

Abstract
Cirrhosis is a high-risk state for hepatocellular carcinoma (HCC) development and represents an opportunity to prevent cancer. In the pre-cancerous state of cirrhosis, there is an accumulation of neoantigens that may be specifically targetable through immunotherapy. We asked if immune checkpoint inhibition could prevent tumorigenesis in a mouse model of diethylnitrosamine (DEN) and carbon tetrachloride (CCl4) induced HCC. We found that initiation of anti-PD-1 therapy prior to tumorigenesis could prevent up to 46% of liver tumors. This significant reduction in tumor burden was accompanied by infiltration of CD4+ T helper and CD8+ cytotoxic T cells into the liver parenchyma. Importantly, Anti-PD-1 therapy did not exacerbate liver dysfunction or worsen overall health in this liver disease model. Given the safety and preservation of quality of life observed with long-term immunotherapy use, an immunotherapy chemoprevention strategy is likely associated with a low risk-to-benefit ratio and high value care in select patients. These results encourage a prevention trial in cirrhotic patients with the highest risk of developing HCC.
Keywords: Cancer Prevention, Hepatocellular Carcinoma, Immune Checkpoint Inhibitors, Liver Cancer, Liver Cirrhosis
Introduction
Primary liver cancer represents a major public health problem, with greater than 850,000 cases diagnosed each year.[1] Among primary liver cancers, hepatocellular carcinoma (HCC) represents approximately 90% of all cases. HCC most often arises in the context of longstanding, progressive liver disease caused by chronic hepatitis B (HBV) infection, chronic hepatitis C (HCV) infection, nonalcoholic steatohepatitis (NASH), or alcoholic liver disease. Over decades, these risk factors can lead to cirrhosis in a subset of patients, and HCC then develops in 5–30% of cirrhosis patients.[2] Once established in a cirrhotic liver, HCC is difficult to treat (5 year survival of 18%) with an incidence-to-mortality ratio approaching 1.[2] Moreover, the majority of HCC patients present with advanced stage disease that cannot be cured.[2]
Cirrhosis represents a premalignant state, and the decades-long latency of progression from cirrhosis to HCC offers a large window for intervention. Therefore, cancer prevention could be the most effective approach for reducing liver cancer burden and mortality, especially if creative and novel strategies to select the highest risk patients from the pool of cirrhosis patients can be devised.[2] Recent work by Hoshida’s group has identified a gene expression signature to predict HCC risk in cirrhosis patients,[3,4] while others have developed risk models using clinical biomarkers to estimate HCC risk.[5,6] These tools could be useful in the clinical setting to identify the highest risk patients that would benefit most from prevention. To date, the only effective chemoprevention strategies proven for HCC are antivirals for viral hepatitis-associated HCC, which is not relevant to a growing proportion of patients with cirrhosis caused by non-viral etiologies.[7]
Immune checkpoint inhibitors (ICIs) have revolutionized the treatment of cancer. Antibody-mediated blockade of the programmed death-1 receptor (PD-1), its ligand programmed death-ligand 1 (PD-L1), and cytotoxic T-lymphocyte antigen 4 (CTLA-4) have demonstrated improved clinical outcomes when administered as first- or second-line therapy in renal cell carcinoma, squamous-cell lung carcinoma, advanced urothelial carcinoma, and advanced melanoma.[8–12] Recent studies have shown that anti-PD-1 therapy can be effective for a subset of advanced HCC patients. Checkmate 040, a Phase I/II clinical trial using nivolumab in advanced HCC, showed an objective response rate of 20% with a manageable safety profile.[8] Similarly, KEYNOTE 224 was a phase II clinical trial using pembrolizumab in patients with previously treated HCC and found an objective response rate of 18%.[13] In addition to these anti-cancer effects, ICIs are generally well tolerated and safe. Importantly, ICIs are associated with durable effects and have demonstrated improved patient outcomes years after initiation of treatment in melanoma, renal cancer, and lung cancer.[14] Yet, no studies have investigated the use of immune checkpoint inhibition to prevent cancer in high-risk cirrhosis patients.
Tumor-specific gene mutations result in novel amino acid sequences that can generate novel peptides, or neoantigens, which can be presented to CD8+ T cells on MHC Class I molecules.[15,16] Cells presenting these non-self neoantigens are eliminated by the immune system, and neoantigen burden is correlated with immune cytolytic activity.[17] Nonetheless, many predicted neoantigens are not depleted in untreated tumors,[18] suggesting that immune evasion mechanisms are present throughout tumorigenesis. Additionally, this indicates neoantigens alone are generally not sufficient for cancer immune rejection and provides a rationale for the use of immune checkpoint inhibitors, even in the earliest stages of cancer development.
It is widely accepted that high mutation and neoantigen loads are associated with greater ICI efficacy in multiple cancer types.[19–21] In addition, loss of neoantigen presentation results in acquired immunotherapy resistance, further demonstrating that neoantigens are required for ICI response.[22] Thus, if present, neoantigens in early pre-neoplastic cells would suggest that subclinical disease may be amenable to treatment with immune checkpoint inhibition. In fact, recent evidence from a study of lung adenocarcinomas demonstrated that cancers with subclonal neoantigens (present in a subset of cells) are less likely to respond to immunotherapies.[23] In contrast, clonal neoantigens (present in all cells) are more likely to elicit a successful immunotherapy response. This suggests that eradicating malignant cells at earlier stages of cancer development may not only be efficacious, but would also prevent further genomic evolution and subclonal outgrowth of neoantigens leading to acquired immunotherapy resistance. However, whether neoantigens play any role in the pre-cancerous state of liver cirrhosis is not known.
Recent work from our lab and others have shown that fibrotic livers carry somatic mutations and that mutational burden correlates with fibrosis stage.[24,25] Since it is unclear if these mutations generate neoantigens and an immune response, we examined genomic data sets generated from human cirrhosis samples in our lab[24] and TCGA data from human HCC patients[26] to assess neoantigen load. Then, we hypothesized that immune checkpoint inhibition in the pre-cancerous, fibrotic liver might enhance T cell mediated elimination of highly mutated, pre-neoplastic or transformed clones bearing neoantigens, pre-empting tumorigenesis. We used the diethylnitrosamine (DEN) and carbon tetrachloride (CCl4) mouse model of mutagenesis, liver damage, and carcinogenesis to test whether anti-PD-1 therapy could thus prevent HCC development.
Materials and Methods
Mutation and Neoantigen Calling
For mouse samples, exome-seq reads were aligned to the GRCm38 genome by BWA-MEM.[27] For human samples, exome-seq reads were aligned to the GRCh38 genome by BWA-MEM.[27] Picard was used to add read group information and Sambamba was used to perform base quality score recalibration and local realignment around Indels. MuTect,[28] VarScan,[29] Shimmer,[30] SpeedSeq,[31] Manta,[32] and Strelka2[33] were used to call SNPs and Indels. A mutation called by ≥ any 3 of these algorithms was retained. Annovar was used to annotate SNPs and Indels.[34] All SNPs and Indels were combined and kept if there were ≥ 7 total (wild-type and variant) reads in the blood sample and ≥ 3 variant reads in the liver sample. Somatic mutations and germline mutations were annotated according to the VAFs in the liver and normal blood samples.
We used the QBRC pipeline for neoantigen calling.[35] We started neoantigen analysis with somatic mutations called by the QBRC mutation calling pipeline. We used only frameshift, non-frameshift, missense, and stoploss mutations that were predicted to lead to protein coding changes. We kept only somatic mutations whose VAFs were < 0.02 (2%) in the blood sample and > 0.05 (5%) in the liver samples. For H2-K, H2-D, we predicted the neoantigens of 8 to 13 amino acids in length. Putative neoantigens with amino acid sequences exactly matching known mouse protein sequences were filtered out. For MHC bindings, IEDB recommended mode (http://tools.iedb.org/main/) was used for prediction of binding affinities. Neoantigens were kept only if the predicted ranks of binding affinities were <2% when compared to an atlas of wild-type peptides. Liver RNA-seq data were aligned to the GRm38 reference genome using the STAR aligner.[36] FeatureCounts was used to summarize gene expression levels.[37] Neoantigens whose corresponding mutations were in genes with expression level <1 RPKM in either the specific exon or the whole transcript were filtered out.
Mice and the DEN + CCl4 Model
Wild-type male and female C3H/HeJ were obtained from the Jackson Laboratory and bred to generate 42 male pups all around the same age. This strain was chosen for its well-known susceptibility to diethylnitrosamine-induced tumors.[38] Diethylnitrosamine (DEN, N0756, Sigma-Aldrich) was dissolved in 0.9% NaCl and injected intraperitoneally at 2 weeks of age at a dose of 25 mg/kg to induce hepatocyte mutagenesis. At weaning age (P21), the mice were randomly assigned to either IgG Control or anti-PD-1 groups. In order to accelerate tumorigenesis and introduce low levels of cell death and fibrosis, we also gave once weekly IP injections of carbon tetrachloride (CCl4) starting at 6 weeks of age. CCl4 (289116, Sigma-Aldrich) was dissolved in corn oil (4%) and injected IP. In vivo grade monoclonal Anti-PD-1 antibody (clone RMP1–14) and isotype control antibody (clone 2A3) were obtained from BioXCell. Mice were injected IP with 175 μg of antibody dissolved in 100 μL dilution buffer every other week starting at 10 weeks of age until 20 weeks of age for a total of 6 doses. When mice were sacrificed, livers were weighed and pictures of the front and back of livers were taken. For quantification of surface tumors, these pictures were randomly assigned numbers by a third party who created a decoding key for group analysis after quantification.
Mice were weighed weekly from 6 weeks of age until the end of the experiment to ensure that the antibody treatment was not affecting overall animal health in either group. Blood was collected from all mice every 4 weeks from 6 weeks of age until the end of the experiment for assessment of liver function through plasma levels of aspartate transaminase (AST), alanine transaminase (ALT), and total bilirubin (Tbil).
Multiplex Immunohistochemistry for Immune Cells
Livers collected from animals sacrificed at 10 weeks of age, 21 weeks of age, and 23 weeks of age were fixed in 4% paraformaldehyde overnight, followed by dehydration in 50% EtOH. Livers were then embedded in paraffin and sectioned by the UT Southwestern Histopathology Core.
Immunohistochemistry (IHC) was performed as previously described.[39] Briefly, slides were warmed in a 60°C oven for 10 min followed by deparaffinization and rehydration. Before antigen retrieval, slides were fixed in 10% neutral buffered formalin for 30 min followed by a PBS wash. Antigen retrieval was performed in antigen retrieval buffer (10 mM Tris-HCl, 1 mM EDTA with 10% glycerol [pH 9]) at 110 °C for 18 min (~4–5ψ). Slides were then allowed to be cooled down to room temperature and were washed once with PBS. Tissue sections were blocked with 2.5% goat serum (Vector Laboratories, S-1012) for 30 min followed by incubation with primary antibody overnight: CD3 (1:2000; Thermo Fisher Scientific, PA1–29547), CD4 (1:2000; Abcam, ab183685), and CD8 (1:4000; Cell Signaling, 98941). Slides were washed three times for 5 min in PBST containing 0.05% Tween20 and 2 mM EDTA and incubated with HRP conjugated secondary Antibody (ImmPRESS; Vector Laboratories, MP-7401) for 30 min on a shaker. Slides were then washed three times for 5 min in PBST. For developing the fluorescence signal, TSA detection system (PerkinElmer) was used. We used OPAL 520 to stain CD3, OPAL 570 to stain CD4, and OPAL 690 to stain CD8. Multiplex staining was performed by stripping the previous antibody in 10 mM citrate buffer (pH 6.2) plus 10% glycerol at 110 °C for 2 min before probing with the next primary Ab. Slides were counter-stained with DAPI and then coverslipped using ProLong Gold mount (no. P36931; Life Technologies).
Imaging and Image Segmentation
Following immunostaining, full liver sections were imaged and scanned using a 20X AxioScan scanning microscope and Zen microscope software, available through the UT Southwestern Whole Brain Microscopy Core Facility. The following channels were used to acquire images: DAPI, AF488 (for CD3), AF555 (for CD4), and AF660 (for CD8). Note that AlexFluor channels were used as the AxioScan scanning microscope and Zen software did not specifically have channels for acquisition of OPAL dyes. This image scanning allowed for a complete, unbiased, and quantifiable characterization of the immune cell population within the entire tissue section, rather than the restricted analysis of a selected frame or area of an image.
In order to quantify cells of interest, image segmentation was performed, using custom-written Fiji and Excel macros, performing the following steps: (1) the CD3 channel was used to threshold and select regions of interest (ROIs), with signal intensity, cell shape, and size taken into account. (2) The maximum intensity within each of these ROIs was measured. (3) A small 2 μm-thick band was drawn around each ROI and mean intensity within this “background” was measured. (4) The ratio of max ROI intensity over mean “background” intensity was calculated, and only ROIs with ratio > 1.7 were considered as CD3-positive cells. (5) The maximum ROI intensities and “background” intensities of these CD3-positive cells were also measured in the CD4 and CD8 channels, respectively, and the same ratio of maximum ROI intensity over mean “background” (i.e. > 1.7) was applied. This procedure allowed us to identify cells that were CD3+ CD4+ and CD3+ CD8+ in each image. In order to account for the different sizes of each section stained and imaged, total surface area of each section was measured. To this aim, the brightness threshold for the CD3 channel was lowered to include the background staining of the entire tissue in order to create an ROI encompassing the entire section, followed by measurement of the total surface area. This value was used to calculate cell density expressed as cells/mm2.
Mutation Analysis of DEN + CCl4 Tumors
At the 23 week sacrifice time point, tumors were carefully and individually macrodissected from non-tumor liver tissue using a disposable scalpel and scissors. Tumors were collected into individual 1.5 mL microtubes and then immediately frozen in dry ice. Genomic DNA was extracted and purified from 17 tumors from thirteen IgG control-treated animals and from 17 tumors from fourteen anti-PD-1-treated animals using the Invitrogen PureLink™ Genomic DNA Mini Kit (Catalog #K182001). Then, PCR primers flanking known, recurrent, point mutations in Hras, Egfr, and Braf in DEN-induced tumors were designed. The primers were synthesized by IDT Technologies. PCR reactions were performed on genomic DNA from the above tumors, and then PCR products were run in 2% agarose gels. Then, the PCR products were isolated after visualization on a UV transilluminator. PCR products were extracted and purified using the Qiagen QIAquick Gel Extraction Kit (Catalog #28704). PCR products were sent to Genewiz for Sanger sequencing.
PCR primers were as follows:
mHras-FWD: atccatcagggtatgagaggtg , mHras-REV: gcatgactgtgtccaggacatt , expected product size 403 bp.
mEgfr-FWD: ggtgtttctgactatcctgg , mEgfr-REV: tgaggactgttgggtgaaagg , expected product size 343 bp.
mBraf-FWD: cagaggacatacgaatctctg , mBraf-REV: gcccttcagtgtatttctcgt , expected product size 353 bp.
Statistics
Variation is indicated using standard error of the mean (SEM) and presented as mean ± SEM. Unless otherwise stated in the figure legends, two-tailed Student’s t tests (two-sample equal variance) were used to test the significance of differences between two groups. Statistical significance is displayed as * (p < 0.05), ** (p < 0.01), *** (p < 0.001).
Mice
All mice were handled in accordance with, and with the approval of the Institutional Animal Care and Use Committee (IACUC) at UT Southwestern. Wild-type male and female C3h/HeJ mice were obtained from the Jackson Laboratory in order to breed the experimental cohorts.
Results
The cirrhotic liver harbors many neoantigens and may respond to immunotherapy
To assess the ability of neoantigens to generate a robust immune response, we measured neoantigen load in cirrhosis and HCC samples. Briefly, whole exome sequencing reads from human cirrhosis or HCC and paired blood samples were used to call mutations. Then, somatic mutations (including frameshift, non-frameshift, missense, and stoploss mutations) predicted to lead to changes in amino acid sequence were used to call neoantigens. Only those neoantigens with >5% variant allele frequency (VAF) in the liver/tumor samples and that ranked in the top 2% of MHC binding affinities were kept. Finally, using RNA-seq data from the same samples, only neoantigens with >1 RPKM were kept.
First, we found that mutations in liver cirrhosis lead to neoantigen production and presentation (Figure 1A). Given that fibrotic livers harbor many more mutations than non-fibrotic livers,[24,25] it is likely that there are also more neoantigens in liver cirrhosis as compared to non-fibrotic liver. Indeed, we found that neoantigen load tends to increase with worsened fibrosis stage (Supplementary Figure S1), which is consistent with our previous finding that mutational burden is highly associated with fibrosis stage.[24] It is also known that higher fibrosis stage is associated with an increased risk of developing HCC.[40,41] Taken together, the cirrhotic liver likely harbors neoantigen-presenting malignant clones in the early stages of cancer development that would therapeutically respond to immunotherapy. As might be expected based on this assumption, we found that the average neoantigen load (Figures 1B and 1C) increases significantly from cirrhosis to clinically detectable HCC.
Figure 1: The cirrhotic liver harbors many neoantigens and may respond to immunotherapy.
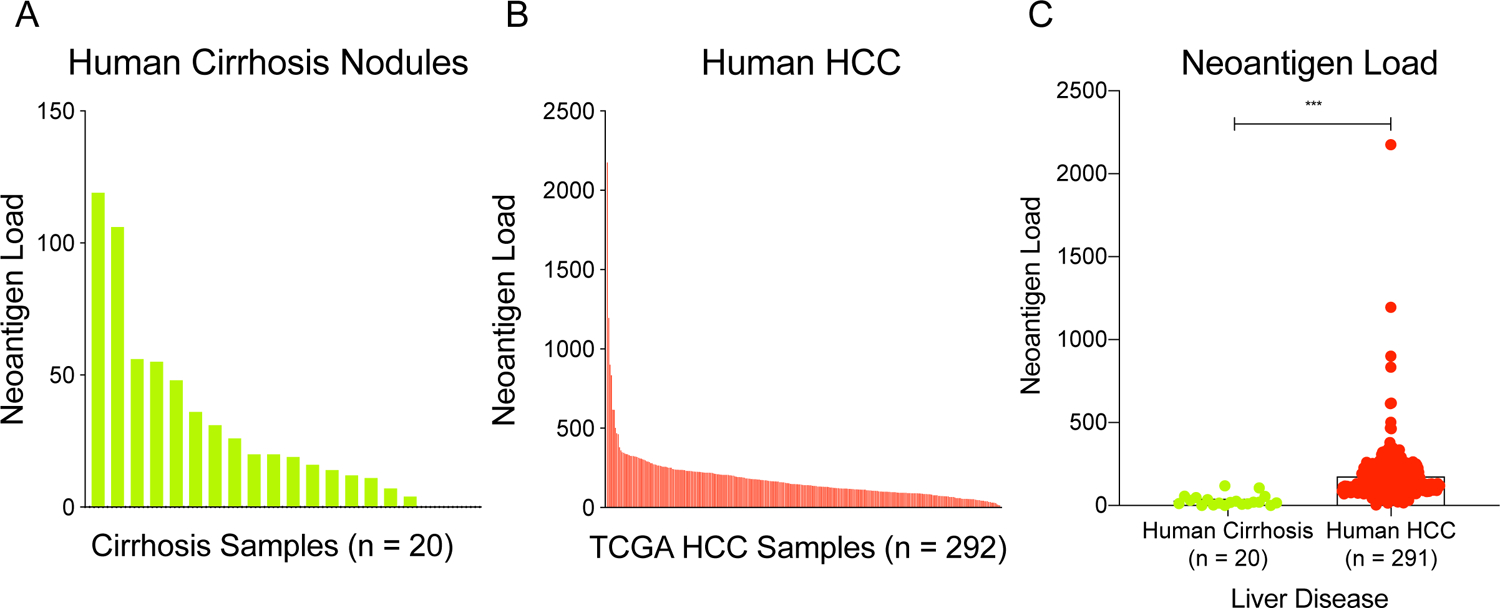
Neoantigen burden was analyzed in (A) human cirrhosis samples and (B) human HCC samples. (C) Predicted neoantigen burden increases from human cirrhosis to HCC (Student’s t-test, two tailed, equal variance). All data are presented as mean ± SEM. ***p < 0.001.
These findings confirm that neoantigens are present in liver cirrhosis, thus making immune checkpoint inhibition a viable preventive strategy. Thus, we hypothesized that in the pre-cancer state of progressive liver disease and cirrhosis, immune checkpoint blockade would be effective in eliminating malignant clones, preempting tumorigenesis. On the other hand, the presence of neoantigens throughout the cirrhotic liver also introduces the possibility of widespread hepatocyte death with immunotherapy, demanding a careful examination of both efficacy and safety.
PD-1 blockade prior to tumorigenesis prevents mutagen induced HCC development
In order to induce liver tumorigenesis and test preventive immunotherapy, we utilized the well-characterized model of DEN + CCl4 induced HCC.[42] DEN is metabolized by hepatocytes to generate free radicals that cause DNA damage and lead to the production of reactive oxygen species (ROS).[43] We reasoned that such widespread damage to DNA would closely model the mutational burden seen in human cirrhosis and HCC.[44] Previous work from our lab and others have characterized the mutational landscape in DEN-induced tumors,[45,46] and our studies found recurrent mutations in several oncogenes and tumor suppressors.[46] However, DEN used alone as a single agent does not recapitulate the chronic damage and liver fibrosis associated with HCC tumorigenesis in patients. Repeated injections of CCl4 are widely used to model liver damage and fibrosis. CCl4 is metabolized by CYP2E1 enzymes in pericentral hepatocytes to generate the trichloromethyl radical, which results in centrilobular hepatocyte necrosis, activation of Kupffer cells and an inflammatory response, and activation of hepatic stellate cells,[42,47–49] causing liver fibrosis similar to that observed in cirrhosis patients. Thus, we combined these two agents in order to model malignant transformation in a background of chronic liver damage and fibrosis.
We induced cancers using DEN + CCl4 in C3h/HeJ strain mice, which are prone to developing liver cancers (Figure 2A). At 10 weeks of age, four animals were sacrificed prior to receiving any antibody treatment, and there were no macroscopic or microscopic tumors (Figure 2B and Supplementary Figure S2). This was important as we sought to test the efficacy of immune checkpoint inhibition in preventing tumor formation, rather than in therapeutically treating existing tumors. Then, starting at 10 weeks of age, mice were given IP injections of either IgG control antibody or anti-PD-1 antibody every other week until 20 weeks of age, for a total of 6 doses (Figure 2A).
Figure 2: PD-1 blockade prior to tumorigenesis prevents mutagen-induced HCC development.
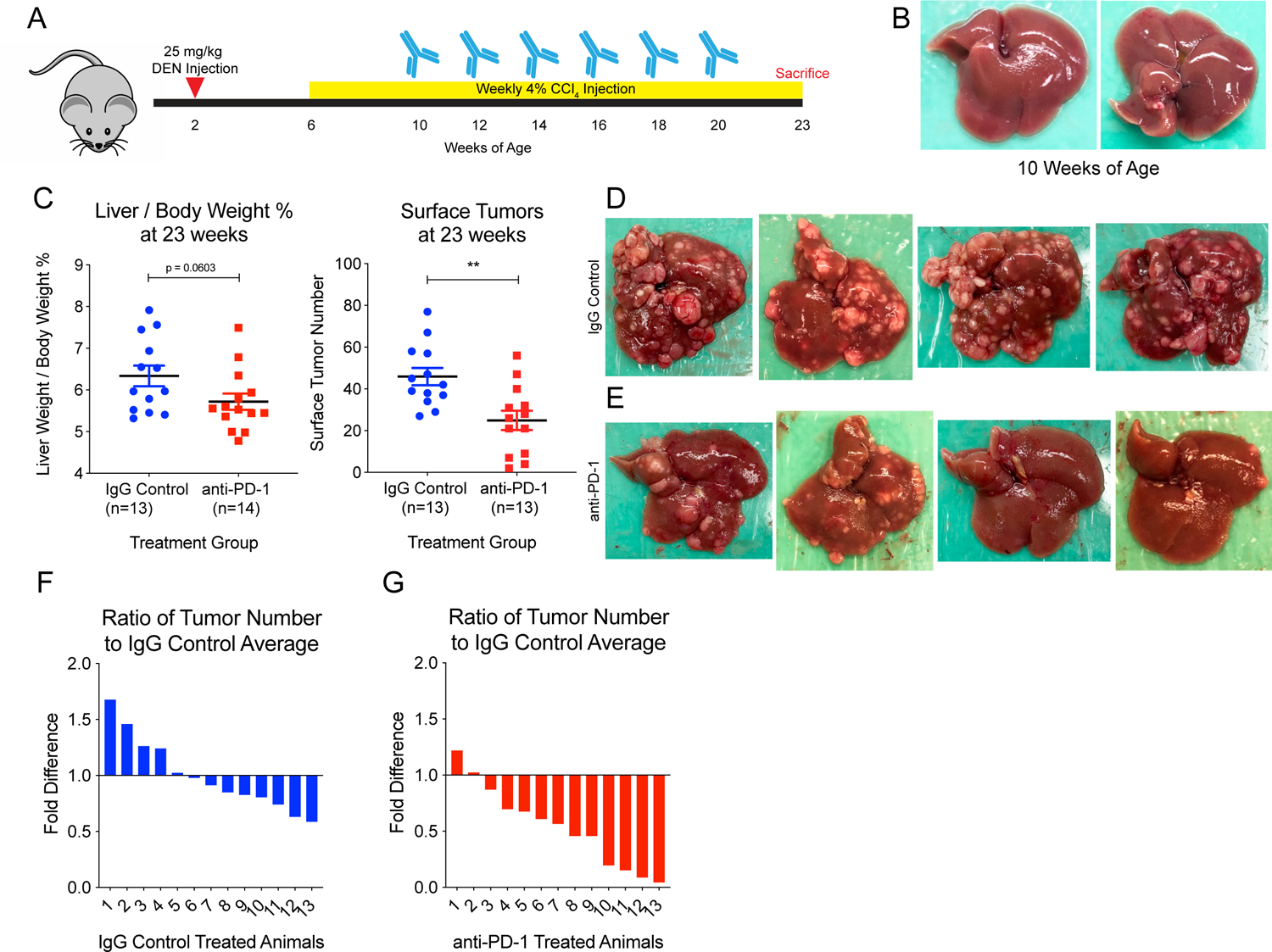
(A) Experimental design. In brief, male WT C3h/HeJ mice were injected IP with 25 mg/kg DEN at 2 weeks of age. Starting at 6 weeks of age, they were injected IP with 4% CCl4 weekly. From 10 to 20 weeks, mice were injected with either IgG Control or anti-PD-1 antibody every other week for a total of 6 doses. Animals were sacrificed at 23 weeks of age. (B) Liver from a mouse sacrificed at 10 weeks of age showing no tumor burden prior to initiation of treatment. (C) Liver over body weight percentage and quantification of surface tumor number at 23 weeks of age after 6 treatments show that anti-PD-1 treated animals exhibit significantly reduced tumor burden. IgG control-treated animals had an average of 45.92 surface tumors, and anti-PD-1-treated animals had an average of 24.92 tumors, a 46% reduction. Representative images of (D) IgG control-treated livers and (E) anti-PD-1-treated livers show that while all IgG control-treated livers carry significant tumor burden, anti-PD-1-treated livers show reduced tumor burden overall, and some livers have minimal tumor burden. Waterfall plots of the ratio of tumors in individual animals to control average (45.92) for (F) IgG control animals and (G) anti-PD-1 animals show that most animals (11/13) in the anti-PD-1 group had fewer tumors than the IgG control average. Two tailed student’s t tests were performed for the data shown in 2C. All data are presented as mean ± SEM. *p < 0.05, **p < 0.01.
At 23 weeks of age, most remaining mice were sacrificed for liver collection and analysis of tumor burden. Anti-PD-1 animals (n=14) had decreased tumor burden compared to IgG control animals (n=13) as assessed by liver to body weight ratios, as well as by surface tumor number. IgG control-treated livers weighed on average 6.3% ± 0.9% of the animal’s body weight and anti-PD-1 treated livers weighed on average 5.7% ± 0.7% of body weight (p = 0.0603), consistent with decreased tumor burden in the anti-PD-1 group (Figure 2C). Furthermore, IgG control-treated animals had an average of 46 ± 14.92 surface tumors and anti-PD-1-treated animals had an average of 25 ± 16.70 surface tumors (p = 0.0025) (Figures 2C, 2D, and 2E). This showed that the anti-PD-1 treatment had a significant effect in preventing the development of 46% of tumors. Notably, four animals in the anti-PD-1-treated group had less than 10 surface tumors, while no animal had less than 27 surface tumors in the IgG control-treated group. Only two animals (15.4%) in the anti-PD-1-treated group had more than the IgG control average of 46 surface tumors (Figures 2F and 2G). It should be noted that three animals in each group were kept alive until around 39 weeks of age to determine if anti-PD-1 injections affected survival, but since no mice in either group died, all six mice were euthanized.
Because serious drug toxicity may offset the benefit of chemoprevention strategies for patients with significant comorbidities, including cirrhosis, we examined metrics of toxicity. Average weights of mice were the same between IgG control-treated and anti-PD-1-treated groups for the duration of the experiment (Figure 3A). AST levels were similar between groups in early time points, but at 14, 18, and 22 weeks of age, AST was lower in anti-PD-1-treated mice compared to IgG control-treated mice (Figure 3B). ALT and total bilirubin levels were similar between groups for the duration of the experiment (Figures 3C and 3D). These results showed that immune checkpoint blockade did not cause major adverse effects on overall health or liver function, even in the setting of established liver damage.
Figure 3: Preventive immunotherapy in a mouse model of HCC does not cause significant adverse effects on liver function.
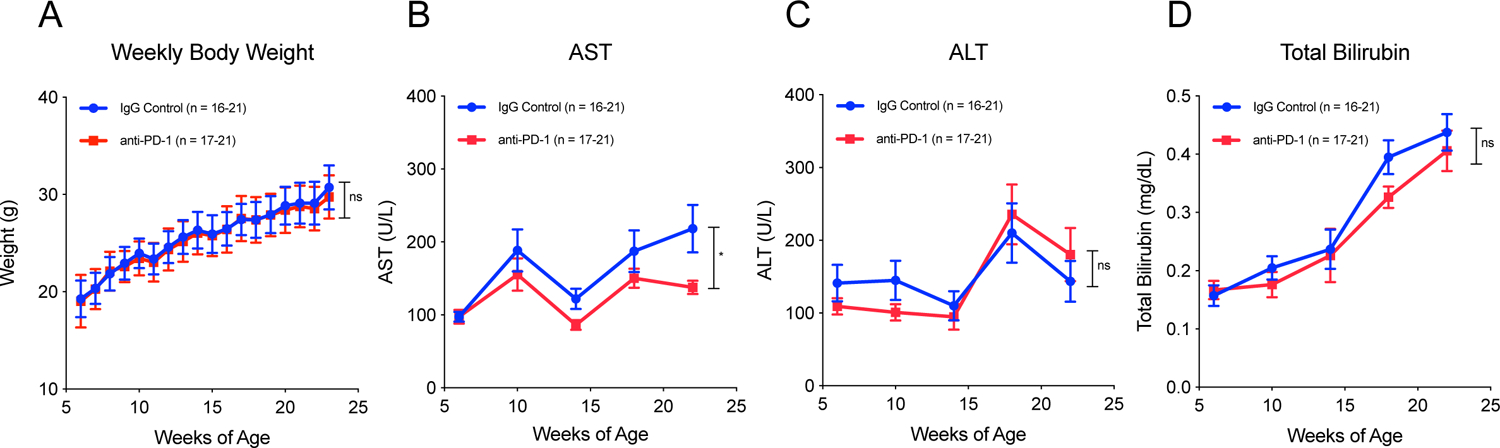
(A) Body weight measured weekly shows no significant difference between groups through the duration of the experiment. (B) AST, (C) ALT, and (D) total bilirubin measured every 4 weeks show similar levels of liver damage in both groups through the duration of the experiment. Two tailed paired t tests were performed for body weight, AST, ALT, and total bilirubin. All data are presented as mean ± SEM. *p < 0.05.
CD4+ T cell and CD8+ T cell tissue infiltration was associated with tumor prevention
The PD-1/PD-L1 interaction between naive T cells and antigen presenting cells results in decreased T cell proliferation, survival, and IL-2 production.[50,51] We hypothesized that treatment with anti-PD-1 antibody would result in an increased T cell response in the normal liver parenchyma. However, we wanted to determine if this would be mediated through CD3+ CD4+ T helper cells or CD3+ CD8+ cytotoxic T cells, or both. CD4+ T cells interact directly through their T cell receptor with antigen presenting cells, such as dendritic cells, and respond appropriately, either by the production of cytokines for recruitment of other immune cells or diminution of the immune response. CD8+ T cell receptors bind to MHC class I complex molecules on all cell types, and if presented with peptides recognized as foreign, induce cell death through molecules such as perforin or granzyme B.[51] It is known that both tumor-infiltrating CD4+ memory T cells and CD8+ cytotoxic T cells are correlated with increased survival in many different cancer types.[52]
We performed multiplex IHC for CD3, CD4, and CD8, and imaged whole liver sections (Figures 4A and 4B AND Supplementary Figures S3A–D). An image segmentation and analysis pipeline (Supplementary Figure S4) was devised to quantify CD3+ cells, CD3+ CD4+ T cells, and CD3+ CD8+ T cells in IgG control and anti-PD-1 livers. We analyzed T cell infiltration into the whole liver parenchyma, rather than in tumors alone, which is more important for prevention in high risk, pre-cancer states via upregulated immune surveillance of malignant clones. The examination of tumor-infiltrating T cells was also precluded by the sparseness of tumors in anti-PD-1 treated animals.
Figure 4: Whole section imaging of liver sections for quantification of immune cells reveals CD4+ T cell and CD8+ T cell tissue infiltration is associated with tumor prevention.
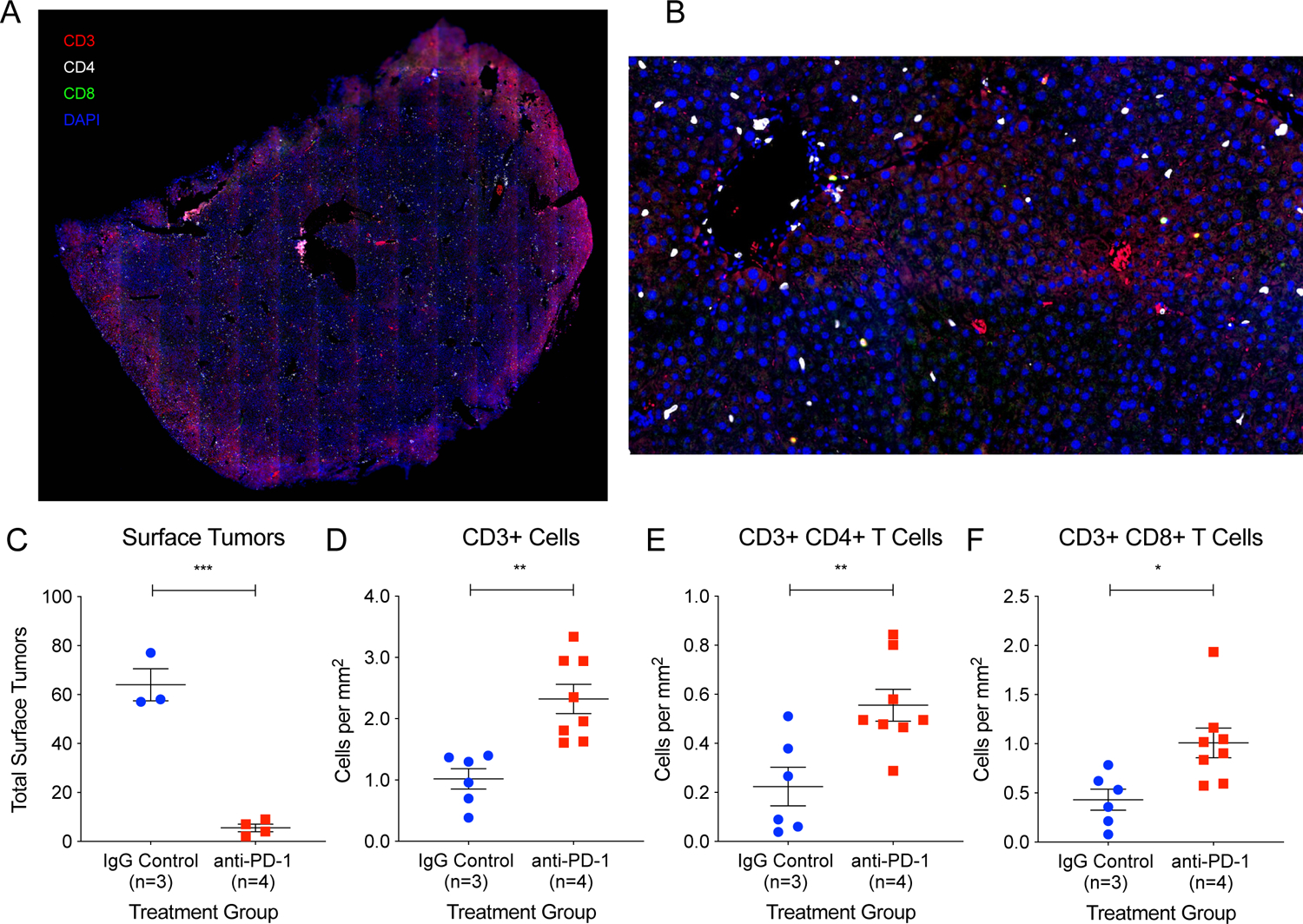
(A) Whole liver section from an anti-PD-1 treated animal immunostained for CD3 (red), CD4 (white), and CD8 (green) imaged using an AxioScan slide scanner microscope system. (B) Inset of (A). (C) Three animals with significant tumor burden in the IgG control group and four animals with minimal tumor burden in the anti-PD-1 group were analyzed using the immunostaining protocol and image analysis pipeline described. Two liver sections from each animal were analyzed for T cell infiltration. The total area analyzed for IgG control liver sections was 225.43 mm2 and for anti-PD-1 liver sections was 281.82 mm2. There were significantly increased infiltrating (D) CD3+ cells, (E) CD3+ CD4+ T cells, and (F) CD3+ CD8+ T cells in the livers of the anti-PD-1 animals. Two tailed student’s t tests were performed for the data shown in 3C-F. All data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
At 21 weeks of age, we randomly selected and sacrificed two IgG control animals and two anti-PD-1 animals in order to examine immune cells immediately after the final doses of antibody injections. We found that these particular animals had similar tumor burden, average of 34 and 37 tumors respectively (Supplementary Figures S5A, S5B, and S5C), suggesting that the two anti-PD-1 animals randomly selected did not exhibit a preventive response. We then found that there was also no difference in CD3+ cells/mm2, CD3+ CD4+ cells/mm2, or CD3+ CD8+ cells/mm2 in the livers of these animals (Supplementary Figures S5D, S5E, and S5F).
To clearly determine if successful tumor prevention was associated with increased T cell infiltration, we then selected and analyzed two sections from two different liver lobes of three control animals (avg. 64 surface tumors) and four anti-PD-1 animals (avg. 4 surface tumors) sacrificed at 23 weeks for the presence of immune cells (Figure 4C). The total area analyzed for IgG control liver sections was 225.43 mm2 and the total area analyzed for anti-PD-1 liver sections was 281.82 mm2. There was a significant increase in CD3+ cells/mm2 (Figure 4D), CD3+ CD4+ cells/mm2 (Figure 4E), and CD3+ CD8+ cells/mm2 (Figure 4F) in the livers of the anti-PD-1 animals. This suggests that anti-PD-1 therapy increased the adaptive immune response and, specifically, increased infiltration of both CD4+ T cells and CD8+ T cells into premalignant liver tissues. However, we did not examine the specific CD4+ T cell subsets (Th1, Th2, Th9, Th17, Th22, Treg, Tfh), that are enriched in anti-PD-1 livers.
Together, these data suggest that upregulation of CD4+ T cells and CD8+ T cells does indeed correlate with the tumor preventive response.
Response to preventive PD-1 blockade is not dependent on specific recurrent mutations
A previous study showed that liver tumorigenesis driven by oncogenic mutant Nras causes senescence and elimination of malignant clones.[53] Another group showed that oncogenic mutant Ctnnb1 in liver tumors imparts resistance to immune checkpoint inhibition.[54] To understand if such mutation-specific mechanisms were at play in determining the response or non-response to anti-PD-1 therapy in our mouse model, we examined oncogenic mutations in these tumors. Previous work has shown that A182 in Hras, T760 in Egfr, and T1910 in Braf are frequently and recurrently mutated in DEN-induced tumors.[45,46] One possibility is the selective elimination of clones carrying these oncogenic driver mutations. If this were the case, the proportion of remaining tumors carrying these oncogenic mutations would be lower in the anti-PD-1 group compared to the control group.
We genotyped these mutations within 34 IgG control or anti-PD-1 treated tumors. In the Hras gene, 24% of IgG control tumors had A182T or A182G mutations, 29% had a C181A mutation, and 47% were wild-type. On the other hand, 12% of anti-PD-1 tumors had A182T or A182G mutations, 35% had C181A mutations, and 53% were wild-type (Figure 5A). In Egfr, 6% of IgG control tumors had a T760A mutation and 94% were wild-type. 12% of anti-PD-1 tumors had a T760A mutation and 88% were wild-type (Figure 5B). In Braf , 41% of IgG Control tumors had a T1910A mutation and 59% were wild-type. On the other hand, 29% of anti-PD-1 tumors had a T1910A mutation and 71% were wild-type (Figure 5C). While there may have been a trend towards reduced A182T/G mutations in Hras and T1910A mutations in Braf in the anti-PD-1-treated animals, there were no statistically significant differences in tumor genotype for the point mutations in any of the three genes assessed. Our results indicate that the response to preventive immunotherapy is not dependent on specific recurrent oncogenic mutations, and thus, this strategy could be applicable to the broader cirrhosis population.
Figure 5: Response to preventive PD-1 blockade is not dependent on specific recurrent mutations.
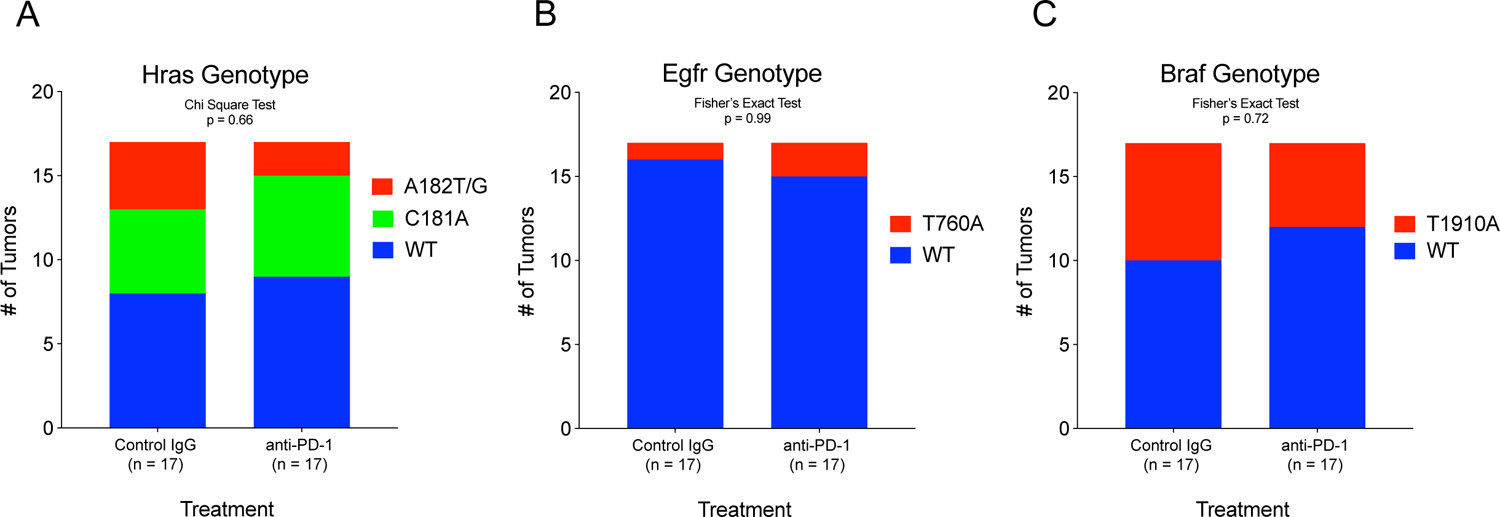
Known recurrent point mutations were sequenced in IgG control and anti-PD-1 tumors. The proportion of tumors bearing point mutations in (A) Hras, (B) Egfr, and (C) Braf were not different between IgG control and anti-PD-1 treated tumors, suggesting that the preventive response was not dependent on specific recurrent oncogenic mutations.
Discussion
The incidence of HCC in the United States is expected to increase as the rates of obesity and NASH rapidly rise.[2] Resection and liver transplantation are reserved for relatively healthy surgical candidates with limited tumor burden, while systemic therapies for advanced HCC extend life by only a few months.[55–58] Regardless of etiology, chronic liver diseases eventually converge onto end-stage cirrhosis before a subset of these patients progress to HCC. Because this decades-long process of liver disease progression offers a window of opportunity to preempt HCC tumorigenesis in a high-risk population, we endeavoured to explore the use of immune checkpoint inhibition in the preventive setting.
There are several strategic advantages of utilizing immunotherapy as chemoprevention. Pharmacokinetic studies of checkpoint inhibitors demonstrate a prolonged half-life (up to 27 days) and little evidence of a correlation between drug efficacy and exposure.[59] This suggests that for the purpose of chemoprevention in patients suspected to have limited cancer burden, such as those with cirrhosis but without clinically detectable HCC, less frequent drug dosing may still be effective. The effectiveness of less frequent dosing strategies with immunotherapies is supported by recent findings in melanoma patients where a single neoadjuvant dose of an immune checkpoint inhibitor was demonstrated to be sufficient to induce complete pathologic responses.[60] Nevertheless, as hepatocytes in the cirrhotic liver will continuously accumulate mutations over time, an intermittent dosing rather than a single dose immunotherapy regimen is likely to be more effective in chemoprevention. In addition, immunotherapies are well tolerated in many patients, including those with significant comorbidities and with a performance status that would preclude other anti-cancer therapies.[61] Importantly, long-term use of ICIs for up to five years in multiple cancer types have not uncovered new safety signals.[14]
Despite the favourable safety profile of ICIs, it is still worth discussing selection of patients at the highest risk of developing HCC, given that only 5–30%[2] of cirrhosis patients go on to develop HCC. Current surveillance protocol is centered around imaging studies, and nodules that are detected are scored using the Liver Imaging Reporting and Data System (LI-RADS) on a scale from LR-1 to LR-5.[62] LI-RADS-3 nodules are defined as “intermediate probability of malignancy” and LI-RADS-4 nodules are “probably HCC.”[62] Interestingly, one group has identified and validated an HCC risk gene expression signature for hepatitis B/C infection, alcohol, and NASH cirrhosis.[3,4] This signature can be used to predict cancer risk within the carcinogenic field in cirrhosis patients. In particular, the “poor prognosis” signature was highly associated with progression to advanced cirrhosis, development of HCC, and survival.[3] Another group has developed HCC risk models for HCV patients who have undergone antiviral treatment,[5] as well as for alcoholic liver disease and non-alcoholic fatty liver disease cirrhosis patients.[6] These models take various clinical biomarkers into account, such as age, gender, race, serum aminotransferase levels, platelet count, and others, in order to stratify patients according to estimated annual HCC risk into low, (<1 % risk per year), medium (1–3% risk per year), and high risk (>3% risk per year). These novel tools, in parallel or in combination with the LI-RADS score, could be invaluable in the clinical setting to select patients most likely to benefit from preventive immunotherapy, while minimizing overall healthcare costs and unnecessary exposure to potential toxicity and side effects. It is worthwhile to note that existing guidelines support the use of HCC screening for patients with cirrhosis or other risk factors for HCC, but no prospective data has ever proven an improvement in survival with biochemical and imaging surveillance.[63] This may be related to the fact that many patients with cirrhosis who develop clinically detectable HCC do not or cannot undergo curative regional or surgical interventions. The ambiguous utility of current HCC screening strategies further reduces the relative risk and increases the potential benefit of an immunotherapy chemoprevention strategy in at-risk persons.
We chose to use the DEN + CCl4 mouse model of fibrosis and HCC.[64–66] This is the best-suited model for a preventive pre-clinical trial because it recapitulates both the high mutational burden of human HCC and the chronic damage that ultimately leads to cirrhosis, which is seen in 80–90% of HCC patients.[2,67] Yet, while DEN tumors meet pathologic criteria for HCC,[42,45] a significant limitation of this model is that on the molecular level, the most frequently recurrent driver mutations (activating mutations in Hras, Egfr, and Braf) are rarely observed in human HCC.[26] Other commonly used mouse models of cancer, which rely on various combinations of tumor suppressor deletion (such as Tp53) and oncogene activation (such as Ctnnb1 and Myc),[54,68–70] more accurately mirror the genetics of human HCC. However, they do not recapitulate the evolutionary course of tumorigenesis from mutant clone to HCC; rather, they reproduce the genetic end state of what is likely a complex, dynamic process. Additionally, genetically-engineered mouse models of HCC do not replicate the surrounding tissue damage and organ dysfunction context from which most HCCs arise. Establishing this tissue context is critical for testing tumor prevention in the pre-malignant field. This is also important because we sought to characterize the toxicity of our preventive intervention in the presence of chronic liver damage. Finally, given the key role of neoantigen presentation in immune surveillance[15,16,21], we felt that it is important to use a model with widespread mutagenesis and chronic liver damage.[24,25] Overall, we believe our model has advantages and disadvantages for testing preventive immunotherapy, and it is clear that additional GEM models of HCCs need to be examined with this regimen.
In our experiment, we show that antibody mediated blockade of the PD-1 and PD-L1 interaction can prevent a significant fraction (46% average reduction) of HCCs. Interestingly, there was a variation in response in that some animals saw moderate reduction and other animals exhibited almost no tumor burden. Further work to understand the mechanisms of resistance and the relative contribution of tumor cell-intrinsic and immune-specific properties to this phenotype could help to uncover the source of this variation. Nevertheless, this model and treatment regimen can now be utilized as a platform to test other drugs or therapies for use in combination with PD-1 blockade.
Overall, the risk reduction we observed in the mouse model certainly makes a trial in select high risk cirrhosis patients worth exploring. However, one point for consideration is that immune checkpoint blockade is well-known to have side effects, most notably autoimmunity. The risk of adding more side effects to the long list of medical problems seen in end-stage liver disease patients must be considered carefully. Our results would predict that immunotherapy, even in the preventive setting, would not exacerbate liver dysfunction, an observation that is consistent with the profile of adverse events observed in clinical trials of PD-1 blockade in HCC patients.[8,13] Hepatocellular carcinoma will increasingly assert itself as a public health challenge, especially as the prevalence of metabolic syndrome increases worldwide. However, as in many medical disciplines, preventive, rather than ex post facto treatment, may more effectively relieve the morbidity and mortality of HCC.
Supplementary Material
Acknowledgments
We would like to thank Helen Hobbs and Teresa Eversole for contributing human liver samples; Sam Wang, Yujin Hoshida, and Amit Singal for comments on the manuscript; John Shelton for histopathology; the CRI Sequencing Core (Jian Xu, Xin Liu) and Admera Health (Yun Zhao) for genomics. T.W. is supported by CPRIT (RP190208). H.Z. is supported by the Pollack Foundation, NCI R01 CA251928, a UT Southwestern Simmons Comprehensive Cancer Center Pilot Award, and a Stand Up To Cancer Innovative Research Grant (SU2C-AACR-IRG 10-16).
Footnotes
Conflicts of Interest:
At the time of publication, H.Z. owned Ionis Pharmaceuticals stock worth less than $5,000. H.Z. has an active collaboration with Alnylam Pharmaceuticals and Twenty-Eight Seven Therapeutics.
References
- 1.Llovet JM, Zucman-Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016;2:16018. [DOI] [PubMed] [Google Scholar]
- 2.Villanueva A Hepatocellular Carcinoma. N Engl J Med. 2019;380:1450–62. [DOI] [PubMed] [Google Scholar]
- 3.Hoshida Y, Villanueva A, Sangiovanni A, Sole M, Hur C, Andersson KL, et al. Prognostic gene expression signature for patients with hepatitis C-related early-stage cirrhosis. Gastroenterology. 2013;144:1024–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakagawa S, Wei L, Song WM, Higashi T, Ghoshal S, Kim RS, et al. Molecular liver cancer prevention in cirrhosis by organ transcriptome analysis and lysophosphatidic acid pathway inhibition. Cancer Cell. 2016;30:879–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ioannou GN, Green PK, Beste LA, Mun EJ, Kerr KF, Berry K. Development of models estimating the risk of hepatocellular carcinoma after antiviral treatment for hepatitis C. J Hepatol. 2018;69:1088–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ioannou GN, Green P, Kerr KF, Berry K. Models estimating risk of hepatocellular carcinoma in patients with alcohol or NAFLD-related cirrhosis for risk stratification. J Hepatol. 2019;71:523–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singh S, Singh PP, Roberts LR, Sanchez W. Chemopreventive strategies in hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2014;11:45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389:2492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Motzer RJ, Tannir NM, McDermott DF, Arén Frontera O, Melichar B, Choueiri TK, et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N Engl J Med. 2018;378:1277–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WE, Poddubskaya E, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373:123–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bellmunt J, de Wit R, Vaughn DJ, Fradet Y, Lee J-L, Fong L, et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N Engl J Med. 2017;376:1015–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320–30. [DOI] [PubMed] [Google Scholar]
- 13.Zhu AX, Finn RS, Edeline J, Cattan S, Ogasawara S, Palmer D, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol. 2018;19:940–52. [DOI] [PubMed] [Google Scholar]
- 14.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Five-Year Survival and Correlates Among Patients With Advanced Melanoma, Renal Cell Carcinoma, or Non-Small Cell Lung Cancer Treated With Nivolumab. JAMA Oncol. 2019;5(10):1411–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. [DOI] [PubMed] [Google Scholar]
- 16.Yarchoan M, Johnson BA, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer. 2017;17:569. [DOI] [PubMed] [Google Scholar]
- 17.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van den Eynden J, Jiménez-Sánchez A, Miller ML, Larsson E. Lack of detectable neoantigen depletion signals in the untreated cancer genome. Nat Genet. 2019;51:1741–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang T, Shi T, Zhang H, Hu J, Song Y, Wei J, et al. Tumor neoantigens: from basic research to clinical applications. J Hematol Oncol. 2019;12:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell. 2017;171:934–949.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med. 2016;375:819–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu M, Lu T, Jia Y, Luo X, Gopal P, Li L, et al. Somatic mutations increase hepatic clonal fitness and regeneration in chronic liver disease. Cell. 2019;177:608–621.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brunner SF, Roberts ND, Wylie LA, Moore L, Aitken SJ, Davies SE, et al. Somatic mutations and clonal dynamics in healthy and cirrhotic human liver. Nature. 2019;574:538–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cancer Genome Atlas Research Network. Electronic address: wheeler@bcm.edu, Cancer Genome Atlas Research Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169:1327–1341.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hansen NF, Gartner JJ, Mei L, Samuels Y, Mullikin JC. Shimmer: detection of genetic alterations in tumors using next-generation sequence data. Bioinformatics. 2013;29:1498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiang C, Layer RM, Faust GG, Lindberg MR, Rose DB, Garrison EP, et al. SpeedSeq: ultra-fast personal genome analysis and interpretation. Nat Methods. 2015;12:966–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32:1220–2. [DOI] [PubMed] [Google Scholar]
- 33.Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–7. [DOI] [PubMed] [Google Scholar]
- 34.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu T, Wang S, Xu L, Zhou Q, Singla N, Gao J, et al. Tumor neoantigenicity assessment with CSiN score incorporates clonality and immunogenicity to predict immunotherapy outcomes. Sci Immunol. 2020;5(44):eaaz3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–30. [DOI] [PubMed] [Google Scholar]
- 38.Diwan BA, Rice JM, Ohshima M, Ward JM. Interstrain differences in susceptibility to liver carcinogenesis initiated by N-nitrosodiethylamine and its promotion by phenobarbital in C57BL/6NCr, C3H/HeNCrMTV- and DBA/2NCr mice. Carcinogenesis. 1986;7:215–20. [DOI] [PubMed] [Google Scholar]
- 39.Sorrelle N, Ganguly D, Dominguez ATA, Zhang Y, Huang H, Dahal LN, et al. Improved Multiplex Immunohistochemistry for Immune Microenvironment Evaluation of Mouse Formalin-Fixed, Paraffin-Embedded Tissues. J Immunol. 2019;202:292–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Toyoda H, Tada T, Yasuda S, Mizuno K, Ito T, Kumada T. Dynamic evaluation of liver fibrosis to assess the risk of hepatocellular carcinoma in patients with chronic hepatitis C who achieved sustained virologic response. Clin Infect Dis. 2020;70:1208–14. [DOI] [PubMed] [Google Scholar]
- 41.Na SK, Song B-C. Development and surveillance of hepatocellular carcinoma in patients with sustained virologic response after antiviral therapy for chronic hepatitis C. Clin Mol Hepatol. 2019;25:234–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heindryckx F, Colle I, Van Vlierberghe H. Experimental mouse models for hepatocellular carcinoma research. Int J Exp Pathol. 2009;90:367–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Verna L, Whysner J, Williams GM. N-nitrosodiethylamine mechanistic data and risk assessment: bioactivation, DNA-adduct formation, mutagenicity, and tumor initiation. Pharmacol Ther. 1996;71:57–81. [DOI] [PubMed] [Google Scholar]
- 44.Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Connor F, Rayner TF, Aitken SJ, Feig C, Lukk M, Santoyo-Lopez J, et al. Mutational landscape of a chemically-induced mouse model of liver cancer. J Hepatol. 2018;69:840–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang S, Zhou K, Luo X, Li L, Tu H-C, Sehgal A, et al. The Polyploid State Plays a Tumor-Suppressive Role in the Liver. Dev Cell. 2018;44:447–459.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yanguas SC, Cogliati B, Willebrords J, Maes M, Colle I, van den Bossche B, et al. Experimental models of liver fibrosis. Arch Toxicol. 2016;90:1025–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Basu S Carbon tetrachloride-induced lipid peroxidation: eicosanoid formation and their regulation by antioxidant nutrients. Toxicology. 2003;189:113–27. [DOI] [PubMed] [Google Scholar]
- 49.Weber LWD, Boll M, Stampfl A. Hepatotoxicity and mechanism of action of haloalkanes: carbon tetrachloride as a toxicological model. Crit Rev Toxicol. 2003;33:105–36. [DOI] [PubMed] [Google Scholar]
- 50.Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21:938–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kang T-W, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479:547–51. [DOI] [PubMed] [Google Scholar]
- 54.Ruiz de Galarreta M, Bresnahan E, Molina-Sánchez P, Lindblad KE, Maier B, Sia D, et al. β-Catenin Activation Promotes Immune Escape and Resistance to Anti-PD-1 Therapy in Hepatocellular Carcinoma. Cancer Discov. 2019;9:1124–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kudo M, Finn RS, Qin S, Han KH, Ikeda K, Piscaglia F, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018;391:1163–73. [DOI] [PubMed] [Google Scholar]
- 56.Bruix J, Qin S, Merle P, Granito A, Huang Y-H, Bodoky G, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389:56–66. [DOI] [PubMed] [Google Scholar]
- 57.Abou-Alfa GK, Meyer T, Cheng AL, El-Khoueiry AB, Rimassa L, Ryoo BY, et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N Engl J Med. 2018;379:54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu AX, Park JO, Ryoo B-Y, Yen C-J, Poon R, Pastorelli D, et al. Ramucirumab versus placebo as second-line treatment in patients with advanced hepatocellular carcinoma following first-line therapy with sorafenib (REACH): a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2015;16:859–70. [DOI] [PubMed] [Google Scholar]
- 59.Centanni M, Moes DJAR, Trocóniz IF, Ciccolini J, van Hasselt JGC. Clinical pharmacokinetics and pharmacodynamics of immune checkpoint inhibitors. Clin Pharmacokinet. 2019;58:835–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang AC, Orlowski RJ, Xu X, Mick R, George SM, Yan PK, et al. A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat Med. 2019;25:454–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kanesvaran R, Cordoba R, Maggiore R. Immunotherapy in Older Adults With Advanced Cancers: Implications for Clinical Decision-Making and Future Research. Am Soc Clin Oncol Educ Book. 2018;38:400–14. [DOI] [PubMed] [Google Scholar]
- 62.Chernyak V, Fowler KJ, Kamaya A, Kielar AZ, Elsayes KM, Bashir MR, et al. Liver Imaging Reporting and Data System (LI-RADS) Version 2018: Imaging of Hepatocellular Carcinoma in At-Risk Patients. Radiology. 2018;289:816–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kanwal F, Singal AG. Surveillance for hepatocellular carcinoma: current best practice and future direction. Gastroenterology. 2019;157:54–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Uehara T, Pogribny IP, Rusyn I. The DEN and CCl4 -Induced Mouse Model of Fibrosis and Inflammation-Associated Hepatocellular Carcinoma. Curr Protoc Pharmacol. 2014;66:14.30.1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tolba R, Kraus T, Liedtke C, Schwarz M, Weiskirchen R. Diethylnitrosamine (DEN)-induced carcinogenic liver injury in mice. Lab Anim. 2015;49:59–69. [DOI] [PubMed] [Google Scholar]
- 66.Romualdo GR, Prata GB, da Silva TC, Fernandes AAH, Moreno FS, Cogliati B, et al. Fibrosis-associated hepatocarcinogenesis revisited: Establishing standard medium-term chemically-induced male and female models. PLoS ONE. 2018;13:e0203879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118–27. [DOI] [PubMed] [Google Scholar]
- 68.Tward AD, Jones KD, Yant S, Cheung ST, Fan ST, Chen X, et al. Distinct pathways of genomic progression to benign and malignant tumors of the liver. Proc Natl Acad Sci USA. 2007;104:14771–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chow EK-H, Fan L, Chen X, Bishop JM. Oncogene-specific formation of chemoresistant murine hepatic cancer stem cells. Hepatology. 2012;56:1331–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen X, Calvisi DF. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am J Pathol. 2014;184:912–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.