ, have been implicated as key effectors of the Hippo pathway in the regulation of cell‐ECM interactions. YAP/TAZ are involved in the mechanotransduction of biophysical cues such as cell shape, 52 substrate stiffness, 53 , 54 and cell density 55 in 2D culture in vitro, and in organ size control 56 and fibrosis 57 in vivo. In 2D MSCs, elevated YAP activity—manifested by high levels of nuclear YAP—has been associated with low cell density, 58 spread cell morphologies, 59 production of type I collagen, 60 and increased proliferation. 58 , 61 On the other hand, mechanotransduction of high cell density and rounded cell morphologies is thought to occur through decreases in YAP activity, which are mediated by phosphorylation of YAP that marks it for cytoplasmic retention, ubiquitination, and proteosomal degradation. 52 , 62 We observed that while 3D aggregation markedly downregulated expression of YAP target genes (CTGF, CYR61) and genes associated with proliferation (MKI67) and ECM synthesis (COL1A1) relative to 2D conditions, day 0 SA aggregates still expressed all three of these genes—as well as collagen I protein—at higher levels compared to HD or FC aggregates (Figures 2C,D and S7). Furthermore, Western blots for phosphoYAP revealed higher YAP phosphorylation in FC aggregates compared to SA aggregates, indicative of higher YAP activity in SA conditions (Figure S8). While the current understanding of MSC mechanosensing in 3D environments is limited, our data are line with general observations from 2D culture that environments of low cell density—which tend to coincide with decreased cell‐cell contact, increased cell area/spreading, and increased actin cytoskeletal tension—are associated with increased YAP/TAZ nuclear localization and, in turn, correlate with increased cell proliferation, synthesis of collagen I, and other hallmarks of fibrosis in cell types of mesenchymal character. 57 These results support the hypothesis that differences in aggregate structure as a function of aggregation method may be interpreted on the cellular level through a YAP‐mediated pathway.
Our observations of trilineage differentiation capacity for 3D MSC aggregates further supported the possibility of differential YAP involvement. In prior studies investigating MSCs in simplified 2D culture models, differential YAP signaling in MSCs instructed trilineage differentiation bias; RhoA‐mediated YAP activation was required for osteogenic differentiation while YAP inhibition led to increased adipogenic differentiation at the expense of osteogenic fates. In our study, although aggregates generated by all three methods demonstrated poor osteogenic differentiation, SA aggregates exhibited stronger Alizarin Red staining (Figure S2C), a lower density of Oil Red O‐positive clusters (Figure S2A), and higher RHOA expression (Figure S7) compared to HD and FC aggregates, mirroring the reported RhoA‐mediated effects of YAP activity on lineage bias during MSC differentiation. 63 While it remains unclear to what degree these changes in YAP‐associated genes are elicited by differences in aggregation kinetics, our data indicate that faster rates of SA aggregation further downregulate expression of CTGF, COL1A1, MKI67 (Figure S9), indicating that the described effects are not simply artifacts of particular aggregation methods. Together, these results support a potential role for YAP signaling in 3D MSC aggregates and suggest that differences in aggregation kinetics may be linked to phenotypic changes via mechanosensitive signaling pathways. Deeper investigation around MSC mechanosensing in 3D is required to understand the relationships between cell packing density, ECM synthesis, and therapeutic function of MSC aggregates.
Based on clear structural differences between aggregates generated via SA vs HD and FC methods, we were interested in whether these differences correlated with distinct changes in MSC production of trophic and immunomodulatory paracrine factors. We chose to additionally compare effects of treatment with the pro‐inflammatory cytokine IFN‐γ on 2D vs 3D MSCs. While IFN‐γ treatment has been widely proposed as a means of priming MSCs' immunomodulatory functions, 64 its effects have not been thoroughly explored in MSC aggregates, partly because 3D aggregation alone was proposed to “self‐activate” MSC immunomodulatory potential even in the absence of exogenous inflammatory signals. 31 , 41 Thus, we were also interested in whether the method of aggregation influenced responsiveness to IFN‐γ, or if 3D “self‐activation” alone was an adequate substitute for cytokine‐based priming of MSCs.
We evaluated paracrine factors previously reported to be strongly induced by 3D aggregation of MSCs (VEGF, 28 , 65 PGE2, 66 , 67 TSG‐6 24 , 41 ). VEGF, a potent pro‐angiogenic factor, was chosen based on its known roles in tissue repair 68 and suggested involvement in M2 macrophage polarization. 69 PGE2 and the enzyme responsible for its synthesis, COX‐2 (PTGS2), have been widely implicated in MSCs' capacity to attenuate pro‐inflammatory M1 macrophage phenotypes 13 and suppress other inflammatory immune cells during injury. 14 TSG‐6 (TNFAIP6) has been shown to reproduce many of the benefits of MSC administration in animal models of corneal inflammation, 70 peritonitis, 71 and myocardial infarction. 72 We also assessed expression of IDO, a molecule proposed to be a major regulator of MSCs' immunosuppressive effects on T cells. 73 Profiling genes associated with these factors revealed stark differences in expression between 2D and 3D MSCs, irrespective of aggregation method (Figure 3A). Under both resting and IFN‐γ‐treated conditions, expression of VEGFA, PTGS2, and TNFAIP6 was markedly upregulated at early time points in SA, HD, and FC aggregates relative to their 2D counterpart (Figure S3). These differences were reflected by measurements of secreted factors in the media, where the amount of PGE2 secreted by aggregates was at least 40‐fold higher than in 2D MSCs (Figure S4).
Notably, day 0 in our studies—72 hours following initial seeding—corresponds to a time point around which HD and FC aggregates are reported to undergo a “compaction” process. MSC aggregate compaction is associated with changes in expression of integrins and cadherins, alterations in cytoskeletal arrangement, and increased activation of genes for cytokines and immune mediators. 24 , 41 , 67 , 74 , 75 Tsai et al demonstrated that robust compaction was associated with upregulation of caspase 3/7 signaling, and that inhibition via the pan‐caspase inhibitor Q‐VD‐OPh not only reduced compaction but also knocked down functional enhancements (CXCR‐4, PGE2 expression) in MSC aggregates. Thus, we surmised that the time of compaction might represent a “burst” in the expression of many genes previously reported to be induced by 3D aggregation of MSCs. This premise would suggest that aggregates forming via different kinetics exhibit distinct temporal profiles of immunomodulatory factor production, depending on the timing and duration of the compaction process. By carrying cultures out to day 7, we identified highly dynamic expression of key genes in HD and FC aggregates. In particular, expression of both VEGFA and PTGS2—as well as secreted PGE2—was high at day 0 and decreased dramatically at days 3 and 7 in HD and FC aggregates, but markedly increased over time in SA aggregates (Figures 3C‐E and S3).
To our knowledge, there have been few reports, if any, that have characterized the duration over which 3D aggregation promotes sustained enhancements in MSC paracrine function. Our findings indicate that—for certain secreted factors like PGE2—conventional aggregation by HD or FC methods induces a short‐lived “burst” of enhanced paracrine function in MSCs, whereas different methods and kinetics of aggregation may change the magnitude and duration of those enhancements. Based on recent reports linking cell‐cell contact‐mediated signaling pathways to improved MSC immunomodulatory function, 76 , 77 future extensions of the current work should investigate whether structural remodeling and alterations in the cytoskeletal network—which are reported to be extensive in conventional MSC aggregates 74 , 78 , 79 —are directly linked to the duration of MSC “self‐activation.” Such studies may inspire development of new strategies to tailor MSC aggregate structure for prolonged paracrine functions in clinical applications.
Our comparison of resting and IFN‐γ‐treated MSCs revealed effects of culture geometry (2D vs 3D) on MSC response to inflammatory stimulus. IDO1—in line with prior studies—was not detectable under resting conditions and, following IFN‐γ treatment, was not significantly affected by 3D culture geometry (Figure S3). TNFAIP6, on the other hand, was not only more highly expressed but also displayed greater induction in response to IFN‐γ in 3D aggregates (Figure 3B). This is notable in the context of overwhelming evidence that immunomodulatory functions of MSCs are not constitutive but rather depend on appropriate licensing by the inflammatory microenvironment. 7 , 40 Indeed, responsiveness to inflammatory cues such as IFN‐γ, TNF‐α, and danger signals in the disease/injury environment are likely to be an important determinant of MSCs' beneficial effects in vivo. 23 It is worth considering that the response of a 3D aggregate to external stimuli is potentially modulated by autocrine signaling from factors produced within the aggregate. For example, multiple studies have found that FC and HD MSC aggregates produce detectable levels of cytokines commonly used to license MSC immunomodulatory function (IFN‐γ, TNF‐α, IL‐1α, IL‐1β), and chemical inhibition of endogenous IL‐1 and TNF‐α signaling in MSC aggregates was shown to abrogate production of PGE2 and other immune mediators. 26 , 41 Our own preliminary work indicates that production of endogenous IFN‐γ and TNF‐α depends on aggregation method (Figure S10); thus, future approaches to engineer the aggregate microenvironment may consider manipulating endogenous cytokine signaling to control MSC phenotype following in vivo delivery. Overall, 3D culture geometries appear to enhance specific MSC paracrine responses to inflammation, which could be an attractive feature for therapeutic applications.
Although our results established a correlation between MSC aggregation kinetics and distinct temporal profiles of paracrine factor production, our initial characterizations of MSC immunomodulatory phenotype were conducted using ~400 μm diameter aggregates across SA, HD, and FC methods (Figure 1D), which restricted our observations to a narrow experimental space. Thus, we next sought to explore whether differences in aggregation kinetics and aggregate size ultimately affected MSCs' capacity to regulate immune cell phenotype. Specifically, we used a DOE approach (Figure 4C) that would enable us to identify not only main effects of each parameter on a multivariate output but also interacting effects between parameters. To avoid potential confounding effects specific to particular aggregation methods, we varied both parameters in SA aggregates, for which we could control aggregation kinetics by varying cycRGDfC density (0.1%, 1%, 10%) and aggregate size by patterning different labile substrate geometries (1.2 mm, 2.4 mm, 5.0 mm circles).
We chose to focus on MSC effects on T cells and macrophages, based on the key roles and complex crosstalk between these cell types in resolving inflammation and determining clinical outcomes of injury and disease. 80 , 81 , 82 Specifically, we quantified MSC aggregates' performance in T‐cell suppression and M2 macrophage polarization assays (Figure 4A,B) to build the DOE model. The model identified significant individual effects of aggregation kinetics and aggregate size on MSC function, where slower aggregation kinetics and larger aggregates correlated with improved immune cell suppression/polarization (Figures 5 and S11). These effects were recapitulated in preliminary studies with MSCs from two additional donors, as large SA aggregates with slow aggregation kinetics were more effective at suppressing T cells and polarizing macrophages toward M2 phenotypes, compared to small SA aggregates that aggregated quickly (Figure 6). Similarly, large FC aggregates outperformed small FC aggregates, and although we could not tune aggregation kinetics via this method, FC aggregates displayed inferior immunomodulatory function compared to their SA counterparts overall (Figure 6). Taken together, the data from two additional donors and across two different aggregation methods corroborated the model prediction that MSC immunomodulatory function can be enhanced by increasing aggregate size and decreasing the rate of aggregation.
The effect of aggregation kinetics identified by the DOE model was somewhat surprising based on our initial findings that IDO1 and PGE2 production—implicated heavily in T‐cell suppression and M2 macrophage polarization, respectively—were either unchanged or significantly lower in SA aggregates compared to FC aggregates (Figures 3D and S3). However, it is likely that the overall effect of MSCs on immune cell function is not fully represented by the limited subset of paracrine mediators that we assessed. In addition, the initial characterization studies were conducted using different aggregation parameters (5% cycRGDfC, 2.4 mm SA aggregates) than the DOE‐selected conditions and represented measurements that were normalized to cell number. As we only controlled for input MSC number in the DOE studies, it is feasible that the identified effects are due to differences in MSC proliferation or viability/apoptosis between aggregate conditions that performed well vs poorly. While we did not explicitly explore the effects of aggregation method and aggregate size on MSC viability in the current study, it is established in the literature that hypoxic cores form in the center of large cell aggregates due to oxygen diffusion limitations at distances of greater than 200 μm, 83 and we have previously shown that aggregation method affects viability in other cell types. For example, embryoid bodies formed by FC showed rapid core necrosis within 4 days of aggregation while no necrosis was observed in size‐matched embryoid bodies formed by SA, which also displayed a lower cell packing density. 32 Reports from other groups have demonstrated increased apoptosis and elevated caspase 3/7 signaling in larger (60 000‐ to 100 000‐cell) MSC aggregates compared to smaller (10 000‐ to 15 000‐cell) aggregates. 24 , 79 Interestingly, recent studies have suggested that the oxygen tension at which necrosis occurs may vary between cell types, and that MSCs appear comparatively resistant to cell death under hypoxic conditions. 79 , 84 As the same pathways proposed to contribute to the paracrine immunomodulatory phenotype of MSCs have been linked to hypoxia, caspase signaling, and the NFκB stress response pathway, 41 , 74 , 85 , 86 , 87 activation of these pathways may transiently promote MSCs' functional effects but could also compromise cell viability and function long term. These possibilities, as well as the potential role of differences in structure and cytoarchitecture of optimal vs suboptimal MSC aggregates, should be tested in future experiments.
Taken together, our approach for using labile substrates to generate MSC aggregates offers possibilities for regulating aspects of cell aggregation that are not accessible by conventional methods. With HD and FC approaches, cell aggregation is a product of cell‐cell interactions that are driven by gravitational sedimentation or centrifugation, and aggregate size is entirely dependent on starting cell number. Thus, these and other conventional methods offer minimal control over aggregation kinetics and spatiotemporal dynamics of aggregation, and are also limited in their ability to dictate structural features of resulting aggregates such as cell packing density. We have shown here that labile substrates enabling control over spatiotemporal elements of aggregation can identify aggregation parameters with dramatic effects on cell therapeutic potential. Importantly, while we focused on varying only two parameters in the current study, labile substrates offer additional versatility inherent in their capacity to control the assembly of cell populations from 2D to 3D. One can envision using this technology to combine features such as spatial patterning, multiple cell types, and soluble factor presentation—all of which are easier to control in 2D culture—with tunable aggregation and control over the 3D cell aggregate microenvironment. Future work will focus on leveraging this broad parameter space offered by our approach to improve the rational design of therapeutic cell aggregates.
5. CONCLUSIONS
In the current study, we characterized MSC aggregates generated via three distinct methods, specifically assessing their typical aggregation kinetics, structure, and trophic and immunomodulatory phenotype. From this analysis, we identified general enhancements to MSC immunomodulatory phenotype resulting from 3D aggregation as well as a potential link between aggregation kinetics, resulting aggregate structure and cell density‐associated mechanotransduction pathways. Exploiting an engineered platform capable of controlling aggregate size and aggregation kinetics, we discovered significant effects of both variables on MSC immunomodulatory function and optimized these variables to generate large, slow‐assembling MSC aggregates with enhanced capacity to suppress T‐cell proliferation and polarize macrophages toward an anti‐inflammatory phenotype. Together, these findings underscore the utility of engineering approaches to control the formation of cell aggregates, and establish aggregation kinetics as a culture parameter that may be manipulated to instruct the structure, phenotype, and therapeutic function of 3D MSC aggregates.
CONFLICT OF INTEREST
A.W.X. declare employment and stock ownership in Catalent Pharma Solutions. W.L.M. is a Cofounder and Chief Scientific Officer at Stem Pharm, Inc. and Dianomi Therapeutics. The other authors declared no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
A.W.X.: conception and design, financial support, collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript; N.A.Z., B.Y.K.B.: collection and/or assembly of data, data analysis and interpretation; W.L.M.: conception and design, financial support, data analysis and interpretation, manuscript writing, final approval of manuscript.
Supporting information
Appendix S1. Supporting Information.
ACKNOWLEDGMENTS
The authors thank Sierra Raglin (UW Department of Surgery Histology Core) for assistance with preparation of histological samples and acknowledge support from staff and the use of equipment at the UW Carbone Cancer Center Flow Cytometry Laboratory (Support Grant P30 CA014520) at UW‐Madison. Chondrogenic induction media and MSCs (donor 2) were kindly provided by Dr Wan‐Ju Li and his laboratory (UW‐Madison, Department of Biomedical Engineering). This work was supported by funding from the National Institutes of Health (R01HL093282 to W.L.M.; Biotechnology Training Program NIGMS 5T32‐GM08349 to A.W.X.), the U.S. Environmental Protection Agency (STAR Grant 83573701 to W.L.M.), and the National Science Foundation (DGE‐1256259 to A.W.X.; DMR‐1306482 to W.L.M.).
Xie AW, Zacharias NA, Binder BYK, Murphy WL. Controlled aggregation enhances immunomodulatory potential of mesenchymal stromal cell aggregates. STEM CELLS Transl Med. 2021;10:1184–1201. 10.1002/sctm.19-0414
Funding information National Science Foundation, Grant/Award Numbers: DMR‐1306482, DGE‐1256259; U.S. Environmental Protection Agency, Grant/Award Number: 83573701; Biotechnology Training Program NIGMS, Grant/Award Number: 5T32‐GM08349; National Institutes of Health, Grant/Award Number: R01HL093282; UW‐Madison, Grant/Award Number: P30 CA014520
DATA AVAILABILITY STATEMENT
The data sets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
REFERENCES
- 1. Kim N, Cho S‐G. Clinical applications of mesenchymal stem cells. Korean J Intern Med. 2013;28:387‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Squillaro T, Peluso G, Galderisi U. Clinical trials with mesenchymal stem cells: an update. Cell Transplant. 2016;25:829‐848. [DOI] [PubMed] [Google Scholar]
- 3. Klyushnenkova E, Mosca JD, Zernetkina V, et al. T cell responses to allogeneic human mesenchymal stem cells: immunogenicity, tolerance, and suppression. J Biomed Sci. 2005;12:47‐57. [DOI] [PubMed] [Google Scholar]
- 4. Aggarwal S. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood. 2005;105:1815‐1822. [DOI] [PubMed] [Google Scholar]
- 5. English K. Mechanisms of mesenchymal stromal cell immunomodulation. Immunol Cell Biol. 2013;91:19‐26. [DOI] [PubMed] [Google Scholar]
- 6. Shi M, Liu Z‐W, Wang F‐S. Immunomodulatory properties and therapeutic application of mesenchymal stem cells. Clin Exp Immunol. 2011;164:1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bernardo ME, Fibbe WE. Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell. 2013;13:392‐402. [DOI] [PubMed] [Google Scholar]
- 8. Zheng G, Ge M, Qiu G, Shu Q, Xu J. Mesenchymal stromal cells affect disease outcomes via macrophage polarization. Stem Cells Int. 2015;2015:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Qi K, Li N, Zhang Z, Melino G. Tissue regeneration: the crosstalk between mesenchymal stem cells and immune response. Cell Immunol. 2018;326:86‐93. [DOI] [PubMed] [Google Scholar]
- 10. Uccelli A, de Rosbo NK. The immunomodulatory function of mesenchymal stem cells: mode of action and pathways. Ann NY Acad Sci. 2015;1351:114‐126. [DOI] [PubMed] [Google Scholar]
- 11. Madrigal M, Rao KS, Riordan NH. A review of therapeutic effects of mesenchymal stem cell secretions and induction of secretory modification by different culture methods. J Transl Med. 2014;12:260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Spaggiari GM, Capobianco A, Abdelrazik H, Becchetti F, Mingari MC, Moretta L. Mesenchymal stem cells inhibit natural killer‐cell proliferation, cytotoxicity, and cytokine production: role of indoleamine 2,3‐dioxygenase and prostaglandin E2. Blood. 2007;111:1327‐1333. [DOI] [PubMed] [Google Scholar]
- 13. Vasandan AB, Jahnavi S, Shashank C, Prasad P, Kumar A, Prasanna SJ. Human mesenchymal stem cells program macrophage plasticity by altering their metabolic status via a PGE2‐dependent mechanism. Sci Rep. 2016;6:38308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Najar M, Raicevic G, Boufker HI, et al. Mesenchymal stromal cells use PGE2 to modulate activation and proliferation of lymphocyte subsets: combined comparison of adipose tissue, Wharton's jelly and bone marrow sources. Cell Immunol. 2010;264:171‐179. [DOI] [PubMed] [Google Scholar]
- 15. Prockop DJ. Concise review: two negative feedback loops place mesenchymal stem/stromal cells at the center of early regulators of inflammation. Stem Cells. 2013;31:2042‐2046. [DOI] [PubMed] [Google Scholar]
- 16. Meisel R, Zibert A, Laryea M, Göbel U, Däubener W, Dilloo D. Human bone marrow stromal cells inhibit allogeneic T‐cell responses by indoleamine 2,3‐dioxygenase‐mediated tryptophan degradation. Blood. 2004;103:4619‐4621. [DOI] [PubMed] [Google Scholar]
- 17. Galipeau J, Sensébé L. Mesenchymal stromal cells: clinical challenges and therapeutic opportunities. Cell Stem Cell. 2018;22:824‐833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Carmen J, Brindley DA, Davie NL, Smith D. Cell therapy manufacturing: identifying and meeting demand. Vertès A, Qureshi N, Caplan A, Babiss L, Stem Cells in Regenerative Medicine. New York, United States: John Wiley & Sons; 2016:49‐68. [Google Scholar]
- 19. Valencic E, Piscianz E, Andolina M, Ventura A, Tommasini A. The immunosuppressive effect of Wharton's jelly stromal cells depends on the timing of their licensing and on lymphocyte activation. Cytotherapy. 2010;12:154‐160. [DOI] [PubMed] [Google Scholar]
- 20. Prasanna SJ, Gopalakrishnan D, Shankar SR, Vasandan AB. Pro‐inflammatory cytokines, IFNγ and TNFα, influence immune properties of human bone marrow and Wharton jelly mesenchymal stem cells differentially. PLoS One. 2010;5(2):e9016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Salem B, Miner S, Hensel NF, et al. Quantitative activation suppression assay to evaluate human bone marrow‐derived mesenchymal stromal cell potency. Cytotherapy. 2015;17:1675‐1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Klinker MW, Marklein RA, Lo Surdo JL, Wei C‐H, Bauer SR. Morphological features of IFN‐γ–stimulated mesenchymal stromal cells predict overall immunosuppressive capacity. Proc Natl Acad Sci USA. 2017;114:E2598‐E2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Polchert D, Sobinsky J, Douglas G, et al. IFN‐γ activation of mesenchymal stem cells for treatment and prevention of graft versus host disease. Eur J Immunol. 2008;38:1745‐1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bartosh TJ, Ylöstalo JH, Mohammadipoor A, et al. Aggregation of human mesenchymal stromal cells (MSCs) into 3D spheroids enhances their antiin fl ammatory properties. Proc Natl Acad Sci USA. 2010;107:13724‐13729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ylostalo JH, Bartosh TJ, Tiblow A, Prockop DJ. Unique characteristics of human mesenchymal stromal/progenitor cells pre‐activated in 3‐dimensional cultures under different conditions. Cytotherapy. 2014;16:1486‐1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Murphy KC, Whitehead J, Falahee PC, Zhou D, Simon SI, Leach JK. Multifactorial experimental design to optimize the anti‐inflammatory and proangiogenic potential of mesenchymal stem cell spheroids. Stem Cells. 2017;35:1493‐1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cheng NC, Wang S, Young TH. The influence of spheroid formation of human adipose‐derived stem cells on chitosan films on stemness and differentiation capabilities. Biomaterials. 2012;33:1748‐1758. [DOI] [PubMed] [Google Scholar]
- 28. Ho SS, Murphy KC, Binder BYK, Vissers CB, Leach JK. Increased survival and function of mesenchymal stem cell spheroids entrapped in instructive alginate hydrogels. Stem Cells Translational Medicine. 2016;5(6):773‐781. 10.5966/sctm.2015-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim J, Ma T. Endogenous extracellular matrices enhance human mesenchymal stem cell aggregate formation and survival. Biotechnol Prog. 2013;29:441‐451. [DOI] [PubMed] [Google Scholar]
- 30. Xu Y, Shi T, Xu A, Zhang L. 3D spheroid culture enhances survival and therapeutic capacities of MSCs injected into ischemic kidney. J Cell Mol Med. 2016;20:1203‐1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ylostalo JH, Bazhanov N, Mohammadipoor A, Bartosh TJ. Production and administration of therapeutic mesenchymal stem/stromal cell (MSC) spheroids primed in 3‐D cultures under xeno‐free conditions. J Vis Exp. 2017;121:1‐13. 10.3791/55126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xie AW, BYK B, Khalil AS, et al. Controlled self‐assembly of stem cell aggregates instructs pluripotency and lineage bias. Sci Rep. 2017;7:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lund ME, To J, O'Brien BA, Donnelly S. The choice of phorbol 12‐myristate 13‐acetate differentiation protocol influences the response of THP‐1 macrophages to a pro‐inflammatory stimulus. J Immunol Methods. 2016;430:64‐70. [DOI] [PubMed] [Google Scholar]
- 34. Genin M, Clement F, Fattaccioli A, Raes M, Michiels C. M1 and M2 macrophages derived from THP‐1 cells differentially modulate the response of cancer cells to etoposide. BMC Cancer. 2015;15:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Del Duca D, Werbowetski T, Del Maestro RF. Spheroid preparation from hanging drops: characterization of a model of brain tumor invasion. J Neurooncol. 2004;67:295‐303. [DOI] [PubMed] [Google Scholar]
- 36. Bloom DD, Centanni JM, Bhatia N, et al. A reproducible immunopotency assay to measure mesenchymal stromal cell‐mediated T‐cell suppression. Cytotherapy. 2015;17:140‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zimmermann JA, Hettiaratchi MH, McDevitt TC. Enhanced immunosuppression of T cells by sustained presentation of bioactive interferon‐γ within three‐dimensional mesenchymal stem cell constructs. Stem Cells Translational Medicine. 2016;6(1):223‐237. 10.5966/sctm.2016-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Crop MJ, Baan CC, Korevaar SS, et al. Inflammatory conditions affect gene expression and function of human adipose tissue‐derived mesenchymal stem cells. Clin Exp Immunol. 2010;162:474‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pourgholaminejad A, Aghdami N, Baharvand H, Moazzeni SM. The effect of pro‐inflammatory cytokines on immunophenotype, differentiation capacity and immunomodulatory functions of human mesenchymal stem cells. Cytokine. 2016;85:51‐60. [DOI] [PubMed] [Google Scholar]
- 40. Krampera M. Mesenchymal stromal cell ‘licensing’: a multistep process. Leukemia. 2011;25:1408‐1414. [DOI] [PubMed] [Google Scholar]
- 41. Bartosh TJ, Ylöstalo JH, Bazhanov N, Kuhlman J, Prockop DJ. Dynamic compaction of human mesenchymal stem/precursor cells into spheres self‐activates caspase‐dependent IL1 signaling to enhance secretion of modulators of inflammation and immunity (PGE2, TSG6, and STC1). Stem Cells. 2013;31:2443‐2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Costa MHG, McDevitt TC, Cabral JMS, da Silva CL, Ferreira FC. Tridimensional configurations of human mesenchymal stem/stromal cells to enhance cell paracrine potential towards wound healing processes. J Biotechnol. 2017;262:28‐39. [DOI] [PubMed] [Google Scholar]
- 43. Ng ES, Davis RP, Azzola L, Stanley EG, Elefanty AG. Forced aggregation of defined numbers of human embryonic stem cells into embryoid bodies fosters robust, reproducible hematopoietic differentiation. Blood. 2005;106:1601‐1603. [DOI] [PubMed] [Google Scholar]
- 44. Mohr JC, Zhang J, Azarin SM, et al. The microwell control of embryoid body size in order to regulate cardiac differentiation of human embryonic stem cells. Biomaterials. 2010;31:1885‐1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Park J, Cho CH, Parashurama N, et al. Microfabrication‐based modulation of embryonic stem cell differentiation. Lab Chip. 2007;7:1018‐1028. [DOI] [PubMed] [Google Scholar]
- 46. Choi YY, Chung BG, Lee DH, Khademhosseini A, Kim JH, Lee SH. Controlled‐size embryoid body formation in concave microwell arrays. Biomaterials. 2010;31:4296‐4303. [DOI] [PubMed] [Google Scholar]
- 47. Xie L, Mao M, Zhou L, Zhang L, Jiang B. Signal factors secreted by 2D and spheroid mesenchymal stem cells and by cocultures of mesenchymal stem cells derived microvesicles and retinal photoreceptor neurons. Stem Cells Int. 2017;2017:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sargent CY, Berguig GY, Kinney MA, et al. Hydrodynamic modulation of embryonic stem cell differentiation by rotary orbital suspension culture. Biotechnol Bioeng. 2010;105:611‐626. [DOI] [PubMed] [Google Scholar]
- 49. Zimmermann JA, Hettiaratchi MH, McDevitt TC. Enhanced immunosuppression of T cells by sustained presentation of bioactive interferon‐γ within three‐dimensional mesenchymal stem cell constructs. Stem Cells Translational Medicine. 2017;6:223‐237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Raghavan S, Mehta P, Horst EN, Ward MR, Rowley KR, Mehta G. Comparative analysis of tumor spheroid generation techniques for differential in vitro drug toxicity. Oncotarget. 2016;7(13):16948‐16961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kinney MA, Sargent CY, McDevitt TC. Temporal modulation of β‐catenin signaling by multicellular aggregation kinetics impacts embryonic stem cell cardiomyogenesis. Stem Cells Dev. 2013;22:2665‐2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dupont S, Morsut L, Aragona M, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179‐183. [DOI] [PubMed] [Google Scholar]
- 53. Musah S, Wrighton PJ, Zaltsman Y, et al. Substratum‐induced differentiation of human pluripotent stem cells reveals the coactivator YAP is a potent regulator of neuronal specification. Proc Natl Acad Sci USA. 2014;111:13805‐13810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yang C, Tibbitt MW, Basta L, Anseth KS. Mechanical memory and dosing influence stem cell fate. Nat Mater. 2014;13:645‐652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hsiao C, Lampe M, Nillasithanukroh S, Han W, Lian X, Palecek SP. Human pluripotent stem cell culture density modulates YAP signaling. Biotechnol J. 2016;11:662‐675. 10.1002/biot.201500374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Low BC, Pan CQ, Shivashankar GV, Bershadsky A, Sudol M, Sheetz M. YAP/TAZ as mechanosensors and mechanotransducers in regulating organ size and tumor growth. FEBS Lett. 2014;588:2663‐2670. [DOI] [PubMed] [Google Scholar]
- 57. Liu F, Lagares D, Choi KM, et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol. 2015;308:L344‐L357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Karystinou A, Roelofs AJ, Neve A, Cantatore FP, Wackerhage H, De Bari C. Yes‐associated protein (YAP) is a negative regulator of chondrogenesis in mesenchymal stem cells. Arthritis Res Ther. 2015;17(1):147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Caliari SR, Vega SL, Kwon M, Soulas EM, Burdick JA. Dimensionality and spreading influence MSC YAP/TAZ signaling in hydrogel environments. Biomaterials. 2016;103:314‐323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Komatsu N, Kajiya M, Motoike S, et al. Type I collagen deposition via osteoinduction ameliorates YAP/TAZ activity in 3D floating culture clumps of mesenchymal stem cell/extracellular matrix complexes. Stem Cell Res Ther. 2018;9:342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhong W, Tian K, Zheng X, et al. Mesenchymal stem cell and chondrocyte fates in a multishear microdevice are regulated by yes‐associated protein. Stem Cells Dev. 2013;22:2083‐2093. [DOI] [PubMed] [Google Scholar]
- 62. Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19:491‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nardone G, De La Cruz JO, Vrbsky J, et al. YAP regulates cell mechanics by controlling focal adhesion assembly. Nat Commun. 2017;8:15321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Galipeau J, Krampera M, Barrett J, et al. International Society for Cellular Therapy prespective on immune functional assays for mesenchymal stromal cells as potency release criterion for advanced phase clinical trials. Cytotherapy. 2016;18:151‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Murphy KC, Fang SY, Leach JK. Human mesenchymal stem cell spheroids in fibrin hydrogels exhibit improved cell survival and potential for bone healing. Cell Tissue Res. 2014;357:91‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ylöstalo JH, Bartosh TJ, Coble K, Prockop DJ. Human mesenchymal stem/stromal cells cultured as spheroids are self‐activated to produce prostaglandin E2 that directs stimulated macrophages into an anti‐inflammatory phenotype. Stem Cells. 2012;30:2283‐2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tsai A‐C, Liu Y, Yuan X, Ma T. Compaction, fusion, and functional activation of three‐dimensional human mesenchymal stem cell aggregate. Tissue Eng Part A. 2015;21:1705‐1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bao P, Kodra A, Tomic‐Canic M, Golinko MS, Ehrlich HP, Brem H. The role of vascular endothelial growth factor in wound healing. J Surg Res. 2009;153:347‐358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wheeler KC, Jena MK, Pradhan BS, et al. VEGF may contribute to macrophage recruitment and M2 polarization in the decidua. PLoS One. 2018;13:1‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Oh JY, Roddy GW, Choi H, et al. Anti‐inflammatory protein TSG‐6 reduces inflammatory damage to the cornea following chemical and mechanical injury. Proc Natl Acad Sci USA. 2010;107:16875‐16880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Choi H, Lee RH, Bazhanov N, Oh JY, Prockop DJ. Anti‐inflammatory protein TSG‐6 secreted by activated MSCs attenuates zymosan‐induced mouse peritonitis by decreasing TLR2/NF‐ B signaling in resident macrophages. Blood. 2011;118:330‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lee RH, Pulin AA, Seo MJ, et al. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti‐inflammatory protein TSG‐6. Cell Stem Cell. 2009;5:54‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tipnis S, Viswanathan C, Majumdar AS. Immunosuppressive properties of human umbilical cord‐derived mesenchymal stem cells: role of B7‐H1 and IDO. Immunol Cell Biol. 2010;88:795‐806. [DOI] [PubMed] [Google Scholar]
- 74. Sart S, Tsai A‐C, Li Y, Ma T. Three‐dimensional aggregates of mesenchymal stem cells: cellular mechanisms, biological properties, and applications. Tissue Eng Part B Rev. 2014;20:365‐380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Egger D, Tripisciano C, Weber V, Dominici M, Kasper C. Dynamic cultivation of mesenchymal stem cell aggregates. Bioengineering. 2018;5:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lee B‐C, Kim H‐S, Shine T‐H, et al. PGE2 maintains self‐renewal of human adult stem cells via EP2‐mediated autocrine signaling and its production is regulated by cell‐to‐cell contact. Sci Rep. 2016;6:26298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Qazi TH, Mooney DJ, Duda GN, Geissler S. Biomaterials that promote cell‐cell interactions enhance the paracrine function of MSCs. Biomaterials. 2017;140:103‐114. [DOI] [PubMed] [Google Scholar]
- 78. Kinney MA, Hookway TA, Wang Y, McDevitt TC. Engineering three‐dimensional stem cell morphogenesis for the development of tissue models and scalable regenerative therapeutics. Ann Biomed Eng. 2014;42:352‐367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Murphy KC, Hung BP, Browne‐Bourne S, et al. Measurement of oxygen tension within mesenchymal stem cell spheroids. J R Soc Interface. 2017;14:20160851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kulkarni OP, Lichtnekert J, Anders HJ, Mulay SR. The immune system in tissue environments regaining homeostasis after injury: is ‘inflammation’ always inflammation? Mediators Inflamm. 2016;2016:2856213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sugimoto MA, Sousa LP, Pinho V, Perretti M, Teixeira MM. Resolution of inflammation: what controls its onset? Front Immunol. 2016;7:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44:450‐462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Grimes DR, Kelly C, Bloch K, Partridge M. A method for estimating the oxygen consumption rate in multicellular tumour spheroids. J R Soc Interface. 2014;11:1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Potier E, Ferreira E, Meunier A, Sedel L, Logeart‐Avramoglou D, Petite H. Prolonged hypoxia concomitant with serum deprivation induces massive human mesenchymal stem cell death. Tissue Eng. 2007;13:1325‐1331. [DOI] [PubMed] [Google Scholar]
- 85. Mercurio F, Manning AM. NF‐κB as a primary regulator of the stress response. Oncogene. 1999;18:6163‐6171. [DOI] [PubMed] [Google Scholar]
- 86. Lawrence T. The nuclear factor NF‐kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Spees JL, Lee RH, Gregory CA. Mechanisms of mesenchymal stem/stromal cell function. Stem Cell Res Ther. 2016;7:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting Information.
Data Availability Statement
The data sets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.