

Abstract
Methane dehydroaromatization (MDA) on Mo/ZSM-5 zeolite catalyst provides a promising route for the direct transformation of natural gas into high-valued chemicals. Understanding the nature of active sites on Mo/ZSM-5 represents one of the challenges to be resolved for technical application of this process. Herein, by using 1H{95Mo} double-resonance solid-state NMR spectroscopy, we identify proximate dual active sites on Mo/ZSM-5 catalyst by direct observation of internuclear spatial interaction between Brønsted acid site and Mo species in zeolite channels. The acidic proton-Mo spatial interaction is correlated with methane conversion and aromatics formation in the MDA process, which is demonstrated to be an important factor in determining the catalyst activity and lifetime. The evolution of olefins and aromatics in Mo/ZSM-5 channels is monitored by detecting their host-guest interactions with both active Mo sites and Brønsted acid sites via 1H{95Mo} double-resonance and two-dimensional 1H-1H correlation NMR spectroscopy, which reveals the intermediate role of olefins in hydrocarbon pool process during the MDA reaction.
Keywords: zeolites, active sites, methane dehydroaromatization, host-gust interaction, solid state NMR
Entry for the Table of Contents
The bifunctional acidic proton-carburized Mo active site in Mo/ZSM-5 zeolite was revealed by probing their spatial proximity using double-resonance solid-state NMR spectroscopy. The spatial interaction of the dual site governs the activity of the catalyst for MDA reaction.
Introduction
Due to the depleting of crude oil and increasing availability of natural gas, direct conversion of methane, the main component of the natural gas, into value-added chemicals is of great academic and industrial interest. Methane dehydromatization (MDA) under non-oxidative condition on Mo and other transition metal (W, Fe, V, Cr, etc.) modified ZSM-5 zeolites has attracted increasing attention owing to the high selectivity to hydrocarbons particularly aromatics[1–4]. However, the MDA reaction is a thermodynamically unfavored process, which necessitates high reaction temperature, typically between 873 and 1073 K, to achieve equilibrium with ca. 10% conversion and up to 80% benzene selectivity[5, 6]. The commercial application of the MDA process is hampered by the rapid deactivation of catalysts[7–9]. Considerable research effort has been invested in improving the MDA catalyst and process with focus on the understanding of the active sites and reaction mechanism[10, 11].
Various spectroscopic techniques including NMR, EXAFS and Raman and theoretical calculations as well as kinetic analysis have been employed to reveal the active Mo-phase on Mo/ZSM-5[12–21]. It is generally accepted partially reduced and/or carburized Mo-oxo species in zeolite channels such as Mo2C and oxycarbidic Mo (MoOxCy) serve as the active sites while the Mo carbides on the external surface are less active. Although the exact structure of the active Mo species is still in debate, previous reports underlined the importance of the bifunctionality of Mo/ZMS-5 for the MDA reaction[22–25]: dehydrogenation and oligomerization of methane occur on active Mo sites forming C2 intermediates such as ethene and acetylene, followed by cyclization producing aromatics and naphthalene on the Brønsted acid sites (BAS) in zeolite. The subsequent transformation of hydrocarbons can be understood by the hydrocarbon pool mechanism that resembles the well-established organocatalytic process in methanol-to-hydrocarbon reaction[26–28]. The hydrocarbon pool can be simply considered as a catalytic scaffold in zeolite, constituted by the organics trapped in zeolite channels or cages, to which the reactant is added and products are removed in a catalytic cycle. The confined aromatics and their derivatives were proposed to play an important role in the hydrocarbon pool mechanism of the MDA reaction. The oxycarbidic Mo species initiates the hydrocarbon pool in the early stage[18, 26], while a close correlation between the benzene formation and the Brønsted acidity was found in the MDA reaction[22]. Nevertheless, how the Mo species and BAS function together to achieve a full activity is still unclear.
Concerning the acidity of zeolite, it was found that Mo-modified silicalite-1 exhibited low MDA activity[29], while methane activation by the BAS solely is negligible and the Mo species is necessary to assist in the formation of benzene[22]. Comparing the component alone, the mixture of Mo carbides with H-ZSM-5 zeolite exhibits higher methane conversion, but the yield of benzene is an order of magnitude less than on Mo/ZSM-5 with both exchanged Mo sites and BAS[22, 30]. The previous investigations manifested the crucial role of co-existence of Mo sites and BAS and their synergistic promotion in the reaction[31].
In addition to structure, the location/distribution of active sites define the catalytic performance of zeolite[32]. It was supposed that only the BAS that interacts with the active Mo sites is required for the MDA reaction and the free BAS exerts detrimental effect on the reaction by causing carbonaceous deposits[33]. The interaction between the two species would thus be a key factor in modulating their catalytic function[30]. Since the formation of different types of Mo species on the zeolite external surface of is often unavoidable, identification of active sites inside zeolite channels is a challenge for spectroscopic characterization.
Solid-state NMR spectroscopy has become a critical tool for characterizing the active sites on heterogeneous catalysts, capable of providing detailed information on correlation and connectivity of distinct nuclei in a local structure[34–38]. In this work, we established a fundamental understanding of the active sites on Mo/ZSM-5 for MDA reaction. The formation of active Mo species is correlated with proximate Brønsted acid site on zeolite. By using 1H{95Mo} double-resonance solid-state NMR spectroscopy, we provide first experimental evidence on the existence of strongly interacted acidic proton-Mo dual site on Mo/ZSM-5. The spatially proximate acidic proton-Mo sites correlate with catalyst activity for the MDA reaction. By detecting the interactions between hydrocarbons and active Mo sites/Brønsted acid sites, the intermediate role of olefins in the hydrocarbon pool process is revealed.
Results and Discussion
Brønsted acidity and distribution of protons on Mo/ZSM-5
Mo/ZSM-5 catalysts with Mo loading of 1~10 wt.% (denoted as 1Mo/ZSM-5~10Mo/ZSM-5) were prepared by incipient wetness impregnation of H-ZSM-5 zeolites (Si/Al=15) (see details in the Supporting Information). 95Mo (94.8%) enriched 5Mo/ZSM-5 samples were prepared for 95Mo MAS NMR and 1H{95Mo} S-RESPDOR NMR experiments. X-ray diffraction (XRD) analysis (Figure S1a) and 27Al MAS NMR spectroscopy (Figure S2) show that the crystal structures of ZSM-5 (MFI type) zeolites are well maintained for all the Mo/ZSM-5 samples and reacted 5Mo/ZSM-5 (Figure S1b). Crystalline MoO3 is detected at Mo loading higher than 5 wt.% (4.7 wt. % from ICP analysis), while the molybdenum species are well dispersed in ZSM-5 at lower Mo loadings. The typical methane conversion and product distribution were observed on the Mo/ZSM-5 samples for MDA reaction at 973 K (Figure S3). The relatively higher aromatics yields (benzene, toluene and naphthalene) indicates the better activity and stability of the 5Mo/ZSM-5 sample. All the following measurements were performed on this sample unless otherwise mentioned specifically.
TEM and N2 physisorption experiments indicate that Mo species exist both inside and outside of the channels of the fresh Mo/ZSM-5 (Tabel S1 and Figure S4–S5). The Brønsted acidity of the fresh Mo/ZSM-5 and the reacted samples was probed by 1H MAS NMR experiments. Three signals at 4.0, 2.6, and 1.7 ppm are observed on the parent H-ZSM-5 and fresh 5Mo/ZSM-5, ascribed to Brønsted acidic protons (SiOHAl, bridging hydroxyl group), extra-framework AlOH, and nonacidic SiOH[39], respectively (Figure 1, left panel). The acidic protons are significantly decreased on 5Mo/ZSM-5 as compared to the parent zeolite, with ca. 70% of them being replaced (Figure S6). This can be accounted for by the formation of exchanged Mo-oxo species on the conjugated base site of BAS inside the channels of ZSM-5. Slight decreasing density of the acidic protons (4.0 ppm) is observed in the reaction before 30 min, corresponding to the early stage of MDA (Figure S3). In this period, MoOx species is reduced by CH4 and transformed into active Mo2C and/or MoOxCy species, which is confirmed by the following 95Mo MAS NMR experiments. The high reaction temperature of 973 K further promotes the migration of the Mo species from the external surface into the channels of zeolite to replace more acidic protons[40, 41] (Figure S5 and S6). In the meantime, two new signals appear at 6.4 and 8.1 ppm, which can be ascribed to the formed olefins and aromatics with the assistance of 2D 1H-13C HETCOR NMR experiment (Figure S7). The prevailing formation of aromatics after 30 min of reaction time is evidenced by the increasing in the intensity of signal at 8.1 ppm. This is accompanied with the further decreasing of acid protons (4.0 ppm) (Figure S6), indicating the participation of BAS in methane aromatization[42]. Two-dimensional (2D) 1H−1H double-quantum-single-quantum (DQ-SQ) MAS NMR was used to characterize the proximity of protons on Mo/ZSM-5, which provides insight into the distribution of the protons of zeolites as well as the hydrocarbon products in zeolite channels. The two auto-correlation peaks at (1.7, 2×1.7) and (4.0, 2×4.0) ppm indicate the proximate silanols and BAS respectively in zeolite. The significant reduction in the peak intensity at 4.0 ppm on the Mo samples manifests the substitution of acidic protons by Mo species, which leads to the decreasing and isolation of the BAS. The proceeding of MDA reaction results in further decline of the proximate BAS due to either the formation of more exchanged Mo species on BAS or the consuming of acidic protons in the MDA. In the meantime, the close proximities of aromatics were observed with reaction, reflected by the increasing intensity in the autocorrelation peak at (8.1, 2×8.1) ppm. This indicates the accumulation of the bulk organic molecules in the confined space of zeolite, which can be correlated with the rapid catalyst deactivation (Figure S3). Our previous investigation on catalyst deactivation showed that the intermolecular spatial proximities/interactions in zeolite channels favor the coupling of aromatics and derivatives to form aphthalene or polyaromatics as precursor to coke[43].
Figure 1.

1H MAS NMR (left) and 1H-1H DQ-SQ (right) MAS NMR spectra (acquired at 9.4 T) of H-ZSM-5, fresh 5Mo/ZSM-5 and reacted 5Mo/ZSM-5 samples for 10–120 min at 973 K.
Proximity between protons and Mo sites on Mo/ZSM-5
Active Mo species on Mo/ZSM-5 was studied by solid-state NMR experiments. Both NMR active Mo nuclei 95Mo and 97Mo are quadrupolar nuclei of spin 5/2. Due to the low gyromagnetic ratio nature, γ(95Mo) =−1.751•107 rad T−1 s−1 and γ(97Mo) = −1.78•107 rad T−1 s−1, sensitivity-enhanced WURST-QCPMG method was employed in combination with ultra-high magnetic field of 35.2 T (1.5 GHz) which shows advantages for high-resolution NMR spectroscopy of half-integer quadrupolar nuclei[44–48]. The 95Mo WURST-QCPMG MAS NMR spectrum acquired at 35.2 T of the fresh 5Mo/ZSM-5 exhibits a broad quadrupolar peak (CQ ≈ 5.5 MHz) with an isotropic chemical shift (δiso) of ca. −35 ppm and an anisotropic chemical shift (CSA) of ca. 320 ppm obtained by fitting the spectrum with consideration of both quadrupolar interaction and CSA (Figure 2, Table 1). According to the δiso, this Mo site could be assigned to MoO3-like species[49], while the larger CQ compared to the bulk MoO3 in orthorhombic phase is indicative of asymmetric local structure, which can be accounted for by the high dispersion of the Mo-oxo species in zeolite channels and on the external surface. After reacting for 60 min, a new and dramatically broad resonance with CQ ≈ 32 MHz and δiso ≈ 800 ppm (± 50 ppm) is detectable in the 95Mo MAS NMR spectrum of 5Mo/ZSM-5–60 sample. The broad 95Mo NMR peak with similar δiso to that of Mo2C compound in orthorhombic phase[49] is most likely due to the formation of the active Mo2C or MoOxCy species in zeolite channels in the MDA. However, the large second-order quadrupolar broadening of the line shape indicates the extreme heterogeneity and structural distortion in the local Mo site. Although the exact Mo specification is not determined, this demonstrates the strong interactions of the active Mo species with the anchoring Brønsted acid sites, which produces large electric field gradient and corresponding CQ on the Mo nuclei.
Figure 2.

Experimental and deconvoluted 95Mo QCPMG MAS NMR spectra (acquired at 35.2 T) of fresh 5Mo/ZSM-5 and 5Mo/ZSM-5 reacted for 60 min at 973 K. Solid (―): experimental, solid (―): simulated, dash (―, ―): deconvoluted.
Table 1.
NMR parameters extracted from 95Mo QCPMG NMR spectra (acquired at 35.2 T) of 5Mo/ZSM-5 and 5Mo/ZSM-5 reacted for 60 min of MDA reaction.
| Sample | Component | δiso(ppm) | CQ(MHz) | η |
|---|---|---|---|---|
| 5Mo/ZSM-5 | Mo-oxo | − 35± 5 | 5.5 | 0.7~1.0 |
| 60min | Mo-oxo | − 35± 5 | 5.5 | 0.7~1.0 |
| Mo2C/MoOxCy | 800± 50 | 32 | 0.9 | |
δiso: isotropic chemical shift, CQ: quadrupolar coupling constant, η: asymmetry factor.
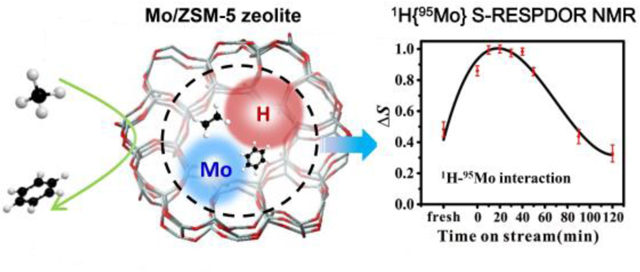
To provide direct experimental evidence on the spatial proximity between protons and active molybdenum species, 1H{95Mo} S-RESPDOR solid-state NMR experiments[50] were performed to measure internuclear dipolar interaction/distances on 5Mo/ZSM-5 reacted for different reaction time at 973 K. The signal of protons in close proximity to Mo atoms will be modulated by 1H-95Mo dipolar interaction upon 95Mo irradiation (S0 spectrum), resulting in decreased 1H signal intensity (S spectrum). The 1H signal from Brønsted acidic protons (4.0 ppm) is observed in the difference spectrum (ΔS= S0-S) of the fresh sample (Figure 3a). This evidences the dipolar interactions and spatial proximity between the acidic protons and Mo species (Mo2C or MoOxCy species) in zeolite channels. The AlOH (2.6 ppm) is also observed, while the SiOH (1.7 ppm) group is invisible in the difference spectrum, due to the absence of close proximities between this species with Mo site. On the reacted samples (30 and 120 min), the proximity between acidic protons (4.0 ppm) and Mo species is still present as demonstrated by the 1H−95Mo dipolar dephasing effect (Figure 3b–3c). Interestingly, the 1H signals from aromatic (8.1 ppm) and olefins (6.4 ppm) also appear in the difference spectra. This observation demonstrates that these hydrocarbons are formed near the active Mo sites in zeolite channels, indicating the catalytic role of the proximate acidic proton-Mo species in the MDA reaction.
Figure 3.

1H{95Mo} S-RESPDOR NMR spectra (acquired at 18.8 T) of (a) fresh 5Mo/ZSM-5, (b) 5Mo/ZSM-5 reacted for 30 min and (c) for 120 min of MAD reaction at 973 K. Recoupling time is 5.12 ms. Normalized ΔS of (d) Brønsted acid, (e) olefins and (f) aromatics versus MDA reaction time. ΔS= S0-S.
Since the concentration of acidic protons shown on the S0 spectrum is decreasing with reaction (Figure S6), we use the dephasing effect ΔS expressed as (S0-S) (where S and S0 represent the signal intensity with and without 95Mo irradiation respectively) to show the evolution of proximate acidic proton-Mo sites with reaction time in the MDA. In principle, for a fixed recoupling time in the upward trend of the buildup curve, ΔS reflects the degree of 1H-95Mo dipolar interaction/spatial proximity between the two species. A significant increase of ΔS for acidic protons on the reacted samples in the initial period (before 30 min) compared to the fresh sample indicates their stronger interactions with Mo species in zeolite. The carburization process promotes the migration of MoO3 from the external surface into zeolite channels (Figure 2) and formation of more proximate acidic proton-Mo sites. We further measured the 1H-95Mo internuclear distance for acidic protons by simulating the 1H{95Mo} S-RESPDOR dephasing built-up curve using analytical formula[50] (Figure 4). The yielded internuclear distance is 4.0±0.3 Å, which is slightly smaller than that of the proximate BAS (ca. 5 Å) in ZSM-5[51]. This close acidic proton-Mo proximity results in the strong spatial interactions between the two species. The generation of proximate acidic proton-Mo site also explains the disappearance of the neighboring BAS in the 1H-1H DQ-SQ NMR (Figure 1). The spatially interacted acidic proton-Mo species endows the Mo/ZSM-5 catalyst with synergistic bifunctionality, which correlates well with the MDA performance (Figure S3). For example, the yield of aromatics (e.g., benzene) follows a similar trend to the evolution of acidic proton-Mo interaction that is built up in the initial period (30 min of TOS) and decreasing at longer reaction time. It is plausible that the MDA proceeds via a concerted pathway enabled by the co-existence of BAS and metal site within molecular distances[52].
Figure 4.

Experimental 1H{95Mo} S-RESPDOR dephasing fraction of Brønsted acidic protons in 5Mo/ZSM-5 as a function of recoupling time. The best fit of the experimental results was determined to be f=0.15 and DIS=120 Hz, corresponding to a 1H-95Mo internuclear distance of 4.0 Å. f denotes the pre-factor parameter and DIS is the heteronuclear dipolar interaction between two spins used in the analytical formula. Sim. and exp. represent simulated and experimental curves, respectively.
The spatial interaction/proximity between Mo and acidic protons reflected by the decay of ΔS is decreasing at longer reaction time for two reasons. First, the acidic protons are consuming with reaction (Figure S6), leading to the decrease of the amount of the proximate acidic proton-Mo sites. Second, the active Mo species may detach from the BAS and form bulk Mo2C on the external surface of zeolite [53] (Figure S5). This would make the Mo species and the acidic protons distantly separated. Note that the ΔS of olefins shows a similar trend (Figure 3e). The larger ΔS is produced by the strong interaction between these species with Mo in the initial reaction stage, a clear indication of forming hydrocarbons around the acidic proton-Mo sites. The subsequent diffusion and consuming of olefins lead to the decreasing of their interactions with the active Mo sites, reflected by the declining ΔS. On the contrary, aromatics exhibit different ΔS variation behavior, increasing with reaction time (Figure 3f), indicative of the closer proximity and stronger interactions between aromatics and Mo sites. The formation and accumulation of aromatics and their derivatives (particularly polyaromatics nearby the Mo sites in zeolite channels) should be the main reason for this trend, which eventually results in catalyst deactivation.
Evolution of hydrocarbon pool species on Mo/ZSM-5
By analogy to methanol conversion on acidic zeolite, olefins and aromatics and derivatives in the MDA reaction may complex with the active sites propagating the catalytic cycles where carbocations or radicals would be involved as intermediates[26, 54]. 2D 1H-1H spin diffusion experiments were performed on the reacted 5Mo/ZSM-5 samples, which allow to correlate the development of the organic compounds with active sites in catalyst, thus giving information on their role in the MDA reaction. Only cross peaks between Brønsted acidic protons (4.0 ppm) and olefins (6.4 ppm) can be observed on the sample reacted for 10 min (Figure 5a), demonstrating the short-range spatial proximity between these protons. Along with the concurrent formation of aromatics (Figure 1 and Figure S3), this experiment indicates that olefins might be firstly produced in the initial stage. The analysis of the gas product shows that ethene dominates the light hydrocarbons before 60 min of reaction time (Table S2). Although the initial C-C bond formation in the MDA is still unclear[55], the observation of the proximity between olefins and acidic protons provides direct evidence for the further dehydrocyclization of olefins to aromatics on BAS in close proximity to Mo site. New cross peak between Brønsted acidic proton (4.0 ppm) and aromatics (8.1 ppm) appears at 30 min of reaction time (Figure 5b), which is absent in the spectrum with short mixing time (50 ms) (Figure S8). This suggests the location of aromatics in a medium-range spatial proximity to the BAS, resulting in the unfavorable polarization-transfer between these species by spin diffusion. Their correlation is enhanced with reaction, reflected by the increasing of intensity in the cross peak (Figure 5c and 5d). The prevailing formation of aromatics leads to the accumulation of aromatics and other organics in zeolite channels, which is demonstrated by the signal broadening. More aromatics produced and distributed around the active acidic proton-Mo sites promotes spin diffusion between proximate protons, enabling the observation of stronger cross peak for aromatics, which is also supported by the above 1H{95Mo} S-RESPDOR NMR experiment (Figure 3f). On the other hand, the correlation of olefins with acidic protons becomes weakened as evidenced by the reduced cross peak intensity (Figure 5b–d). This can be explained by the consuming of olefins in zeolite channels on catalyst deactivation, in agreement with the 1H{95Mo} S-RESPDOR result (Figure 3e). Taken together with the product analysis, these data suggest the intermediate role of olefins in the hydrocarbon pool process during the MDA reaction: olefins produced in the initial reaction period are expected to link the methane reactant and aromatics product. Although the proximate aromatics and acidic protons would facilitate olefins production via the aromatics-based reaction cycle as established in the hydrocarbon pool mechanism[27, 28], the formation of polyaromatics would be favored on the BAS, leading to the formation of catalyst deactivating species such as naphthalene.
Figure 5.

2D 1H-1H spin diffusion NMR spectra (acquired at 9.4 T) of 5Mo/ZSM-5 reacted at 973 K for (a) 10 min, (b) 30 min, (c) 60 min and (d) 120 min. Mixing time is 300 ms.
The determination of the active sites and their functions benefits a better understanding of the MDA reaction mechanism. Probing the spatial proximity between the acidic proton-Mo sites allows for observation and determination of the complex reaction center, constituted by the strongly interacted Mo2C/MoOxCy and Brønsted acid sites which reside on the neighboring conjugated base sites in Mo/ZSM-5 channels. The catalytic activity for methane conversion and the production of aromatics are well correlated with the interaction strength/spatial proximity between the acidic proton-Mo sites. This suggests that a concerted reaction route for methane activation and transformation would likely occur in the MDA due to the synergistic effect generated by the proximate acidic-Mo sites. Moreover, the proximity of the acidic proton-Mo sites is shown to be a key factor in determining the catalyst lifetime as the migration of Mo species to the zeolite external surface leads to quick catalyst deactivation. In the meantime, the remained BAS in zeolite channels is deactivated by polyaromatics. Light olefins such as ethene mostly generated in the initial stage of the MDA are found to strongly interact with the proximate acidic proton-Mo sites and subsequently transform into aromatics. From the host-guest point of view, the intermediate role of olefins for the formation of aromatics in the proposed hydrocarbon pool mechanism is ascertained.
Conclusions
The active sites on Mo/ZSM-5 zeolite for methane dehydroaromatization reaction were investigated by solid state NMR spectroscopy. The spatial interaction between the Brønsted acid sites and active Mo sites in zeolite channels has been identified on Mo/ZSM-5 catalyst, providing direct evidence on the synergistic bifunctionality of the catalyst for the MDA reaction. The strongly interacted acidic proton-Mo site correlates with the products formation and catalyst lifetime, which shows the important implication in modulating the catalytic MDA performance by taking advantage of the proximity of the acidic proton-Mo sites at molecular level. The hydrocarbon pool process in MDA is revealed by the analysis of the host-guest interactions between the trapped olefins/aromatics and Brønsted acid sites associated with Mo sites. Light olefins dominant in the early reaction stage and having strong interactions with the proximate acidic proton-Mo sites are connected with the formation of aromatics. The reduction of the acidic proton-Mo proximity in zeolite channels and increasing interactions between aromatics/their derivatives and Brønsted acid sites evidence catalyst deactivation. The results presented herein provide new insights into the understanding of the active sites on Mo/ZSM-5 catalyst as well as the hydrocarbon pool chemistry in MDA reaction.
Supplementary Material
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants 21733013, U1932218, 22061130202, 21872170), key program for frontier science of the Chinese Academy of Sciences (QYZDB-SSW-SLH027). A part of the NMR measurement was performed at the National High Magnetic Field Laboratory (http://www.nationalmaglab.org) supported by the National Science Foundation of the US through NSF/DMR-1644779 and the State of Florida. Development of the SCH magnet and NMR instrumentation was supported by NSF (DMR-1039938 and DMR-0603042) and NIH P41 GM122698.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Conflict of interest
The authors declare no conflict of interest.
References
- [1].Wang L, Tao L, Xie M, Xu G, Huang J, Xu Y, Cata. Lett. 1993, 21, 35–41. [Google Scholar]
- [2].Spivey JJ, Hutchings G, Chem. Soc. Rev. 2014, 43, 792–803. [DOI] [PubMed] [Google Scholar]
- [3].Weckhuysen BM, Wang D, Rosynek MP, Lunsford JH, J. Catal. 1998, 175, 347–351. [Google Scholar]
- [4].Weckhuysen BM, Wang D, Rosynek MP, Lunsford JH, J. Catal. 1998, 175, 338–346. [Google Scholar]
- [5].Xu Y, Bao X, Lin L, J. Catal. 2003, 216, 386–395. [Google Scholar]
- [6].Ismagilov ZR, Matus EV, Tsikoza LT, Energy Environ. Sci. 2008, 1, 526–541. [Google Scholar]
- [7].Cao Z, Jiang H, Luo H, Baumann S, Meulenberg WA, Assmann J, Mleczko L, Liu Y, Caro J, Angew. Chem. Int. Ed. 2013, 52, 13794–13797; Angew. Chem. 2013, 125, 14039–14042. [DOI] [PubMed] [Google Scholar]
- [8].Kosinov N, Coumans FJ, Uslamin E, Kapteijn F, Hensen EJ, Angew. Chem. Int. Ed. 2016, 55, 15086–15090; Angew. Chem. 2016, 128, 15310–15314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Morejudo SH, Zanón R, Escolástico S, Yuste-Tirados I, Malerød-Fjeld H, Vestre PK, Coors WG, Martínez A, Norby T, Serra JM, Kjølseth C, Science 2016, 353, 563–566. [DOI] [PubMed] [Google Scholar]
- [10].Vollmer I, Yarulina I, Kapteijn F, Gascon J, ChemCatChem 2019, 11, 39–52. [Google Scholar]
- [11].Kosinov N, Hensen EJM, Adv. Mater. 2020, 32, 2002565. [DOI] [PubMed] [Google Scholar]
- [12].Liu H, Shen W, Bao X, Xu Y, J. Mol. Catal. A 2006, 244, 229–236. [Google Scholar]
- [13].Zheng H, Ma D, Bao X, Hu JZ, Kwak JH, Wang Y, Peden CHF, J. Am. Chem. Soc. 2008, 130, 3722–3723. [DOI] [PubMed] [Google Scholar]
- [14].Lezcano-González I, Oord R, Rovezzi M, Glatzel P, Botchway SW, Weckhuysen BM, Beale AM, Angew. Chem. Int. Ed. 2016, 55, 5215–5219; Angew. Chem. 2016, 128, 5301–5305; [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Li G, Vollmer I, Liu C, Gascon J, Pidko EA, ACS Catal. 2019, 9, 8731–8737. [Google Scholar]
- [16].Vollmer I, van der Linden B, Ould-Chikh S, Aguilar-Tapia A, Yarulina I, Abou-Hamad E, Sneider YG, Olivos Suarez AI, Hazemann JL, Kapteijn F, Gascon J, Chem. Sci. 2018, 9, 4801–4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Vollmer I, Ould-Chikh S, Aguilar-Tapia A, Li G, Pidko E, Hazemann J-L, Kapteijn F, Gascon J, J. Am. Chem. Soc. 2019, 141, 18814–18824. [DOI] [PubMed] [Google Scholar]
- [18].Vollmer I, Kosinov N, Szécsényi Á, Li G, Yarulina I, Abou-Hamad E, Gurinov A, Ould-Chikh S, Aguilar-Tapia A, Hazemann J-L, Pidko E, Hensen E, Kapteijn F, Gascon J, Catal J. 2019, 370, 321–331. [Google Scholar]
- [19].Razdan NK, Bhan A, Catal J. 2020, 389, 667–676. [Google Scholar]
- [20].Ding W, Li S, D Meitzner G, Iglesia E, J. Phys. Chem. B 2001, 105, 506–513. [Google Scholar]
- [21].Agote-Arán M, Fletcher RE, Briceno M, Kroner AB, Sazanovich IV, Slater B, Rivas ME, Smith AWJ, Collier P, Lezcano-González I, Beale AM, ChemCatChem 2020, 12, 294–304. [Google Scholar]
- [22].Liu S, Wang L, Ohnishi R, Ichikawa M, J. Catal. 1999, 181, 175–188. [Google Scholar]
- [23].Liu W, Xu Y, J. Catal. 1999, 185, 386–392. [Google Scholar]
- [24].Han SJ, Kim SK, Hwang A, Kim S, Hong D-Y, Kwak G, Jun K-W, Kim YT, Appl. Catal. B 2019, 241, 305–318. [Google Scholar]
- [25].Vollmer I, Mondal A, Yarulina I, Abou-Hamad E, Kapteijn F, Gascon J, Appl. Catal. A 2019, 574, 144–150. [Google Scholar]
- [26].Kosinov N, Wijpkema ASG, Uslamin E, Rohling R, Coumans FJAG, Mezari B, Parastaev A, Poryvaev AS, Fedin MV, Pidko EA, Hensen EJM, Angew. Chem. Int. Ed. 2018, 57, 1016–1020; Angew. Chem. 2018, 130, 1028–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Olsbye U, Svelle S, Bjørgen M, Beato P, Janssens TVW, Joensen F, Bordiga S, Lillerud KP, Angew. Chem. Int. Ed. 2012, 51, 5810–5831; Angew. Chem. 2012, 124, 5910–5933. [DOI] [PubMed] [Google Scholar]
- [28].Wang C, Xu J, Deng F, ChemCatChem 2020, 12, 965–980. [Google Scholar]
- [29].Kosinov N, Coumans FJAG, Uslamin EA, Wijpkema ASG, Mezari B, Hensen EJM, ACS Catal. 2017, 7, 520–529. [Google Scholar]
- [30].Liu H, Bao X, Xu Y, J. Catal 2006, 239, 441–450. [Google Scholar]
- [31].Chen L, Lin L, Xu Z, Li X, Zhang T, J. Catal. 1995, 157, 190–200. [Google Scholar]
- [32].Buurmans ILC, Weckhuysen BM, Nat. Chem. 2012, 4, 873–886. [DOI] [PubMed] [Google Scholar]
- [33].Liu H, Li Y, Shen W, Bao X, Xu Y, Catal. Today 2004, 93–95, 65–73. [Google Scholar]
- [34].Xu J, Wang Q, Deng F, Acc. Chem. Res. 2019, 52, 2179–2189. [DOI] [PubMed] [Google Scholar]
- [35].Copéret C, Liao W-C, Gordon CP, Ong T-C, J. Am. Chem. Soc. 2017, 139, 10588–10596. [DOI] [PubMed] [Google Scholar]
- [36].Hunger M, Catal Rev Sci Eng 1997, 39, 345–393. [Google Scholar]
- [37].Jiang Y, Huang J, Dai W, Hunger M, Solid State Nucl. Magn. Reson. 2011, 39, 116–141. [DOI] [PubMed] [Google Scholar]
- [38].Wang Z, Li T, Jiang Y, Lafon O, Liu Z, Trébosc J, Baiker A, Amoureux J-P, Huang J, Nat. Commun. 2020, 11, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hunger M, Solid State Nucl. Magn. Reson. 1996, 6, 1–29. [DOI] [PubMed] [Google Scholar]
- [40].Ma D, Zhang W, Shu Y, Liu X, Xu Y, Bao X, Catal. Lett. 2000, 66, 155–160. [Google Scholar]
- [41].Liu H, Shen W, Bao X, Xu Y, Appl. Catal. A 2005, 295, 79–88. [Google Scholar]
- [42].Ma D, Shu Y, Zhang W, Xiuwen Han, Xu Y, Bao X, Angew. Chem. Int. Ed. 2000, 39, 2928–2931; Angew. Chem. 2000, 112, 3050–3053. [PubMed] [Google Scholar]
- [43].Wang C, Hu M, Chu Y, Zhou X, Wang Q, Qi G, Li S, Xu J, Deng F, Angew. Chem. Int. Ed. 2020, 59, 7198–7202; Angew. Chem. 2020, 132, 7265–7269. [DOI] [PubMed] [Google Scholar]
- [44].Dorn RW, Cendejas MC, Chen K, Hung I, Altvater NR, McDermott WP, Gan Z, Hermans I, Rossini AJ, ACS Catal. 2020, 10, 13852–13866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Chen K, Horstmeier S, Nguyen VT, Wang B, Crossley SP, Pham T, Gan Z, Hung I, White JL, J. Am. Chem. Soc. 2020, 142, 7514–7523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wang Q, Li W, Hung I, Mentink-Vigier F, Wang X, Qi G, Wang X, Gan Z, Xu J, Deng F, Nat. Commun. 2020, 11, 3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Martins V, Xu J, Wang X, Chen K, Hung I, Gan Z, Gervais C, Bonhomme C, Jiang S, Zheng A, Lucier BEG, Huang Y, J. Am. Chem. Soc. 2020, 142, 14877–14889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gan Z, Hung I, Wang X, Paulino J, Wu G, Litvak IM, Gor’kov PL, Brey WW, Lendi P, Schiano JL, Bird MD, Dixon IR, Toth J, Boebinger GS, Cross TA, J. Magn. Reson. 2017, 284, 125–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bastow TJ, Solid State Nucl. Magn. Reson. 1998, 12, 191–199. [DOI] [PubMed] [Google Scholar]
- [50].Chen L, Wang Q, Hu B, Lafon O, Trébosc J, Deng F, Amoureux J-P, Phys. Chem. Chem. Phys. 2010, 12, 9395–9405. [DOI] [PubMed] [Google Scholar]
- [51].Bernauer M, Tabor E, Pashkova V, Kaucký D, Sobalík Z, Wichterlová B, Dedecek J, J. Catal. 2016, 344, 157–172. [Google Scholar]
- [52].Iglesia E, Barton DG, Biscardi JA, Gines MJL, Soled SL, Catal. Today 1997, 38, 339–360. [Google Scholar]
- [53].Agote-Arán M, Kroner AB, Islam HU, Sławiński WA, Wragg DS, Lezcano-González I, Beale AM, ChemCatChem 2019, 11, 473–480. [Google Scholar]
- [54].Wang C, Wang Q, Xu J, Qi G, Gao P, Wang W, Zou Y, Feng N, Liu X, Deng F, Angew. Chem. Int. Ed. 2016, 55, 2507–2511; Angew. Chem. 2016, 128, 2553–2557. [DOI] [PubMed] [Google Scholar]
- [55].Çağlayan M, Lucini Paioni A, Abou-Hamad E, Shterk G, Pustovarenko A, Baldus M, Chowdhury AD, Gascon J, Angew. Chem. Int. Ed. 2020, 59, 16741–16746; Angew. Chem. 2020, 132, 16884–16889. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.