

Abstract
Multiple sclerosis (MS) is an autoimmune inflammatory disease of the CNS driven by the inflammatory activity of peripheral immune cells recruited to the CNS and by CNS-resident glial cells. MS pathogenesis has been linked to both genetic and environmental factors. In addition, the commensal flora have been shown to modulate immune processes relevant to MS pathogenesis. We discuss the effects of the gut microbiota on T cells and glial cells, and their relevance for the control of inflammation and neurodegeneration in MS. A better understanding of the gut–CNS axis will shed new light on the mechanisms of disease pathogenesis, and may help to guide the development of efficacious therapies for MS.
The Gut–CNS Axis in Multiple Sclerosis
The concept of a ‘gut–CNS axis’ linking the gut and the CNS was put forward more than 200 years ago when physicians acknowledged that the gastrointestinal and mental status were somehow connected [1]. Recent advances in neuroimmunology, gastroenterology, and microbiology support this concept and have provided several mechanistic insights, which we discuss below in the context of MS.
MS is an autoimmune disease which targets the CNS [2]. Genetic and environmental factors, as well as the microbiome, play important roles in MS pathogenesis (Box 1). Indeed, recent studies suggest that the course of MS is influenced by the effects of the commensal microbiome on the gut–CNS axis. Although specific microorganisms, cell types, and metabolites that participate in this gut–CNS axis have been identified, little is known about the underlying mechanisms. Identifying these mechanisms may guide the development of novel therapeutic approaches for MS and other neurologic disorders. In this review we discuss our current understanding of the role of the gut–CNS axis in MS and the mechanisms involved.
Box 1. MS Is a Multifactorial Inflammatory Disease Targeting the CNS.
MS is the most prevalent chronic inflammatory disease of the CNS, and affects more than 2 million people worldwide. Most MS patients initially experience relapsing-remitting (RRMS) neurological symptoms at between 20 and 40 years of age [2]. Within 10 years from disease onset, ~30–40% of MS patients transition into secondary progressive MS (SPMS) that is characterized by irreversible and progressive accumulation of neurological disability [135]. Various genetic and environmental factors have been linked to MS development. For example, genetic studies have identified SNPs that are associated with adaptive immunity in MS [136–138]. However, the concordance rate for monozygotic twins is ~30%, highlighting the role of environmental factors in disease development. Multiple environmental factors associated to MS have been identified in epidemiological studies, including geographic latitude, smoking, obesity, and infection by viruses such as Epstein–Barr virus [139,140]. In addition, the gut microbiome has been identified as an important factor contributing to MS pathogenesis by modulating the gut–CNS axis, and likely contributes to the increased MS prevalence in developed countries [141].
Autoimmune Pathogenesis of MS: Role of T Cells and Glia
Genetic and immunologic studies in MS and its animal model experimental autoimmune encephalomyelitis (EAE, see Glossary), as well as the clinical success of therapies targeting the immune system, have established T cell autoimmunity as an important contributor to MS pathogenesis.
Approximately 100 billion nonregenerative neurons form complex circuits in the CNS, and these are difficult to repair following damage. Consequently, inflammatory responses that could potentially trigger neurodegeneration are tightly controlled. Following their maturation in the thymus, CD4+ T cells circulate systemically as ‘naïve’ T cells that lack effector function. Despite the elimination of self-reactive clones in the thymus, the mature T cell repertoire includes clones that are reactive against self-antigens present in myelin and other CNS structures. When naïve T cells are stimulated by antigens presented by professional antigen-presenting cells (APCs), cytokines and other signals provided by the microenvironment in lymphoid tissues promote the differentiation of T cells into multiple subsets. Each T cell subset is characterized by the expression of specific transcription factors, cytokines, and immune-regulatory and tissue-homing molecules that contribute to physiological functions of adaptive immunity, such as combating pathogenic microorganisms or regulating the immune response to prevent immunopathology.
In the context of MS, myelin-reactive T cells with a proinflammatory phenotype, particularly type 1 (Th1) and type 17 (Th17) T helper cells, gain access to the CNS where they are reactivated to initiate tissue damage [3,4]. Conversely, regulatory T cells (Tregs) such as Foxp3+ or Tr1 cells limit CNS inflammation [5–12]. Thus, the balance between myelin-reactive effector and Tregs is thought to control CNS lesion formation in MS. Other cell types such as CD8+ T cells, γδ T cells, B cells, natural killer T (NKT) cells, and mucosa-associated invariant T (MAIT) cells may also participate in the control of CNS inflammation [13–17]. This balance between pro- and anti-inflammatory immune elements is continuously influenced by the gut microbiome. In the following we provide a brief overview of recruited and CNS-resident cell types involved in inflammation and neurodegeneration in MS.
T cells play important roles in MS pathogenesis, particularly during the relapsing-remitting MS (RRMS) phase of the disease (Box 1 for overview of MS and disease phases). However, CNS-resident glial cells, especially astrocytes and microglia, also contribute to MS pathogenesis [9,18–25], particularly during the secondary progressive MS (SPMS) phase of the disease [2]. Indeed, the ‘plaques’ that characterize MS – focal lesions of active demyelination, neurodegeneration, and leukocyte infiltration – are characterized by the accumulation of activated microglia and reactive astrocytes [26,27]. The gut microbiota has also emerged as an environmental regulator of astrocytic and microglial function.
Microglia are brain-resident macrophages that originate from yolk sac precursors that seed the CNS during fetal development [28]. Under homeostatic conditions, microglia participate in synapse maturation and myelinogenesis, secrete neurotrophic factors, phagocytose debris, and generally survey the CNS environment searching for pathogenic insults. However, in the context of CNS inflammation, microglia produce inflammatory cytokines and chemokines, and upregulate co-stimulatory molecules such as MHC class I/II, suggesting their involvement in the recruitment and activation of T cells, monocytes, and other leukocytes, although the role of these molecules in microglia activation during CNS pathology has not been fully characterized [28–30]. In this respect, deletion of MHC class II molecules in microglia does not affect the development of acute EAE, suggesting that microglia participate in MS pathology through mechanisms other than regulating the T cell response [31]. For example, microglia induce proinflammatory and neurotoxic activities in astrocytes during T cell-mediated autoimmune inflammation [21,32,33].
Astrocytes are the most abundant glial cells in the CNS, and play active roles in development, ion homeostasis, pH and neurotransmitter regulation, control of synaptic function, and brain metabolism, as well as in regulating blood flow and the blood–brain barrier (BBB) [34,35].
Astrocytes have also been identified as key players in pro- and anti-inflammatory processes [20,34,36,37]. Astrocytes form the ‘glia limitans’ that lines all interfaces between the CNS paren-chyma and the pia matter, and that regulates the access of immune cells and small molecules to the CNS [38]. Indeed, astrocytes produce various chemokines such as CCL2 and CXCL10 that recruit T cells, monocytes, and other immune cells to the CNS [39,40]. Astrocytes also control BBB stability and function by producing molecules such as vascular endothelial growth factor (VEGF) and nitric oxide (NO) to facilitate leukocyte extravasation. Astrocytes produce proinflammatory cytokines such as tumor necrosis factor (TNF)-α, interleukin (IL)-6, and IL-1β, as well as the immunoregulatory cytokines IL-10, IL-27, and transforming growth factor (TGF)-β, to boost or suppress inflammation, respectively [34]. In addition, ‘reactive’ astrocytes driven by STAT3 [41] respond to CNS damage by forming an astrocytic scar that limits inflammation and damage [42–44]. However, reactive astrocytes can also adopt a neurotoxic phenotype in response to soluble factors secreted by microglia, and this is thought to contribute to the pathogenesis of MS and other neurologic diseases [32]. Finally, astrocytes may also limit neuronal damage by their ability to support the metabolic needs of neurons, and also through the production of neurotrophic factors [18,45–47]. Thus, astrocytes have complex proinflammatory/neurotoxic and anti-inflammatory/neuroprotective roles during CNS inflammation.
In summary, complex interactions between CNS-resident and recruited peripheral cells control inflammation and neurodegeneration in MS. The activity of each of these cell types, as well as their interactions, are controlled by the microbiome. Thus, changes in the commensal flora are likely to impact on multiple aspects of MS pathology.
The Gut Microbiota in MS and EAE
Mammals coevolved with a vast number of commensal gut microbiota. It is estimated that, in humans, the internal surface area of the gastrointestinal tract harbors >100 trillion microbial cells belonging to >1000 bacterial species [48]. Thus, it is not surprising that the microbiome plays important roles in multiple aspects of physiology, including the regulation of immune system development and function [49]. Indeed, perturbations in commensal communities (referred to as dysbiosis) have been linked to multiple inflammatory conditions [50].
The development of new sequencing technologies has enabled the analysis of bacterial 16S rRNA sequences to identify components of the microbiota without the need to culture them. This technological development has facilitated comprehensive investigation of the gut microbiome in MS (Table 1) [51–59] and EAE.
Table 1.
Microbiome Studies in MSa
| Country/region | Subjects | Methods for 16S rRNA sequencing | Gut microbiota changes in MS | Immunological findings | Refs |
|---|---|---|---|---|---|
| Japan | 20 RRMS 40 HCs |
Roche 454, V1–V2 regions | ↓ Clostridial species (such as Faecalibacterium prausnitzii) belonging to Clostridia clusters XIVa and IV and Bacteroidetes | N/A | [51] |
| North America | Children under 18 18 RRMS 17 HCs |
Illumina MiSeq, V4 region | ↑ Desulfovibrionaceae ↓ Lachnospiraceae ↓ Ruminococcaceae |
N/A | [52] |
| North America | 60 RRMS 43 HCs |
Roche 454, V3–V5 regions Illumina MiSeq, V4 region |
↑ Methanobrevibacter ↑ Akkermansia ↓ Butyricimonas |
Bacterial changes in MS correlated with inflammatory gene expression in peripheral blood T cells and monocytes | [53] |
| North America | 31 RRMS 36 HCs |
Illumina MiSeq, V3–V5 regions | ↑ Pseudomonas ↑ Mycoplana ↑ Haemophilus ↑ Blautia ↑ Dorea ↓ Parabacteroides ↓ Adlercreutzia ↓ Prevotella |
N/A | [54] |
| North America | Children under 18 15 RRMS 9 HCs |
Illumina MiSeq, V4 region | N/A | Bacterial richness positively, and Bacteroidetes abundance inversely, correlated with Th17 in peripheral blood of MS. Fusobacteria correlated with regulatory T cells in HCs | [55] |
| Germany | Monozygotic twins 34 MS (3 CIS, 22 RRMS, 7 SPMS, 2 PPMS) 34 HCs (counterparts) |
Roche 454, V3–V5 regions | ↑Akkermansia muciniphila | Higher incidence of spontaneous EAE upon MS fecal transfer. IL-10 reduction from peripheral blood CD4+ T cells in MS compared with HCs | [56] |
| North America | 71 RRMS (untreated) 71 HCs |
Earth Microbiome Project standard protocol, V4 region | ↑ Akkermansia muciniphila ↑ Acinetobacter calcoacetius ↓ Parabacteroides distasonis |
Exacerbated EAE by MS fecal transfer to GF mice compared with HC. Decreased IL-10+ regulatory T cells in mesenteric lymph nodes of MS fecal transferred EAE mice. | [57] |
Symbols and abbreviations: ↑ increased abundance; ↓ decreased abundance; CIS, clinically isolated symptom; EAE, experimental autoimmune encephalomyelitis; GF, germ-free; HC, healthy control; PPMS, primary progressive MS; N/A, not available; RRMS, relapsing-remitting MS; SPMS, secondary progressive MS.
Alterations in the gut environment can augment or suppress EAE development [60–63]. Early studies showed that depletion of the gut microbiota with broad-spectrum oral antibiotics leads to suppression of EAE [61,63]. Thereafter, studies using germ-free (GF) mice showed that components of the gut microbiota can promote CNS autoimmune inflammation. Indeed, using a spontaneous EAE model based on transgenic myelin-reactive T cell receptor (TCR) mice, it was shown that spontaneous EAE does not develop in GF mice [60]. Another group reported the identification of segmented filamentous bacteria (SFB) as major inducers of Th17 cell differentiation [64]. It was also shown that actively induced EAE is diminished in GF mice, but is restored by gut colonization with SFB [65]. Of note, most SFB-induced Th17 cells react with SFB antigens, highlighting the role of microbial antigens in effector T cell induction in the gut [66]. Indeed, following colonization with SFB, SFB-specific CD4+ T cells are primed to become Th17 cells in the mesenteric lymph nodes (MLNs) [67]. SFB may activate CNS-reactive T cells via crossreactivity between SFB and so far uncharacterized CNS antigens, or by mechanisms of bystander activation [68]. Following priming in MLNs, SFB-specific Th17 cells may directly reach the CNS via the systemic circulation, or recirculate to the CNS after trafficking to intestinal tissue (Figure 1, Key Figure). Interestingly, gut nociceptor neurons, that are usually associated with protective reflexes, were recently shown to control SFB levels [69], suggesting that their activity may regulate indirectly the generation of CNS-reactive Th17 cells.
Conversely, the commensal flora has also been shown to promote the differentiation of regulatory T cells via the production of multiple metabolites. For example, polysaccharide A (PSA) produced by the human colonic commensal Bacteroides fragilis promotes the expansion of anti-inflammatory IL-10-producing CD4+ T cells via Toll-like receptor 2 (TLR2) activation, in co-operation with TLR1 and Dectin1 signaling [70–76]. However, the specific mechanisms involved in the amelioration of EAE by PSA-induced IL-10+CD4+ T cells remain to be identified. PSA-driven immunoregulation also involves the ectoenzyme CD39, which plays a central role in the control of CNS inflammation by modulating purinergic signaling in APCs and T cells [11,72,73]. Short-chain fatty acids (SCFAs), a main bacterial fermentation product of dietary fiber especially in the colon [77], also induce Tregs [78] in the colon and in the small intestine [79], and the latter class has been shown to suppress CNS inflammation [79]. Relatedly, both in humans and mice, clostridial strains in the colon have been shown to induce Tregs through a mechanism linked to the production of SCFAs [80,81]. These findings suggest an important role for the commensal flora in the control of pro- and anti-inflammatory T cells that target the CNS and contribute to MS pathogenesis.
Studies in MS patients further corroborate the involvement of the commensal flora in the pathogenesis of the disease. In a study using 16S rRNA pyrosequencing to analyze 40 healthy control (HC) and 20 MS patients, the authors detected moderate dysbiosis in MS patients, with significant decreases in Bacteroidetes and the Clostridia clusters XIVa and IV [51,82] (Table 1). Similarly, a study of pediatric MS patients detected decreased levels of the Clostridia Faecalibacterium prausnitzii [52]. Interestingly, decreased abundance of F. prausnitzii was associated with higher recurrence of ileal Crohn’s disease [83]. Another study, analyzing 43 HC and 60 MS patients, detected increased abundances of Methanobrevibacter and Akkermansia, and decreased Butyricimonas spp. [53]. These changes were correlated with variations in the expression of genes involved in dendritic cell maturation, interferon signaling, and NF-κB signaling pathways in circulating T cells and monocytes. However, little is known about the mechanisms linking the gut microbiome to specific immune processes relevant to the pathogenesis of MS.
Two recent studies attempted to link the gut microbiome in MS to immune processes relevant to disease pathogenesis. One study examined 34 monozygotic twin pairs discordant for MS, and detected increased Akkermansia spp. in MS twins compared with their nondiseased counterpart [56]. A second, related study used myelin-specific TCR transgenic mice, that spontaneously develop EAE when kept under specific pathogen free (SPF) but not under GF conditions [60]. The authors demonstrated that fecal microbiota transplants (FMT) from MS twins induce a higher incidence of spontaneous EAE than transplants from healthy twins, concomitant with reduced IL-10 production in splenic CD4+ T cells [56]. These findings suggest that components of the gut MS microbiome boost T cell-dependent CNS autoimmunity. However, these results should be carefully interpreted because it is not clear to what extent the commensal human bacteria transplanted into mice recapitulate the physiological interactions between the microbiota and the human immune system.
In related studies, fecal samples from 71 HC and 71 MS patients were examined; the authors detected increased abundance of Akkermansia spp., and also of Acinetobacter, in MS [57,59]. Of note, antibodies reactive with Acinetobacter were previously reported to be elevated in MS serum [84]. Moreover, bacterial extracts from MS stool samples, relative to their counterparts isolated from HC subjects, showed an impaired ability to promote the differentiation of Tregs in peripheral blood mononuclear cell (PBMC) cultures [57]. In addition, individual culture extracts of Akkermansia muciniphila and Acinetobacter calcoaceticus boosted the differentiation of Th1 cells and suppressed Treg differentiation under T cell polarizing conditions in vitro [57]. The authors also found that extracts of Parabacteroides distasonis, a gut commensal whose abundance is decreased in MS patients, enhance the differentiation of IL-10+ Tregs under Treg-polarizing conditions. These results suggest that components of the MS gut microbiota may boost the polarization of proinflammatory T cells. However, it should be kept in mind that these studies use bacterial extracts and in vitro PBMCs; future studies should evaluate the physiological interactions between live bacteria and mucosal cells such as epithelial cells and specialized intestinal APCs [85].
Finally, gut microbiota transplants from MS patients into GF mice worsened EAE induced by active immunization [57]. Multiple mechanisms are likely to contribute to these effects, and these may include functional alterations in intestinal APCs [11,85–90] and/or direct effects of microbial metabolites such as PSA [71,73], aryl hydrocarbon receptor (AHR) agonists [91,92], and SCFAs [62] on T cells, eventually impacting on T cell activation, polarization, and migration. The antigen specificity of the microbiota-altered T cells in vivo and their modes of action remain to be determined. Taken together, these findings suggest that the MS microbiome promotes pathogenic T cell responses, but has an impaired ability to induce IL-10+ Tregs.
Of note, Akkermansia spp. were found to be abundant in MS stool samples in three different cohorts (Table 1) [53,56,57]. Akkermansia is an oval-shape, nonmotile, strictly anaerobic bacterium that has previously been linked to obesity [93]. Interestingly, it was recently reported that the levels of the miRNA miR-30d are increased in MS and EAE feces [94]. Moreover, miR-30d increases Akkermansia abundance in the gut, and Akkermansia promotes the expansion of Tregs that suppress CNS inflammation [94].
Although changes in some commensal bacteria were detected consistently across different MS microbiota studies, many of the results vary between the different reports. These discrepancies may reflect differences in the genetic backgrounds and lifestyles of the patient cohorts being analyzed. Additional confounding factors may be the methods used for sample collection, preservation, and processing, as well as the procedures used for 16S rRNA sequencing. Efforts to establish uniform methods for microbiome analysis will help to overcome these discrepancies [95–97]. In addition, future studies should also investigate the microbiome associated with different regions of the gastrointestinal tract because each region harbors a distinct microbiome that is controlled by specific regulatory mechanisms, and may have different effects on the immune system [98].
Key Figure
Role of T Cells and Glial Cells in the Gut–CNS Axis in the Context of Multiple Sclerosis (MS)
Figure 1.
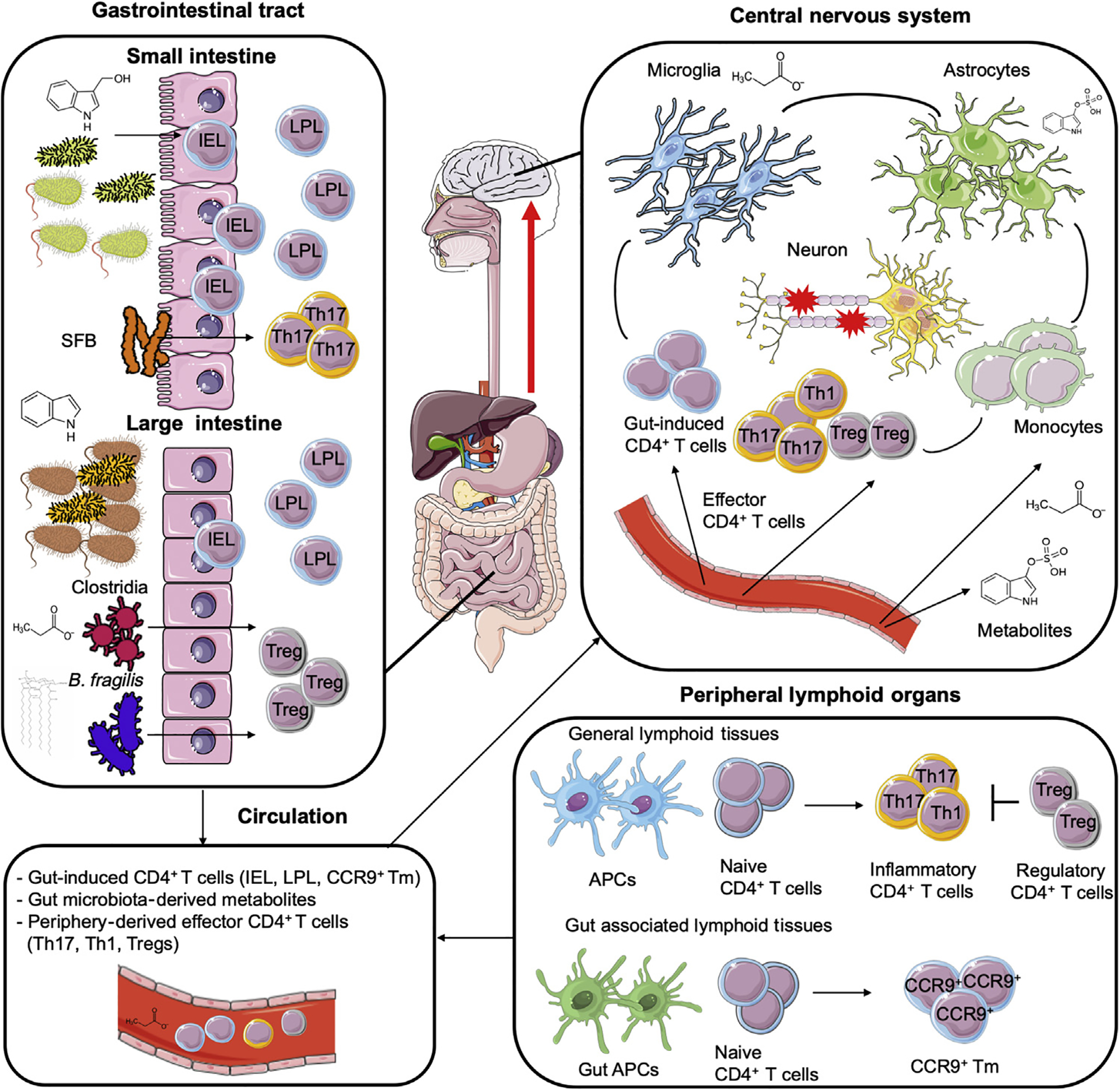
In MS, naive CD4+ T cells are polarized into proinflammatory phenotypes (Th1 and Th17 T helper cells) in peripheral lymphoid organs (bottom right). Proinflammatory CD4+ T cells, which overcome regulatory mechanisms such as regulatory T cell (Treg) suppression, traffic to the CNS and initiate inflammation. Antigen-presenting cells (APCs) in gut-associated lymphoid tissue (GALT) present gut-derived antigens and promote the differentiation of CCR9+ memory CD4+ T (CCR9+ Tm) cells. CCR9+ Tm cells circulate systemically (bottom left), and reach the CNS (top right) to suppress inflammation; they also recirculate to the gut to become CD4+ intraepithelial lymphocytes (IELs) and CD4+ lamina propria lymphocytes (LPLs). Small intestine CD4+ IEL cells migrate to the CNS and suppress inflammation, and their differentiation depends on the AHR agonist indole-3-carbinol (I3C) and the gut microbiota. The terminal differentiation of CCR9+ Tm cells into IEL and LPL cells depends on the microbiota. Some bacteria preferentially induce specific Th subtypes. For example, segmented filamentous bacteria (SFB) induce Th17 cell differentiation in the ileum, and Clostridia strains and Bacteroides fragilis favor Treg differentiation in the colon. In the figure, gut T cells (CD4+ IELs and LPLs) and CCR9+ Tm cells are depicted as ‘gut-induced T cells’. Metabolites controlled by the commensal flora such as short-chain fatty acids or tryptophan derivatives may reach the CNS directly or after further processing in other organs. For example, bacterially produced indole is further metabolized to indoxyl-3-sulfate (I3S) in the liver. Once in the CNS, microbe-controlled metabolites can modulate the activity of astrocytes and microglia.
Cells Participating in the Gut–CNS Axis in MS
To investigate the Gut–CNS axis in MS, it is central to identify the mechanisms that link the gut microenvironment to CNS inflammation. Based on the important roles of CD4+ T cells in MS, the effects of the gut microbiome on T cells are likely to play major roles in MS pathogenesis [99]. Indeed, the gut microbiota and the diet provide multiple antigens and small molecules which can mimic self-antigens [84,100,101] and stimulate innate immunity [85], potentially promoting the activation of autoreactive T cells.
Studies in mice have indicated that CD4+ T cells modulated by the gut microbiome link the gut environment and CNS inflammation in MS [102,103] (Figure 1). Using myelin oligodendrocyte glycoprotein (MOG)-specific TCR transgenic mice, myelin-specific CD4+ T cells were reported to manifest an activated phenotype in the small intestinal epithelium as induced intraepithelial lymphocytes (IELs) that are driven by the gut microbiota. These CD4+ IELs proliferate in MLNs and the gut, but not in other lymphoid organs, suggesting that they react with intestinal antigens. Furthermore, CD4+ IELs showed increased expression of AHR signaling pathway-related genes. Indeed, CD4+ IEL number was increased following feeding with the AHR agonist indole-3-carbinol (I3C). Strikingly, although these gut-induced T cells have a Th17 cell phenotype, they migrate to the CNS where they arrest inflammation through a LAG3-dependent mechanism. Indeed, previous studies of parabiotic mice detected recirculation of CD4+ IELs and lamina propria CD4+ T cells [104], and analysis of mice harboring photoconvertible reporters showed that gut T cells traffic to secondary lymphoid organs and the CNS [105–107], where they can have direct effects on inflammation [60,65,108]. Taken together, these studies show that the intestinal environment favors the induction of myelin-reactive CD4+ T cells that suppress CNS inflammation.
Despite the observations discussed above, the recirculation of the gut-resident T cells (IELs and lamina propria lymphocytes, LPLs) requires further investigation. For example, because previous studies relied on the use of photoconvertible reporter mice [105–107], the migration of T cells from gut tissue to the CNS should be further confirmed with reliable markers to identify gutenvironment experienced T cells in clinical samples. To investigate gut-tropic CD4+ T cells in humans, a recent study focused on the C-C chemokine receptor type 9 (CCR9) [109,110]. Interactions between CCR9 and its ligand CCL25 in the small intestine epithelium are crucial for T cell migration to the gut [111]. CCR9 is upregulated by conventional CD4+ T cells after activation in gut-associated lymphoid tissues, such as Peyer’s patches and MLNs, in which APCs are educated under the influence of microbial products and other modulators in the gut microenvironment [85,112]. After activation, CCR9+ CD4+ T cells acquire a memory phenotype and recirculate through the peripheral blood to the small intestine for terminal differentiation into gut IELs and LPLs (Figure 1). Therefore, CCR9+CD4+ T cells can be considered to be precursors of gut-resident T cells, and may possibly also contain recirculating CD4+ IELs because all IELs express CCR9 [113]. In the peripheral blood of SPMS patients, reduction in CCR9+ memory T (Tm) cells was detected, as well as the adoption of a proinflammatory Th17 phenotype [109,110]. CCR9+ Tm cells expressed high levels of c-MAF, produced anti-inflammatory IL-4 and IL-10, expressed the CNS-homing receptor CCR6, and upregulated LAG3 in cerebrospinal fluid during MS relapses. Of note, c-MAF mediates the production of IL-10 triggered by AHR activation in T cells [5,7,8,12], and was recently found to contribute to Treg-driven immune tolerance in the gut [114]. In addition, the frequency of CCR9+ Tm cells in peripheral blood is reportedly influenced by aging and alterations in the gut microbiota. Indeed, the increase in circulating CCR9+ Tm cells induced by treatment with antibodies to mucosal vascular addressin cell adhesion molecule 1 (MAdCAM-1) or antibiotics alleviated EAE. Although inhibition of myelin-specific Th17 cell trafficking and proliferation in the colon is also thought to suppress EAE [115], these findings suggest that CCR9+ Tm cells participate in the control of CNS inflammation, and that changes in their frequency or function contribute to MS pathogenesis.
Studies on CD4+ IEL and CCR9+ T cells illustrate one of multiple mechanisms by which the gut–CNS axis controls CNS inflammation, specifically by promoting the generation of anti-inflammatory cells in the gut that migrate to the CNS to limit inflammation (Figure 1). Therefore, the gut may offer an avenue for therapeutic manipulation of the balance between pro-and anti-inflammatory T cells in immunological diseases by using probiotics or alternative approaches. These therapies could potentially activate additional anti-inflammatory cell populations such as B cells [116]. Indeed, it was recently reported that IgA+ plasma cells originating in the gut migrate to the CNS to limit inflammation via the production of IL-10 [17].
Effects of the Microbiome on CNS-Resident Cells
Alterations in the gut microbiome have been described in a variety of neurologic disorders, including disorders in which peripheral immune cells are not generally thought to play a central role in disease pathogenesis. These include social behavior disorders [117], Parkinson’s disease (PD) [118], and amyotrophic lateral sclerosis (ALS) [119]. It seems likely that the involvement of the gut microbiome in these disorders is via direct effects of the microbiome on CNS-resident cells, rather than through intermediary changes in peripheral immune cells, as discussed earlier in the context of MS. Indeed, metabolomic studies identified multiple circulating metabolites that are controlled by the microbiome [120], some of which may cross the BBB and thereby participate in the gut–CNS axis (Figure 1).
Microglia are among the CNS-resident cell types that respond to changes in the gut microbiome. One study, for instance, described an immature microglia phenotype in GF and antibiotic-treated mice [121]. In particular, the authors detected increased expression of CSF1R, F4/80, and CD31, molecules whose expression is usually reduced during development. Histopathological analyses detected longer microglial processes and increased numbers of segments, branching, and terminal points in GF mice. These phenotypically immature microglia showed reduced inflammatory responses following challenge with lipopolysaccharide or lymphocytic choriomeningitis virus, suggesting a priming role for the commensal flora in microglial responses to microbial challenge [121]; the role of these immature microglia in the context of autoimmunity remains incompletely understood. Of note, SCFA oral administration partially reversed the immature phenotype of microglia, suggesting that SCFA-producing gut microbiota may play a role in controlling microglial responses during CNS inflammation. A study using α-synuclein (α-Syn)-overexpressing mice, which model multiple aspects of PD, reported that absence of the gut microbiota reduces motor dysfunction, microglial activation, and inflammatory cytokine production in the CNS; these effects were reversed by SCFA administration [118]. Thus, microbial SCFAs seem to support homeostatic microglial function. However, SCFAs also act on peripheral immune cells such as Tregs [79], which have been shown to regulate the activity of CNS-resident cells [9,122], suggesting complex direct and indirect effects of commensal metabolites on CNS-resident cells [77].
A recent study in Sod1 transgenic mice, a model of familiar ALS, reported that Akkermansia muciniphila ameliorates whereas Ruminococcus torques and Parabacteroides distasonis exacerbate ALS-like symptoms in this model [119]. Furthermore, Sod1 mice treated with A. muciniphila accumulate A. muciniphila-associated nicotinamide in the CNS, and systemic supplementation with nicotinamide improves motor symptoms and gene expression patterns in the spinal cord of Sod1 mice.
AHR is another mediator between the gut microbiome and its effects on CNS-resident cells. AHR is a ligand-activated transcription factor that integrates environmental, dietary, microbial, and metabolic cues to control complex transcriptional programs in a ligand- and cell type-specific manner [92]. Tryptophanase (TnAse) expressed in commensal bacteria catalyzes the conversion of dietary tryptophan to indole, which is used in the liver as a precursor for the synthesis of the AHR agonist indoxyl-3-sulfate (I3S) [123,124]; additional microbial metabolites capable of regulating AHR activity have also been identified. Our group recently reported that AHR signaling triggered by gut microbial metabolites regulates microglia and astrocyte function in the context of CNS inflammation [21,23]. In particular, AHR limits NF-κB-driven proinflammatory responses in astrocytes through a SOCS2- dependent mechanism, and Ahr deletion in astrocytes exacerbates EAE [37]. Oral administration of TnAse or AHR-activating tryptophan metabolites recapitulated some of the anti-inflammatory effects of the gut microbiome in astrocytes. Similar anti-inflammatory effects of AHR were also detected in microglia [33]. Moreover, modulation of AHR signaling by microbial metabolites was found to control microglial production of TGF-α and VEGF-β, which regulate astrocyte responses in EAE. Together with reports of reduced AHR agonistic activity in serum samples from MS patients [37,125,126], these findings suggest that AHR operates as a checkpoint molecule that limits the proinflammatory activities of CNS-resident cells in response to microbial cues. Interestingly, this immunomodulatory pathway is exploited by glioblastoma to dampen antitumor immunity [127,128], and also by Zika virus to evade antiviral immunity and cause CNS pathology [129]. Taken together, these findings illustrate how the gut–CNS axis controls CNS-resident cells via the activity of microbial immunomodulatory metabolites that reach the CNS.
Concluding Remarks and Future Perspectives
The multiple mechanisms by which the gut–CNS axis controls CNS inflammation identify guttargeting approaches as novel avenues for therapeutic intervention in MS and other neurologic diseases. The targeting of ‘oral tolerance’ [130], the phenomenon by which oral administration of myelin antigens suppresses CNS inflammation, was one of the first attempts to therapeutically target the gut–CNS axis (Box 2). Several clinical studies have investigated the therapeutic potential of probiotics (Box 2), but the lack of consistency in study design and the limited number of studies preclude clear conclusions regarding the efficacy of this approach [131]. However, it was recently reported that probiotic administration suppressed peripheral inflammatory responses in MS patients, as shown by decreased expression of co-stimulatory and MHC class II molecules in monocytes and dendritic cells [132]. Deeper understanding of the mechanisms by which the gut microbiota controls CNS inflammation, in combination with the development of engineered microorganisms optimized for their anti-inflammatory and neuroprotective activities, will guide the development of probiotic-based approaches to MS and other neurologic disorders.
Box 2. Potential Therapies Targeting the Gut–CNS Axis.
Oral Tolerance
Involves suppression of the immune response directed against antigens fed orally. This mechanism may have evolved to prevent aberrant immune reactivity to food. Although effective in experimental models of autoimmunity including EAE [130], this approach has not yet been successfully applied for the treatment of MS.
Probiotics and Prebiotics
‘Probiotics’ are live microorganisms (e.g., bacteria) that are intended to provide health benefits when consumed or applied to the body. The most commonly used probiotics now belong to the genera Lactobacillus and Bifidobacterium. ‘Prebiotics’ are nondigestible food components that selectively stimulate the growth or activity of beneficial microorganisms.
Fecal Microbiota Transplantation (FMT)
First described as ‘yellow soup’ derived from human feces that was used in 4th Century China to treat food poisoning and diarrhea. In 1958, investigators in Denver first reported the successful administration of feces by enema to treat fulminant life-threatening pseudomembranous enterocolitis [142], an infectious disease caused by Clostridium difficile that is often accompanied by dysbiosis after antibiotic treatments. Fifty years later, a clinical study was conducted to evaluate the effects of fecal transplantation on C. difficile infection. Duodenal infusion of gut microbiota from healthy donors to recurrent C. difficile-infected patients outperformed the cure rate of the standard treatment, vancomycin (81% vs 31%)i [143]. Microbiota diversity in fecal samples, that is usually reduced in C. difficile-infected patients, was also restored in the fecal infusion group, suggesting that FMT corrects dysbiosis. FMT capsules administered by the oral route are now used in clinical trials. Although the frequency of adverse events associated with FMT is low, the severity of some of the rare complications raises concerns about the approach, and it was recently reported that two patients who had undergone FMT died as a result of complications associated with extended-spectrum β-lactamase-producing Escherichia coli bacteremiaii,iii [133].
Oral Antibiotics
Oral antibiotics modulate the gut microbiota, and have been shown in experimental models to alleviate EAE [37,61,63,109]. However, the doses and spectra of the antibiotics used in EAE are high and broad. Thus, further studies should develop approaches to target specific components of the gut microbiota without causing dysbiosis.
Diet-Based Microbiota Therapies
The diet has strong effects on the gut microbiota [134,144,145]. However, the diet–microbiota–host crosstalk is extremely complex, making it difficult to develop dietary interventions to treat CNS inflammation.
FMT is used for the treatment of dysbiosis (Box 2), and represents another candidate approach to target dysbiosis in MS. Although adverse events associated with FMT are rare, it should be kept in mind that this approach has been linked to serious adverse events such as life-threatening infection by donor-derived antibiotic-resistant bacteriai,ii,iii [133]. Alternative approaches for targeting dysbiosis and the expansion of disease-promoting microbiota in MS may involve the use of miRNAs [94], antibiotics [61,63], or dietary interventions [134] as stand-alone therapies or in combination with FMT. However, our limited understanding of the connection between the diet, the gut microbiota, and CNS inflammation limits successful therapeutic targeting of the gut–CNS axis in neurologic disorders (see Outstanding Questions). Progress in these areas will guide the development of novel therapies for MS and other disorders affecting the CNS.
Outstanding Questions.
What factors are responsible for the high prevalence of MS in developed countries? Do gut microbiome-dependent mechanisms contribute to it?
Does the dysbiosis detected in MS patients play a causal role in the disease, or is it merely a consequence of it? Which are the crucial commensal bacteria that promote or limit MS development?
Which cellular mechanisms participate in the gut–CNS axis in MS?
Which immunomodulatory microbial metabolites reach the CNS?
Can one therapeutically target the gut–CNS axis as a treatment avenue for MS?
Highlights.
MS is an autoimmune inflammatory disease characterized by CNS inflammation and damage to myelin.
T cells, B cells, and CNS-resident cells including astrocytes and microglia contribute to MS pathogenesis.
MS pathogenesis has been linked to genetic and environmental factors, as well as to the gut microbiome.
The microbiome modulates the differentiation and function of peripheral immune cells that control CNS inflammation.
Microbial metabolites that reach the CNS can modulate the activity of resident glial cells such as astrocytes and microglia.
Acknowledgments
A.K. was supported by a Uehara Memorial Foundation Overseas Research Fellowship and by the Japan Society for the Promotion of Science (JSPS) Overseas Research Fellowship. F.J.Q. is supported by grants NS102807, ES02530, ES029136, and AI126880 from the National Institutes of Health, RG4111A1 and JF2161-A-5 from the National Multiple Sclerosis Society, and PA-1604-08459 from the International Progressive MS Alliance.
Glossary
- Adaptive immunity
immune response mediated by antigen-specific receptors in T cells and B cells.
- Amyotrophic lateral sclerosis (ALS)
a progressive neurodegenerative disease which usually affects upper and lower motor neurons, causing muscle weakness and eventually loss of muscle control.
- Antigen
a molecule capable of inducing a specific immune response.
- Chemokines
a family of cytokines which induce chemotaxis towards producing cells.
- Co-stimulatory molecules
cellsurface molecules expressed by antigen-presenting cells (APCs) to modulate T cell activation
- Cytokines
secreted proteins that are involved in modulating the immune response.
- Dendritic cells
specialized APCs that process and present antigens in MHC molecules together with co-stimulatory molecules to induce T cell activation.
- Experimental autoimmune encephalitis (EAE)
a preclinical animal model of multiple sclerosis (MS) induced by immunization with myelin antigens and adjuvant or by the transfer of myelin-specific T cells.
- Induced intraepithelial lymphocytes (IELs)
T cells in the gut epithelium that are generated by the activation of naïve T cells in gut-associated lymphoid tissues. By contrast, natural IELs are activated in the thymus.
- MHC class I/II
major histocompatibility complex (MHC) molecules that bind to antigenderived peptides and present them to CD8+ (MHC class I) and CD4+ T cells (MHC class II) to induce T cell activation via interactions with their T cell receptors.
- Mucosal vascular addressin cell adhesion molecule 1 (MadCAM-1)
a mucosal endothelial adhesion molecule that preferentially interacts with leukocyte α4β7 integrin.
- Parkinson’s disease (PD)
a progressive neurological disorder that usually affects movement coordination. Lewy bodies composed of aggregated α-synuclein are detected in the brain of patients, and are thought to be associated with loss of dopaminergic neurons.
- Short-chain fatty acids (SCFAs)
fatty acids with fewer than six carbon atoms that are produced by the fermentation of dietary fibers by anaerobic intestinal microbiota.
- 16S rRNA
the RNA component of the 30S small subunit of a prokaryotic ribosome. 16S rRNA sequencing is used for phylogenetic studies.
- T cell autoimmunity
a T cell-mediated immune response directed against self-antigens that is associated with autoimmune diseases such as MS, type 1 diabetes, and rheumatoid arthritis.
Footnotes
References
- 1.Miller I (2018) The gut–brain axis: historical reflections. Microb. Ecol. Health Dis 29, 1542921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reich DS et al. (2018) Multiple sclerosis. N. Engl. J. Med 378, 169–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Korn T and Kallies A (2017) T cell responses in the central nervous system. Nat. Rev. Immunol 17, 179–194 [DOI] [PubMed] [Google Scholar]
- 4.Quintana FJ (2019) Myeloid cells in the central nervous system: so similar, yet so different. Sci. Immunol 4, eaaw2841. [DOI] [PubMed] [Google Scholar]
- 5.Apetoh L et al. (2010) The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat. Immunol 11, 854–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farez MF et al. (2015) Melatonin contributes to the seasonality of multiple sclerosis relapses. Cell 162, 1338–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gandhi R et al. (2010) Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3+ regulatory T cells. Nat. Immunol 11, 846–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mascanfroni ID et al. (2015) Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-alpha. Nat. Med 21, 638–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mayo L et al. (2016) IL-10-dependent Tr1 cells attenuate astrocyte activation and ameliorate chronic central nervous system inflammation. Brain 139, 1939–1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quintana FJ et al. (2010) An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. U. S. A 107, 20768–20773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takenaka MC et al. (2016) Regulation of the T cell response by CD39. Trends Immunol. 37, 427–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu HY et al. (2011) In vivo induction of Tr1 cells via mucosal dendritic cells and AHR signaling. PLoS One 6, e23618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saligrama N et al. (2019) Opposing T cell responses in experimental autoimmune encephalomyelitis. Nature 572, 481–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Malik S et al. (2016) The emerging roles of gamma-delta T cells in tissue inflammation in experimental autoimmune encephalomyelitis. Front. Immunol 7, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamamura T et al. (2007) Understanding the behavior of invariant NKT cells in autoimmune diseases. J. Neuroimmunol 191, 8–15 [DOI] [PubMed] [Google Scholar]
- 16.Chiba A et al. (2018) Mucosal-associated invariant T cells in autoimmune diseases. Front. Immunol 9, 1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rojas OL et al. (2019) Recirculating intestinal IgA-producing cells regulate neuroinflammation via IL-10. Cell 177, 492–493 [DOI] [PubMed] [Google Scholar]
- 18.Chao CC et al. (2019) Metabolic control of astrocyte pathogenic activity via cPLA2-MAVS. Cell 179, 1483–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farez MF et al. (2009) Toll-like receptor 2 and poly(ADP-ribose) polymerase 1 promote central nervous system neuroinflammation in progressive EAE. Nat. Immunol 10, 958–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mayo L et al. (2014) Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat. Med 20, 1147–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rothhammer V et al. (2018) Microglial control of astrocytes in response to microbial metabolites. Nature 557, 724–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rothhammer V et al. (2017) Sphingosine 1-phosphate receptor modulation suppresses pathogenic astrocyte activation and chronic progressive CNS inflammation. Proc. Natl. Acad. Sci.U. S. A 114, 2012–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rothhammer V et al. (2016) Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med 22, 586–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wheeler MA et al. (2020) MAFG-driven astrocytes promote CNS inflammation. Nature 578, 593–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wheeler MA et al. (2019) Environmental control of astrocyte pathogenic activities in CNS inflammation. Cell 176, 581–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zrzavy T et al. (2017) Loss of ‘homeostatic’ microglia and patterns of their activation in active multiple sclerosis. Brain 140, 1900–1913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sofroniew MV and Vinters HV (2010) Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Voet S et al. (2019) Microglia in central nervous system inflammation and multiple sclerosis pathology. Trends Mol. Med 25, 112–123 [DOI] [PubMed] [Google Scholar]
- 29.Jordao MJC et al. (2019) Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 363, eaat7554. [DOI] [PubMed] [Google Scholar]
- 30.Wolf Y et al. (2018) Microglial MHC class II is dispensable for experimental autoimmune encephalomyelitis and cuprizoneinduced demyelination. Eur. J. Immunol 48, 1308–1318 [DOI] [PubMed] [Google Scholar]
- 31.Colonna M and Butovsky O (2017) Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol 35, 441–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liddelow SA et al. (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vainchtein ID et al. (2020) Astrocytes and microglia: in sickness and in health. Trends Neurosci. 43, 144–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sofroniew MV (2015) Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci 16, 249–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quintana FJ (2017) Astrocytes to the rescue! Glia limitans astrocytic endfeet control CNS inflammation. J. Clin. Invest 127, 2897–2899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ponath G et al. (2018) Enhanced astrocyte responses are driven by a genetic risk allele associated with multiple sclerosis. Nat. Commun 9, 5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rothhammer V et al. (2015) Control of autoimmune CNS inflammation by astrocytes. Semin. Immunopathol 37, 625–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Engelhardt B and Coisne C (2011) Fluids and barriers of the CNS establish immune privilege by confining immune surveillance to a two-walled castle moat surrounding the CNS castle. Fluids Barriers CNS 8, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moreno M et al. (2014) Conditional ablation of astroglial CCL2 suppresses CNS accumulation of M1 macrophages and preserves axons in mice with MOG peptide EAE. J. Neurosci 34, 8175–8185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mills Ko E et al. (2014) Deletion of astroglial CXCL10 delays clinical onset but does not affect progressive axon loss in a murine autoimmune multiple sclerosis model. J. Neuroinflammation 11, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Herrmann JE et al. (2008) STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J. Neurosci 28, 7231–7243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bush TG et al. (1999) Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron 23, 297–308 [DOI] [PubMed] [Google Scholar]
- 43.Myer DJ et al. (2006) Essential protective roles of reactive astrocytes in traumatic brain injury. Brain 129, 2761–2772 [DOI] [PubMed] [Google Scholar]
- 44.Toft-Hansen H et al. (2011) Inhibition of reactive astrocytosis in established experimental autoimmune encephalomyelitis favors infiltration by myeloid cells over T cells and enhances severity of disease. Glia 59, 166–176 [DOI] [PubMed] [Google Scholar]
- 45.Hayakawa K et al. (2016) Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535, 551–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mason S (2017) Lactate shuttles in neuroenergetics – homeostasis, allostasis and beyond. Front. Neurosci 11, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anderson MA et al. (2016) Astrocyte scar formation aids central nervous system axon regeneration. Nature 532, 195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guinane CM and Cotter PD (2013) Role of the gut microbiota in health and chronic gastrointestinal disease: understanding a hidden metabolic organ. Ther. Adv. Gastroenterol 6, 295–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Geva-Zatorsky N et al. (2017) Mining the human gut microbiota for immunomodulatory organisms. Cell 168, 928–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Honda K and Littman DR (2016) The microbiota in adaptive immune homeostasis and disease. Nature 535, 75–84 [DOI] [PubMed] [Google Scholar]
- 51.Miyake S et al. (2015) Dysbiosis in the gut microbiota of patients with multiple sclerosis, with a striking depletion of species belonging to clostridia XIVa and IV clusters. PLoS One 10, e0137429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tremlett H et al. (2016) Gut microbiota in early pediatric multiple sclerosis: a case–control study. Eur. J. Neurol 23, 1308–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jangi S et al. (2016) Alterations of the human gut microbiome in multiple sclerosis. Nat. Commun 7, 12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen J et al. (2016) Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci. Rep 6, 28484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tremlett H et al. (2016) Associations between the gut microbiota and host immune markers in pediatric multiple sclerosis and controls. BMC Neurol. 16, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Berer K et al. (2017) Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc. Natl. Acad. Sci. U. S. A 114, 10719–10724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cekanaviciute E et al. (2017) Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc. Natl. Acad. Sci. U. S. A 114, 10713–10718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tremlett H et al. (2017) The gut microbiome in human neurological disease: a review. Ann. Neurol 81, 369–382 [DOI] [PubMed] [Google Scholar]
- 59.Probstel AK and Baranzini SE (2018) The role of the gut microbiome in multiple sclerosis risk and progression: towards characterization of the ‘MS microbiome’. Neurotherapeutics 15, 126–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Berer K et al. (2011) Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature 479, 538–541 [DOI] [PubMed] [Google Scholar]
- 61.Yokote H et al. (2008) NKT cell-dependent amelioration of a mouse model of multiple sclerosis by altering gut flora. Am.J. Pathol 173, 1714–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haghikia A et al. (2015) Dietary fatty acids directly impact central nervous system autoimmunity via the small intestine. Immunity 43, 817–829 [DOI] [PubMed] [Google Scholar]
- 63.Ochoa-Reparaz J et al. (2009) Role of gut commensal microflora in the development of experimental autoimmune encephalomyelitis. J. Immunol 183, 6041–6050 [DOI] [PubMed] [Google Scholar]
- 64.Ivanov II et al. (2009) Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee YK et al. (2011) Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. U. S. A 108, 4615–4622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang Y et al. (2014) Focused specificity of intestinal TH17 cells towards commensal bacterial antigens. Nature 510, 152–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sano T et al. (2015) An IL-23R/IL-22 circuit regulates epithelial serum amyloid A to promote local effector Th17 responses. Cell 163, 381–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Berer K and Krishnamoorthy G (2014) Microbial view of central nervous system autoimmunity. FEBS Lett. 588, 4207–4213 [DOI] [PubMed] [Google Scholar]
- 69.Lai NY et al. (2020) Gut-innervating nociceptor neurons regulate Peyer’s patch microfold cells and SFB levels to mediate Salmonella host defense. Cell 180, 33–49 e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ochoa-Reparaz J et al. (2010) Central nervous system demyelinating disease protection by the human commensal Bacteroides fragilis depends on polysaccharide A expression. J. Immunol 185, 4101–4108 [DOI] [PubMed] [Google Scholar]
- 71.Ochoa-Reparaz J et al. (2010) A polysaccharide from the human commensal Bacteroides fragilis protects against CNS demyelinating disease. Mucosal Immunol. 3, 487–495 [DOI] [PubMed] [Google Scholar]
- 72.Wang Y et al. (2014) A commensal bacterial product elicits and modulates migratory capacity of CD39+ CD4 T regulatory subsets in the suppression of neuroinflammation. Gut Microbes 5, 552–561 [DOI] [PubMed] [Google Scholar]
- 73.Wang Y et al. (2014) An intestinal commensal symbiosis factor controls neuroinflammation via TLR2-mediated CD39 signalling. Nat. Commun 5, 4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mazmanian SK et al. (2008) A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 453, 620–625 [DOI] [PubMed] [Google Scholar]
- 75.Round JL et al. (2011) The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science 332, 974–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Erturk-Hasdemir D et al. (2019) Symbionts exploit complex signaling to educate the immune system. Proc. Natl. Acad. Sci. U. S. A 116, 26157–26166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dalile B et al. (2019) The role of short-chain fatty acids in microbiota–gut–brain communication. Nat. Rev. Gastroenterol. Hepatol 16, 461–478 [DOI] [PubMed] [Google Scholar]
- 78.Furusawa Y et al. (2013) Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450 [DOI] [PubMed] [Google Scholar]
- 79.Haghikia A et al. (2015) Dietary fatty acids directly impact central nervous system autoimmunity via the small intestine. Immunity 43, 817–829 [DOI] [PubMed] [Google Scholar]
- 80.Atarashi K et al. (2011) Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331, 337–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Atarashi K et al. (2013) Treg induction by a rationally selected mixture of clostridia strains from the human microbiota. Nature 500, 232–236 [DOI] [PubMed] [Google Scholar]
- 82.Sato W and Yamamura T (2019) Multiple sclerosis: possibility of a gut environment-induced disease. Neurochem. Int 130, 104475. [DOI] [PubMed] [Google Scholar]
- 83.Sokol H et al. (2008) Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. U. S. A 105, 16731–16736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hughes L (2003) Cross-reactivity between related sequences found in Acinetobacter sp., Pseudomonas aeruginosa, myelin basic protein and myelin oligodendrocyte glycoprotein in multiple sclerosis. J. Neuroimmunol 144, 105–115 [DOI] [PubMed] [Google Scholar]
- 85.Varol C et al. (2010) Securing the immune tightrope: mononuclear phagocytes in the intestinal lamina propria. Nat. Rev. Immunol 10, 415–426 [DOI] [PubMed] [Google Scholar]
- 86.Mascanfroni ID et al. (2013) IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nat. Immunol 14, 1054–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Quintana FJ et al. (2015) Role and therapeutic value of dendritic cells in central nervous system autoimmunity. Cell Death Differ. 22, 215–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Takenaka MC et al. (2016) Norepinephrine controls effector T cell differentiation through beta2-adrenergic receptor-mediated inhibition of NF-kappaB and AP-1 in dendritic cells. J. Immunol 196, 637–644 [DOI] [PubMed] [Google Scholar]
- 89.Takenaka MC and Quintana FJ (2017) Tolerogenic dendritic cells. Semin. Immunopathol 39, 113–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yeste A et al. (2016) Tolerogenic nanoparticles inhibit T cell-mediated autoimmunity through SOCS2. Sci. Signal 9, ra61. [DOI] [PubMed] [Google Scholar]
- 91.Gutierrez-Vazquez C and Quintana FJ (2018) Regulation of the immune response by the aryl hydrocarbon receptor. Immunity 48, 19–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rothhammer V and Quintana FJ (2019) The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol 19, 184–197 [DOI] [PubMed] [Google Scholar]
- 93.Xu Y et al. (2020) Function of Akkermansia muciniphila in obesity: interactions with lipid metabolism, immune response and gut systems. Front. Microbiol 11, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu S et al. (2019) Oral administration of miR-30d from feces of MS patients suppresses MS-like symptoms in mice by expanding Akkermansia muciniphila. Cell Host Microbe 26, 779–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Baranzini SE (2018) Insights into microbiome research 2: experimental design, sample collection, and shipment. Mult. Scler 24, 1419–1420 [DOI] [PubMed] [Google Scholar]
- 96.Baranzini SE (2018) Insights into microbiome research. 1. How to choose appropriate controls for a microbiome study in MS? Mult. Scler 24, 1278–1279 [DOI] [PubMed] [Google Scholar]
- 97.Baranzini SE (2019) Insights into microbiome research. 6. The role of consortia in studying the role of microbes in health and disease. Mult. Scler 25, 336–337 [DOI] [PubMed] [Google Scholar]
- 98.Martinez-Guryn K et al. (2019) Regional diversity of the gastrointestinal microbiome. Cell Host Microbe 26, 314–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hohlfeld R et al. (2016) The search for the target antigens of multiple sclerosis, part 1: autoreactive CD4+ T cells as pathogenic effectors and therapeutic targets. Lancet Neurol. 15, 198–209 [DOI] [PubMed] [Google Scholar]
- 100.Varrin-Doyer M et al. (2012) Aquaporin 4-specific T cells in neuromyelitis optica exhibit a Th17 bias and recognize Clostridium ABC transporter. Ann. Neurol 72, 53–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Stefferl A et al. (2000) Butyrophilin, a milk protein, modulates the encephalitogenic T cell response to myelin oligodendrocyte glycoprotein in experimental autoimmune encephalomyelitis. J. Immunol 165, 2859–2865 [DOI] [PubMed] [Google Scholar]
- 102.Kadowaki A et al. (2016) Gut environment-induced intraepithelial autoreactive CD4+ T cells suppress central nervous system autoimmunity via LAG-3. Nat. Commun 7, 11639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kadowaki A (2017) Gut environment favors the induction of intraepithelial myelin-reactive CD4+ T cells that inhibit central nervous system autoimmunity through LAG-3. Clin. Exp. Neuroimmunol 8, 11–12 [Google Scholar]
- 104.Suzuki S et al. (1998) Low level of mixing of partner cells seen in extrathymic T cells in the liver and intestine of parabiotic mice: its biological implication. Eur. J. Immunol 28, 3719–3729 [DOI] [PubMed] [Google Scholar]
- 105.Schmidt TH et al. (2013) CXCR4 promotes B cell egress from Peyer’s patches. J. Exp. Med 210, 1099–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Morton AM et al. (2014) Endoscopic photoconversion reveals unexpectedly broad leukocyte trafficking to and from the gut. Proc. Natl. Acad. Sci. U. S. A 111, 6696–6701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Benakis C et al. (2016) Commensal microbiota affects ischemic stroke outcome by regulating intestinal γδ T cells. Nat. Med 22, 516–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Horai R et al. (2015) Microbiota-dependent activation of an autoreactive T cell receptor provokes autoimmunity in an immunologically privileged site. Immunity 43, 343–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kadowaki A et al. (2019) Gut microbiota-dependent CCR9+CD4+ T cells are altered in secondary progressive multiple sclerosis. Brain 142, 916–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kadowaki A (2019) Gut microbiota-dependent CCR9+CD4+ T cells are reduced and altered to an inflammatory phenotype in secondary progressive multiple sclerosis. Clin. Exp. Neuroimmunol 10, 211–212 [Google Scholar]
- 111.Guy-Grand D et al. (2013) Origin, trafficking, and intraepithelial fate of gut-tropic T cells. J. Exp. Med 210, 1839–1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Iwata M et al. (2004) Retinoic acid imprints gut-homing specificity on T cells. Immunity 21, 527–538 [DOI] [PubMed] [Google Scholar]
- 113.Zabel BA et al. (1999) Human G protein-coupled receptor GPR-9–6/CC chemokine receptor 9 is selectively expressed on intestinal homing T lymphocytes, mucosal lymphocytes, and thymocytes and is required for thymus-expressed chemokinemediated chemotaxis. J. Exp. Med 190, 1241–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Xu M et al. (2018) c-MAF-dependent regulatory T cells mediate immunological tolerance to a gut pathobiont. Nature 554, 373–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Duc D et al. (2019) Disrupting myelin-specific Th17 cell gut homing confers protection in an adoptive transfer experimental autoimmune encephalomyelitis. Cell Rep. 29, 378–390 [DOI] [PubMed] [Google Scholar]
- 116.Ochoa-Reparaz J et al. (2010) Induction of a regulatory B cell population in experimental allergic encephalomyelitis by alteration of the gut commensal microflora. Gut Microbes 1, 103–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mayer EA et al. (2015) Gut/brain axis and the microbiota. J. Clin. Invest 125, 926–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sampson TR et al. (2016) Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 167, 1469–1480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Blacher E et al. (2019) Potential roles of gut microbiome and metabolites in modulating ALS in mice. Nature 572, 474–480 [DOI] [PubMed] [Google Scholar]
- 120.Wikoff WR et al. (2009) Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. U. S. A 106, 3698–3703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Erny D et al. (2015) Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci 18, 965–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ito M et al. (2019) Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature 565, 246–250 [DOI] [PubMed] [Google Scholar]
- 123.Schroeder JC et al. (2010) The uremic toxin 3-indoxyl sulfate is a potent endogenous agonist for the human aryl hydrocarbon receptor. Biochemistry 49, 393–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zelante T et al. (2013) Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 39, 372–385 [DOI] [PubMed] [Google Scholar]
- 125.Rothhammer V et al. (2017) Dynamic regulation of serum aryl hydrocarbon receptor agonists in MS. Neurol. Neuroimmunol. Neuroinflamm 4, e359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rothhammer V et al. (2018) Detection of aryl hydrocarbon receptor agonists in human samples. Sci. Rep 8, 4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gabriely G and Quintana FJ (2020) Role of AHR in the control of GBM-associated myeloid cells. Semin. Cancer Biol 64, 13–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Takenaka MC et al. (2019) Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat. Neurosci 22, 729–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Giovannoni F et al. AHR is a Zika virus host factor and a candidate target for antiviral therapy. Nat. Neurosci (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Weiner HL et al. (2011) Oral tolerance. Immunol. Rev 241,241–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wolvers D et al. (2010) Guidance for substantiating the evidence for beneficial effects of probiotics: prevention and management of infections by probiotics. J. Nutr 140, 698S–712S [DOI] [PubMed] [Google Scholar]
- 132.Tankou SK et al. (2018) A probiotic modulates the microbiome and immunity in multiple sclerosis. Ann. Neurol 83, 1147–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.DeFilipp Z et al. (2019) Drug-resistant E. coli bacteremia transmitted by fecal microbiota transplant. N. Engl. J. Med 381, 2043–2050 [DOI] [PubMed] [Google Scholar]
- 134.David LA et al. (2014) Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mahad DH et al. (2015) Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 14, 183–193 [DOI] [PubMed] [Google Scholar]
- 136.International Multiple Sclerosis Genetics, C et al. (2011) Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476, 214–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sawcer S et al. (2014) Multiple sclerosis genetics. LancetNeurol. 13, 700–709 [DOI] [PubMed] [Google Scholar]
- 138.Consortium, I.M.S.G (2019) Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 365, eaav7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Belbasis L et al. (2015) Environmental risk factors and multiple sclerosis: an umbrella review of systematic reviews and meta- analyses. Lancet Neurol. 14, 263–273 [DOI] [PubMed] [Google Scholar]
- 140.Rothhammer V and Quintana FJ (2016) Environmental control of autoimmune inflammation in the central nervous system. Curr. Opin. Immunol 43, 46–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bach J (2002) The effect of infections and susceptibility to autoimmune and allergic diseases. N. Engl. J. Med 347, 911. [DOI] [PubMed] [Google Scholar]
- 142.Eiseman B et al. (1958) Fecal enema as an adjunct in the treatment of pseudomembranous enterocolitis. Surgery 44, 854–859 [PubMed] [Google Scholar]
- 143.van Nood E et al. (2013) Duodenal infusion of donor feces for recurrent Clostridium difficile. N. Engl. J. Med 368, 407–415 [DOI] [PubMed] [Google Scholar]
- 144.Zmora N et al. (2019) You are what you eat: diet, health and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol 16, 35–56 [DOI] [PubMed] [Google Scholar]
- 145.Claesson MJ et al. (2012) Gut microbiota composition correlates with diet and health in the elderly. Nature 488, 178–184 [DOI] [PubMed] [Google Scholar]