

Abstract
Focal dystonias are the most common forms of isolated dystonia; however, the etiopathophysiological signatures of disorder penetrance and clinical manifestation remain unclear. Using an imaging genetics approach, we investigated functional and structural representations of neural endophenotypes underlying the penetrance and manifestation of laryngeal dystonia in families, including 21 probands and 21 unaffected relatives, compared to 32 unrelated healthy controls. We further used a supervised machine-learning algorithm to predict the risk for dystonia development in susceptible individuals based on neural features of identified endophenotypes. We found that abnormalities in prefrontal-parietal cortex, thalamus, and caudate nucleus were commonly shared between patients and their unaffected relatives, representing an intermediate endophenotype of laryngeal dystonia. Machine learning classified 95.2% of unaffected relatives as patients rather than healthy controls, substantiating that these neural alterations represent the endophenotypic marker of dystonia penetrance, independent of its symptomatology. Additional abnormalities in premotor-parietal-temporal cortical regions, caudate nucleus, and cerebellum were present only in patients but not their unaffected relatives, likely representing a secondary endophenotype of dystonia manifestation. Based on alterations in the parietal cortex and caudate nucleus, the machine learning categorized 28.6% of unaffected relative as patients, indicating their increased lifetime risk for developing clinical manifestation of dystonia. The identified endophenotypic neural markers may be implemented for screening of at-risk individuals for dystonia development, selection of families for genetic studies of novel variants based on their risk for disease penetrance, or stratification of patients who would respond differently to a particular treatment in clinical trials.
Keywords: Dystonia, Endophenotypes, Brain imaging
1. Introduction
Focal dystonias are the most common forms of isolated dystonia; however, their etiopathophysiology remains largely unknown. Among these, laryngeal dystonia (LD) is a task-specific dystonia characterized by focal, involuntary contractions of laryngeal muscles selectively affecting speech production. A familial history of dystonia in up to 25% of LD patients strongly suggests the presence of underlying genetic causes (Blitzer et al., 1998; Guiry et al., 2019; Kirke et al., 2015). Nonetheless, traditional genetic studies in LD have thus far failed to identify a disorder-causative mutation due, in part, to reduced penetrance, clinical heterogeneity, and the overall rare incidence of dystonia. On the other hand, recent neuroimaging studies in patients with familial vs. sporadic LD have been more successful in disclosing the interplay between putative genetic factors and neural changes contributing to the pathophysiology of this disorder (Battistella et al., 2016; Bianchi et al., 2017; Fuertinger and Simonyan, 2017). Specifically, these studies showed that LD genotype-related structural abnormalities involve cortical regions responsible for sensory and phonological processing, while widespread functional brain disorganization is instigated by thalamo-prefrontal and parietal aberrations. Although these findings clarified the genetic trends in the pathophysiology of LD, the neural representations underlying the penetrance and clinical manifestation of this disorder remain largely unknown.
In this regard, it is important to evaluate the development and contribution of neural endophenotypes of dystonia. Typically, intermediate endophenotypes are expressed in both manifesting and non-manifesting mutation carriers, and secondary endophenotypes are found only in the disease state (Hutchinson et al., 2013). We propose that, although the genetic disease burden within families is impossible to assess in the absence of a known LD-specific causative mutation, the identification of neuroimaging endophenotypes within the dystonia families may help guide elucidation of the pathophysiological contributors to its penetrance and manifestation and allow the future development of objective tools for the dystonia risk assessment in susceptible individuals.
In this study, we investigated neural endophenotypic traits of structural and functional brain alterations in patients with familial LD compared to their unaffected (asymptomatic) relatives and unrelated healthy controls using high-resolution structural MRI with voxel-based morphometry (VBM) and functional magnetic resonance imaging (fMRI) during LD-symptomatic speech production. We examined the relationship between neural alterations and LD clinical characteristics to determine regional changes associated with the disorder symptomatology. Finally, we employed a supervised machine-learning algorithm to predict LD penetrance and the risk of dystonia manifestation in LD families based on the neural features of identified endophenotypes. We hypothesized that unaffected relatives, similar to their affected patients but in contrast to unrelated healthy controls, are carriers of a subset of LD-characteristic neural alterations within the sensorimotor circuitry, which represent an intermediate endophenotype underlying the penetrance of LD. We further hypothesized that LD patients exhibit an additional set of changes in brain regions controlling sensorimotor integration and planning of motor behavior that distinguishes them from their unaffected relatives; these alterations represent a secondary endophenotype underlying clinical manifestation of this disorder. We postulated that the neural endophenotypic markers may be jointly used for a predictive assessment of the risk for LD development in susceptible individuals.
2. Materials and methods
2.1. Study participants
A total of 74 subjects participated in the study, including 21 patients with familial LD (mean age 56.2 ± 15.8 years; 19 females/2 males), 21 blood-related unaffected, entirely asymptomatic relatives of these patients (mean age 48.5 ± 16.0; 17 females/4 males), and 32 blood-unrelated healthy control subjects (mean age 50.2 ± 11.0; 20 females/12 males) (Table 1). A total of 21 families participated in this study; one unaffected relative per each familial LD patient was included to assure the matched distribution of LD phenotypes and putative genotypes. The family history of dystonia in the study cohort is detailed in Table 1; the sample pedigrees are shown in Fig. 1-I and Fig. 3-II. Four unaffected relatives from four different families were obligate carriers based on analysis of their family history.
Table 1.
Demographics of the study participants.
| Familial LD | Unaffected Relatives | Healthy Controls | |
|---|---|---|---|
| Number of participants | 21 | 21 | 32 |
| Age (years; mean ± st. dev.) | 56.2 ± 15.8 | 48.5 ± 16.0 | 50.2 ± 11.0 |
| Sex (female/male) | 19/2 | 17/4 | 20/12 |
| Familial relationship | proband |
in relation to proband, 1 unaffected member per family 2 parents 9 children 9 siblings 1 first cousin once removed |
none |
| Family history of dystonia (in relation to proband) | 15 families: 2 affected | none | |
| 4 families: 3 affected | |||
| 2 families: 4 affected | |||
| _______________ | |||
| 8 families: parent/child | |||
| 5 families: sibling | |||
| 2 families: half-sibling | |||
| 2 families: grandparent | |||
| 1 family: aunt | |||
| 3 families: first cousin | |||
| 1 family: granduncle | |||
| 1 family: grandnephew | |||
| 1 family: first cousin once removed | |||
| 1 family: second cousin | |||
| Dystonia type | 15 adductor: 6 abductor | N/A | N/A |
| Symptom Onset (years; mean ± st. dev.) | 40.3 ± 17.3 | N/A | N/A |
| Symptom Duration (years; mean ± st. dev.) | 15.9 ± 10.9 | N/A | N/A |
| Genetic status | Negative for TOR1A/DYT1, TUBB4A/DYT4, THAP1/DYT6, GNAL/DYT25, KMT2B/DYT28 and GNAO1 mutations | ||
| Language | Native English | ||
| Cognitive status | Mini-Mental State Examination ≥27 | ||
Fig. 1.
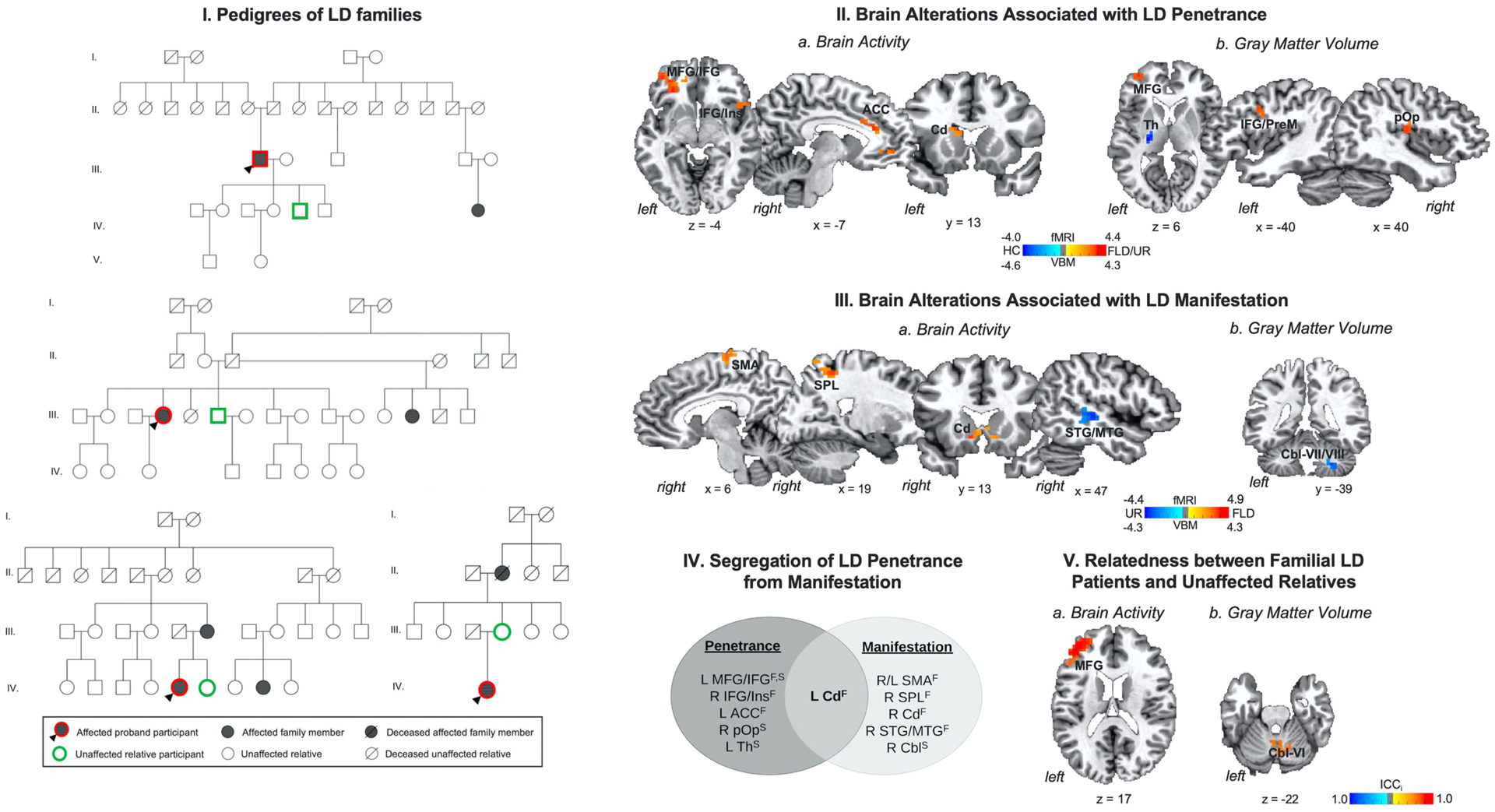
(I) Example pedigrees of families of patients with laryngeal dystonia (LD). (II) Brain alterations associated with LD penetrance are based on differences in (a) brain activity and (b) gray matter volume in familial LD patients and their unaffected relatives vs. healthy controls. (III) Brain alterations associated with LD manifestation are based on differences in (a) brain activity and (b) gray matter volume in familial LD patients vs. their unaffected relatives. (IV) Schematic of functional and structural alterations segregating LD penetrance from manifestation. (V) Relatedness between patients with familial LD and their unaffected relatives as a function of (a) brain activity and (b) gray matter volume. Brain alterations are shown on a series of axial, sagittal or coronal brain slices in the AFNI standard Talairach-Tournoux space. The color bar indicates the z-statistics in (II, III) and the intraclass correlation index (ICCi) in (V). Abbreviations: ACC - anterior cingulate cortex, Cbl - cerebellum, Cd - caudate nucleus, F – functional alteration, FLD – familial laryngeal dystonia patients, HC – healthy controls, ICCi – intraclass correlation index, IFG - inferior frontal gyrus, Ins – insula, L – left, MFG - middle frontal gyrus, MTG - middle temporal gyrus, PrM - premotor cortex, pOp - parietal operculum, R – right, SMA - supplementary motor area, SPL - superior parietal lobule, STG - superior temporal gyrus, S – structural alteration, Th – thalamus, UR – unaffected relatives.
Fig. 3.
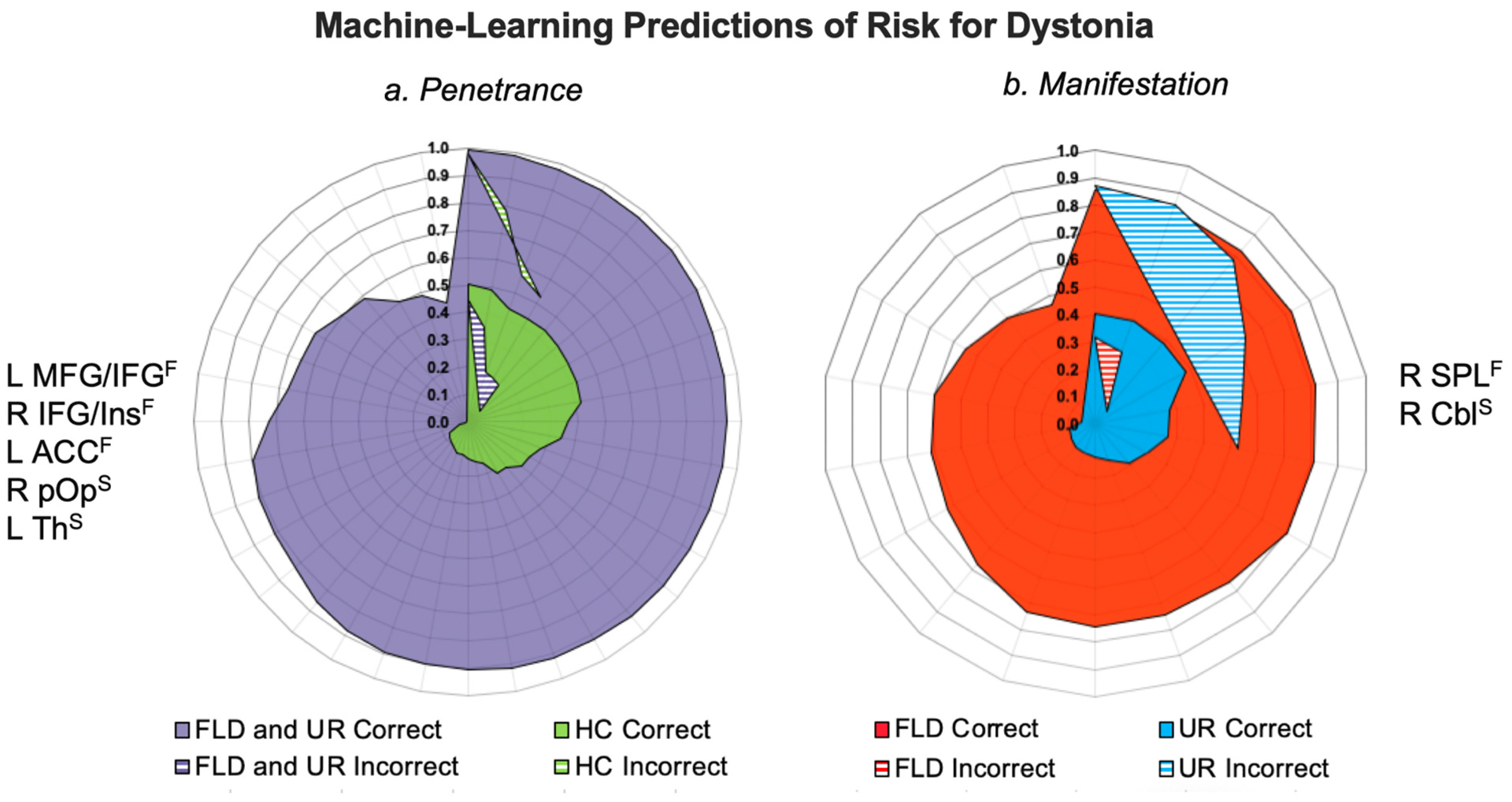
(I) Machine-learning predictions of risk for dystonia relevant to disorder (a) penetrance and (b) manifestation. Radial diagrams depict posterior probability of the subject-specific classification score [range 0.0–1.0]. Corresponding neural alterations used in each classification are shown on the left or right side of the diagrams. Solid areas indicate correctly classified subjects; dashed areas indicate incorrectly identified subjects. (II) Pedigrees of LD families where obligate carriers participated as unaffected relatives and were assigned to the same class as LD patients by the support vector machine. Abbreviations: ACC - anterior cingulate cortex, Cbl - cerebellum, F – functional alteration, FLD – familial laryngeal dystonia patients, HC – healthy controls, IFG - inferior frontal gyrus, Ins – insula, L – left, MFG - middle frontal gyrus, pOp - parietal operculum, R – right, SPL - superior parietal lobule, S – structural alteration, Th – thalamus, UR – unaffected relatives.
No participant had any past or present history of any neurological (except for familial LD in the patient group), psychiatric, or laryngeal problems. None were carriers of either the verified isolated dystonia gene mutations (TOR1A/DYT1, TUBB4A/DYT4, THAP1/DYT6, GNAL/DYT25) or KMT2B/DYT28 and GNAO1 mutations, as confirmed by whole-exome sequencing. The age of LD onset was 40.3 ± 17.3 years, which is in line with previous reports in this disorder (Blitzer et al., 2018; de Lima Xavier and Simonyan, 2019; Guiry et al., 2019). Therefore, unaffected relatives were recruited with the mean age of 48.5 ± 16.0 years to reduce the possibility of being too young to exhibit LD symptoms but capture the range of a lifetime risk of dystonia development. Twenty patients received botulinum toxin injections at least once to manage LD symptoms. All patients receiving botulinum toxin treatment participated in the study at least three months after their last injection when they were entirely symptomatic. None of the participants were on any centrally acting medications. All participants were native English speakers and had normal cognitive status based on Mini-Mental State Examination. There were no significant differences in age (two-sample independent t-test p ≥ 0.12) and sex (chi-square test χ2 = 5.83, p ≥ 0.05) between the groups.
All participants gave written informed consent for study participation, which was approved by the Institutional Review Board of Mass General Brigham. Data from some patients with familial LD and healthy controls were used in our previous studies (Battistella et al., 2016; Bianchi et al., 2017; Fuertinger and Simonyan, 2017; Termsarasab et al., 2016; Valeriani and Simonyan, 2020).
2.2. Image acquisition
Whole-brain images were acquired on a 3.0 Tesla Siemens Skyra scanner equipped with a 32-channel head coil. Functional MRI data were obtained using a gradient-weighted echo planar imaging pulse sequence and an event-related sparse-sampling design, which minimized the scanning artifacts due to orofacial movements during speech production (effective TR = 10.6 s, including 8.6 s for stimulus delivery and task production and 2.0 s for volume acquisition; TE = 30 ms; FA = 90◦; FOV = 240 × 240 mm2; voxel size = 3.5 mm × 3.5 mm; slice thickness = 4 mm). To minimize movements during scanning, the subject’s head was tightly cushioned, and all subjects were instructed to remain motionless throughout scanning. The speech-production task included the repetition of a series of LD symptom-provoking sentences, as described earlier (e.g., (Fuertinger and Simonyan, 2017)). The sample sentences were delivered acoustically via an MR-compatible headphone for 3.6 s while the subject fixated on a cross. Next, an arrow cued the subject to produce the sentence during a 5-s interval, which was followed by a 2-s brain image acquisition when the subject again fixated on the cross. The resting condition without any auditory or visual stimuli was used as a baseline, during which the subject fixated on the cross. Task and resting conditions were pseudorandomized; four scanning sessions were acquired, consisting of 24 task and 16 resting conditions. The subjects’ head was tightly cushioned within the head coil to restrict movements, and all subjects were monitored during the scanning session for possible motions as well as for the correctness and completeness of tasks via a two-way communication system.
A high-resolution T1-weighted MR image was acquired in each subject using a 3D magnetization prepared rapid acquisition gradient echo sequence (3D-MPRAGE: TR = 7.5 ms, TE = 2 ms, TI = 819 ms, FA = 8°, FOV = 210 × 210 mm2, voxel size = 1.0 mm3, 172 slices) and used as an anatomical reference for fMRI data as well as for VBM analysis.
2.3. Data preprocessing
Functional data analysis was performed using AFNI software. Briefly, functional images were despiked, aligned with the anatomical dataset, spatially normalized to the AFNI standard Talairach space, smoothed with a 4-mm Gaussian filter, and scaled by voxelwise mean. For motion correction, six motion parameter estimates were included as covariates of no interest, and three quadratic polynomials were used to model baseline drifts for each imaging run. Head motion was additionally corrected by applying TR censoring, which excluded TR pairs where the Euclidean norm of the motion derivative exceeded 1.0 mm based on simulations of motion artifacts in the presence of slow effective TR. Further censoring excluded those TRs when more than 10% of the auto-masked brain were marked as outliers. Because, in some cases, outliers may capture residual motion that is missed by the motion parameters, this combined approach ensured the stringent exclusion of TRs affected potentially by motion artifacts. Following motion correction, a single regressor associated with the speech task was convolved with a canonical hemodynamic response function and entered into a multiple regression model to predict the BOLD response.
The CAT12 toolbox of SPM12 software was used for VBM analysis. Following the standard pipeline, the T1-weighted images were normalized, segmented into gray and white matter using standard tissue probability maps, spatially normalized to the same AFNI standard Talairach space, and smoothed using a 4-mm Gaussian kernel. Image quality was examined by visual inspection and using the quality check modules of the CAT12 toolbox.
2.4. Statistical analysis
To determine the neural correlates of the intermediate endophenotype underlying LD penetrance, between-group differences in brain activity and gray matter volume were examined in a combined group of 21 familial LD patients and 21 unaffected relatives vs. 32 healthy controls. To identify neural changes of the secondary endophenotype underlying LD manifestation, between-group differences in brain activity and gray matter volume were examined in 21 familial LD patients vs. 21 unaffected relatives. Using the hierarchical mixed-effects model,
where yij is the effect of ith subject in family j, the fixed effects a were associated with the three groups coded through xij (familial LD patients, unaffected relatives, healthy controls), and the two sets of random effects were considered: one set captured the variance across all families z0j, while the other set modeled the variance of those families with two members z1j relative to all families. Because of its trend toward significance, sex was modeled as a nuisance covariate embedded in xij, and εij was the residual term. With the Gaussianity assumptions of , , εij~N(0,σ2), the relatedness between the members of LD families was calculated as a ratio of two variances relative to total variance and defined as an intraclass correlation index, . The resultant statistical maps of between-group comparisons were thresholded at family-wise error (FWE)-corrected p ≤ 0.05, with the voxelwise threshold p ≤ 0.01 and the minimum cluster size threshold 858 mm3.
2.5. Clinical correlates of neural alterations
Clinical information on the symptom onset and duration was obtained during neurological and laryngeal evaluations. Symptom severity was assessed using the Burke-Fahn-Marsden Dystonia Rating Scale (BFM-DRS). In addition, voice and speech were recorded during the production of sustained vowels, repeated syllables, and a set of 20 symptom-provoking speech sentences in LD patients. Symptom severity was perceptually quantified by calculating the number of dystonic voice breaks per sentence (Ludlow et al., 2008). LD-associated harshness and breathiness of voice quality were assessed using a visual analog scale (0 - no symptoms, 100 - most severe symptoms), as described earlier (Rumbach et al., 2017).
To examine the relationship between brain alterations and the clinical features in familial LD patients, Spearman rank correlation co-efficients were computed between whole-brain voxelwise activity, gray matter volume, LD symptom severity, duration, and age of onset, respectively, using AFNI software. The significance level was set at FWE-corrected p ≤ 0.05, with minimum RS ≥ 0.59, voxelwise p ≤ 0.005 and the minimum cluster size 858 mm3.
2.6. Machine-learning prediction of risk for LD penetrance and manifestation
A supervised machine-learning algorithm, support vector machine (SVM), was implemented to predict the risk for dystonia development in LD families. Two SVMs were trained as binary classifiers of (1) a combined group of familial LD patients and unaffected relatives vs. healthy controls to examine the penetrance of disease, and (2) familial LD patients vs. their unaffected relatives to assess the risk for dystonia manifestation, respectively. For this, the mean signal of significant structural and functional clusters derived from the hierarchical mixed-effects models of corresponding between-group comparisons (familial LD patients + unaffected relatives vs. healthy controls and familial LD patients vs. their unaffected relatives, Table 2) were extracted and used as input features of the SVMs. For each predictive SVM model, a 10-fold cross-validation procedure was used, which randomly shuffled and then partitioned the given dataset into a training set to train the model and a test set to evaluate it. For each cross-validation, 90% of dataset was used for model training and 10% was used for model testing. This procedure was iteratively repeated 10 times (folds), where each subject was used for validation exactly once. As commonly employed in machine-learning studies, including those in dystonia patients (Battistella et al., 2016; Li et al., 2017; Valeriani and Simonyan, 2020), the SVM accuracy was computed as the percentage of correctly classified samples, and the posterior probability of the classification score was calculated for each subject. The MATLAB 2018a (Mathworks, MA) was used for SVM computations.
Table 2.
Brain functional and structural abnormalities in familial LD patients and their unaffected relatives.
| Brain Region | Cluster Peak Coordinates x y z | Cluster Size mm3 | Cluster Peak Z-value |
|---|---|---|---|
| Familial LD and Unaffected Relatives vs. Healthy Controls | |||
| Functional activity during speech production | |||
| L middle frontal gyrus extending to inferior frontal gyrus | −37, 45, −4 | 2401 | 4.2 |
| R inferior frontal gyrus extending to insula | 47, 17, −8 | 1286 | 4.4 |
| L anterior cingulate cortex | −12, 31, 20 | 1243 | 4.0 |
| L caudate nucleus | −12, 17, 13 | 858 | 3.9 |
| Gray matter volume | |||
| L middle frontal gyrus | −37, 52, 3 | 643 | 3.9 |
| L inferior frontal gyrus extending to premotor cortex | −40, 3, 34 | 643 | 3.7 |
| R parietal operculum | 40, −18, 17 | 643 | 4.0 |
| L thalamus (motor, premotor and parietal subdivisions) | −23, −18, 6 | 1029 | −4.6 |
| Familial LD vs. Unaffected Relatives | |||
| Functional activity during speech production | |||
| R/L supplementary motor area (area 6) | 9, −11, 62 | 1243 | 4.9 |
| R superior parietal lobule | 19, −50, 48 | 1329 | 4.1 |
| L/R caudate nucleus | −9, 13, −4 | 2315 | 4.4 |
| R superior and middle temporal gyrus | 47, −25, −1 | 1115 | −4.4 |
| Gray matter volume | |||
| R cerebellum (lobules VII/VIII) | 26, −39, −39 | 1201 | −4.0 |
L – left; R – right
3. Results
Abnormalities in brain activity and gray matter volume in patients with familial LD compared to healthy subjects were reported in our previous studies (Battistella et al., 2016; Bianchi et al., 2017; Fuertinger and Simonyan, 2017; Termsarasab et al., 2016). Additional information on structural and functional brain organization in unaffected relatives of LD patients compared to healthy controls can be found in Supplementary Material. The current study focused on the examination of neural representations of LD endophenotypic traits as predictors of dystonia penetrance and manifestation in familial LD patients and their unaffected relatives.
3.1. Neural correlates of the intermediate endophenotype underlying LD penetrance
Compared to unrelated healthy controls, amilial LD patients and their unaffected relatives shared commonly increased activity during speech production in the left middle frontal gyrus (MFG) extending to inferior frontal gyrus (IFG), right IFG extending to the insula, left anterior cingulate cortex (ACC), and left caudate nucleus at FWE-corrected p ≤ 0.05 (Fig. 1-IIa, Table 2). Additionally, both familial LD patients and their unaffected relatives, compared to healthy controls, showed commonly increased gray matter volume in the left MFG and IFG extending to the premotor cortex and right parietal operculum and commonly decreased gray matter volume in the left thalamus, encompassing its motor, premotor and parietal subdivisions, at FWE-corrected p ≤ 0.05 (Fig. 1-IIb, Table 2).
3.2. Neural correlates of the secondary endophenotype underlying LD manifestation
Compared to their unaffected relatives, familial LD patients had distinctly increased activity during speech production in the bilateral supplementary motor area (SMA), right superior parietal lobule (SPL), bilateral caudate nucleus, and decreased activity in the right superior/middle temporal gyrus (STG/MTG) at FWE-corrected p ≤ 0.05 (Fig. 1-IIIa, Table 2). Distinctly reduced gray matter volume in familial LD patients vs. their unaffected relatives was found in the right cerebellar lobules VII/VIII at FWE-corrected p ≤ 0.05 (Fig. 1-IIIb, Table 2).
As shown in Fig. 1-IV, identified functional and structural brain changes strictly segregated the neural correlates of dystonia penetrance from those of LD manifestation. Only the left caudate nucleus was found to be commonly associated with both disorder penetrance and manifestation. In addition, neural correlates of relatedness between family members were found in the activity of left MFG (ICCi = 0.95) and gray matter volume of the cerebellar lobule VI (ICCi = 0.82) (Fig. 1-V).
3.3. Neural correlates of LD symptomatology
No significant correlations were found between LD duration, age of onset, and symptom severity (all corrected p ≥ 0.24). However, the age of LD onset was significantly and negatively correlated with brain activity in the right IFG extending to insula (RS = −0.79, p = 0.00002), suggesting that patients who develop LD at a younger age have higher increases of brain activity in these regions (Fig. 2a). The duration of LD was significantly and positively correlated with brain activity in the right STG/MTG (RS = 0.77, p = 0.00004), indicating that patients at the later stages of their disorder have higher activity in these regions potentially due to compensatory mechanisms developed over the lifetime of disorder (Fig. 2b). Finally, the severity of LD symptoms assessed using the BFM-DRS was significantly and positively correlated with gray matter volume in the cerebellar lobules VII/VIII, suggesting that cerebellar abnormalities increase in the presence of more severe symptomatology (RS = 0.75, p = 0.00008) (Fig. 2c).
Fig. 2.
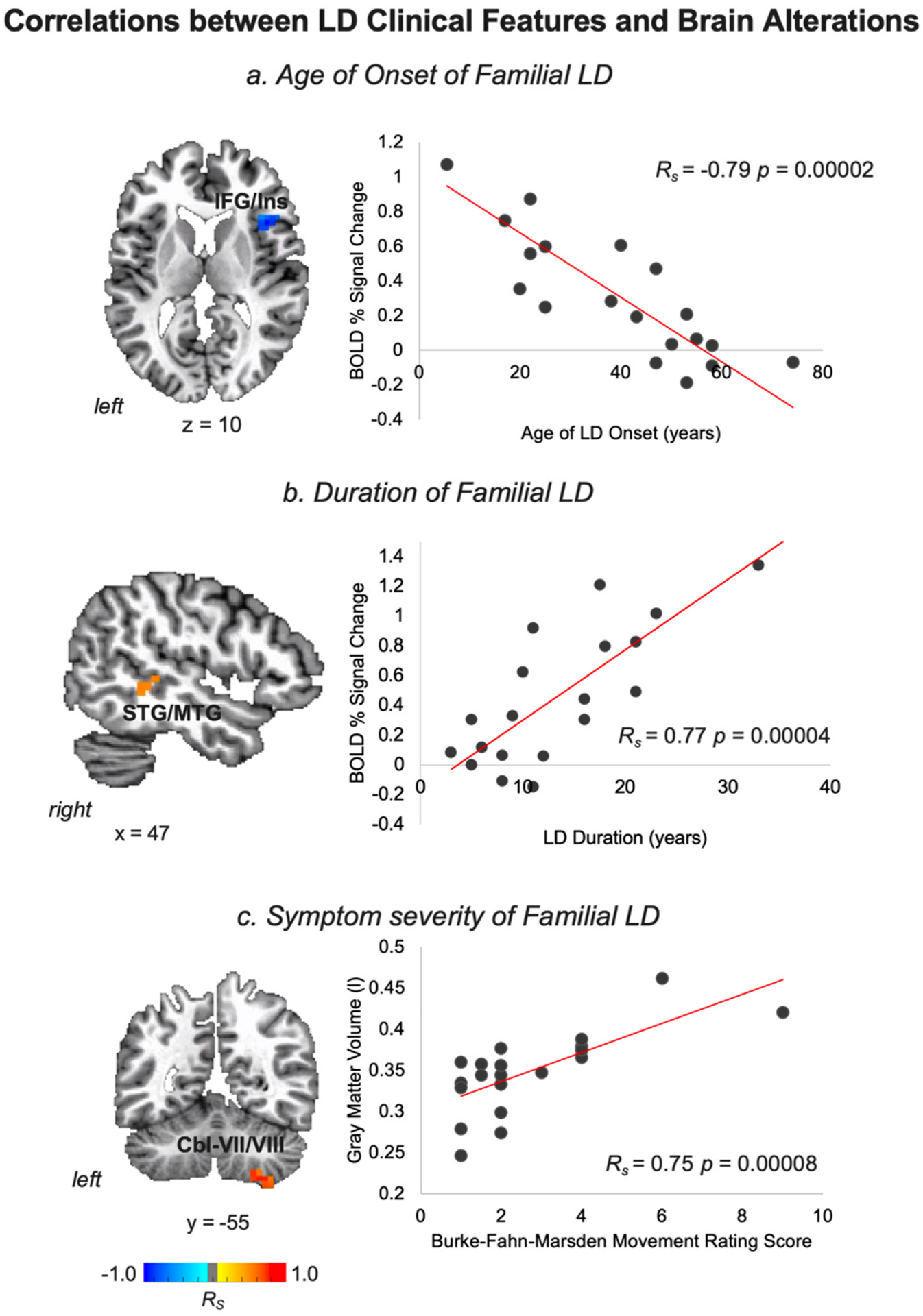
Correlations between LD clinical features and brain alterations are depicted in a series of axial, sagittal, or coronal brain slices in the AFNI standard Talairach-Tournoux space and the corresponding scatter plots. Abbreviations: Cbl - cerebellum, IFG - inferior frontal gyrus, Ins – insula, LD - laryngeal dystonia, MTG - middle temporal gyrus, STG - superior temporal gyrus. The color bar indicates Spearman rank correlation coefficients.
3.4. Predictive risk of LD development
To predict the risk of dystonia penetrance, the SVM classifier included all identified clusters of structural and functional alterations in a combined group of familial LD patients and their unaffected relatives vs. healthy controls as input features (Fig. 1-II, Table 2). Using three clusters of functional abnormalities in the left IFG/MFG, right IFG/insula, left ACC and two clusters of structural abnormalities in the right parietal operculum and left thalamus, the SVM achieved the highest overall classification accuracy of 86.5% by correctly separating 87.5% (28 out of 32) of healthy controls from 85.7% of familial LD patients (16 out of 21) and their unaffected relatives (20 out of 21) (Fig. 3-a).
To predict the risk of dystonia manifestation, the SVM classifier included all identified structural and functional clusters of neural alterations in familial LD patients vs. their unaffected relatives as input features (Fig. 1-III, Table 2). Using one functionally abnormal cluster in the right SPL and one structurally abnormal cluster in the right cerebellar lobule VII/VIII, the SVM had the highest overall classification accuracy of 78.6% by correctly identifying 85.7% (18 out of 21) of familial LD patients and 71.4% (15 out 21) of unaffected relatives. SVM classified 28.6% (6 out of 21) of unaffected relatives as familial LD patients. Among unaffected relatives classified as LD patients, three individuals were obligate carriers of yet unknown dystonia gene mutation, constituting 75% (3 out of 4) of all obligate carriers in tnis study cohort. This finding points to the validity of the SVM for detecting the genetic status based on the neural endophenotypic traits.
4. Discussion
The causative factors in the development of isolated focal dystonia in general and LD in particular remain unclear. Here, we identified a subset of structural and functional brain alterations in patients with familial LD and their unaffected, asymptomatic relatives that likely represent the endophenotypic traits underlying the penetrance and manifestation of this disorder. We found that a subset of these alterations may be successfully utilized as neural markers for the predictive assessment of the risk for dystonia penetrance in families and symptom manifestation in susceptible individuals.
Related to the intermediate endophenotypic trait of dystonia penetrance, common alterations in prefrontal-parietal cortical regions, thalamus, and caudate nucleus were found in both familial LD patients and their unaffected relatives compared to healthy controls. The role of abnormal prefrontal-parietal axis in the pathophysiology of focal dystonias has been recently highlighted by several studies (e.g., (Battistella et al., 2016; Fuertinger and Simonyan, 2018; Gallea et al., 2016; Maguire et al., 2020)). Specifically, it has been suggested that prefrontal-parietal alterations may represent a common trend across patients with different forms of task-specific focal dystonia and contribute to alterations of modality-specific hierarchical processing within the sensorimotor network (Bianchi et al., 2019). Among these regions, functional and structural changes in MFG and ACC may predispose to abnormalities in higher-order executive functions relevant to learning and coordination of the correct sequences of complex motor behaviors and their conflict and error monitoring (Arrighi et al., 2016; Holroyd and Coles, 2002). To that end, previous studies have shown that patients with LD and other forms of focal dystonia have various subclinical aberrations of executive control, for example, delayed reaction time to initiate a motor sequence and elevated temporal discrimination thresholds (Bradley et al., 2012; Simonyan et al., 2013; Termsarasab et al., 2016). The latter has been linked to altered brain activity and gray matter volume of MFG and ACC in LD patients (Termsarasab et al., 2016) and also found to be abnormal in unaffected relatives of dystonia patients (Kimmich et al., 2014), pointing to potential genetic underpinnings of these alterations. Notably, in this study, we found that brain activity in the left MFG shows a strong association between familial LD patients and their unaffected relatives, suggesting that a subset of functional changes contributing to dystonia penetrance may be explained by the existing predisposition due to kinship between the family members.
Other commonly abnormal regions in patients with familial LD and their unaffected relatives included the IFG extending to the premotor cortex, insula, and parietal operculum. These regions are necessary for the multilevel integration of sensorimotor information for speech motor planning and the monitoring of internal movement representations in preparation for speech production (Fuertinger et al., 2015; Price, 2012). Altered insular connectivity was previously shown to contribute to the large-scale disorganization of structural and functional networks in focal dystonias (Battistella et al., 2017; Hanekamp and Simonyan, 2020) and partake in the formation of LD genotype-specific functional changes (Battistella et al., 2016). In an earlier study, the IFG was found to be one of the few brain regions, along with the STG/MTG and cerebellum, where abnormally increased activity was coupled with abnormally increased gray matter volume (Simonyan and Ludlow, 2012). Interestingly, we found significant correlations between alterations in these brain regions and LD clinical characteristics. Understanding the modulatory influences of dystonia symptomatology on a structure-function relationship in these regions may be useful for the development of objective outcome measures of centrally acting treatment response.
Decreased gray matter volume of the left thalamus (its motor, premotor and parietal subdivisions) and increased activity of the left caudate nucleus were identified as subcortical neural correlates of dystonia penetrance. Thalamic abnormalities in LD patients have been previously attributed to altered speech motor planning and execution (Termsarasab et al., 2016), aberrant entrainment of learning reinforcement of motor actions (Fuertinger and Simonyan, 2018), and proposed as critical contributors to LD genotypical associations of the functional connectome (Fuertinger and Simonyan, 2017). The thalamus establishes a wide-ranging output projection system with the prefrontal-parietal cortex and likely represents a relay station of abnormal information transfer between the basal ganglia, cerebellum, and cortical output regions.
As a basal-ganglia input structure into the thalamus, the involvement of caudate nucleus is particularly interesting because alterations in this region were the only ones to be associated with both dystonia penetrance and manifestation. The role of caudate nucleus within the goaldirected control system has been well delineated in the pathophysiology of dystonia (Simonyan, 2018). Relevant to impaired speech production in LD, a failure of caudate nucleus to suppress unintended responses and appropriately engage in error feedback monitoring during speaking (Price, 2010; Simonyan et al., 2013) emerges as a critical hereditary trait underlying disorder penetrance that further deteriorates with clinical manifestation of dystonic symptoms.
Taken together, the presence of abnormalities in prefrontal-parietal cortex, thalamus, and caudate nucleus in both patients with familial LD and their unaffected relatives likely points to the intermediate endophenotype of dystonia penetrance. It is, therefore, not surprising that these regions also constituted the predictive marker of dystonia penetrance, with the overall 86.5% accuracy. Notably, machine-learning categorization of 95.2% of unaffected relatives together with familial LD patients rather than healthy controls further supports the presence of common neural alterations as an endophenotypic marker of dystonia penetrance, independent of its symptomatology.
On the other hand, the fact that these neural alterations are commonly present in entirely asymptomatic relatives of LD patients suggests that symptom manifestation may, in part, be triggered due to the development of secondary endophenotypes that involve additional functional and structural abnormalities. In line with this assumption, we found that familial LD patients, compared to their unaffected relatives, exhibit distinct abnormalities in SMA, SPL, STG/MTG, and cerebellum. We propose that these regional alterations, together with more extended abnormalities in caudate nucleus, constitute the secondary endophenotype of LD, the development of which is necessary for dystonia manifestation in families.
As discussed above, prefrontal-parietal abnormalities play an important role in the pathophysiology of task-specific focal dystonias. Altered premotor-parietal activity and functional connectivity have been previously linked with the polygenic risk of dystonia (Putzel et al., 2018) and the extrinsic risk for the development of symptoms in familial LD patients (de Lima Xavier and Simonyan, 2019). Structurally, the SMA and SPL have been shown to contribute to LD genotype-specific changes in cortical thickness (Bianchi et al., 2017) and to represent significant regional alterations of the LD-specific structural connectome (Hanekamp and Simonyan, 2020).
Similarly, cerebellar changes have been widely implicated in the development of dystonia, both in patients with the genetic forms of disorder and corresponding animal models. Reduced integrity of white matter pathways of the cerebellar lobule VI has been reported in DYT1 and DYT6 manifesting careers (Argyelan et al., 2009). Substantiating these findings, an ex vivo study in the heterozygous DYT1 knock-in mouse model showed reduced structural connectivity of the cerebellothalamic pathway in mutants compared to the wild type (Ulug et al., 2011). An in vivo study in the conditional knock-out mice of DYT1 protein torsinA also found deficits of free water diffusivity and widespread increases in functional connectivity involving the cerebellum (DeSimone et al., 2017). Our findings of a strong relationship in the gray matter volume of cerebellar lobule VI between familial LD patients and their unaffected relatives and distinct alterations of the cerebellar lobules VII/VIII in familial LD patients are in line with these previous reports. Moreover, a positive correlation between symptom severity and gray matter volume of cerebellar lobule VII/VIII further suggests its modulatory involvement in LD symptomatology. The importance of cerebellar and parietal alterations in dystonia pathophysiology is evident from their contribution as the predictive markers of LD manifestation showing an 85.7%-accuracy in classifying familial LD patients. Equally important, these regional markers categorized 28.6% of unaffected relatives as familial LD patients, pointing to their increased lifetime risk for developing clinical manifestation of dystonia. This predicted rate of the risk for developing dystonia by (yet) unaffected relatives of familial LD patients is similar to the previously reported rate of age-adjusted lifetime risk of dystonia manifestation in asymptomatic carriers of DYT1 mutation, with the range of 11.4% to 26%, depending on the degree of relatedness with the manifesting carrier (Bressman et al., 1989; Risch et al., 1990). Future longitudinal studies are warranted in this cohort of unaffected relatives of LD patients to follow their trends to phenoconversion to LD or other forms of isolated dystonia.
In summary, patients with familial history of dystonia share common features of structural and functional alterations with their unaffected relatives, which likely represent the intermediate endophenotype underlying disorder penetrance and encompass prefrontal-parietal, basal ganglia, and thalamic regions. Building on these hereditary brain changes, an additional set of alterations within premotor-parietal and temporal cortices, caudate nucleus, and cerebellum comprise the secondary endophenotype, the development of which appears to be necessary for the manifestation of dystonic symptoms in predisposed individuals. Importantly, these endophenotypic neural markers might be successful in screening of at-risk individuals for dystonia development, selection of LD families based on their initial risk for dystonia penetrance in genetic studies of novel variants, and potential stratification of patients who would respond differently to a particular treatment in clinical trials. Future studies in larger cohorts are warranted to further validate the identified machine-learning markers of LD penetrance and manifestations and to extend a similar approach to other forms of dystonia.
Supplementary Material
Acknowledgments
We thank Davide Valeriani, PhD, for his assistance with the machine-learning analysis. We thank Dr. Steven J. Frucht, MD, and Dr. Andrew Blitzer, MD, DDS, for patient referral and clinical evaluations. This study was funded by the National Institute on Deafness and Other Communication Disorders R01DC011805 grant to KS.
Footnotes
Declaration of Competing Interest
The authors report no conflict of interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2020.105223.
References
- Argyelan M, et al. , 2009. Cerebellothalamocortical connectivity regulates penetrance in dystonia. J. Neurosci 29, 9740–9747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrighi P, et al. , 2016. EEG Theta dynamics within frontal and parietal cortices for error processing during reaching movements in a prism adaptation study altering visuo-motor predictive planning. PLoS One 11, e0150265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battistella G, et al. , 2016. Cortical sensorimotor alterations classify clinical phenotype and putative genotype of spasmodic dysphonia. Eur. J. Neurol 23, 1517–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battistella G, et al. , 2017. Isolated focal dystonia as a disorder of large-scale functional networks. Cereb. Cortex 27, 1203–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi S, et al. , 2017. Phenotype- and genotype-specific structural alterations in spasmodic dysphonia. Mov. Disord 32, 560–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi S, et al. , 2019. Functional and structural neural bases of task specificity in isolated focal dystonia. Mov. Disord 34, 555–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitzer A, et al. , 1998. Botulinum toxin management of spasmodic dysphonia (laryngeal dystonia): a 12 year experience in more than 900 patients. Laryngoscope. 108, 1435–1441. [DOI] [PubMed] [Google Scholar]
- Blitzer A, et al. , 2018. Phenomenology, genetics, and CNS network abnormalities in laryngeal dystonia: a 30-year experience. Laryngoscope. 128 (Suppl. 1), S1–S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley D, et al. , 2012. Temporal discrimination thresholds in adult-onset primary torsion dystonia: an analysis by task type and by dystonia phenotype. J. Neurol 259, 77–82. [DOI] [PubMed] [Google Scholar]
- Bressman SB, et al. , 1989. Idiopathic dystonia among Ashkenazi Jews: evidence for autosomal dominant inheritance. Ann. Neurol 26, 612–620. [DOI] [PubMed] [Google Scholar]
- de Lima Xavier L, Simonyan K, 2019. The extrinsic risk and its association with neural alterations in spasmodic dysphonia. Parkinsonism Relat. Disord 65, 117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSimone JC, et al. , 2017. Forebrain knock-out of torsinA reduces striatal free-water and impairs whole-brain functional connectivity in a symptomatic mouse model of DYT1 dystonia. Neurobiol. Dis 106, 124–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuertinger S, Simonyan K, 2017. Connectome-wide phenotypical and genotypical associations in focal dystonia. J. Neurosci 37, 7438–7449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuertinger S, Simonyan K, 2018. Task-specificity in focal dystonia is shaped by aberrant diversity of a functional network kernel. Mov. Disord. Dec 33 (12), 1918–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuertinger S, et al. , 2015. The functional connectome of speech control. PLoS Biol. 13, e1002209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallea C, et al. , 2016. Impairment of a parieto-premotor network specialized for handwriting in writer’s cramp. Hum. Brain Mapp 37, 4363–4375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiry S, et al. , 2019. A separation of innate and learned vocal behaviors defines the symptomatology of spasmodic dysphonia. Laryngoscope. 129, 1627–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanekamp S, Simonyan K, 2020. August 15. The large-scale structural connectome of task-specific focal dystonia. Hum. Brain Mapp 41 (12), 3253–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holroyd CB, Coles MGH, 2002. The neural basis of human error processing: reinforcement learning, dopamine, and the error-related negativity. Psychol. Rev 109, 679–709. [DOI] [PubMed] [Google Scholar]
- Hutchinson M, et al. , 2013. The endophenotype and the phenotype: temporal discrimination and adult-onset dystonia. Mov. Disord 28, 1766–1774. [DOI] [PubMed] [Google Scholar]
- Kimmich O, et al. , 2014. Temporal discrimination, a cervical dystonia endophenotype: penetrance and functional correlates. Mov. Disord 29, 804–811. [DOI] [PubMed] [Google Scholar]
- Kirke D, et al. , 2015. Alcohol responsiveness in laryngeal dystonia: a survey study. Neurology. 262, 1548–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, et al. , 2017. Alterations of resting-state fMRI measurements in individuals with cervical dystonia. Hum. Brain Mapp 38, 4098–4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludlow CL, et al. , 2008. Research priorities in spasmodic dysphonia. Otolaryngol. Head Neck Surg 139, 495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire F, Reilly RB, Simonyan K, et al. , 2020. May. Normal Temporal Discrimination in Musician’s Dystonia Is Linked to Aberrant Sensorimotor Processing. Mov. Disord 35 (5), 800–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price CJ, 2010. The anatomy of language: a review of 100 fMRI studies published in 2009. Ann. N. Y. Acad. Sci 1191, 62–88. [DOI] [PubMed] [Google Scholar]
- Price CJ, 2012. A review and synthesis of the first 20 years of PET and fMRI studies of heard speech, spoken language and reading. Neuroimage. 62, 816–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putzel GG, et al. , 2018. Polygenic risk of spasmodic dysphonia is associated with vulnerable sensorimotor connectivity. Cereb. Cortex 28, 158–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risch NJ, et al. , 1990. Segregation analysis of idiopathic torsion dystonia in Ashkenazi Jews suggests autosomal dominant inheritance. Am. J. Hum. Genet 46, 533–538. [PMC free article] [PubMed] [Google Scholar]
- Rumbach AF, et al. , 2017. An open-label study of sodium oxybate in spasmodic dysphonia. Laryngoscope. 127, 1402–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonyan K, 2018. Neuroimaging applications in dystonia. Int. Rev. Neurobiol 143, 1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonyan K, Ludlow CL, 2012. Abnormal structure-function relationship in spasmodic dysphonia. Cereb. Cortex 22, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonyan K, et al. , 2013. Abnormal striatal dopaminergic neurotransmission during rest and task production in spasmodic dysphonia. J. Neurosci 33, 14705–14714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Termsarasab P, et al. , 2016. Neural correlates of abnormal sensory discrimination in laryngeal dystonia. Neuroimage Clin. 10, 18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulug AM, et al. , 2011. Cerebellothalamocortical pathway abnormalities in torsinA DYT1 knock-in mice. Proc. Natl. Acad. Sci. U. S. A 108, 6638–6643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valeriani D, Simonyan K, 2020. October 20. A microstructural neural network biomarker for dystonia diagnosis identified by a DystoniaNet deep learning platform. Proc. Natl. Acad. Sci. U. S. A 117 (42), 26398–26405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.