

Bacteriophages are rapidly shared among hosts and act as key genetic regulators for establishing chronic infections.
Abstract
Interactions between bacteria, their close competitors, and viral parasites are common in infections, but understanding of these eco-evolutionary dynamics is limited. Most examples of adaptations caused by phage lysogeny are through the acquisition of new genes. However, integrated prophages can also insert into functional genes and impart a fitness benefit by disrupting their expression, a process called active lysogeny. Here, we show that active lysogeny can fuel rapid, parallel adaptations in establishing a chronic infection. These recombination events repeatedly disrupted genes encoding global regulators, leading to increased cyclic di-GMP levels and elevated biofilm production. The implications of prophage-mediated adaptation are broad, as even transient members of microbial communities can alter the course of evolution and generate persistent phenotypes associated with poor clinical outcomes.
INTRODUCTION
“A non-specific parasite, to which partial immunity has been acquired, is a powerful natural weapon.” (1)
“But this weapon, if captured by the victim and re-deployed, can engender victory.” (this study)
We often begin study and treatment of chronic bacterial infections much as we might start watching movies already an hour underway—we do our best to understand the current scene despite missing much of the character and plot development. By the time an infection is recognized, hundreds of pathogen generations may have passed, during which the population may have evolved along cryptic paths that cannot be retraced. Consequently, the earliest factors governing the fate of a new bacterial infection remain unclear (2).
Because many infections arise from a single clonal population, longitudinal isolates from patients have yielded invaluable insight into how individual bacteria initially adapt and then persist in infections (3–9). By studying a time series of isolates or populations, the selective pressures experienced by pathogens can often be inferred retrospectively by determining what genetic and phenotypic traits were selected in the infecting population (10–13). What many of these studies missed, however, were the initial selective pressures and eco-evolutionary interactions between the pathogen and close competitors as the microbial community is established. If we were instead able to watch the movie from the beginning, we can understand why certain pathogens outcompete others and become successful colonizers in the new host.
One potential axis of competition between bacterial lineages is lysogenic bacteriophage that reactivates to become lytic (14–19). It has long been appreciated that interactions among competing species can be mediated by their shared predators or parasites (20), a process known as apparent competition. In the case of integrated prophage, reactivation during the stress of competition is often lethal to the host, but sibling clones with the dormant prophage remain resistant and benefit from the high mortality of susceptible competing populations lacking the phage. Thus, the fitness cost of bearing prophage can be overcome at a population level by attacks of the active phage against competitors.
The possibility that lysogenic phage may mediate fitness when initiating infection became evident in our recent study of Pseudomonas aeruginosa experimental chronic wound infections (21). Six different strains of P. aeruginosa were inoculated into a porcine full-thickness thermal injury wound model to determine which strain(s) were superior competitors and how they genetically adapted to the infection. The PA14 and PAO1 strains repeatedly prevailed in independent infections, and PA14 hyperbiofilm-forming variants became detectable within days and persisted for weeks. These variants were caused by single mutations in the wsp pathway that induced hyperbiofilm formation by increasing levels of the key signal cyclic di-GMP (guanosine 3′,5′-monophosphate) (22). Some variants also gained CRISPR spacer insertions that conferred immunity to a temperate phage present in a co-inoculated strain (21). Therefore, we hypothesized that prophages mediate competition by killing sensitive competitors, selecting for resistant lineages, and generating new mutant genotypes that may be adaptive in the host environment. Here, we find that the genomes of winning clones contain multiple mutations caused by these lysogenic phages and other mobile genetic elements. In many cases, prophages were the sole means of adaptation over the course of the infection, and their speed and multiplicity of actions revise our understanding of the genetics of rapid evolution during infection.
RESULTS
P. aeruginosa RSCVs are selected in porcine chronic wound infections
Given a mixture of bacterial strains added in equal fractions to a defined environment, picking the winner remains a daunting challenge. To determine which P. aeruginosa strain was competitively superior in a chronic infection, we used a porcine full-thickness thermal injury wound model that was co-inoculated with six different strains of P. aeruginosa and followed the infection for 28 days (Fig. 1A). We previously reported the general outcomes of the infection (21) but describe some of the relevant data here for clarity. The starting inoculum was 108 colony-forming units/wound, with an equal starting distribution of P. aeruginosa strains PA14-1 and PAO1-B11 (burn wound isolates and model organisms), B23-2 (wound isolate), CF18-1 (nonmucoid cystic fibrosis isolate), MSH10-2 (water isolate), and S54485-1 (urinary tract infection isolate). By day 3 and continuing throughout the duration of the experiment, strains PA14 and PAO1 became predominant, while the other strains remained at or below the detection limit (<0.1%), based on neutral genetic markers. We used heritable differences in colony morphologies to screen for evolved variants over the course of the infection. The primary novel phenotype was the rugose small colony variant (RSCV), which accounted for up to 2% of the total P. aeruginosa population screened. The RSCV phenotype is well known to arise in chronic infections (23–25) and have a hyperbiofilm phenotype due to the overproduction of exopolysaccharides Pel and Psl (22, 25, 26) and the protein adhesin CdrA (27). RSCVs are associated with persistence and worse clinical outcomes (26, 28). Following evolution in the wound environment, we sequenced both RSCV and non-RSCV isolates to identify the causative mutations of the RSCV phenotype. We used the standard approach of variant calling by mapping short, accurate reads (Illumina) to the ancestral genome. All of the RSCVs derived from PA14 had one of two mutations in the wsp operon (21), an important genetic locus for cyclic di-GMP production and biofilm regulation in P. aeruginosa (22, 29). Unexpectedly, however, no single-nucleotide polymorphisms (SNPs) or short insertion/deletion (indel) mutations were detected among the majority of PAO1-derived RSCV isolates (12 of 17 sequenced RSCVs had no mutations; table S1) or in most (16 of 28) PAO1 non-RSCV isolates sequenced (table S2).
Fig. 1. PAO1 RSCVs isolated from porcine burn wounds have a hyperbiofilm phenotype.
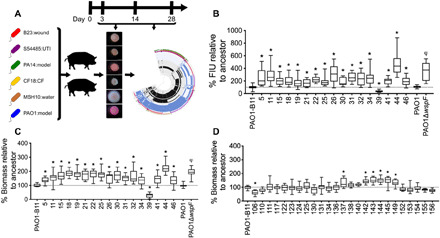
(A) Summary of the experimental design. Six P. aeruginosa strains were inoculated into 12 wounds on two pigs, and biopsies were taken on days 3, 14, and 28 after infection. RSCVs were isolated and subjected to sequencing and analysis. (B) A cyclic di-GMP reporter was electroporated into PAO1 RSCVs that were selected for sequencing. Cyclic di-GMP levels were measured as green fluorescence and reported as fluorescence intensity units (FIUs) normalized to the ancestor strain, which was set at 100%. FIU was normalized to optical density. N = 3, each with four technical replicates. Biofilms of PAO1 (C) RSCVs and (D) non-RSCVs that were selected for sequencing were grown in a 96-well plate for 4 hours. Biofilm biomass was quantified by crystal violet. Biofilm biomass was expressed as a percentage, relative to the ancestor strain, which was set to 100%, N = 4, each with four technical replicates. *P < 0.05 compared to the PAO1-B11 ancestor [one-way analysis of variance (ANOVA) with Tukey’s post hoc test], and φP < 0.05 compared to PAO1 (Student’s t test). Numbers on x axis denote isolate number.
PAO1 RSCVs acquire adaptive mutations that increase biofilm production
Of the five PAO1 RSCV isolates with detectable mutations, only two (RSCV-41 and RSCV-44) had mutations in genes previously implicated in the RSCV phenotype. RSCV-44 had a SNP in wspF (T➔G, I68S), and RSCV-41 had a SNP in retS (A➔C, T443P) (table S1). RetS (regulator of exopolysaccharide and type III secretion) is a sensor kinase that regulates the switch between acute and chronic infection and, when inactivated, stimulates biofilm production through elevated cyclic di-GMP levels (30–32). Introduction of wspF and retS wild-type alleles in trans complemented the RSCV colony phenotype of these isolates (fig. S1A). This confirms that the SNPs in wspF in RSCV-44 and retS in RSCV-41 were responsible for the RSCV phenotype of these two isolates. The remaining three isolates with identified mutations (RSCV-11, RSCV-25, and RSCV-32; table S1) had SNPs in genes hypothesized to be unrelated to the RSCV phenotype, as introduction of the wild-type allele did not complement the variant colony phenotype (fig. S1B). That left 15 of 17 sequenced RSCVs with no obvious genetic cause for the RSCV phenotype.
The absence of SNPs or short indels in the PAO1 RSCV isolates was perplexing, given their heritable RSCV phenotypes (Fig. 1A). Using the same approach, we previously identified mutational parallelism in the wsp operon in RSCVs derived from strain PA14 (21). Furthermore, we confirmed that the heritable variant colony morphologies of PAO1-derived RSCVs were accompanied by elevated levels of cyclic di-GMP and significant hyperbiofilm formation compared to the ancestor (Fig. 1, B and C). In contrast, the majority (22 of 28) of the PAO1-derived non-RSCVs recovered over the course of the infection did not have significantly elevated levels of biofilm production (Fig. 1D), and no identifiable nonsynonymous mutations were found in 21 of 28 non-RSCV isolates (table S2).
Newly acquired prophages are responsible for the RSCV phenotype
Reference-based mutation calling is largely blind to horizontal gene transfer (HGT) because any sequences not in the reference genome are unmapped and are not reported. We therefore hypothesized that the evolved PAO1 RSCV phenotypes resulted from HGT events between co-inoculated strains. To discover possible HGT events, we aligned each reference and isolate genome to one another and cataloged the presence or absence of homologous genomic regions. We discovered fragments of exogenous DNA ranging from 7 to 70 kb incorporated into nearly all of the PAO1 genomes isolated from the porcine wounds, both RSCV and non-RSCV (Fig. 2). These foreign DNA sequences all mapped to mobile genetic elements, primarily prophages, found in one of the four other co-inoculated P. aeruginosa strains that eventually became undetectable in the wounds (table S3). The amplification and mobilization of prophages, along with the demise of their donor genotypes, show that transiently colonizing strains can substantially alter the genomes and frequencies of numerically dominant strains.
Fig. 2. Genome alignments highlight the acquisition of mobile genetic elements by PAO1 isolates.

Map of the pangenome containing the ancestral co-inoculated P. aeruginosa strains and the derived PAO1 isolates sequenced. Black bars indicate genome sequences belonging to the non-RSCV isolates, and blue bars indicate sequences belonging to the RSCV isolates. The six co-inoculated strains are in red (B23), purple (S54485), green (PA14), yellow (CF18), orange (MSH10), and dark blue (PAO1). The solid block of bars to the left represents the core P. aeruginosa genome shared by all strains. The zoomed-in area to the right represents part of the accessory genome that was largely absent in the PAO1 ancestor and acquired during evolution in the porcine wound. The presence of these bars in the accessory genome of the recovered PAO1 isolates indicates recently acquired mobile genetic elements.
The newly integrated prophages in PAO1 isolates originated from either the co-inoculated B23 or MSH10 strains. By day 3, these donor strains fell to at or below the detection limit from amplicon sequencing and were never detected by culturing methods (21), suggesting that the prophage was activated and lysogenized the PAO1 population early in the infection. The prophage integrated in MSH10 is closely related to the F116 (Podoviridae) phage (33), is one of two predicted prophages in its genome (table S4), and was found in 82% of sequenced RSCVs and in 64% of non-RSCVs (table S3). In addition, a prophage closely related to the Mu-like JBD24 (Siphoviridae) phage (34, 35) was identified in the B23 genome (table S4) and was detected in 100% of the RSCVs and in 50% of non-RSCVs (table S3).
We predicted that these newly acquired prophage contributed to the RSCV phenotype. Through long-read sequencing, we determined that many prophage genes, particularly from phage JBD24, were repeatedly inserted into loci that could explain the RSCV phenotype (Table 1). Mutations (phage insertions and SNPs) in retS (PA4856) were found in the majority of RSCVs sequenced (10 of 17) and consisted of at least four separate mutational events across two animals and three different wounds (table S5). All phage insertions into retS appeared to originate from the co-inoculated P. aeruginosa strain B23 (Table 1). This parallelism of HGT events at the gene level indicates positive selection on the resulting phenotype.
Table 1. Mutations and mobile genetic elements in PAO1 RSCV isolates.
Wound numbers 1 and 2 are from pig 1, and wounds 3 and 4 are from pig 2. bp, base pair.
| Sample | Driver mutation | Secondary mutation | ||||
| Day | Wound no. | Isolate no. | Gene | Mutation/phage | Gene | Mutation/phage |
| 14 | 4 | 5 | retS | JBD24* | PA3825/tRNA-leu | F10† |
| 11 | dipA | JBD24 | tRNA-leu (PA1796) | F116‡ | ||
| PA4095 | JBD24 | |||||
| PA0069 | JBD24 | |||||
| PA2402 | JBD24 | |||||
| pelC | JBD24 | |||||
| mmsR | JBD24 | |||||
| pyeM | JBD24 | |||||
| PA4681 | JBD24 | |||||
| mgtA | JBD24 | |||||
| betT1 | JBD24 | |||||
| sbcD | T14P | |||||
| 15 | dipA | JBD24 | tRNA-leu (PA1796) | F116 | ||
| PA3825/tRNA-leu | F10 | |||||
| 18 | dipA | JBD24 | tRNA-leu (PA1796) | F116 | ||
| PA3825/tRNA-leu | F10 | |||||
| 19 | dipA | JBD24 | ||||
| 46 | dipA | JBD24 | fapE | JBD25§ | ||
| 1 | 21 | retS | JBD24 | tRNA-leu (PA1796) | F116 | |
| 22 | retS | JBD24 | tRNA-leu (PA1796) | F116 | ||
| 25 | retS | JBD24 | tRNA-leu (PA1796) | F116 | ||
| PA0209 | Δ55 bp | |||||
| 26 | retS | JBD24 | tRNA-leu (PA1796) | F116 | ||
| 30 | retS | JBD24 | tRNA-leu (PA1796) | F116 | ||
| 31 | retS | JBD24 | tRNA-leu (PA1796) | F116 | ||
| lysR | PA14_35710 | |||||
| phzS | JBD24 | |||||
| 32 | retS | JBD24 | tRNA-leu (PA1796) | F116 | ||
| lysR | PA14_35710 | |||||
| dnaX | +CGAGCC | |||||
| PA5201 | T139P | |||||
| 34 | retS | JBD24 | tRNA-leu (PA1796) | F116 | ||
| 3 | 33 | retS | JBD24 | |||
| 35 | retS | JBD24 | ||||
| 39 | fliR | JBD24 | tRNA-leu (PA1796) | F116 | ||
| PA3825/tRNA-leu | F10 | |||||
| vgrG4 | JBD24 | |||||
|
MFS transporter (PA3595) |
JBD24 | |||||
| 28 | 2 | 41 | retS | T443P | tRNA-leu (PA1796) | F116 |
| PA3825/tRNA-leu | F10 | |||||
| 3 | 44 | wspF | I68S | PA2942/PA2943 | C→T | |
| pilQ | +GT | |||||
To test whether phage-mediated inactivation of retS caused the RSCV phenotype, rather than by producing a novel phenotype from a phage-encoded gene, we introduced the wild-type retS allele in trans. This restored the RSCV colony phenotype to the ancestral morphology (Fig. 3). However, introduction of wild-type retS in trans had no effect on the ancestral PAO1 strain, or on an engineered wspF deletion mutant (PAO1ΔwspF) (fig. S2), demonstrating allele specificity. Therefore, loss-of-function retS mutations that derepress a signaling cascade and elevate cyclic di-GMP production are hypothesized to be an important early adaptation in wound infections.
Fig. 3. Disruption of genes involved in cyclic di-GMP regulation by prophage insertion is responsible for the PAO1 RSCV phenotype.
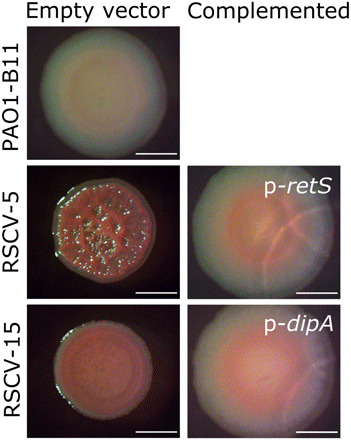
Representative PAO1 RSCVs were complemented by introducing the wild-type copy of the gene disrupted by prophage insertion. Parent strains (empty vector; pUCP18) and complemented strains were grown on Vogel-Bonner minimal medium, and colony morphology was assessed. RSCV-5 was selected as a representative retS-disrupted RSCV, and RSCV-15 was selected as a representative dipA-disrupted RSCV. PAO1-B11 is the ancestor strain that the RSCVs evolved from. Scale bars, 2 mm.
In addition to the recurring retS mutations, phage insertions into other genes responsible for the RSCV phenotype were observed. Five isolates (RSCV-11, RSCV-15, RSCV-18, RSCV-19, and RSCV-46; all from the same wound) gained a prophage insertion in dipA (PA5017) (Table 1). DipA (dispersion-induced phosphodiesterase A) is responsible for initiating biofilm dispersion. DipA deletion mutants have increased exopolysaccharide production, dampened swimming and swarming motility, and enhanced initial attachment (36, 37). Complementation of RSCV-15 and RSCV-46 with the wild-type dipA allele in trans restored the colony morphology to the ancestral type, demonstrating that prophage insertions in this gene caused the RSCV phenotype (Fig. 3 and fig. S1A). Here, again, the prophage-mediated adaptation was not caused by the addition of new genes but rather by the disruption of an existing regulatory gene, which is a common initial route to increased fitness in new environments (38–40). Furthermore, competition between strains in chronic wounds can generate eco-evolutionary dynamics, where induced prophage can not only infect and kill susceptible competitors and favor resistant types but also produce new genotypes that are adapted to the phage-laden environment.
Mutations in dipA, retS, and wspF explained the phenotype of 16 of 17 PAO1 RSCVs, leaving only one unexplained genotype-phenotype link. This remaining isolate, RSCV-39, had the variant colony phenotype but was significantly impaired in biofilm production (Fig. 1B) and had reduced levels of cyclic di-GMP (Fig. 1C). This isolate had a prophage insertion in fliR (Table 1), which is involved in flagella biosynthesis. A recent study linked mutations in flagellar biosynthesis pathways to the RSCV phenotype, but only when the mutants were grown on a solid surface (41). We therefore repeated the cyclic di-GMP quantification of RSCV-39 by comparing levels in planktonic and surface grown cells. Consistent with the previous study (41), RSCV-39 only displayed elevated levels of cyclic di-GMP when grown on a surface (fig. S3). These results, together with previous observations of flagella-mediated RSCVs, suggest that the disruption of fliR by prophage insertion is responsible for the RSCV phenotype in RSCV-39.
All of the sequenced PAO1 RSCV isolates, and 50% of the PAO1 non-RSCV isolates, also acquired a putative conjugative plasmid from the strain S54485. This plasmid had not been annotated in the S54485 genome, but circularized in our assemblies and shares homology with plasmid sequences from Burkholderia pseudomallei (CP009154) (42) and an Acinetobacter baumannii clinical isolate from Thailand (ERS1930304). The vast majority of the predicted proteins encoded on this plasmid were annotated as hypothetical, and the role that this plasmid may play in host adaptation is under further investigation.
Acquisition of new prophages provides immunity to phage reinfection
Given that prophages can provide resistance to phage superinfection (43), we hypothesized that the newly acquired prophages in the PAO1 wound isolates could provide immunity to phages isolated from the co-inoculated P. aeruginosa strains. To test this hypothesis, we isolated the JBD24-like and F116-like phages from strains B23 and MSH10, respectively, because these were most commonly integrated into the PAO1 isolates. Phages isolated from both strains were able to inhibit the growth of the ancestral PAO1 strain (Fig. 4 and fig. S4A) and the engineered PAO1ΔwspF mutant (fig. S5). However, the growth of RSCV-5 and RSCV-44, containing mobile elements from B23 and S54485, and RSCV-15 and RSCV-41, containing mobile elements from B23, MSH10, and S54485 (Fig. 2), was unaffected by the isolated phages, demonstrating acquired immunity (Fig. 4A and fig. S4, B to E). Note that RSCV-5 and RSCV-44 did not acquire genetic elements from MSH10 (Fig. 2) but nonetheless became resistant to MSH10-derived phages (Fig. 4A), suggesting cross-resistance. However, unlike RSCVs, non-RSCVs display immunity to the isolated temperate phages in a lysogen-dependent manner (Fig. 4B and fig. S4, F to H).
Fig. 4. PAO1 wound isolates are immune to phage infection.
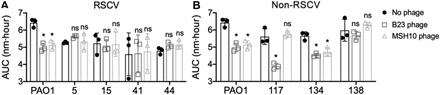
Representative PAO1 (A) RSCV and (B) non-RSCV wound isolates were grown in planktonic culture with phage isolated from B23 (open squares) or MSH10 (open triangles) for 16 hours, and optical density was measured every 30 min. Data are presented as area under the curve (AUC) of the growth curves depicted in fig. S4. *P < 0.05 compared to no phage control (solid circle) (one-way ANOVA with Tukey’s post hoc test); ns, no significant difference. N = 3, each with three technical replicates. Data presented as means ± SD, with the individual data points reflecting the mean of each biological replicate.
Extreme fitness advantages of evolved RSCV-causing mutations
Having established that activated prophage both selected and produced the isolated variants, we asked whether fitness effects of lysogeny extended beyond resistance to superinfection. The regulatory genes disrupted by prophage alter phenotypes like RSCV, but new genes encoded by the prophage could also influence fitness. Measuring fitness, even in vitro, can be predictive of pathogen success in the host (44). Isolates were competed against the lacZ-marked ancestral strain over 48 hours in biofilm and planktonic conditions. Both types of retS mutants, whether the T443P substitution (RSCV-41) or a prophage insertion mutant (RSCV-5), were significantly more fit, outcompeting the PAO1 ancestor in both planktonic and biofilm conditions (Fig. 5A). This was also the case for the dipA prophage insertion mutant (RSCV-15) (Fig. 5A).
Fig. 5. PAO1 RSCVs have increased fitness relative to the ancestral PAO1.
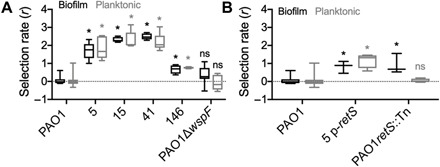
PAO1 strains were competed against the ancestral parent tagged with lacZ for 48 hours in both biofilm (black) and planktonic (gray) conditions for 48 hours. Fitness of the competing strain was determined by calculating the selection rate (r). (A) Fitness of representative PAO1 wound isolates relative to the ancestral PAO1 strain. PAO1ΔwspF competed against the isogenic PAO1 parent was used as a representative wsp mutant instead of RSCV-44 due to the secondary SNP in pilQ in this isolate (table S1). N = 5. (B) To separate the contributions of the retS mutation and the presence of newly acquired prophage to the increased fitness phenotype, RSCV-5 containing the retS complementing plasmid (p-retS) and a PAO1 retS transposon mutant were completed against their parent PAO1 strains. N = 3. *P < 0.05 compared to the PAO1 pairwise competition (one-way ANOVA with Tukey’s post hoc test).
To distinguish effects of retS mutations from effects of prophage acquisition, we competed PAO1 against a transposon mutant of retS [PAO1retS::Tn (45)] and against RSCV-5 complemented with wild-type retS allele in trans (RSCV-5 p-retS). PAO1retS::Tn was more fit than the ancestor only in biofilm conditions; however, RSCV-5 p-retS was significantly more fit in both planktonic and biofilm conditions, which demonstrates benefits of lysogeny beyond disrupting retS (Fig. 5B). Furthermore, a mutant that acquired several mobile genetic elements, but no observable gene disruptions or variant phenotypes (non–RSCV-146; table S6), outcompeted PAO1 (Fig. 5A), although to a lesser extent than RSCV-5 (fig. S7). Therefore, the enhanced competitive fitness of PAO1 RSCVs compared to the ancestor appears to result from combined effects of the RSCV hyperbiofilm phenotype and the horizontally acquired mobile genetic elements themselves (Fig. 5B).
Given the focused selection on wsp mutants of PA14 (21), we were surprised that homologous mutants were not found in PAO1. We therefore competed PAO1ΔwspF against the isogenic PAO1 parent in both planktonic and biofilm conditions. Contrary to the high fitness advantage of ΔwspF observed in PA14 (21), PAO1ΔwspF exhibited no fitness advantage even in biofilm growth conditions (Fig. 5A). These results contrast with the high selective advantage of retS and dipA mutants had over the PAO1 ancestor (Fig. 5). Furthermore, in direct pairwise competition, RSCV-5 (the retS prophage insertion mutant) was significantly more fit than PAO1ΔwspF (fig. S7B), again suggesting why PAO1 retS mutants may be selected in vivo over PAO1 wsp mutants.
The relative fitness values of the evolved RSCV isolates indicate extreme competitive advantages that could indicate active exclusion or killing of the competitor. Specifically, the ancestral PAO1 strain was rapidly outcompeted and became undetectable after 24 to 48 hours in many of these fitness assays. We also observed zones of inhibitions around the RSCV isolates when plated adjacent to the ancestor (fig. S8), suggesting active killing of the ancestral strain by the RSCV isolates. To test this hypothesis, we plated colonies of evolved RSCV isolates next to the ancestor and filmed their interactions while growing over time (movies S1 to S4). These movies indicated that both the retS T443P SNP (RSCV-41) and the retS prophage insertion mutant (RSCV-5) outcompeted the ancestral strain by direct inhibition. The inhibition by the insertion mutant, RSCV-5, appears to be through phage lysis. In support of this, we found that this newly acquired prophage in RSCV-5 can be induced to kill the ancestral strain (fig. S6). However, the mechanisms of the observed direct inhibition are still under investigation.
DISCUSSION
For any given set of bacterial strains, the fittest in a given environment is usually unpredictable, regardless of available data. But a growing literature shows that bacterial fitness is often at the mercy of, and empowered by, prophage (46, 47). These enemy cargoes harm the carrier when activated but can kill nonlysogenized competitors, providing significant competitive advantages to resistant genotypes. In this study, when mixed populations of P. aeruginosa strains were added to model chronic wounds, the earliest dynamics and adaptations occurred in response to prophage, but not in the typical manner where lysogens prevail because they resist superinfection. Rather, the four strains that contributed at least four mobile elements to this strain mixture (Fig. 2 and table S4) were swiftly outcompeted, as these elements were selected for CRISPR-mediated resistance in some clones of PA14 (21) and produced an assortment of new lysogens in the PAO1 background. Newly lysogenized PAO1 variants also acquired novel adaptations to the wound environment beyond phage resistance, including hyperbiofilm traits that may enhance resistance to host defenses (28). The ecological theory of apparent competition can be summarized as “the enemy of your enemy is your friend,” for example, when one host species carrying a pathogen that it evolved to tolerate outcompetes another species that is more susceptible. The PAO1 variants characterized here upend this dynamic by capturing prophages that generate adaptations via the genes they disrupt and the defenses to superinfection that they encode.
Here, we demonstrate that transient ecological interactions alter the course of an infection through prophage movement and subsequent selection on prophage-disrupted genetic targets. Prophages that emerged from strains that were rapidly outcompeted provided substantial fitness benefits to the winning strains and increased the mutation supply upon which selection could act. We identify the causes of at least part of that enhanced competitiveness as effects of the genetic loci disrupted by inserted prophage. PAO1-derived RSCVs with prophage insertions in retS were routinely isolated from the porcine chronic wounds (Table 1). By disrupting the RetS signaling cascade that regulates biofilm formation and virulence (30, 31), the RSCVs could outcompete other ancestral strains in the wound environment. This discovery has two major consequences: (i) Short-term exposure to closely related strains can alter the evolutionary trajectory of bacterial populations, and (ii) selection for hyperbiofilm formation is almost certainly adaptive in an infection, as has been previously hypothesized (21, 25, 48, 49).
Many chronic infections begin clonally and temporally diversify as the infecting strain adapts to new niches in the host (3, 4, 50–52). This has been elegantly demonstrated for cystic fibrosis lung isolates, where the phylogenies indicate a single originating colonizing strain, giving rise to multiple coexisting lineages (6, 10, 50). However, our findings here suggest an alternative outcome: Several competing strains may infect a host and dictate the evolutionary fate of the predominant strain. In this model, the infection would still appear clonal because an invading or co-infecting strain would be outcompeted by the more fit, abundant strain [a process that only takes a few days at most according to our results (21)]. However, these transient co-infecting strains may significantly and cryptically alter the targets of selection. Given that patients are exposed to many opportunistic pathogens in the environment and in clinical settings during treatment (53–58), this scenario seems possible and is still consistent with the ultimately clonal appearance of chronic infections. An additional piece of evidence supporting our hypothesis is that most of the sequenced isolates from chronic infections have many genomic regions that appear to be recently acquired mobile genetic elements, including phage (19, 59). This is usually thought to be from competition in the environmental reservoir. This may still be the case, but it could also be due to previous competition within the host that drove early adaptations in the predominant strain.
Prophages are often maintained in the bacterial chromosome because they not only provide resistance to phage superinfection (43) but also can enhance competitiveness in many environments including in vivo (59). In contrast to the better understood path of lysogenic conversion where the bacterium gains a fitness advantage from a horizontally acquired gene, active lysogeny involves prophage insertion into a gene or regulatory region that inactivates that gene, creating a sort of genetic switch (60). In this study, active lysogeny disrupted different regulators of biofilm formation and improved fitness in vitro and in vivo. Mutations in retS that increase biofilm formation and type VI secretion are thought to be adaptive in chronic infections (9, 13), as this signaling cascade is a central switch between acute and chronic infection (30, 31). Other examples of active lysogeny include prophages causing mutator phenotypes in Streptococcus pyogenes by disrupting mutL (61, 62), prophages disrupting com genes required for Listeria virulence and survival in phagosomes (47, 63), and the classic example of prophage regulating sporulation cycle components (64–68). Given the potentially reversible nature of active lysogeny and technical difficulty in observing these events, it may be that active lysogeny is more common than previously appreciated, especially in environments where competition among related strains is prevalent. Because active lysogeny contributes to a growing list of pathogenic traits, from virulence to sporulation and, now, hyperbiofilm formation, the mechanisms and scope of active lysogeny demand further inquiry.
There is a growing body of work demonstrating that mobile genetic elements are a significant source of mutation driving increased biofilm formation in infections. In a patient with chronic P. aeruginosa pulmonary disease, insertion sequences of the ISL3 family disrupted genes involved in flagellar function, type IV pili, and other genetic loci that increased virulence of the mutants (69). Davies et al. (70) observed in vitro a similar mechanism of phage-mediated adaptation to what we report here in vivo. In that study, an evolution experiment with PAO1 was conducted with added temperate phages, some of which inserted in targets relating to biofilm formation or motility, including dipA (70). Furthermore, it has long been known that adding phages to biofilms can select for small-colony variants and that active phages have been observed in many chronic infection isolates (71–73). Our work synthesizes these findings by showing how competing strains can provide a source of the infecting phages, which, in turn, produce newly infected genotypes that are selected in vivo to enhance biofilm production and maintain the prophage in the population.
Two main takeaways emerge that advance our understanding of the evolutionary forces and selected traits within opportunistic infections. The first is that mutations that increase cyclic di-GMP levels and therefore increase biofilm formation are adaptive in wounds. This result adds to a growing body of evidence from patients that biofilm formation is adaptive in chronic infections (25, 49). Our study shows that different strains of P. aeruginosa may take different routes to higher cyclic di-GMP levels, where the convergent evolution of a hyperbiofilm phenotype may be caused by mutations in wsp, retS, dipA, or one of the other diguanylate cyclases or phosphodiesterases (74–77). The prevalence of these mutations that increase cyclic di-GMP across many different hosts and studies strongly supports the idea that these are beneficial mutations, but rarely are they the most abundant mutation in the host infection (21, 78). An explanation for their prevalence but low abundance could be negative frequency–dependent selection, where RSCVs are more fit when rare but disfavored at high frequency (79, 80). Nonetheless, the constellation of routes to RSCVs and their relative rarity may make clinical targeting of this phenotype challenging. The second takeaway from this work is that interstrain, and likely interspecies, interactions can significantly alter the evolutionary dynamics of an infection. Dormant or extinct strains that were initially present at equal ratios in the wounds provided prophage that infected and altered genes in the eventual winners, producing new genotypes and phenotypes for selection to act upon in addition to the hyperbiofilm phenotype.
Ultimately, the work presented here should inspire a broader view of how natural selection operates in an infection, where transient interactions in the microbial community and the contributions of mobile genetic elements can define the evolutionary-genetic course of an infection. These multistrain or species interactions can happen early in the infection, in a window of time often missed by current surveillance methods. Therefore, catching the movie in the middle is not sufficient to understand how the characters behaved in the end. Instead, initial pathogen adaptations in chronic infections provide crucial plot development that needs to be examined more closely to better anticipate the rest of the story.
Acknowledgments
We thank S. B. Chaney for assistance with wound biopsies, D. Snyder and MIGS for genome sequencing, and A. Santos-Lopez for critical discussions. Funding: E.S.G. was funded by an American Heart Association Career Development Award (19CDA34630005). D.J.W. was funded by the NIH (R01AI134895 and R01AI143916). C.W.M. and V.S.C. were funded by the NIH (U01AI124302 and R33HL137077). Author contributions: C.W.M., E.S.G., D.J.W., and V.S.C. conceptualized the study. C.W.M., E.S.G., and C.L. were responsible for data generation and data curation. C.W.M. and E.S.G. wrote the original drafts, and all authors reviewed and edited subsequent writing. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials except the following: All Illumina and Nanopore sequences have been deposited in the NCBI Sequence Read Archive (SRA) to the BioProject PRJNA633671 under accession numbers SAMN14968233 to SAMN14968292. Code for sequence processing can be found at https://github.com/sirmicrobe/pig_wound_manuscripts/.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/29/eabh1489/DC1
REFERENCES AND NOTES
- 1.Haldane J. B. S., Disease and evolution. Ric. Sci. 19, 68–76 (1949). [Google Scholar]
- 2.Brockhurst M. A., Experimental evolution can unravel the complex causes of natural selection in clinical infections. Microbiology 161, 1175–1179 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Struelens M. J., Schwam V., Deplano A., Baran D., Genome macrorestriction analysis of diversity and variability of Pseudomonas aeruginosa strains infecting cystic fibrosis patients. J. Clin. Microbiol. 31, 2320–2326 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith E. E., Buckley D. G., Wu Z., Saenphimmachak C., Hoffman L. R., D’Argenio D. A., Miller S. I., Ramsey B. W., Speert D. P., Moskowitz S. M., Burns J. L., Kaul R., Olson M. V., Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc. Natl. Acad. Sci. U.S.A. 103, 8487–8492 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coutinho C. P., de Carvalho C. C. C. R., Madeira A., Pinto-de-Oliveira A., Sá-Correia I., Burkholderia cenocepacia phenotypic clonal variation during a 3.5-year colonization in the lungs of a cystic fibrosis patient. Infect. Immun. 79, 2950–2960 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Silva I. N., Santos P. M., Santos M. R., Zlosnik J. E. A., Speert D. P., Buskirk S. W., Bruger E. L., Waters C. M., Cooper V. S., Moreira L. M., Long-term evolution of Burkholderia multivorans during a chronic cystic fibrosis infection reveals shifting forces of selection. mSystems 1, e00029-16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Honsa E. S., Cooper V. S., Mhaissen M. N., Frank M., Shaker J., Iverson A., Rubnitz J., Hayden R. T., Lee R. E., Rock C. O., Tuomanen E. I., Wolf J., Rosch J. W., RelA mutant Enterococcus faecium with multiantibiotic tolerance arising in an immunocompromised host. MBio 8, e02124-16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosa R. L., Johansen H. K., Molin S., Convergent metabolic specialization through distinct evolutionary paths in Pseudomonas aeruginosa. MBio 9, e00269-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bartell J. A., Sommer L. M., Haagensen J. A. J., Loch A., Espinosa R., Molin S., Johansen H. K., Evolutionary highways to persistent bacterial infection. Nat. Commun. 10, 629 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lieberman T. D., Michel J.-B., Aingaran M., Potter-Bynoe G., Roux D., Davis M. R., Skurnik D., Leiby N., LiPuma J. J., Goldberg J. B., McAdam A. J., Priebe G. P., Kishony R., Parallel bacterial evolution within multiple patients identifies candidate pathogenicity genes. Nat. Genet. 43, 1275–1280 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Folkesson A., Jelsbak L., Yang L., Johansen H. K., Ciofu O., Høiby N., Molin S., Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: An evolutionary perspective. Nat. Rev. Microbiol. 10, 841–851 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Fothergill J. L., Neill D. R., Loman N., Winstanley C., Kadioglu A., Pseudomonas aeruginosa adaptation in the nasopharyngeal reservoir leads to migration and persistence in the lungs. Nat. Commun. 5, 4780 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Marvig R. L., Sommer L. M., Molin S., Johansen H. K., Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat. Genet. 47, 57–64 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Buckling A., Rainey P. B., Antagonistic coevolution between a bacterium and a bacteriophage. Proc. R. Soc. B Biol. Sci. 269, 931–936 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brockhurst M. A., Buckling A., Rainey P. B., The effect of a bacteriophage on diversification of the opportunistic bacterial pathogen, Pseudomonas aeruginosa. Proc. R. Soc. B Biol. Sci. 272, 1385–1391 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paterson S., Vogwill T., Buckling A., Benmayor R., Spiers A. J., Thomson N. R., Quail M., Smith F., Walker D., Libberton B., Fenton A., Hall N., Brockhurst M. A., Antagonistic coevolution accelerates molecular evolution. Nature 464, 275–278 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gómez P., Buckling A., Bacteria-phage antagonistic coevolution in soil. Science 332, 106–109 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Koskella B., Brockhurst M. A., Bacteria–phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol. Rev. 38, 916–931 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tariq M. A., Everest F. L. C., Cowley L. A., Wright R., Holt G. S., Ingram H., Duignan L. A. M., Nelson A., Lanyon C. V., Perry A., Perry J. D., Bourke S., Brockhurst M. A., Bridge S. H., De Soyza A., Smith D. L., Temperate bacteriophages from chronic Pseudomonas aeruginosa lung infections show disease-specific changes in host range and modulate antimicrobial susceptibility. mSystems 4, e00191-18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holt R. D., Lawton J. H., The ecological consequences of shared natural enemies. Annu. Rev. Ecol. Syst. 25, 495–520 (1994). [Google Scholar]
- 21.Gloag E. S., Marshall C. W., Snyder D., Lewin G. R., Harris J. S., Santos-Lopez A., Chaney S. B., Whiteley M., Cooper V. S., Wozniak D. J., Pseudomonas aeruginosa interstrain dynamics and selection of hyperbiofilm mutants during a chronic infection. MBio 10, e01698-19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hickman J. W., Tifrea D. F., Harwood C. S., A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc. Natl. Acad. Sci. U.S.A. 102, 14422–14427 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Häussler S., Tümmler B., Weißbrodt H., Rohde M., Steinmetz I., Small-colony variants of Pseudomonas aeruginosa in cystic fibrosis. Clin. Infect. Dis. 29, 621–625 (1999). [DOI] [PubMed] [Google Scholar]
- 24.Häußler S., Lehmann C., Breselge C., Rohde M., Claßen M., Tümmler B., Vandamme P., Steinmetz I., Fatal outcome of lung transplantation in cystic fibrosis patients due to small-colony variants of the Burkholderia cepacia complex. Eur. J. Clin. Microbiol. Infect. Dis. 22, 249–253 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Evans T. J., Small colony variants of Pseudomonas aeruginosa in chronic bacterial infection of the lung in cystic fibrosis. Future Microbiol. 10, 231–239 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Starkey M., Hickman J. H., Ma L., Zhang N., Long S. D., Hinz A., Palacios S., Manoil C., Kirisits M. J., Starner T. D., Wozniak D. J., Harwood C. S., Parsek M. R., Pseudomonas aeruginosa rugose small-colony variants have adaptations that likely promote persistence in the cystic fibrosis lung. J. Bacteriol. 191, 3492–3503 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borlee B. R., Goldman A. D., Murakami K., Samudrala R., Wozniak D. J., Parsek M. R., Pseudomonas aeruginosa uses a cyclic-di-GMP-regulated adhesin to reinforce the biofilm extracellular matrix. Mol. Microbiol. 75, 827–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pestrak M. J., Chaney S. B., Eggleston H. C., Dellos-Nolan S., Dixit S., Mathew-Steiner S. S., Roy S., Parsek M. R., Sen C. K., Wozniak D. J., Pseudomonas aeruginosa rugose small-colony variants evade host clearance, are hyper-inflammatory, and persist in multiple host environments. PLOS Pathog. 14, e1006842 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O’Connor J. R., Kuwada N. J., Huangyutitham V., Wiggins P. A., Harwood C. S., Surface sensing and lateral subcellular localization of WspA, the receptor in a chemosensory-like system leading to c-di-GMP production. Mol. Microbiol. 86, 720–729 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Francis V. I., Waters E. M., Finton-James S. E., Gori A., Kadioglu A., Brown A. R., Porter S. L., Multiple communication mechanisms between sensor kinases are crucial for virulence in Pseudomonas aeruginosa. Nat. Commun. 9, 2219 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moscoso J. A., Mikkelsen H., Heeb S., Williams P., Filloux A., The Pseudomonas aeruginosa sensor RetS switches type III and type VI secretion via c-di-GMP signalling. Environ. Microbiol. 13, 3128–3138 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Goodman A. L., Kulasekara B., Rietsch A., Boyd D., Smith R. S., Lory S., A signaling network reciprocally regulates genes associated with acute infection and chronic persistence in Pseudomonas aeruginosa. Dev. Cell 7, 745–754 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Byrne M., Kropinski A. M., The genome of the Pseudomonas aeruginosa generalized transducing bacteriophage F116. Gene 346, 187–194 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Bondy-Denomy J., Pawluk A., Maxwell K. L., Davidson A. R., Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 493, 429–432 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hulo C., Masson P., Le Mercier P., Toussaint A., A structured annotation frame for the transposable phages: A new proposed family “Saltoviridae” within the Caudovirales. Virology 477, 155–163 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Roy A. B., Petrova O. E., Sauer K., The phosphodiesterase DipA (PA5017) is essential for Pseudomonas aeruginosa biofilm dispersion. J. Bacteriol. 194, 2904–2915 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mattingly A. E., Kamatkar N. G., Morales-Soto N., Borlee B. R., Shrout J. D., Multiple environmental factors influence the importance of the phosphodiesterase DipA upon Pseudomonas aeruginosa Swarming. Appl. Environ. Microbiol. 84, e02847-17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cooper V. S., Long-term experimental evolution in Escherichia coli. X. Quantifying the fundamental and realized niche. BMC Evol. Biol. 2, 12 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hottes A. K., Freddolino P. L., Khare A., Donnell Z. N., Liu J. C., Tavazoie S., Bacterial adaptation through loss of function. PLOS Genet. 9, e1003617 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mhatre E., Snyder D. J., Sileo E., Turner C. B., Buskirk S. W., Fernandez N. L., Neiditch M. B., Waters C. M., Cooper V. S., One gene, multiple ecological strategies: A biofilm regulator is a capacitor for sustainable diversity. Proc. Natl. Acad. Sci. U.S.A. 117, 21647–21657 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harrison J. J., Almblad H., Irie Y., Wolter D. J., Eggleston H. C., Randall T. E., Kitzman J. O., Stackhouse B., Emerson J. C., Mcnamara S., Larsen T. J., Shendure J., Hoffman L. R., Wozniak D. J., Parsek M. R., Elevated exopolysaccharide levels in Pseudomonas aeruginosa flagellar mutants have implications for biofilm growth and chronic infections. PLOS Genet. 16, e1008848 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johnson S. L., Baker A. L., Chain P. S., Currie B. J., Daligault H. E., Davenport K. W., Davis C. B., Inglis T. J. J., Kaestli M., Koren S., Mayo M., Merritt A. J., Price E. P., Sarovich D. S., Warner J., Rosovitz M. J., Whole-genome sequences of 80 environmental and clinical isolates of Burkholderia pseudomallei. Genome Announc. 3, e01282-14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bondy-Denomy J., Qian J., Westra E. R., Buckling A., Guttman D. S., Davidson A. R., Maxwell K. L., Prophages mediate defense against phage infection through diverse mechanisms. ISME J. 10, 2854–2866 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sommer M. O. A., Munck C., Toft-Kehler R. V., Andersson D. I., Prediction of antibiotic resistance: Time for a new preclinical paradigm? Nat. Rev. Microbiol. 15, 689–696 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Jacobs M. A., Alwood A., Thaipisuttikul I., Spencer D., Haugen E., Ernst S., Will O., Kaul R., Raymond C., Levy R., Chun-Rong L., Guenthner D., Bovee D., Olson M. V., Manoil C., Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 100, 14339–14344 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.S. F. Fitzgerald, N. Lupolova, S. Shaaban, T. J. Dallman, D. Greig, L. Allison, S. C. Tongue, J. Evans, M. K. Henry, T. N. McNeilly, J. L. Bono, D. L. Gally, Prophage-dependent recombination drives genome structural variation and phenotypic heterogeneity in Escherichia coli O157:H7. bioRxiv 2020.12.02.407981 [Preprint]. 2 December 2020. 10.1101/2020.12.02.407981. [DOI]
- 47.Pasechnek A., Rabinovich L., Stadnyuk O., Azulay G., Mioduser J., Argov T., Borovok I., Sigal N., Herskovits A. A., Active lysogeny in Listeria Monocytogenes is a bacteria-phage adaptive response in the mammalian environment. Cell Rep. 32, 107956 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Donlan R. M., Costerton J. W., Biofilms: Survival mechanisms of clinically relevant microorganisms. Clin. Microbiol. Rev. 15, 167–193 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bjarnsholt T., The role of bacterial biofilms in chronic infections. APMIS Suppl. 121, 1–51 (2013). [DOI] [PubMed] [Google Scholar]
- 50.Burns J. L., Gibson R. L., McNamara S., Yim D., Emerson J., Rosenfeld M., Hiatt P., McCoy K., Castile R., Smith A. L., Ramsey B. W., Longitudinal assessment of Pseudomonas aeruginosa in Young Children with Cystic Fibrosis. J. Infect. Dis. 183, 444–452 (2001). [DOI] [PubMed] [Google Scholar]
- 51.Yang L., Jelsbak L., Marvig R. L., Damkiær S., Workman C. T., Rau M. H., Hansen S. K., Folkesson A., Johansen H. K., Ciofu O., Høiby N., Sommer M. O. A., Molin S., Evolutionary dynamics of bacteria in a human host environment. Proc. Natl. Acad. Sci. U.S.A. 108, 7481–7486 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jorth P., Staudinger B. J., Wu X., Hisert K. B., Hayden H., Garudathri J., Harding C. L., Radey M. C., Rezayat A., Bautista G., Berrington W. R., Goddard A. F., Zheng C., Angermeyer A., Brittnacher M. J., Kitzman J., Shendure J., Fligner C. L., Mittler J., Aitken M. L., Manoil C., Bruce J. E., Yahr T. L., Singh P. K., Regional isolation drives bacterial diversification within cystic fibrosis lungs. Cell Host Microbe 18, 307–319 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Panagea S., Winstanley C., Walshaw M. J., Ledson M. J., Hart C. A., Environmental contamination with an epidemic strain of Pseudomonas aeruginosa in a Liverpool cystic fibrosis centre, and study of its survival on dry surfaces. J. Hosp. Infect. 59, 102–107 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Casey J. A., Curriero F. C., Cosgrove S. E., Nachman K. E., Schwartz B. S., High-density livestock operations, crop field application of manure, and risk of community-associated methicillin-resistant Staphylococcus aureus infection, Pennsylvania, USA. JAMA Intern. Med. 173, 1980–1990 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pham T. M., Kretzschmar M., Bertrand X., Bootsma M.; COMBACTE-MAGNET Consortium , Tracking Pseudomonas aeruginosa transmissions due to environmental contamination after discharge in ICUs using mathematical models. PLOS Comput. Biol. 15, e1006697 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kramer A., Schwebke I., Kampf G., How long do nosocomial pathogens persist on inanimate surfaces? A systematic review. BMC Infect. Dis. 6, 130 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Loveday H. P., Wilson J. A., Kerr K., Pitchers R., Walker J. T., Browne J., Association between healthcare water systems and Pseudomonas aeruginosa infections: A rapid systematic review. J. Hosp. Infect. 86, 7–15 (2014). [DOI] [PubMed] [Google Scholar]
- 58.Kizny Gordon A. E., Mathers A. J., Cheong E. Y. L., Gottlieb T., Kotay S., Walker A. S., Peto T. E. A., Crook D. W., Stoesser N., The hospital water environment as a reservoir for carbapenem-resistant organisms causing hospital-acquired infections—A systematic review of the literature. Clin. Infect. Dis. 64, 1435–1444 (2017). [DOI] [PubMed] [Google Scholar]
- 59.Winstanley C., Langille M. G. I., Fothergill J. L., Kukavica-Ibrulj I., Paradis-Bleau C., Sanschagrin F., Thomson N. R., Winsor G. L., Quail M. A., Lennard N., Bignell A., Clarke L., Seeger K., Saunders D., Harris D., Parkhill J., Hancock R. E. W., Brinkman F. S. L., Levesque R. C., Newly introduced genomic prophage islands are critical determinants of in vivo competitiveness in the Liverpool epidemic strain of Pseudomonas aeruginosa. Genome Res. 19, 12–23 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Feiner R., Argov T., Rabinovich L., Sigal N., Borovok I., Herskovits A. A., A new perspective on lysogeny: Prophages as active regulatory switches of bacteria. Nat. Rev. Microbiol. 13, 641–650 (2015). [DOI] [PubMed] [Google Scholar]
- 61.Scott J., Thompson-Mayberry P., Lahmamsi S., King C. J., McShan W. M., Phage-associated mutator phenotype in group A streptococcus. J. Bacteriol. 190, 6290–6301 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Scott J., Nguyen S. V., King C. J., Hendrickson C., McShan W. M., Phage-like Streptococcus pyogenes chromosomal islands (SpyCI) and mutator phenotypes: Control by growth state and rescue by a SpyCI-encoded promoter. Front. Microbiol. 3, 317 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rabinovich L., Sigal N., Borovok I., Nir-Paz R., Herskovits A. A., Prophage excision activates Listeria competence genes that promote phagosomal escape and virulence. Cell 150, 792–802 (2012). [DOI] [PubMed] [Google Scholar]
- 64.Stragier P., Kunkel B., Kroos L., Losick R., Chromosomal rearrangement generating a composite gene for a developmental transcription factor. Science 243, 507–512 (1989). [DOI] [PubMed] [Google Scholar]
- 65.Kunkel B., Losick R., Stragier P., The Bacillus subtilis gene for the development transcription factor sigma K is generated by excision of a dispensable DNA element containing a sporulation recombinase gene. Genes Dev. 4, 525–535 (1990). [DOI] [PubMed] [Google Scholar]
- 66.Takemaru K.-c., Mizuno M., Sato T., Takeuchi M., Kobayashi Y., Complete nucleotide sequence of a skin element excised by DNA rearrangement during sporulation in Bacillus subtilis. Microbiology 141, 323–327 (1995). [DOI] [PubMed] [Google Scholar]
- 67.Abe K., Yoshinari A., Aoyagi T., Hirota Y., Iwamoto K., Sato T., Regulated DNA rearrangement during sporulation in Bacillus weihenstephanensis KBAB4. Mol. Microbiol. 90, 415–427 (2013). [DOI] [PubMed] [Google Scholar]
- 68.Abe K., Kawano Y., Iwamoto K., Arai K., Maruyama Y., Eichenberger P., Sato T., Developmentally-regulated excision of the SPβ prophage reconstitutes a gene required for spore envelope maturation in Bacillus subtilis. PLOS Genet. 10, e1004636 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sentausa E., Basso P., Berry A., Adrait A., Bellement G., Couté Y., Lory S., Elsen S., Attrée I., Insertion sequences drive the emergence of a highly adapted human pathogen. Microb. Genom. 6, mgen000265 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Davies E. V., James C. E., Williams D., O’Brien S., Fothergill J. L., Haldenby S., Paterson S., Winstanley C., Brockhurst M. A., Temperate phages both mediate and drive adaptive evolution in pathogen biofilms. Proc. Natl. Acad. Sci. U.S.A. 113, 8266–8271 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Webb J. S., Thompson L. S., James S., Charlton T., Tolker-Nielsen T., Koch B., Givskov M., Kjelleberg S., Cell death in Pseudomonas aeruginosa biofilm development. J. Bacteriol. 185, 4585–4592 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Webb J. S., Lau M., Kjelleberg S., Bacteriophage and phenotypic variation in Pseudomonas aeruginosa biofilm development. J. Bacteriol. 186, 8066–8073 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kirov S. M., Webb J. S., O’May C. Y., Reid D. W., Woo J. K. K., Rice S. A., Kjelleberg S., Biofilm differentiation and dispersal in mucoid Pseudomonas aeruginosa isolates from patients with cystic fibrosis. Microbiology 153, 3264–3274 (2007). [DOI] [PubMed] [Google Scholar]
- 74.Kulesekara H., Lee V., Brencic A., Liberati N., Urbach J., Miyata S., Lee D. G., Neely A. N., Hyodo M., Hayakawa Y., Ausubel F. M., Lory S., Analysis of Pseudomonas aeruginosa diguanylate cyclases and phosphodiesterases reveals a role for bis-(3′-5′)-cyclic-GMP in virulence. Proc. Natl. Acad. Sci. U.S.A. 103, 2839–2844 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ha D.-G., O’Toole G. A., c-di-GMP and its effects on biofilm formation and dispersion: A Pseudomonas aeruginosa review. Microbiol. Spectr. 3, MB-0003-2014 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Valentini M., Filloux A., Biofilms and cyclic di-GMP (c-di-GMP) signaling: Lessons from Pseudomonas aeruginosa and other bacteria. J. Biol. Chem. 291, 12547–12555 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Maunders E., Welch M., Matrix exopolysaccharides; the sticky side of biofilm formation. FEMS Microbiol. Lett. 364, fnx120 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Drenkard E., Ausubel F. M., Pseudomonas biofilm formation and antibiotic resistance are linked to phenotypic variation. Nature 416, 740–743 (2002). [DOI] [PubMed] [Google Scholar]
- 79.Rainey P. B., Travisano M., Adaptive radiation in a heterogeneous environment. Nature 394, 69–72 (1998). [DOI] [PubMed] [Google Scholar]
- 80.Kim W., Levy S. B., Foster K. R., Rapid radiation in bacteria leads to a division of labour. Nat. Commun. 7, 10508 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rybtke M. T., Borlee B. R., Murakami K., Irie Y., Hentzer M., Nielsen T. E., Givskov M., Parsek M. R., Tolker-Nielsen T., Fluorescence-based reporter for gauging cyclic di-GMP levels in Pseudomonas aeruginosa. Appl. Environ. Microbiol. 78, 5060–5069 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schweizer H. P., Escherichia-Pseudomonas shuttle vectors derived from pUC18/19. Gene 97, 109–112 (1991). [DOI] [PubMed] [Google Scholar]
- 83.Hoang T. T., Kutchma A. J., Becher A., Schweizer H. P., Integration-proficient plasmids for Pseudomonas aeruginosa: Site-specific integration and use for engineering of reporter and expression strains. Plasmid 43, 59–72 (2000). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/29/eabh1489/DC1