

A designed protein complex processively copies one strand of a plasmid with errors to speed up molecular evolution in vivo.
Abstract
Extensive exploration of a protein’s sequence space for improved or new molecular functions requires in vivo evolution with large populations. But disentangling the evolution of a target protein from the rest of the proteome is challenging. Here, we designed a protein complex of a targeted artificial DNA replisome (TADR) that operates in live cells to processively replicate one strand of a plasmid with errors. It enhanced mutation rates of the target plasmid up to 2.3 × 105–fold with only a 78-fold increase in off-target mutagenesis. It was used to evolve itself to increase error rate and increase the efficiency of an efflux pump while simultaneously expanding the substrate repertoire. TADR enables multiple simultaneous substitutions to discover functions inaccessible by accumulating single substitutions, affording potential for solving hard problems in molecular evolution and developing biologic drugs and industrial catalysts.
INTRODUCTION
Evolutionary innovation of new protein functions is central to Darwinian adaptation. For example, bacterial efflux pumps evolved into antibiotic resistance proteins (1). Natural evolution depends in complex ways on population size, mutation rate, and the shape of the adaptive landscape. Because evolution in nature occurs over decades or longer, it is difficult to study and also difficult to apply to making new enzymes for medicine and industry. The tool to solve this problem would target enhanced mutagenesis to specific loci in vivo, such as a plasmid that carries gene(s) for the protein(s) of interest. The in vivo approach creates larger populations with mutation combinations beyond that accessible by in vitro directed evolution. One milliliter of overnight bacterial culture carries cells in billions—the theoretical size of mutant library, whereas directed evolution experiments, which rely on in vitro chemistry of error-prone polymerase chain reaction (PCR), use no more than several million variants by pooling colonies from many electroporations (2). Targeting specific loci prevents deleterious mutations elsewhere from obscuring beneficial mutations on the target. Enhanced mutagenesis speeds up evolution from decades to hours.
The ideal in vivo mutagenesis tool would (i) target a region no less than a gene and show low off-target mutagenesis, (ii) have a high mutation rate that can be turned on and off and include all types of nucleotide substitutions, and (iii) be easy to use and not limit the type of trait that can be evolved. Previous in vivo mutagenesis tools lack one or more of these features (table S1). Some of these tools repurpose complex natural systems (3–12), whose intrinsic complexity limits functions of the tools. The requirement of coupling the target function to the expression of phage protein (7) limits the traits that can be evolved, and the inability to switch mutagenesis on/off (4, 6, 7, 9) complicates identification of beneficial mutations. Other tools are simple fusions between a targeting protein (RNA polymerase or Cas9) and a mutagenesis protein (cytosine deaminase or error-prone DNA polymerase) (13–15). Limitations of these tools are either a narrow mutagenesis region of several dozen base pairs (13, 14) and/or creation of only 1 of the 12 possible types of mutations (C to U) (14, 15). Therefore, no tools currently satisfy all three requirements.
Here, we report targeted in vivo mutagenesis using a three-protein complex of a viral nickase, bacterial Rep helicase, and an error-prone DNA polymerase to copy one strand of a target plasmid with errors. This targeted artificial DNA replisome (TADR) fulfills the three requirements of targeting, mutagenesis, and trait flexibility defined above. It minimizes interference with natural cellular processes by its simplicity but is complex enough to target a plasmid carrying full-length genes of interest with the full spectrum of mutations.
RESULTS
Designing a replisome in live cells to accelerate protein evolution
Most mutations originate from errors during copying of DNA by a multiprotein complex known as the DNA replisome. In the bacterium Escherichia coli, this protein complex consists of 18 components (16), but that of bacteriophage PhiX174 uses only 3 components (17): phage protein CisA, bacterial Rep helicase, and bacterial DNA polymerase III. CisA initiates the copying of bacteriophage DNA by binding to a 30–base pair (bp) initiation sequence, nicking one DNA strand, and recruiting Rep helicase of host cell (18). The helicase unwinds the double-stranded DNA from the nick site. Bacterial DNA polymerase III diffuses to the nick site and adds nucleotides to the free hydroxyl group to synthesize a new strand. This minimal bacteriophage replisome provides the starting point for the design of an artificial replisome with an enhanced mutation rate targeted to a plasmid.
We hypothesized that fusing an error-prone DNA polymerase to Rep helicase would create an error-prone DNA replisome (Fig. 1, A and B). The close proximity of the error-prone DNA polymerase would favor its use in the replisome complex, resulting in a high mutation rate (Fig. 1C; detailed discussions in Materials and Methods, “Biophysical rationale of building an artificial replisome”). This error-generating replisome required nontrivial coordination between the two molecular motors of helicase and DNA polymerase (Fig. 1D and fig. S1). If DNA polymerase is slower than Rep helicase, the unwound single-strand DNA will build up between the two motors, promoting destruction by nucleases abundant in the cell and thus causing collapse of the replisome. If DNA polymerase is faster than Rep helicase, a collision between them might dislodge the helicase. Single-molecule studies showed that both motors tolerated collisions: Rep helicase maintained a nearly constant speed when subjected to external forces (19), while T7 DNA polymerase slowed down (Fig. 1D) (20). The speeds intersected predicting that applying an external force on T7 DNA polymerase could slow it down so that both motors act at the same speed. As Rep helicase unwinds the DNA, the faster T7 DNA polymerase bumps into it. This bumping creates an external force that does not affect Rep helicase, but slows down T7 DNA polymerase, leading to the coordinated action of both.
Fig. 1. A targeted error-prone artificial DNA replisome.
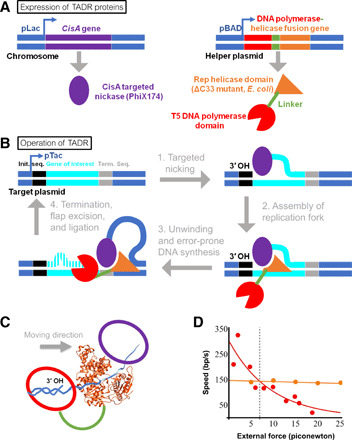
(A) The two components of TADR, cisA targeted nickase from phage PhiX174 and the fusion of T5 DNA polymerase from phage T5 and Rep helicase from bacterium E. coli, are expressed from chromosome and helper plasmid, respectively. A mutant Rep helicase is used with deletion of 33 C-terminal amino acid residues. The two parallel bars denote double-stranded DNA; purple oval, CisA protein; orange wedge, Rep helicase domain of the fusion protein; red circle, DNA polymerase domain; green line, linker. Promoters are indicated with arrows and the names. The chromosomal rep of the host cell was knocked out. (B) Proposed steps of in vivo targeted mutagenesis by an artificial replisome. See details in the text. Color coding is as per (A). Black, gray, and cyan blocks indicate the initiation sequence, the termination sequence, and the gene(s) of interest, respectively. Dashes mark nascent DNA, and the bulge marks the mutation introduced from synthesis. (C) Proposed arrangement of proteins in the artificial replisome. The proposed location of the CisA protein (purple) relative to the ribbon diagram of the x-ray structure of Rep helicase (PDB: 1AUU) is based on a structure of a homologous protein complex (45). The DNA polymerase (red circle) is fused via a linker (green curve) to the N terminus of Rep helicase. Arrow indicates direction of the replisome translocating on DNA. (D) External force has little effect on the speed of DNA unwinding by Rep helicase (orange) but slows T7 phage DNA polymerase (red). Experimental data were retrieved from (19, 20) and fitted by a linear and an exponential model, respectively. Dashed line indicates the intersection of the two speed profiles, marking the force required for both enzymes to act at the same speed.
Our initial attempt of building TADR with T7 DNA polymerase failed because the active site was too close to the linker site at the C terminus (details in Supplementary Text, “Choice of DNA polymerase”). We chose T5 as the replacement over the previously used E. coli DNA polymerase I (3, 13). T5 is a fast polymerase like T7 (21), while the alternative E. coli DNA polymerase I is slower than Rep (20), and thus not expected to form a stable replisome according to Fig. 1D. The active site of T5 polymerase lies farther from the C terminus than in T7 polymerase (table S5) (21), suggesting that the fusion protein would maintain polymerase activity.
A flexible 81–amino acid linker connected the N terminus of Rep helicase to the C terminus of T5 DNA polymerase (Fig. 1A and fig. S2B). This DNA polymerase carried three amino acid substitutions (D164A, E166A, and A593R) to increase its error rate. Substitution of these residues increased the error rate in homologous polymerases (table S2) (3, 22).
We further hypothesized that the enhanced mutagenesis might be restricted to the gene(s) of interest by adding the initiation and termination sequences for CisA before and after the gene(s) of interest, respectively (23), but experiments below indicate that enhanced mutagenesis occurred on the entire target plasmid and read-through occurred with high frequency. Copying a template DNA strand creates a new DNA strand annealed to the template strand, while the nontemplate strand is unbound as a flap (Fig. 1B). This flap is naturally excised (24), and the resulting nick ligated (25). If an error-prone polymerase carries out the copying, then the copied DNA likely contains a mutation. This copying process repeats to introduce additional mutations. As will be detailed later, targeting was highly selective for the plasmid over the chromosome but targeting was poorly selective within the target plasmid.
Three DNA elements were added to E. coli to create a targeted error-prone artificial replisome: a constitutively expressed, chromosomal CisA gene, a target plasmid, and a helper plasmid (Fig. 1, A and B). The target plasmid contained the gene(s) of interest between the CisA initiation and termination sequences. To quantify the increase in mutagenesis, we used a gene encoding kanamycin resistance protein [aminoglycoside phosphotransferase (3′)-IIIa]. This gene, named kanR*, carried a single-nucleotide substitution (A785C), which encodes an E262A amino acid substitution that inactivates the kanamycin resistance (26). Mutations that reversed this substitution would restore gene function, allowing the cells to grow in the presence of kanamycin. Frequency of resistant colony-forming unit (CFU) per cell plated revealed the mutagenic capacity. The helper plasmid contained a gene encoding the fusion of the error-prone T5 DNA polymerase to Rep helicase under the control of a T7 promoter. (The host cell also contained a chromosomal gene to express the T7 RNA polymerase and a deletion of the chromosomal Rep gene to eliminate potential competition with the fusion protein.)
Individual components of TADR are functional in vivo
In vivo activity of CisA was measured by its ability to nick DNA at the CisA initiation site. Bacteria containing constitutively expressed, chromosomal CisA gene and target plasmid carrying kanR* (but no helper plasmid) were propagated for multiple generations. The target plasmid was extracted and separated by gel electrophoresis into the supercoiled, linear, and nicked forms (Fig. 2A). Substantial nicking of the target plasmid occurred when both CisA and its initiation sequence were present, consistent with CisA protein being active. When either CisA protein or its initiation sequence was absent, less nicking occurred. The small amount of untargeted nicking is likely nonspecific hydrolysis during the alkaline lysis step of the plasmid extraction (27).
Fig. 2. Testing in vivo functionality of TADR components.
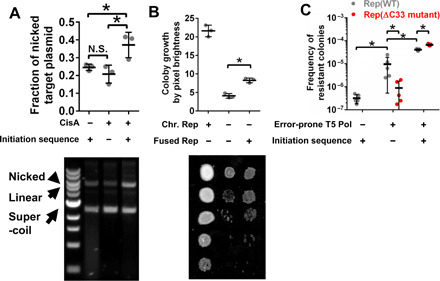
(A) CisA nicking of the target plasmid. Picture of gel electrophoresis is shown in the bottom with different species of plasmid marked. Quantification of the nicked fraction by image analysis is shown on the top. Star indicates statistical significance with α of 0.05, Student’s t test, and n = 3 independently prepared and measured biological replicates. N.S., not significant. Error bars, SD. (B) Rep fused to DNA polymerase to alleviate sensitivity of rep- cells to nalidixic acid. Picture of colony growth of serially diluted cultures is shown (dilution factor of 10, starting from no dilution). Colony growths of 10-fold dilution (second row from the top) were quantified by brightness. An expanded picture with three replicates is shown in fig. S3. Medium: LB supplemented with nalidixic acid (2.5 μg/ml). Statistics is as per (A). Error bar, SD. (C) Mutagenesis on the target plasmid. After overnight liquid growth of cells to full density, during which mutations also accumulated, cultures from each sample were plated on LB supplemented with kanamycin (30 μg/ml). Colonies were counted after 16 hours of incubation. The frequency of kanamycin-resistant colonies per cell plated is reported. Statistics is as per (A) except n = 5 independently prepared and measured biological replicates. Error bar, SD.
The Rep helicase activity of the T5 DNA polymerase–Rep fusion was measured by its ability to alleviate growth retardation caused by nalidixic acid in a strain lacking endogenous Rep helicase (28). Bacteria normally tolerate low levels of nalidixic acid, but those lacking endogenous Rep helicase grow more slowly because nalidixic acid inhibits DNA gyrase and topoisomerase. Introducing a helper plasmid, which added expression of the fusion protein, into rep- bacteria partially alleviated the nalidixic acid inhibition of growth (Fig. 2B and fig. S3), which confirmed that the fusion protein maintained Rep helicase activity.
The DNA polymerase activity of the fusion protein was measured by its capacity to mutate kanR* and restore kanamycin resistance. The bacteria contained all three elements of the targeted error-prone artificial replisome: a constitutively expressed, chromosomal CisA gene; the target plasmid carrying the kanR* gene flanked by the initiation and termination sequences; and the helper plasmid expressing the fusion protein. These cells were picked from a transformation plate, propagated in liquid for 10 generations to accumulate mutations, and then plated onto kanamycin-containing medium for selection. Figure 2C shows that the treatment (third column, gray dots) increased 128-fold than the control with wild-type T5 DNA polymerase (first column, gray dots) in the frequency of kanamycin-resistant colonies and 4.2-fold than the control whose target plasmid was without initiation sequence (second column, gray dots). These results demonstrated qualitatively that the fusion protein provided error-prone DNA polymerase activity, which was able to distinguish nontarget from target as marked by initiation sequence. Nevertheless, the performance characteristics of the tool, i. e., on-target mutagenic activity and selectivity (on- over off-target mutagenic activity), needed to be optimized for applications in in vivo targeted mutagenesis (Table 1).
Table 1. Determinants of mutation rate and targeting selectivity of TADR, and their corresponding strategies for optimization.
The measured effects from applying these strategies are also given. N/A, not applicable. CisA has evolved in nature to bind initiation sequence tightly with high specificity.
| Determinants |
Optimization strategies |
Effects (fold increase) |
|
| Mutation rate | Expression level of T5 DNA polymerase |
Self-evolve TADR to increase error rate |
3.2 |
| Expression level of the fusion protein |
Modify 5′UTR to increase transcription of the fusion gene |
150 | |
| Targeting selectivity |
Binding affinity of cisA to initiation sequence |
N/A | |
| Rep helicase and thus its fused error-prone T5 DNA polymerase colocalize with innate replisome, causing off-target mutations |
Adopt RepΔC33 mutant to eliminate colocalization to reduce off-target mutations |
16 | |
| Encounter of error-prone T5 DNA polymerase with innate replisome by random diffusion, causing off-target mutations |
Reduce concentration (copy number per cell) of chromosome and thus innate replisome by growing cells in minimal medium to reduce off-target mutations |
89, 45* |
*Off-target mutation rate used to calculate selectivity was measured by spontaneous rifampicin (89) or streptomycin (45) resistance.
Self-evolution of TADR for increase in mutation rate
To optimize performance of TADR and as a proof-of-concept experiment, the error-prone T5 DNA polymerase was subjected to self-evolution, which increased the mutation rate 3.2-fold [Fig. 3A; analysis of variance (ANOVA), α = 0.05, P = 0.033, n = 5]. To mutagenize the gene encoding T5 DNA polymerase, initiation and termination sequences were inserted on helper plasmid to flank the polymerase portion of the fusion gene. The termination sequence was in-frame to maintain protein expression of the fusion protein. TADR cells with only this helper plasmid but no target plasmid were cultured for the T5 DNA polymerase to mutate its own gene. Target plasmid with kanR* was then introduced by transformation for selection. To select for increased mutation rate, we reduced the incubation time before selection for kanamycin resistance (Materials and Methods, fig. S4). This approach identified a variant DNA polymerase, which, in separate experiments, increased the number of kanamycin-resistant CFU 3.2-fold (Fig. 3A). This polymerase variant carried a single I308V substitution (Fig. 3B, blue residue). This substitution is unlikely to change the accuracy of the polymerase, but may stabilize it or increase its expression, thereby increasing the error frequency. This substitution lies in the exonuclease (proofreading) domain, which is already inactivated by two substitutions (D164A and E166A; Fig. 3A, pink residues) (22), and also lies outside the catalytic pocket (8.8 Å from D164), so it is unlikely to affect catalytic activity. The change in shape of the hydrophobic side chain from isoleucine to valine may alter the protein stability or increase protein expression, thereby increasing the observed error frequency.
Fig. 3. Development and characterization of TADR.
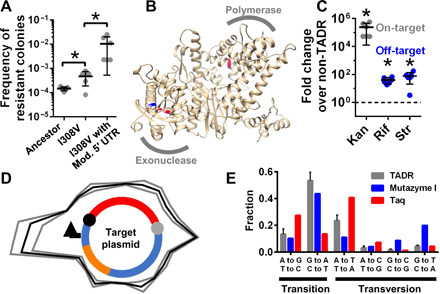
(A) Mutagenic capacity of TADR with different versions of the helper plasmid. The conditions were as per Fig. 2C. RepΔC33 was used. Star indicates statistical significance with α of 0.05, Student’s t test, and n = 5 or 6 independently prepared and measured biological replicates. Error bar, SD. (B) Structure of T5 DNA polymerase was modeled using Robetta server (46). The evolved I308V was blue, and mutations in the ancestor were magenta. (C) Performance of TADR was measured by its fold change in on- and off-target mutations over non-TADR. Non-TADR: Wild-type E. coli with the target plasmid carrying kanR*. TADR: TADR cells with the same target plasmid and the helper plasmid carrying RepΔC33, I308V, and modified 5′ UTR. The on-target fold change was measured using kanamycin-resistant colonies; the off-target fold change was measured using streptomycin- and rifampicin-resistant colonies separately. Statistics is as per (A) except n = 6 independently prepared and measured biological replicates. Error bar, SD. N.S., not significant. Dashed line indicates the level of non-TADR. (D) Mutational density across the whole target plasmid. The red fragment indicates the gene(s) of interest; orange, origin of plasmid replication; black ball, initiation sequence; gray ball, termination sequence; arrow, promoter. Black curve depicts the average mutational density of three biological replicas; gray curves, SD. (E) Mutational spectrum of TADR (n = 3 independently prepared and measured biological replicates; error bar, SD) and other error-prone DNA polymerases.
Maximizing on-target and minimizing off-target mutagenesis
A stem-loop was designed into 5′ untranslated region (5′ UTR) upstream the gene encoding the fusion protein and increased the mutation rate 150-fold (Fig. 3A), likely by increasing the amount of fusion protein, whose expression level was calculated to increase 37-fold (table S3).
Because TADR uses Rep helicase from E. coli, it may cause off-target mutations in the chromosome, where Rep helicase normally participates in DNA replication (29). Consistent with this expectation, removal of the initiation sequence for CisA decreased mutagenesis only 4.2-fold (Fig. 2C). To improve the targeting, we deleted the 33 C-terminal amino acids of Rep helicase in the fusion protein. These amino acids bind Rep helicase to the innate replisome (30). The fusion containing RepΔC33 showed a 16-fold increase in selectivity. Removal of the initiation sequence now decreased mutagenesis 67-fold (Fig. 2C, red dots, third over second column).
To further increase selectivity, we reasoned that the error-prone T5 DNA polymerase in the fusion protein, even with RepΔC33, could bump into innate replisomes by random diffusion and thus mutating the chromosome. So far, the experiments were all conducted in rich medium [Luigi-Bertani (LB) broth], where a single cell contains multiple copies of the chromosome, which replicated continuously throughout the cell cycle (31). The resulting many copies of innate replisomes provide sites where the error-prone T5 DNA polymerase could mutate the chromosome. We hypothesized that reducing the number of innate replisomes would suppress off-target mutagenesis. One method is growing cells in minimal medium, where each cell has only one copy of the chromosome and where chromosomal replication occurs in only a fraction of the cell cycle (32). The lowered number of innate replisomes would minimize their exposure to the error-prone T5 DNA polymerase.
Cells of the previous control to measure nontargeted mutagenesis (Fig. 2C, second column), whose target plasmid was without initiation sequence, failed to grow in minimal medium, while those of the treatment, with initiation sequence, grew normally. A similar pattern was seen in LB and discussed in detail in Supplementary Text, “Toxicity of the target plasmid without initiation sequence.” In consequence, a new control was needed to measure off-target mutagenesis.
Spontaneous resistance to rifampicin and streptomycin is an established trait to allow measurement of mutation rate on chromosome (13, 33). Because chromosomal and plasmid-borne traits differ in type and in the number of copies, mutation rate of a mutagenic system as measured by resistance to an antibiotic was normalized to that of its nonmutagenic counterpart as done previously by Camps et al. (3). The resulting fold change adjusts for the different genetic underpinnings of resistance to different markers and thus provides a proxy for universal mutation rate (3).
Figure 3C shows that the engineered and evolved TADR, when induced in minimal medium for mutagenesis, had high activity and selectivity for mutations on target plasmid. The on-target mutagenesis of TADR increased 2.37 × 105–fold compared to that of non-TADR baseline. The off-target mutagenesis increased slightly: 39.7- and 77.5-fold for rifampicin and streptomycin resistance, respectively, corresponding to selectivities of 5970- and 3060-fold (obtained by dividing 2.37 × 105 by 39.7 and 77.5). The agreement between the increases in off-target mutagenesis measured by different antibiotics (ANOVA, n = 6, P = 0.16) confirms that the normalization method allows comparison of mutation rate of different traits on plasmid versus on chromosome.
To test if the mutagenic activity could be suppressed, glucose was added to minimal medium in which TADR cells were grown—glucose suppresses transcription from the promoters for cisA and the fusion protein by catabolite repression. The mutagenic activity was lowered by 112-fold with the presence of glucose than without (fig. S5).
NGS confirms the designed mechanisms for high performance of TADR
Next-generation sequencing (NGS) revealed increased mutagenesis throughout the plasmid with two hotspots. (Figure 3D shows mutational density defined as the number of mutations normalized to the total mutations of the sample per 200-bp window across the entire plasmid; a total of 264 point mutations were called in the three samples; see details in Materials and Methods and figs. S6 and S7.) Both hotspots coincide with sites of transcription initiation or termination and can be explained by molecular collision–induced hypermutagenesis (see Supplementary Text, “NGS identifies mutational hotspots explicable by molecular collision”).
The two hotspots together only account for 11.5% of the target plasmid. We now focus on the remaining 88.5%. The region defined by the flanking initiation and termination sequences [the gene(s) of interest] had 1.75-fold higher mutations per base pair (ANOVA, n = 3, P = 0.01) than the plasmid backbone. Hence, TADR replisome reads through the termination sequence 57% of the time. Within the region from initiation to termination sequences, mutational density did not decline as tested by linear regression across this region of 1596 bp (bin size, 100 bp; P = 0.77). This high processivity and the targeting—although with substantial read-throughs and a selectivity of 1.75—are expected from the design in Fig. 1. The high mutation rate throughout the plasmid demonstrates the ability to mutagenize multiple genes simultaneously.
The mutational spectrum of TADR is similar to those of Taq DNA polymerase and commercial error-prone DNA polymerase widely used for directed evolution (mutazyme I, Agilent Technologies) with a bias for transitions of G to A and C to T (Fig. 3E). The ratio of transitions over transversions did not differ between the mutational hotspots and the rest of the plasmid (ANOVA, n = 3, P = 0.35). Overall, NGS confirmed that the designed replisome copied one strand of target plasmid with error, yielding a mutational spectrum similar to model error-prone DNA polymerases.
TADR evolves new molecular functions with large mutational step size
We evolved a tetracycline efflux pump [encoded by tetA(C)] to confer resistance to an analog, tigecycline. This tetracycline derivative, used as a last resort antibiotic, contains a large substituent that hinders the evolution of resistance (fig. S8) (34). TADR cells carrying both helper plasmid and a target plasmid containing the tetA(C) gene were grown in liquid culture to accumulate mutations and then selected by plating on tigecycline-supplemented medium. After two rounds of mutagenesis selection, the growth rate in tigecycline (8 ng/μl) increased up to 16-fold compared to the ancestor (wild-type pump) (ANOVA, α = 0.05, n = 5, P < 0.005). All five mutants isolated with increased resistance to tigecycline maintained or increased their resistance to tetracycline (Fig. 4A; details in fig. S9). For example, mutant 1-1 increased resistance to both tetracycline and tigecycline as compared to the ancestor. This ability to resist both antibiotics indicates that expanding the function of the tetracycline efflux pump did not require a trade-off between resistance to tetracycline and to tigecycline.
Fig. 4. Evolutionary innovation by TADR: Optimizing an efflux pump with expanded substrate repertoire and reduced cellular toxicity.
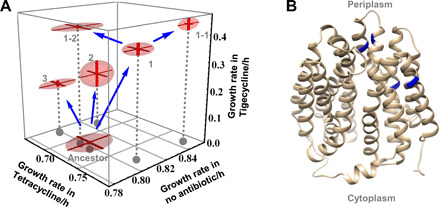
(A) Trajectories of phenotypic adaptation. TADR with RepΔC33, 5′ UTR–modified, and I308V was used. Starting from the ancestor, three parallel populations were subjected to the first round of mutagenesis selection on LB supplemented with tigecycline (4 ng/μl). One big colony was selected from each population and numbered 1, 2, and 3. Number 1 gave bigger colonies than the other two and thus was used as the ancestor for a second round of mutagenesis selection on LB supplemented with tigecycline (8 ng/μl). One big colony was selected from each of two populations and numbered 1-1 and 1-2. The efflux pump gene from five isolates was each PCR-amplified and reintroduced to the unevolved backbone of target plasmid. This treatment purged the potential complicating effect of adaptive mutations that occurred off-target during evolution. The reconstructed evolved target plasmids, along with the ancestor, were transformed into cells, and growth rates in no antibiotic, tetracycline (8 ng/μl), and tigecycline (8 ng/μl) were measured. Each dot was an average of five independently prepared and measured biological replicates; error bar, SE. (B) Structure of the efflux pump was modeled using Phyre2 server (47) with the intensive mode. Residues mutated during evolution were marked blue.
Amino acid substitutions in the efflux pump occurred at the opening of the channel that directly contacted substrates and in peripheral locations that did not (Fig. 4B, blue residues; mutant genotypes in table S4). The first round of selection yielded double-substitution variants from all three parallel populations. Mutants 1 to 3 all contained the I235F substitution at the opening of the channel, as well as another substitution, which differed between the three. Previous experiments showed that I235F confers tigecycline resistance (34), but mutant 1 had resistance more than 1.8-fold those of mutants 2 and 3. Hence, the second substitution of S312F in mutant 1 conferred further resistance to tigecycline in addition to I235F. Hence, TADR was able to identify beneficial mutations of more than a single substitution in one round. This large step size of exploring the sequence space enables overcoming fitness valleys, where the individual substitutions are not beneficial but their combination is (35). The best mutant (Fig. 4A, mutant 1-1) not only evolved the highest level of resistance to tigecycline and increased resistance to tetracycline but also unexpectedly grew 6.2% faster than the wild type in the absence of any antibiotic (ANOVA, n = 5, α = 0.05, P = 0.023). These results suggested that the wild-type efflux pump was far from optimality. TADR achieved evolutionary innovation by expanding the substrate repertoire while reducing cellular toxicity of an efflux pump.
To specifically test for the ability to simultaneously introduce two substitutions, we designed a double reversion assay, where the target plasmid carries the gene for chloramphenicol resistance with two stop codons in the middle of the open reading frame. Emergence of chloramphenicol resistance requires simultaneous mutation of both stop codons to compatible amino acids. This double substitution requires finding one in hundreds of thousand possible double substitution variants. Chloramphenicol-resistant colonies emerged from a single cycle of mutagenesis selection, and Sanger sequencing confirmed that the two synthetic stop codons were both mutated to amino acid–encoding codons (fig. S10). Hence, TADR was able to identify beneficial mutations of more than a single substitution in one round. This large step size of exploring the sequence space enables overcoming fitness valleys, where the individual substitutions are not beneficial but their combination is (35).
DISCUSSION
Our results first confirmed the design of a TADR in the activities of its individual components and then demonstrated high performance after optimization (Table 1): TADR directed high mutagenesis to target plasmid to find specific single or double mutations of a gene while avoiding mutagenesis on the chromosome.
The TADR is complex enough to meet varied requirements of functionality. TADR replicated DNA processively as designed based on biophysical principles, thus the large target window, and was directed by the specific interaction between CisA and a 30-bp initiation sequence, thus the selectivity. This interaction alone was not enough for high selectivity because only 4.2-fold fewer mutations occurred without the initiation sequence. Improvements by adding genetic (using the RepΔC33 mutant) and physiological (inducing mutations in minimal medium) changes resulted in high selectivity between plasmid and chromosome.
TADR is simple enough to be modular. One feature of this modularity is that TADR can be almost shut off (lowered by 112-fold), while the cell and the target plasmid continue propagating (fig. S5). The advantage is simpler isolation of the beneficial genetic construct. After a signal is detected in screening, the mutant virus or plasmid needs to be amplified in the host cell and purified to obtain the genetic construct. Amplification requires dozens of generations. If additional mutations occur during this amplification, they can confound the result. Tools that rely on virus-cell interaction or plasmid propagation cannot be turned off. An early system of in vivo evolution is similar to TADR (3) in that both rely on targeting error-prone DNA polymerase to a plasmid. But the early system is not modular: While its DNA polymerase (polA from E. coli) synthesizes ColE1 plasmid with error, this enzyme also participates in chromosomal replication. As a result, the selectivity of this system is 15.3-fold lower than that of TADR (390 versus 5970; the off-target mutation rate used to calculate selectivity was measured with rifampicin resistance in both systems).
Two other potential benefits of modularity are the ability to move TADR into other cell types and even cell-free systems. TADR is likely to work in yeast and mammalian cells because it does not depend on specific processes within the E. coli cell. CisA-mediated phage replication was reconstituted in cell-free systems (17), suggesting that TADR may also work outside cells.
The ability to identify, in a single round, beneficial double mutants makes TADR superior to error-prone PCR (which rarely report these mutants) in producing genetic diversity. But this superiority relies on the screening with life-and-death selection. Without this efficient screening, error-prone PCR might still be better for experiments where mutant proteins need to be characterized individually. This is because TADR currently does not guarantee that every colony it generates before screening carries a mutation on the gene of interest. Further increasing mutation rate of TADR can make it superior to error-prone PCR in any circumstances.
TADR could be further optimized. Despite good selectivity between plasmid and chromosome mutagenesis (Fig. 3C), TADR shows poor selectivity within the plasmid (Fig. 3D). The plasmid does not contain other genes that could contribute to adaptation, so these mutations have no major effect. One minor effect is slowing down the evolution of the gene(s) of interest because deleterious mutations outside the gene(s) of interest obscure beneficial mutations within. As mutational density of these two regions have the same order of magnitude, this slow down should not prevent evolution. On the positive side, this low selectivity demonstrates that mutation of multiple genes forming a biosynthetic pathway will likely be possible. The reason for the low selectivity is low termination efficiency (43%). In the future, adding additional copies of the termination sequence is likely to improve termination efficiency, reducing mutations outside the gene(s) of interest, as this strategy has been successfully applied before (15). The fusion protein and CisA are currently expressed from helper plasmid and chromosome, respectively. Expressing them from the same locus in chromosome will increase genetic stability of TADR (by eliminating helper plasmid). Alternatively, expressing them in the same plasmid will provide convenience for installment in other bacterial strains.
MATERIALS AND METHODS
Strains and materials
TADR was implemented in wild-type E. coli K12 strain MG1655 from the Coli Genetic Stock Center at Yale. The MM294 derivative of this strain from the same center was used for the construction and propagation of plasmids. The CisA gene was PCR-amplified from PhiX174 RF1 DNA (Thermo Fisher Scientific). Bacteria were grown in liquid LB broth (10 g of NaCl2, 10 g of tryptone, and 5 g of yeast extract per liter; Fisher BioReagents) or its agar plates (15 g of agar per liter; Fisher BioReagents). Cells were recovered from electroporation in super optimal broth with catabolite repression (SOC medium) (20 g of tryptone, 5 g of yeast extract, 4.8 g of MgSO4, 3.603 g of dextrose, 0.5 g of NaCl, and 0.186 g of KCl per liter; MP Biomedicals). Self-evolution of TADR experiment and characterization of the optimized TADR were carried out in minimal medium [1 g of (NH4)2SO4, 7 g of K2HPO4, 2 g of KH2PO4, 0.1 g of MgSO4, 0.5 g of sodium citrate, 4 g of glycerol, 2 g of yeast synthetic dropout medium supplements (Sigma) per liter]. Antibiotics were added accordingly: kanamycin (30 ng/μl; Fisher Scientific), ampicillin (50 ng/μl; IBI Scientific), chloramphenicol (15 ng/μl; Sigma), nalidixic acid (2.5 ng/μl; Fisher Biotech), tetracycline (8 ng/μl; Fisher Scientific), and tigecycline (4 or 8 ng/μl; Neta Scientific). Phire Hot Start II DNA Polymerase (Thermo Fisher Scientific) was used for PCRs; QIAprep Spin Miniprep Kit (Qiagen) was used for purifying plasmid; GeneJET Gel Extraction Kit (Thermo Fisher Scientific) was used for purifying PCR products; FastDigest BsaI restriction enzyme (Thermo Fisher Scientific) was used for digesting DNA; Rapid DNA Ligation Kit (both from Thermo Fisher Scientific) was used for ligation; and GeneJET Gel Extraction Kit and DNA Cleanup Micro Kit were used for purifying ligation products for electroporation.
Biophysical rationale of building an artificial replisome
The artificial replisome must simultaneously unwind DNA and copy the exposed strand to prevent degradation of the exposed single-stranded DNA, which causes genetic instability. In addition, DNA synthesis must avoid premature termination before reaching the termination sequence. Connecting Rep helicase and T5 DNA polymerase with a flexible linker increases the local effective concentrations to an estimated 0.2 to 0.6 mM (36) to achieve both of these goals. The CisA protein recruits Rep helicase and binds and stabilizes it. Rep helicase unwinds DNA at 144 bp/s (19), so DNA synthesis must initiate at least that fast to proceed simultaneously. The bimolecular association rate constant of T7 DNA polymerase (kon) for primed DNA substrate is 3.6 × 107 M−1 s−1 (37), and we assume that T5 DNA polymerase associates at a similar rate. If the local effective concentration of polymerase is 0.2 to 0.6 mM, then the rate of association is 7200 to 22,000 s−1, which far exceeds the rate of unwinding. Subsequent DNA synthesis by T5 DNA polymerase starts without a pause (38) and is faster than unwinding by Rep helicase (19). Faster polymerase would bump into the slower helicase, but single-molecule studies show that both proteins tolerate mechanical forces without being displaced (Fig. 1D and fig. S1) (19, 20). The previous linking of similar polymerase to a slower helicase allowed them to proceed simultaneously along the DNA. To ensure that the linker does not hinder T5 DNA polymerase, we used an intrinsically disordered peptide linker, human α-synuclein, which allows diffusion of two proteins along the linker at a rate comparable to the diffusion of free proteins (39).
The increased effective concentration of the polymerase also promotes its rebinding to the DNA when it occasionally dissociates. An enhanced mutagenesis construct that relied on free polymerase copied only short stretches of DNA (13). The linking of polymerase to helicase promotes rebinding and resumption of DNA synthesis, thereby maintaining high processivity and permitting a wide window for enhanced mutagenesis. These properties were confirmed by NGS characterization of target plasmids mutagenized by TADR (Fig. 3D).
Construction of TADR
The host cells for TADR were a modified strain of E. coli strain MG1655, where the chromosomal araB gene was replaced with the gene for T7 RNA polymerase and the chromosomal lacZ gene was replaced with the gene for CisA. The gene for T7 RNA polymerase was PCR-amplified from E. coli strain BL21(DE3) (Coli Genetic Stock Center at Yale). One kilobase of DNA was PCR-amplified from each of upstream and downstream araB gene in the chromosome of E. coli strain MG1655. These three PCR products were integrated into a single fragment using the technique of fusion PCR (40). This fragment served as a template to replace the chromosomal araB gene of the strain MG1655 with the gene for T7 RNA transcriptase using the technique of lambda red recombineering (41). In this strain, the gene for T7 RNA transcriptase was under the native pBAD promoter, and the gene for the fusion protein carried on plasmid could be expressed using T7 promoter with high expression level and stringent repression (by adding 0.2% glucose to medium). In the same manner, the lacZ gene of this strain was replaced with the gene for CisA. Chromosomal rep gene was knocked out using P1 transduction from a Keio collection null mutant strain, and the marker was cured (42). The resulting strain provided host cells for TADR.
The helper plasmid expresses the linked Rep helicase and T5 DNA polymerase under the control of T7 promoter. The gene for T5 DNA polymerase was PCR-amplified from T5 phage lysate, the gene for Rep helicase was PCR-amplified from the genomic DNA of strain MG1655, and a DNA fragment encoding a linker—a 20–amino acid glycine-rich peptide concatenated to an intrinsically disordered peptide from human α-synuclein (61 amino acids, codon-optimized for E. coli) (fig. S2B)—was chemically synthesized using the gBlock service of Integrated DNA Technologies. These three fragments were fused using fusion PCR into a gene that encodes a single polypeptide with T5 DNA polymerase and Rep helicase fused through the linker. This fragment was then inserted into a plasmid derived from pKD46 (Coli Genetic Stock Center at Yale, #7669) with an ampicillin resistance marker in the backbone using standard technique of molecular cloning (43). The original pKD46 did not grow at 37°C, and a mutant was selected from growth at this temperature and used in this study. The fused gene was expressed under T7 promoter. Several versions of this plasmid were made: one carried a gene for wild-type T5 DNA polymerase and the other carried an error-prone version with three point mutations. Also, the helper plasmids with RepΔC33 and 5′ UTR–modified were using the same method.
To construct the target plasmid, fusion PCR was used to make an operon where the constitutive Tac promoter expressed the kanR* gene together with the gene for green fluorescent protein. The two genes were flanked by an upstream initiation sequence and a downstream termination sequence. The operon was inserted into plasmid pACYC184 (Addgene, #37033) with a chloramphenicol resistance marker in the backbone. Another version of this plasmid was also made that did not carry the initiation sequence.
To construct the target plasmid for evolution in tigecycline, the tetA(C) gene encoding the tetracycline efflux pump was PCR-amplified from plasmid pBR322 (New England Biolabs) and inserted into the target plasmid just described, replacing the gene for green fluorescent protein. The full system of TADR is schematically shown in fig. S2A.
Nicking of target plasmid by CisA
Three transformations were carried out as shown in Fig. 2A: the intact target plasmid into cells without the chromosomal gene for CisA, the target plasmid without initiation and termination sequences into cells with the chromosomal gene for CisA, and the intact target plasmid into cells with the chromosomal gene for CisA. Transformant colonies were selected on LB agar plates supplemented with chloramphenicol (15 ng/μl) and inoculated to liquid LB cultures with chloramphenicol. After overnight growth in 37°C shaken at 225 rounds per minute (the same growth conditions from here on unless specified otherwise), 1 ml of culture was purified and eluted to 40 μl of elution buffer, and 5 μl was used for electrophoresis (0.8% agarose gel, 110 V, 40 min; Bio-Rad Mini-Sub Cell GT Systems). An image was taken using UVP BioDoc-It Imaging System and analyzed using ImageJ to calculate fraction of the nicked target plasmid with the following equation: brightness of the band for nicked DNA (brightness of the band for nicked DNA + brightness of the band for supercoiled DNA + brightness of the band for linear DNA).
Helicase activity of T5 DNA polymerase–Rep fusion protein
Assay of sensitivity to nalidixic acid was used (28). Three cell lines were prepared. TADR cells modified to have intact chromosomal rep, TADR cells (rep-), and TADR cells (rep-) transformed with helper plasmid (expressing the fusion protein). They were grown in liquid LB to full density [ampicillin (50 ng/μl) was added to the last cell line]. Sample from each culture was serially diluted with a dilution factor of 10, and 5 μl from each dilution was spotted on LB agar plate supplemented with nalidixic acid (2.5 ng/μl). After 16 hours of incubation at 37°C, a picture of the plate was taken using imageRUNNER ADVANCE C5250 Copier (Canon), and the background was subtracted using ImageJ for enhanced visual effect.
Increased reversion rate of inactivated kanamycin resistance gene
KanR* reversion assay was used. Three transformations were carried out, all into the host cell for TADR, as shown in Fig. 2C: the helper plasmid with wild-type T5 polymerase cotransformed with an intact target plasmid; the helper plasmid with the error-prone T5 polymerase mutant, with the target plasmid without initiation sequence; and the helper plasmid with the error-prone T5 polymerase mutant, with an intact target plasmid. Transformant colonies were selected on LB agar plates supplemented with chloramphenicol (15 ng/μl) and ampicillin (50 ng/μl) and inoculated to a liquid LB cultures with chloramphenicol and ampicillin. After overnight growth, 50 μl of culture from each sample was plated on LB plates supplemented with kanamycin (30 ng/μl). After 16 hours of incubation at 37°C, colonies were counted. For experiments with more mutagenic TADR constructs as in Fig. 3A, less volumes of culture were plated for countable number of colonies.
Self-evolution of TADR for higher mutation rate
A schematic of the steps is shown in fig. S4. The helper plasmid was modified so that initiation sequence was added at the 5′ UTR of the T5 DNA polymerase gene and that termination sequence was added in the linker region as an extension of the linker without altering the encoding frame for T5 DNA polymerase and Rep helicase. This helper plasmid was transformed into TADR cells and plated on agar plate of minimal medium supplemented with ampicillin (50 ng/μl). After incubation at 37°C for 24 hours, a single colony was inoculated to liquid minimal medium with ampicillin to allow the T5 DNA polymerase gene to be mutagenized. After ~24 hours of growth, cells grew to mid-log, were treated for competency, and were electroporated with ~500 ng of the target plasmid with kanR* following standard protocol (43). Right after electropulsing, SOC medium (1 ml) was added immediately to the 50-μl slurry of competent cells. This mix was incubated at 37°C and shaken for 5 min before plating onto minimal medium plate supplemented with chloramphenicol (15 ng/μl), ampicillin (50 ng/μl), and kanamycin (30 ng/μl). After incubation at 37°C for 16 hours, typically a dozen colonies showed up; a few of the biggest colonies were inoculated into liquid LB medium supplemented with chloramphenicol (15 ng/μl ) and ampicillin (50 ng/μl). Not all of them grew to full density, and for those that did, plasmid mixes (of the helper and target) were purified and transformed into a fresh stock of the host cell for TADR and plated on LB with kanamycin (30 ng/μl). This step checked if the kanamycin resistance was carried on the target plasmid (true positive). For the plasmid mixes that gave rise to kanamycin-resistant colonies and thus passed the check, they were treated by FastDigest PvuII (Thermo Fisher Scientific), which only cut the target but not the helper plasmids, and purified using the GeneJET Gel Extraction Kit. This treatment resulted in purified evolved helper plasmids, which were then subjected to the procedures of kanR* reversion assay to quantify mutagenic activity. For the helper plasmids that showed increased mutagenic activity compared to the ancestor, they were Sanger-sequenced (Genewiz) in the entire region of the fused gene to identify mutations.
NGS characterization of mutant library generated by TADR
TADR cells were transformed with the target plasmid with kanR* and the helper plasmid with RepΔC33, modified 5′ UTR, and the error-prone T5 DNA polymerase mutant I308V (the most mutagenic version of TADR). Colonies were selected on minimal medium supplemented with ampicillin and chloramphenicol. Wild-type E. coli cells (MG1655) were transformed with the target plasmid as negative control, and colonies were selected on minimal medium supplemented with chloramphenicol. Three colonies from each transformation were separately grown up in minimal medium with appropriate antibiotics to full density. Plasmids were purified. Plasmid (600 ng) from each sample was submitted to the Microbial Genome Sequencing Center (MiGS) in Pittsburgh for sequencing using Illumina NextSeq 550 with paired end reads of 150 bp. Each sample was sequenced with the coverage of 20,000 to 70,000 to capture rare variants. Galaxy platform was used to process the data and map reads to the reference sequence of the target plasmid. The raw data were trimmed to filter out low-quality reads so that all reads had quality scores larger than 20. VarScan (version 2.4.4) (44) package was used to call variants. None of the variants from the three negative control samples exceeded 0.14%, and thus, 0.14% was used as a cutoff for point mutations. The three treatment samples contained a total of 264 point mutations above this cutoff value. The few in-dels called in the treatment samples did not meet the threshold for statistical significance and were discarded. The 264 point-mutation variants were further analyzed to plot mutational density across the target plasmid (Fig. 3D) and measure the mutational spectrum (Fig. 3E).
Evolution of tetA for resistance to tigecycline
The target plasmid with the tetA(C) gene was cotransformed with the helper plasmid carrying RepΔC33, modified 5′ UTR, and mutation I308V into the host cell for TADR. Transformant colonies were selected on LB agar plates with chloramphenicol (15 ng/μl) and ampicillin (50 ng/μl) and inoculated to a liquid LB culture with chloramphenicol and ampicillin. After overnight growth, 50 μl of culture from each sample was plated on LB plates with tigecycline (4 ng/μl). After 16 hours of incubation at 37°C, several dozen colonies typically showed up, and the biggest a few were inoculated into liquid LB with chloramphenicol. After overnight culture, plasmids were purified, digested with FastDigest AflII (Thermo Fisher Scientific), which cut only the helper but not the target plasmids, and purified using the GeneJET Gel Extraction Kit. This treatment resulted in purified evolved target plasmids. They were cotransformed with helper plasmid carrying modified 5′ UTR and mutation I308V into the host cell for TADR for a second round of experimental evolution and this time were selected on LB plates with tigecycline (8 ng/μl). The resulting plasmids were purified and Sanger-sequenced.
Five evolved plasmids were selected based on their big colony size on tigecycline plates during experimental evolution. Their tetA(C) gene was each PCR-amplified and inserted into the backbone of the unevolved target plasmid to purge any serendipitous off-target mutations accumulated during evolution. The resulting reconstructed evolved plasmids were transformed into TADR cells, and the growth rates were measured in the absence of antibiotic, in tetracycline, and in tigecycline using SpectraMax Plus 384 Microplate Reader [200-μl volume, optical density at 600 nm (OD 600), 37°C, shaken for 500 s followed by 100 s still continuously, one reading every 600 s, 16 hours in total; Molecular Devices]. The readings of OD 600 were analyzed using JMP software (SAS): For the growth curve of each sample, the optical density values were log-transformed, and the slope of the linear part was calculated as the corresponding growth rate.
Acknowledgments
We thank K. Zhou from the University of Colorado Boulder for discussion on the design of the linker, V. Noireaux from the University of Minnesota for donating PhiX174 genomic DNA, and L. Peng for discussion on design of TADR. Funding: This work was supported by the Seed Fund of the BioTechnology Institute at the University of Minnesota. Author contributions: X.Y. conceived the project, designed and implemented TADR, and collected and analyzed data. X.Y., J.K., M.T., and R.J.K. discussed the results and wrote the manuscript. The manuscript was read, edited, and approved by all authors. Competing interests: X.Y., R.J.K., and M.T. are inventors on a pending patent related to this work filed by the University of Minnesota (no. 63/150,374 , filed on 17 February 2021). The authors declare no other competing interests. Data and materials availability: TADR components (the target and helper plasmids and the bacterial strain) can be provided by R.J.K. pending scientific review and a completed material transfer agreement. Requests for the TADR components should be submitted to R.J.K. Raw NGS sequencing data are available at the NCBI Sequence Read Archive (PRJNA672220). All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/29/eabg8712/DC1
REFERENCES AND NOTES
- 1.Lupo A., Coyne S., Berendonk T. U., Origin and evolution of antibiotic resistance: The common mechanisms of emergence and spread in water bodies. Front. Microbiol. 3, 18 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gaudelli N. M., Komor A. C., Rees H. A., Packer M. S., Badran A. H., Bryson D. I., Liu D. R., Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Camps M., Naukkarinen J., Johnson B. P., Loeb L. A., Targeted gene evolution in Escherichia coli using a highly error-prone DNA polymerase I. Proc. Natl. Acad. Sci. U.S.A. 100, 9727–9732 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berman C. M., Papa L. J. III, Hendel S. J., Moore C. L., Suen P. H., Weickhardt A. F., Doan N. D., Kumar C. M., Uil T. G., Butty V. L., Hoeben R. C., Shoulders M. D., An adaptable platform for directed evolution in human cells. J. Am. Chem. Soc. 140, 18093–18103 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crook N., Abatemarco J., Sun J., Wagner J. M., Schmitz A., Alper H. S., In vivo continuous evolution of genes and pathways in yeast. Nat. Commun. 7, 13051 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.English J. G., Olsen R. H. J., Lansu K., Patel M., White K., Cockrell A. S., Singh D., Strachan R. T., Wacker D., Roth B. L., VEGAS as a platform for facile directed evolution in mammalian cells. Cell 178, 748–761.e17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Esvelt K. M., Carlson J. C., Liu D. R., A system for the continuous directed evolution of biomolecules. Nature 472, 499–503 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nyerges Á., Csörgő B., Draskovits G., Kintses B., Szili P., Ferenc G., Révész T., Ari E., Nagy I., Bálint B., Vásárhelyi B. M., Bihari P., Számel M., Balogh D., Papp H., Kalapis D., Papp B., Pál C., Directed evolution of multiple genomic loci allows the prediction of antibiotic resistance. Proc. Natl. Acad. Sci. U.S.A. 115, E5726–E5735 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ravikumar A., Arrieta A., Liu C. C., An orthogonal DNA replication system in yeast. Nat. Chem. Biol. 10, 175–177 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Yi X., Kazlauskas R., Travisano M., Evolutionary innovation using EDGE, a system for localized elevated mutagenesis. PLOS ONE 15, e0232330 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erdogan M., Fabritius A., Basquin J., Griesbeck O., Targeted in situ protein diversification and intra-organelle validation in mammalian cells. Cell Chem. Biol. 27, 610–621.e5 (2020). [DOI] [PubMed] [Google Scholar]
- 12.Simon A. J., Morrow B. R., Ellington A. D., Retroelement-based genome editing and evolution. ACS Synth. Biol. 7, 2600–2611 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Halperin S. O., Tou C. J., Wong E. B., Modavi C., Schaffer D. V., Dueber J. E., CRISPR-guided DNA polymerases enable diversification of all nucleotides in a tunable window. Nature 560, 248–252 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Hess G. T., Frésard L., Han K., Lee C. H., Li A., Cimprich K. A., Montgomery S. B., Bassik M. C., Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat. Methods 13, 1036–1042 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moore C. L., Papa L. J. III, Shoulders M. D., A processive protein chimera introduces mutations across defined DNA regions in vivo. J. Am. Chem. Soc. 140, 11560–11564 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fernandez-Leiro R., Conrad J., Scheres S. H. W., Lamers M. H., cryo-EM structures of the E. coli replicative DNA polymerase reveal its dynamic interactions with the DNA sliding clamp, exonuclease and τ. eLife 4, e11134 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eisenberg S., Griffith J., Kornberg A., phiX174 cistron A protein is a multifunctional enzyme in DNA replication. Proc. Natl. Acad. Sci. U.S.A. 74, 3198–3202 (1977). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Denhardt D. T., Dressler D. H., Hathaway A., The abortive replication of phix174 DNA in a recombination-deficient mutant of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 57, 813–820 (1967). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arslan S., Khafizov R., Thomas C. D., Chemla Y. R., Ha T., Engineering of a superhelicase through conformational control. Science 348, 344–347 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maier B., Bensimon D., Croquette V., Replication by a single DNA polymerase of a stretched single-stranded DNA. Proc. Natl. Acad. Sci. U.S.A. 97, 12002–12007 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andraos N., Tabor S., Richardson C., The highly processive DNA polymerase of bacteriophage T5. J. Biol. Chem. 279, 50609–50618 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Morrison A., Bell J. B., Kunkel T. A., Sugino A., Eukaryotic DNA polymerase amino acid sequence required for 3′--5′ exonuclease activity. Proc. Natl. Acad. Sci. U.S.A. 88, 9473–9477 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fluit A. C., Baas P. D., Jansz H. S., Termination and reinitiation signals of bacteriophage φX174 rolling circle DNA replication. Virology 154, 357–368 (1986). [DOI] [PubMed] [Google Scholar]
- 24.Lyamichev V., Brow M., Dahlberg J., Structure-specific endonucleolytic cleavage of nucleic acids by eubacterial DNA polymerases. Science 260, 778–783 (1993). [DOI] [PubMed] [Google Scholar]
- 25.Uphoff S., Reyes-Lamothe R., Garza de Leon F., Sherratt D. J., Kapanidis A. N., Single-molecule DNA repair in live bacteria. Proc. Natl. Acad. Sci. U.S.A. 110, 8063–8068 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thompson P. R., Schwartzenhauer J., Hughes D. W., Berghuis A. M., Wright G. D., The COOH terminus of aminoglycoside phosphotransferase (3′)-IIIa is critical for antibiotic recognition and resistance. J. Biol. Chem. 274, 30697–30706 (1999). [DOI] [PubMed] [Google Scholar]
- 27.Clemson M., Kelly W. J., Optimizing alkaline lysis for DNA plasmid recovery. Biotechnol. Appl. Biochem. 37, 235–244 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Henderson M. L., Kreuzer K. N., Functions that protect Escherichia coli from tightly bound DNA-protein complexes created by mutant EcoRII methyltransferase. PLOS ONE 10, e0128092 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Syeda A. H., Wollman A. J. M., Hargreaves A. L., Howard J. A. L., Brüning J. G., McGlynn P., Leake M. C., Single-molecule live cell imaging of Rep reveals the dynamic interplay between an accessory replicative helicase and the replisome. Nucleic Acids Res. 47, 6287–6298 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guy C. P., Atkinson J., Gupta M. K., Mahdi A. A., Gwynn E. J., Rudolph C. J., Moon P. B., van Knippenberg I. C., Cadman C. J., Dillingham M. S., Lloyd R. G., McGlynn P., Rep provides a second motor at the replisome to promote duplication of protein-bound DNA. Mol. Cell 36, 654–666 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hiraga S., Ichinose C., Niki H., Yamazoe M., Cell cycle–dependent duplication and bidirectional migration of seqA-associated DNA–protein complexes in E. coli. Mol. Cell 1, 381–387 (1998). [DOI] [PubMed] [Google Scholar]
- 32.Reyes-Lamothe R., Sherratt D. J., The bacterial cell cycle, chromosome inheritance and cell growth. Nat. Rev. Microbiol. 17, 467–478 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Lee H., Popodi E., Tang H., Foster P. L., Rate and molecular spectrum of spontaneous mutations in the bacterium Escherichia coli as determined by whole-genome sequencing. Proc. Natl. Acad. Sci. U.S.A. 109, E2774–E2783 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Linkevicius M., Sandegren L., Andersson D. I., Potential of tetracycline resistance proteins to evolve tigecycline resistance. Antimicrob. Agents Chemother. 60, 789–796 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weinreich D. M., Delaney N. F., DePristo M. A., Hartl D. L., Darwinian evolution can follow only very few mutational paths to fitter proteins. Science 312, 111–114 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Li M., Cao H., Lai L., Liu Z., Disordered linkers in multidomain allosteric proteins: Entropic effect to favor the open state or enhanced local concentration to favor the closed state? Protein Sci. 27, 1600–1610 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo G., Wang M., Konigsberg W. H., Xie X. S., Single-molecule and ensemble fluorescence assays for a functionally important conformational change in T7 DNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 104, 12610–12615 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Christian T. D., Romano L. J., Rueda D., Single-molecule measurements of synthesis by DNA polymerase with base-pair resolution. Proc. Natl. Acad. Sci. U.S.A. 106, 21109–21114 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grupi A., Haas E., Segmental conformational disorder and dynamics in the intrinsically disordered protein α-synuclein and its chain length dependence. J. Mol. Biol. 405, 1267–1283 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Szewczyk E., Nayak T., Oakley C. E., Edgerton H., Xiong Y., Taheri-Talesh N., Osmani S. A., Oakley B. R., Fusion PCR and gene targeting in Aspergillus nidulans. Nat. Protoc. 1, 3111–3120 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Datsenko K. A., Wanner B. L., One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saragliadis A., Trunk T., Leo J. C., Producing gene deletions in Escherichia coli by P1 transduction with excisable antibiotic resistance cassettes. J. Vis. Exp. 58267 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.M. Green, J. Sambrook, Molecular Cloning (Cold Spring Harbor Laboratory Press, 2012). [Google Scholar]
- 44.Koboldt D. C., Zhang Q., Larson D. E., Shen D., McLellan M. D., Lin L., Miller C. A., Mardis E. R., Ding L., Wilson R. K., VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carr S. B., Phillips S. E. V., Thomas C. D., Structures of replication initiation proteins from staphylococcal antibiotic resistance plasmids reveal protein asymmetry and flexibility are necessary for replication. Nucleic Acids Res. 44, 2417–2428 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim D. E., Chivian D., Baker D., Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 32, W526–W531 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kelley L. A., Mezulis S., Yates C. M., Wass M. N., Sternberg M. J. E., The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kumar J. K., Tabor S., Richardson C. C., Role of the C-terminal residue of the DNA polymerase of bacteriophage T7. J. Biol. Chem. 276, 34905–34912 (2001). [DOI] [PubMed] [Google Scholar]
- 49.Amblar M., de Lacoba M. G. A., Corrales M. A., López P., Biochemical analysis of point mutations in the 5′-3′ exonuclease of DNA polymerase I of Streptococcus pneumoniae: Functional and structural implications. J. Biol. Chem. 276, 19172–19181 (2001). [DOI] [PubMed] [Google Scholar]
- 50.Pomerantz R. T., O’Donnell M., What happens when replication and transcription complexes collide? Cell Cycle 9, 2537–2543 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meng C. A., Fazal F. M., Block S. M., Real-time observation of polymerase-promoter contact remodeling during transcription initiation. Nat. Commun. 8, 1178 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Labib K., Hodgson B., Replication fork barriers: Pausing for a break or stalling for time? EMBO Rep. 8, 346–353 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murat P., Guilbaud G., Sale J. E., DNA polymerase stalling at structured DNA constrains the expansion of short tandem repeats. Genome Biol. 21, 209 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Banerjee K., Kolomeisky A. B., Igoshin O. A., Elucidating interplay of speed and accuracy in biological error correction. Proc. Natl. Acad. Sci. U.S.A. 114, 5183–5188 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kang W., Ha K. S., Uhm H., Park K., Lee J. Y., Hohng S., Kang C., Transcription reinitiation by recycling RNA polymerase that diffuses on DNA after releasing terminated RNA. Nat. Commun. 11, 450 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Selzer G., Tomizawa J. I., Specific cleavage of the p15A primer precursor by ribonuclease H at the origin of DNA replication. Proc. Natl. Acad. Sci. U.S.A. 79, 7082–7086 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lima W. F., Crooke S. T., Binding affinity and specificity of Escherichia coli RNase H1: Impact on the kinetics of catalysis of antisense Oligonucleotide−RNA hybrids. Biochemistry 36, 390–398 (1997). [DOI] [PubMed] [Google Scholar]
- 58.Zuker M., Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Espah Borujeni A., Cetnar D., Farasat I., Smith A., Lundgren N., Salis H. M., Precise quantification of translation inhibition by mRNA structures that overlap with the ribosomal footprint in N-terminal coding sequences. Nucleic Acids Res. 45, 5437–5448 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/29/eabg8712/DC1