

Foxc2 sustains adult lymphatic vascular specialization and coordinates organ-specific lymph flow, gut microbiota, and inflammation.
Abstract
The mechanisms maintaining adult lymphatic vascular specialization throughout life and their role in coordinating inter-organ communication to sustain homeostasis remain elusive. We report that inactivation of the mechanosensitive transcription factor Foxc2 in adult lymphatic endothelium leads to a stepwise intestine-to-lung systemic failure. Foxc2 loss compromised the gut epithelial barrier, promoted dysbiosis and bacterial translocation to peripheral lymph nodes, and increased circulating levels of purine metabolites and angiopoietin-2. Commensal microbiota depletion dampened systemic pro-inflammatory cytokine levels, corrected intestinal lymphatic dysfunction, and improved survival. Foxc2 loss skewed the specialization of lymphatic endothelial subsets, leading to populations with mixed, pro-fibrotic identities and to emergence of lymph node–like endothelial cells. Our study uncovers a cross-talk between lymphatic vascular function and commensal microbiota, provides single-cell atlas of lymphatic endothelial subtypes, and reveals organ-specific and systemic effects of dysfunctional lymphatics. These effects potentially contribute to the pathogenesis of diseases, such as inflammatory bowel disease, cancer, or lymphedema.
INTRODUCTION
The lymphatic vasculature is indispensable for the maintenance of plasma and tissue volume, immunosurveillance, and absorption of dietary fat. The importance of lymphatic vessels (LVs) for human physiology and pathology and their implication in some of the most common diseases, such as cancer and cardiovascular and neurodegenerative diseases, are only beginning to be appreciated (1, 2). Therefore, progress in this rapidly expanding field critically depends on increased molecular knowledge of both basic principles of lymphatic vascular organization and function as well as on the pathophysiological consequences of lymphatic dysfunction.
The mature lymphatic vascular network comprises blind-ended capillaries that take up interstitial fluid and cells and collecting LVs (collLVs) that transport the lymph to lymph nodes (LNs) and then back to blood circulation. In agreement with their functions, lymphatic capillaries and collLVs are structurally distinct. Capillary lymphatic endothelial cells (capLECs) display discontinuous cell-cell junctions and scarce basement membrane and have no mural cell coverage. CollLVs are lined by elongated LECs with continuous cell junctions and are surrounded by basement membrane and contractile smooth muscle cells (SMCs). CollLVs contain numerous intraluminal bileaflet valves, which subdivide collLVs into structural units, called lymphangions, and prevent lymph backflow by ensuring the unidirectional lymph transport (3).
Capillary, lymphangion, and valve LECs (vLECs) display specialized molecular profiles (4, 5). CapLECs express molecules facilitating trafficking of immune cells (CCL21A and LYVE1) (6, 7), while lymphangion LECs produce higher levels of basement membrane components and SMC chemoattractant platelet-derived growth factor β (PDGFβ) (8, 9). In line with their exposure to high mechanical stress, vLECs express elevated levels of the mechanosensitive transcription factors FOXC2, GATA2, and NFATC1 (8, 10, 11). CollLVs are physically continuous with the LN lymphatic vascular network. Recent single-cell transcriptomic profiling identified various LN LEC types with distinct molecular features that reflect their specialized roles in immune cell trafficking, antigen presentation, and establishment of tolerance (12).
Specialization of adult LVs into collectors and capillaries is crucial for the functionality of the entire lymphatic vascular bed (3). Multiple regulators of collLV patterning and, especially, lymphatic valve development have been identified (4, 5). However, how the hierarchical organization and function of lymphatic vasculature are maintained throughout life is unknown, as is the role of organ-specific environment in this process. Furthermore, while loss of lymphatic capillaries leads to local tissue fibrosis, inflammation, and impaired immune response (13, 14), the biological consequences of collLV dysfunction beyond defective tissue drainage remain elusive.
Lymph flow–induced forkhead transcription factor FOXC2 controls formation of lymphatic valves and specialization of collLVs during embryonic and postnatal development (5, 8, 15). Inactivating mutations in FOXC2 underlie the lymphedema-distichiasis syndrome (LDS) (16), a human hereditary disease of LVs. LDS is characterized by late-onset swelling of legs as a result of defective collLVs and valve function and lymph reflux (17). While lymph stasis and swelling of extremities are features of all hereditary lymphedemas (16), LDS patients uniquely develop ectopic peritoneal lymphoid nodules (17), indicating that loss of FOXC2 leads to organ-specific immunological defects.
Here, we analyzed mice with LEC-specific inactivation of Foxc2 to evaluate the impact of defective collLV function on adult homeostasis. Using whole-mount imaging and single-cell transcriptomics, we establish the atlas of adult mesenteric LEC subtypes and show a key role of Foxc2 in the maintenance of their molecular identities and functions. By global analysis of tissues and investigation of gut microbiota and blood metabolites, we also uncover the importance of collLVs in maintaining both intestinal health and long-range communication of the intestine with lungs.
RESULTS
FOXC2 is necessary for the transport function of the adult lymphatic vasculature
Embryonic and postnatal lymphatic vLECs express high levels of FOXC2, whereas lymphangion LECs have intermediate FOXC2 expression [(5, 8, 11) and fig. S1A]. We investigated FOXC2 expression pattern in adult LVs using a whole-mount approach. Unlike in developing lymphatics, FOXC2 protein was very low in lymphangion LECs of large afferent collLVs, whereas FOXC2high cells were detected among valve leaflet and sinus LECs. FOXC2 was also expressed in small collecting/pre-collecting LVs (precollLVs) in a proportion of leaflet and sinus LECs and in scattered lymphangion LECs (Fig. 1A). Adult collLVs thus have a more restricted pattern of FOXC2 expression as compared to embryonic or early postnatal lymphatics (5, 11), suggesting that FOXC2 may be especially important in the maintenance of small collecting and precollLVs.
Fig. 1. Foxc2 loss disrupts transport function of adult lymphatics.
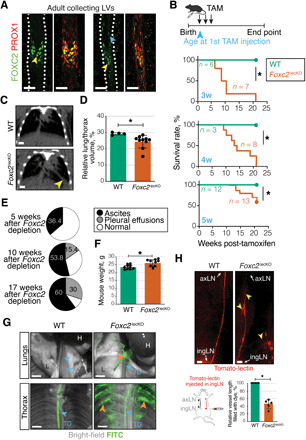
(A) FOXC2 in adult mesenteric LVs. Green, FOXC2; red, PROX1. Scale bars, 50 μm. Yellow arrowheads, FOXC2high vLECs; blue arrowhead, FOXC2+ lymphangion LECs. (B) Kaplan-Meier curve of WT or Foxc2lecKO mice survival. Tamoxifen administration at 3 weeks (*P < 0.001; n = 6 WT; n = 7 Foxc2lecKO), 4 weeks (*P < 0.01; n = 3 WT; n = 8 Foxc2lecKO), and 5 weeks of age (*P < 0.01; n = 12 WT; n = 13 Foxc2lecKO). (C) Computed tomography (CT) image of WT and Foxc2lecKO lungs. Yellow arrowhead, fluid accumulation. Scale bar, 0.25 cm. Tamoxifen administration at 5 weeks, analysis at 22 weeks. (D) Normalized WT and Foxc2lecKO lung volume. *P < 0.05; n = 4 WT; n = 10 Foxc2lecKO. (E) Ascites and pleural effusions at 5 weeks (n = 11), 10 weeks (n = 26), and 17 weeks (n = 10) after Foxc2 inactivation. (F) Increased weight of Foxc2lecKO mice. *P < 0.05; n = 10 WT; n = 9 16-week depleted Foxc2lecKO. (G) Backflow into lungs and intercostal Foxc2lecKO LVs following intraperitoneal injection of fluorescein isothiocyanate (FITC)–dextran (green). Orange arrowheads, backflow; blue arrowheads, thoracic duct (TD), lung (L), and heart (H). Scale bars, 2 mm. (H) Backflow in efferent skin Foxc2lecKO collecting vessel. Tomato-lectin injection into the inguinal LN (ingLN). Yellow arrowheads, backflow/lymph stasis. axLN, axillary LN. Scale bars, 2 mm. *P < 0.0001; n = 7 WT; n = 5 Foxc2lecKO mice. Data are means ± SD.
In accordance with its expression pattern, early postnatal Foxc2 inactivation led to disorganization of major collLVs and was lethal within a few days (5). To study whether FOXC2 plays a role in the maintenance of the adult lymphatic vascular network, we analyzed Foxc2fl/fl;Prox1-CreERT2 (Foxc2lecKO) mice (5), in which LEC-specific loss of Foxc2 was induced at different time points after birth. Analysis of sorted LECs by reverse transcription quantitative polymerase chain reaction (RT-qPCR) and protein staining of tissues confirmed efficient depletion of Foxc2 (fig. S1, B to D). Foxc2 deletion in young adult and adult mice extended the survival compared to newborn Foxc2lecKO pups (5), with some mice surviving over 20 weeks after tamoxifen administration (Fig. 1B and fig. S1, E and F). Chronic Foxc2 deficiency initially led to the development of peritoneal ascites, with the majority of mice displaying increased lung vessel permeability (fig. S1G) and pleural effusion at later time points (Fig. 1, C to E). In agreement with these findings, Foxc2lecKO mice had increased body weight compared to their wild-type (WT) counterparts (Fig. 1F).
To further analyze LV functionality, we injected high–molecular weight fluorescein isothiocyanate (FITC)–dextran intraperitoneally and monitored its appearance in the thoracic cavity. Dextran rapidly accumulated in the thoracic duct of WT animals (Fig. 1G), highlighting the active role of LVs in surveying the peritoneal cavity. However, the dye filled multiple branches of intercostal LVs and was present in lungs of Foxc2lecKO, but not WT, mice, indicating extensive backflow and rerouting of lymph in the thoracic cavity (Fig. 1G). Similarly, lymph backflow was observed in dermal collLVs following intranodal injection of labeled Tomato lectin (Fig. 1H). In summary, FOXC2 chronic deficiency in adult mice leads to an initial phenotype in the peritoneal cavity and results in lethality due to fluid accumulation in the thoracic cavity.
Foxc2 deficiency leads to organ-specific immunological response
Pleural effusion was a late symptom of Foxc2 deficiency, observed the earliest after 10 weeks of Foxc2 inactivation in 5-week-old mice (Fig. 1E). In contrast, one-third of animals developed chyle stasis in the gut and ascites after only 5 weeks of tamoxifen administration (Fig. 1E), indicating that dysfunction of peritoneal LVs precedes other lymphatic vascular beds. To further investigate the accompanying organ changes, we compared the transcriptomes of WT and Foxc2lecKO mesenteries and lungs 10 weeks after Foxc2 deletion. Unbiased pathway analysis using MSigDB database (18) demonstrated a significant enrichment in immunity-related Gene Ontology (GO) terms, such as “adaptive immune response,” “B cell proliferation,” and “response to interferon gamma (IFNγ)” in the mesentery, but not lungs, of Foxc2lecKO mice (Fig. 2A and table S1). The transcriptomics data were further confirmed by flow cytometry analysis of immune cells, which demonstrated an increased proportion of mature B cells, plasma cells, and T cells in the peritoneal, but not pleural washes from mice short-term depleted for Foxc2 compared to WT mice (Fig. 2B and fig. S2A). Within the mature B cell population, we observed an increased number of conventional B2 cells and a tendency for increase in B1a cells (fig. S2, E and F). No differences in the number of peritoneal macrophages, monocytes, neutrophils, or eosinophils were observed (fig. S2, E and F). Lymphocyte accumulation in peritoneal, but not pleural, cavity of Foxc2lecKO mice was also observed at a later time point (16 weeks after Foxc2 depletion) (Fig. 2C). A proportion of long-term depleted Foxc2lecKO mice presented increased blood levels of immunoglobulin M (IgM; fig. S3), in agreement with increased peritoneal B cell number and activation.
Fig. 2. Foxc2 deficiency generates organ-specific immune response.
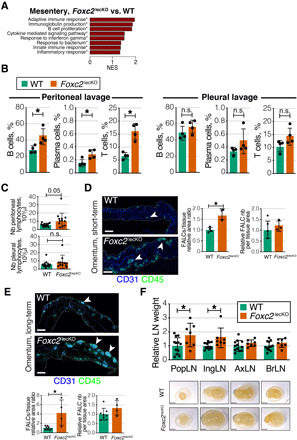
(A) Pathways enriched in transcriptomes of 10-week depleted Foxc2lecKO versus WT mesenteries. n = 3 WT; n = 4 Foxc2lecKO. NES, normalized enrichment score. Asterisks: Adjusted P value ≤ 0.05. (B) Increased percentage of CD45+B220+CD19+ B cells, CD45+B220+CD19+GL7−CD138+ plasma cells, and CD45+CD90+CD19− T cells in the peritoneal, but not pleural cavity of Foxc2lecKO mice. *P ≤ 0.05; n = 4 WT; n = 4 Foxc2lecKO. n.s., not significant. (C) Total lymphocyte number in the peritoneal and pleural cavities of WT and long-term depleted Foxc2lecKO mice. *P < 0.05; n = 14 WT; n = 12 Foxc2lecKO. (D) Increased size, but not number, of omental FALCs after short-term, 5-week Foxc2 depletion. Green, CD45; blue, CD31. Scale bars, 400 μm. *P < 0.05; n = 4 WT; n = 3 Foxc2lecKO. (E) Increased size, but not number, of omental FALCs after long-term Foxc2 depletion. Green, CD45; blue, CD31. Scale bars, 400 μm. *P < 0.01; n = 8 WT; n = 4 Foxc2lecKO. (F) Normalized weights of popliteal (popLN), inguinal (ingLN), axillary (axLN), and brachial (BrLN) LNs 16 weeks after Foxc2 deletion. *P < 0.05; n = 12 WT; n = 7 Foxc2lecKO mice. Scale bars, 2 mm. Data are means ± SD.
Fat-associated lymphoid clusters (FALCs) are tertiary lymphoid tissues associated with visceral fat. They survey body cavities for contaminants and act as first-line responders during cavity inflammation (19, 20). To analyze the status of peritoneal FALCs, we stained the omentum of Foxc2lecKO and WT mice for the global hematopoietic cell marker CD45. Omental FALCs were strongly enlarged in both short- and long-term Foxc2-depleted mice (Fig. 2, D and E, and fig. S2, B and C). Only a tendency toward increased size and number of mesenteric FALCs was observed in Foxc2lecKO mice (fig. S4). This is probably due to a more prominent disruption of omental LVs in Foxc2lecKO mice (see below) and a role of omental FALCs as filters for the peritoneal fluid components (20). Defective lung lymphatic function in Clec2−/− mice is known to generate abundant bronchus-associated lymphoid tissues (21). However, Foxc2lecKO mice developed these lung tertiary lymphoid tissues only at a late stage, which agrees with the delayed onset of pulmonary dysfunction (fig. S2D). Together, these data designate ongoing inflammation in the peritoneal cavity as an early and lasting consequence of lymphatic Foxc2 deficiency.
Defective capillary lymphatic vascular function leads to LN hypoplasia due to reduced delivery of antigens and antigen-presenting cells and absence of lymph flow (22). The arrest of embryonic collLV development is known to prevent LN expansion (23). However, the effects of defective collLV function on adult LNs are not fully understood (24). Therefore, we analyzed the size of LNs in Foxc2lecKO and WT mice. Unexpectedly, inguinal and popliteal LNs were significantly larger in Foxc2lecKO mice, while the size of axillary and brachial LNs was comparable in Foxc2lecKO and WT animals (Fig. 2F). Mesenteric LN (mLN) was not altered in Foxc2lecKO mice (fig. S5). This result may be explained by the fact that the mLN filters the intestinal lymph, and therefore, it is likely intrinsically protected against inflammatory insults such as bacteria (25). Mesenteric lymphadenopathy has been described only in models receiving a high dose of pathogen insult (26). On the other hand, intestinal lymph likely partially bypasses the mLN due to leakage from mesenteric LVs and by redirection of flow. Together, our results indicate that chronic deficiency in Foxc2 impairs adult lymphatic draining function and leads to selective peritoneal inflammation and enlargement of lower body peripheral LNs, thus indicating organ-specific alterations upon FOXC2 loss.
Foxc2 inactivation disrupts hierarchical organization of the lymphatic network in an organ-specific manner
We next investigated the impact of Foxc2 inactivation on the lymphatic vascular network patterning in different organs. In agreement with its expression pattern, Foxc2 depletion led to degeneration and shortening of valve leaflets in skin and mesenteric LVs 16 weeks after tamoxifen administration (Fig. 3, A and B), as determined by staining for the valve leaflet marker laminin-α5 (27). However, the total number of valve sites in both tissues was not altered (fig. S6, A and B), indicating that Foxc2 ablation affected only partly the valve architecture, which is in agreement with its restricted expression pattern in valve leaflet and sinus adult LVs compared to younger animals (Fig. 1A). Lymphangion LECs in both dermal and mesenteric collLVs acquired a more rounded shape, and in a proportion of mice, dilated collectors with oak leaf–shaped LECs were observed (fig. S6, C to E).
Fig. 3. Foxc2 inactivation disrupts hierarchical organization of lymphatic network in an organ-specific manner.
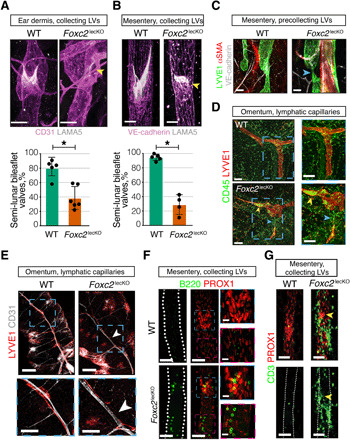
(A) Lymphatic valve leaflet degeneration in dermal vessels of 16-week depleted adult Foxc2lecKO mice ears. Pink, CD31; white, LAMA5. Yellow arrowhead, degenerated valve. Scale bars, 30 μm. Quantification of valves with mature semi-lunar shape in WT and Foxc2lecKO mice. *P < 0.001; n = 5 WT; n = 6 Foxc2lecKO. (B) Lymphatic valve leaflet degeneration in mesenteric vessels of adult Foxc2lecKO mice 16 weeks after tamoxifen administration. Staining for VE-cadherin (pink) and LAMA5 (white). Scale bars, 30 μm. Quantification of valves with mature semi-lunar shape in WT and Foxc2lecKO mice. *P < 0.001; n = 5 WT; n = 4 Foxc2lecKO. (C) Thinning of lymphatic precollecting vessels in mesentery of long-term depleted Foxc2lecKO mice. Green, LYVE1; red, αSMA; white, VE-cadherin. Blue arrow, vessel thinning. Scale bars, 50 μm. (D) Immune cell accumulation in omental lymphatic capillaries of Foxc2lecKO mice. Red, LYVE1; green, CD45. Yellow arrowhead, immune cells; blue arrowhead, LV disruption. Scale bars, 100 μm. (E) Omental lymphatic capillary disruption in Foxc2lecKO mice. Red, LYVE1; white, CD31. n = 6 WT; n = 4 Foxc2lecKO mice. White arrowheads, the point of LV disruption. Scale bars, 200 and 100 μm in magnified pictures. (F) B cells accumulate within LVs of Foxc2lecKO mice. Red, PROX1; green, B220. Blue square, accumulation of B cells within lymphatic valve sinuses. Pink square, accumulation of B cells within lymphangion. Scale bars, 50 and 20 μm for left and right images. (G) T cells around defective mesenteric lymphatic valves in Foxc2lecKO mice. Red, PROX1; green, CD3. Yellow arrowhead, defective valve. Scale bars, 50 μm.
Patterning of capillaries was similar in the skin of WT and Foxc2lecko mice (fig. S6F). In contrast, we observed marked changes in the organization of LVs in the peritoneal cavity. In mesentery and omentum, we observed thinning of precollLVs, which at times were completely regressed, leading to isolated lymphatic capillary “islands,” with this phenotype being especially striking in the omentum of Foxc2lecKO mice (Fig. 3, C to E). This strong phenotype in omental lymphatics may be due to a direct effect of Foxc2 deficiency as, unlike in other organs, we observed expression of FOXC2 in some omental capLECs (fig. S7). Given increased intraperitoneal infiltration of T and B lymphocytes in Foxc2lecKO mice (Fig. 2B), we analyzed the location of immune cells in relation to LVs. Intralymphatic B220+ B and CD3+ T cells were rarely detected in WT mice. However, we observed an accumulation of T and especially B lymphocytes, which frequently formed clusters in the Foxc2lecKO omental and mesenteric lymphatic capillaries (Fig. 3D and fig. S6G). Although dynamic imaging analysis is further necessary, stagnation of immune cells may be a result of decreased lymph flow. Immune cell accumulation was observed even in completely isolated lymphatic islands (Fig. 3D), suggesting that they continue to produce chemoattractive cues. We also observed immune cell aggregates inside and outside of mesenteric collLVs in Foxc2lecKO, but not WT, mice (Fig. 3, F and G, and fig. S6H). A selective intra- and peri-lymphatic distribution of lymphocytes seemed to occur in the knockout (KO) collLVs. Valves and, to a lesser extent, other regions of mesenteric collLVs in Foxc2lecKO mice harbored intralymphatic aggregates of B cells (Fig. 3F and fig. S6, I and K), whereas perivascular T cells accumulated around disrupted valve areas (Fig. 3G and fig. S6J).
Collectively, these data demonstrate that FOXC2 maintains lymphatic valve integrity in all investigated adult collLVs. They also show that loss of Foxc2 leads to disruption of hierarchical lymphatic vascular organization and immune cell trafficking in an organ-specific manner. Notably, in peritoneal cavity tissues, in addition to valve loss, we observed regression of small collecting and precollLVs, resulting in disruption of the communication between lymphatic capillaries and large collLVs. Intriguingly, B cells accumulated within LVs, whereas T cells appeared to be attracted to defective valve areas, thus suggesting distinct effects of Foxc2 deficiency on these two types of lymphocytes.
Intestinal lymphangiectasia, disruption of intestinal epithelial barrier, and gut microbiota translocation in Foxc2lecKO mice
One puzzling macroscopic phenotype of Foxc2lecKO mice was the selective enlargement of inguinal and popliteal LNs (Fig. 2F). Further analyses confirmed increased inguinal and popliteal LN cellularity (Fig. 4A) and revealed LN hypertrophy with relatively higher proportion of B cells (Fig. 4A and fig. S8, A to D), while no differences were observed in the immune populations of spleen and bone marrow of Foxc2lecKO and WT mice (fig. S8B). Together, these results pointed to persistent activation of selected peripheral LNs in Foxc2lecKO mice.
Fig. 4. Intestinal lymphangiectasia and gut epithelial barrier disruption in Foxc2lecKO mice.
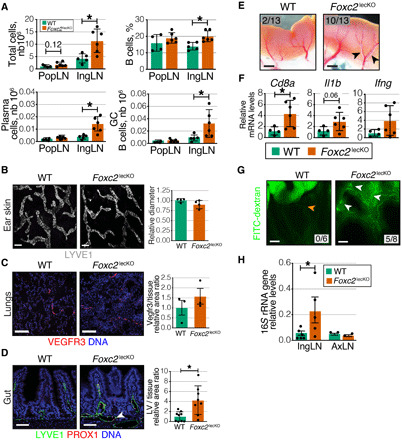
(A) Increased average cell count and frequency of B cells, plasma cells number, and germinal center (GC) B cells in popliteal (popLN) and inguinal (ingLN) LNs of 16-week depleted Foxc2lecKO mice. *P ≤ 0.05; popLN, n = 4 WT, n = 6 Foxc2lecKO; ingLN, n = 5 WT, n = 6 Foxc2lecKO. (B) Normal ear lymphatic capillary organization in WT and 16-week depleted Foxc2lecKO mice. White, LYVE1. Capillary diameter normalized to WT mice. n.s.; n = 4 WT, n = 4 Foxc2lecKO. Scale bars, 50 μm. (C) Comparable lung LV organization in WT and 16-week depleted Foxc2lecKO mice. Red, vascular endothelial growth factor receptor 3 (VEGFR3); blue, 4′,6-diamidino-2-phenylindole (DAPI). VEGFR3+ area normalized to tissue area and WT values. n.s. n = 4 WT, n = 4 Foxc2lecKO. Scale bars, 400 μm. (D) Intestinal lymphangiectasia in 16-week depleted Foxc2lecKO mice. Green, LYVE1; red, PROX1; blue, DAPI. Scale bars, 50 μm. LV area normalized to tissue area and WT values. *P < 0.01; n = 9 WT; n = 8 Foxc2lecKO. (E) Chyle stasis in 16-week depleted Foxc2lecKO mice. Scale bars, 1 mm. (F) Increased CD8a in Foxc2lecKO intestine. RT-qPCR for the indicated transcripts. *P < 0.05; n = 4 WT, n = 6 Foxc2lecKO. (G) Gavaged FITC-dextran fills mesenteric LVs in Foxc2lecKO mice (white arrow), but not in WT mice (orange arrow). n = 6 WT, n = 8 Foxc2lecKO mice. Scale bars, 0.5 cm. (H) Bacterial translocation to Foxc2lecKO LNs. 16S rRNA levels in ingLNs and axLNs of WT and Foxc2lecKO mice, normalized to gut values. *P < 0.05; n = 6 WT, n = 5 Foxc2lecKO; axLN, n.s., n = 2 WT, n = 2 Foxc2lecKO.
Although inguinal and popliteal LNs do not drain the intestine or peritoneal cavity, we reasoned that loss of lymphatic valves reroutes intestinal lymph, carrying bacteria or bacteria-derived products to peripheral LNs. To study this question, we first analyzed the impact of Foxc2 inactivation on intestinal LVs. The intestinal lymphatic network comprises lacteals, the lymphatic capillaries that transport dietary lipids from intestinal villi, and submucosal intestinal LVs, draining intestinal lymph to mesenteric collLVs (28). Lacteals, but not lung or skin lymphatic capillaries, were significantly enlarged in Foxc2lecKO mice, and the intestinal submucosal lymphatic vascular network was dilated and filled with chyle, indicating lymph stasis (Fig. 4, B to E). Moreover, transcripts for T cell marker Cd8a and, in a proportion of mice, for pro-inflammatory cytokines Il1b and Ifng were up-regulated in the intestine of Foxc2lecKO mice (Fig. 4F), indicating increased inflammation.
The intestinal epithelial barrier allows uptake of nutrients and water while preventing the entry of microorganisms and toxins. Inflammation is a major trigger for increased intestinal permeability and development of infections, inflammatory bowel disease, and food allergies [reviewed in (25)]. To study the status of the intestinal epithelial barrier, we gavaged Foxc2lecKO and WT mice with low–molecular weight FITC-dextran and assessed mesenteric LVs 1 hour later. As expected, the fluorescent signal was confined to the intestinal lumen of WT mice, demonstrating an intact intestinal barrier (Fig. 4G). However, FITC-dextran accumulated in mesenteric LVs and the surrounding fat in five of eight Foxc2lecKO mice (Fig. 4G), indicating intestinal barrier disruption. To study this in more detail, we analyzed the organization of the intestinal epithelial layer in Foxc2lecKO and WT mice by staining for the junctional proteins E-cadherin and ZO-1. We observed focal areas of thin intestinal epithelium in the gut of Foxc2lecKO mice (fig. S9A). Such phenotype was described previously in models of intestinal injury and represents the sites of epithelial loss covered by the neighboring enterocytes in the process of ongoing mucosal repair (29). These areas lacked ZO-1 expression, indicating locally compromised epithelial barrier (fig. S9B). We also assessed the expression of the antimicrobial peptide Reg3β, which is up-regulated upon gut bacteria colonization and in response to intestinal inflammation or infection to protect the epithelial mucosal surface (30). Increased levels of Reg3β were observed in the crypts of Foxc2lecKO mice (fig. S9C), indicating that crypts of Foxc2lecKO mice were subjected to increased inflammation.
To investigate the underlying mechanism, we assessed the effects of intestinal lymph on the junctional integrity of intestinal epithelial cells in vitro. We incubated transformed mouse intestinal epithelial cells (31) with freshly collected mouse intestinal lymph, and we assessed the integrity of the cell-cell junctions by staining for E-cadherin and ZO-1. We used dextran sulfate sodium (DSS) known to directly cause intestinal epithelial damage (32) as a positive control. We found that after 16 hours of incubation and similar to DSS, addition of lymph induced loss of cell-to-cell contact, internalization of E-cadherin, and focal loss of ZO-1 staining (fig. S10, A and B). This result indicates that the prolonged contact with intestinal lymph may directly disrupt the intestinal barrier integrity.
A disrupted intestinal barrier can lead to translocation of gut bacteria; therefore, we evaluated the presence of bacterial DNA in the LNs of Foxc2lecKO or WT mice by PCR for the 16S ribosomal RNA (rRNA) gene. Bacterial DNA was detected in inguinal, but not axillary, LNs in Foxc2lecKO mice, while WT LNs were sterile (Fig. 4H). Analysis of B cell populations in the intestinal lamina propria revealed an increased frequency of IgA+ plasma cells in Foxc2lecKO mice (fig. S11, A and B), suggesting an augmented humoral response to chronic bacterial exposure. Collectively, these results indicate that loss of lymphatic Foxc2 impairs normal intestinal barrier function and establishes an abnormal communication of intestine with peripheral LNs.
Foxc2 deficiency alters the gut microbiota and blood metabolome
We next asked whether dysfunctional peritoneal and intestinal lymphatics affect the intestinal microbiota. By performing 16S rRNA gene sequencing analysis of fecal samples, we observed that Foxc2 inactivation increased the proportion of mice harboring Epsilonbacteraeota (Fig. 5A and fig. S12, A and B). Within the phylum Epsilonbacteraeota, Helicobacter hepaticus was significantly more abundant in Foxc2lecKO mice (Fig. 5B), indicating that disrupted lymphatic function induces gut dysbiosis. H. hepaticus is a well-studied bacterium known to be implicated in lower bowel inflammation (33), thus indicating that lymphatic vascular dysfunction promotes expansion of colitogenic bacteria.
Fig. 5. Foxc2 loss modifies gut microbiome and increases circulating levels of purine metabolites.
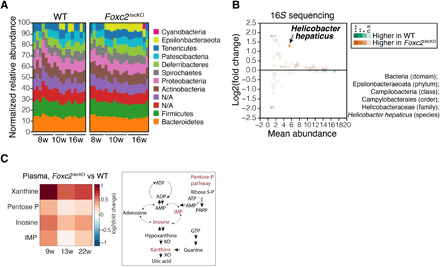
(A) A higher proportion of Foxc2lecKO mice harbor members of the phylum Epsilonbacteraeota after 2 months of Foxc2 depletion. Stacked bar plots: relative bacterial amplicon sequence variant (ASV) abundance at the phylum level 8 weeks (n = 7 WT, n = 6 Foxc2lecKO), 10 weeks (n = 4 WT, n = 8 Foxc2lecKO), and 16 weeks (n = 7 WT, n = 7 Foxc2lecKO) after Foxc2 deletion. (B) Increased abundance of H. hepaticus in Foxc2lecKO mice. MA plot: The mean relative bacterial species abundance versus the log-transformed fold change of bacterial species relative abundance in the feces of WT and 8- to 16-week depleted Foxc2lecKO mice. Circles, species with calculated fold change abundance; triangles, species with infinite fold difference due to zero abundance. Color intensity proportional to the relative species abundance. n = 18 WT, n = 21 Foxc2lecKO. (C) Increased blood xanthine levels in Foxc2lecKO mice. Heatmap: Fold change of induced (red) or reduced (blue) metabolites in Foxc2lecKO versus WT mice. n = 6 WT, n = 7 Foxc2lecKO mice. Metabolites ordered by m/z (mass/charge ratio), adjusted P < 0.05. On the right, purine metabolic pathway is depicted.
To study the systemic impact of chronic Foxc2 deficiency, we analyzed blood metabolites of Foxc2lecKO and WT mice using untargeted metabolomics. We observed higher xanthine levels in the blood of Foxc2lecKO mice compared to their WT counterparts. Inosine and inosine monophosphate (IMP), precursors of purine biosynthesis pathways, were also significantly up-regulated in Foxc2lecKO mice (Fig. 5C and table S2). Together, these results show that chronic Foxc2 deficiency and impaired lymph transport set in motion a cascade of both local and systemic pathological changes that affect whole-body homeostasis.
Microbiota depletion rescues the intestinal and systemic phenotypes of Foxc2lecKO mice
To determine the contribution of the altered gut microbiome to the Foxc2lecKO mice phenotype, we depleted gut microbiota by oral broad-spectrum antibiotic treatment 8 weeks after Foxc2 inactivation. 16S rRNA gene sequencing confirmed the depletion of the majority of bacteria phyla (fig. S12C). Notably, treatment with antibiotics significantly improved Foxc2lecKO mouse survival (Fig. 6A). Antibiotic treatment reduced the number of mice developing pleural effusion and submucosal chyle stasis (Fig. 6, B and C). Further analyses established that oral antibiotics also reduced intestinal dilation of capillary LVs (lymphangiectasia) and enlargement of omental FALCs as well as inguinal and popliteal LNs (Fig. 6, D to F). Last, analysis of plasma metabolites demonstrated reduction of xanthine, inosine, and IMP levels upon antibiotic treatment (fig. S12D). As previously described (34, 35), levels of primary bile acids such as cholate and taurocholic acid, as well as cholesterol were increased in plasma upon antibiotic treatment. Because sphingolipids are known to be produced by microbiota (36), their absence may lead to the observed reduced levels of plasma ceramide in antibiotic-treated Foxc2lecKO mice (fig. S12D).
Fig. 6. Microbiota depletion rescues the phenotype of Foxc2lecKO mice.
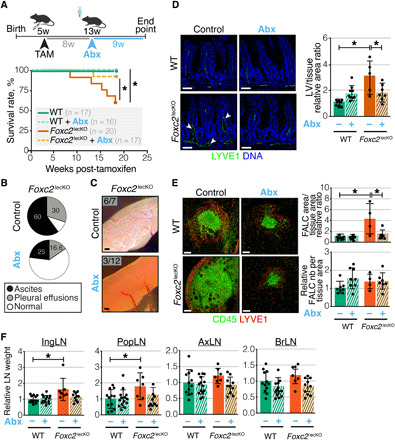
(A) Antibiotics improve Foxc2lecKO mice survival. Treatment scheme and Kaplan-Meier survival curve of untreated and antibiotic-treated (Abx) WT mice and Foxc2lecKO mice *P < 0.05; n = 17 WT, n = 16 WT + Abx, n = 20 Foxc2lecKO, n = 17 Foxc2lecKO + Abx. (B) Ascites and pleural effusions in untreated and Abx-treated Foxc2lecKO mice. n = 10 Foxc2lecKO, n = 12 Foxc2lecKO + Abx. (C) Microbiota depletion rescues chyle stasis of Foxc2lecKO mice. Bright-field pictures, n = 7 Foxc2lecKO, n = 12 Foxc2lecKO + Abx. Scale bars, 1 mm. (D) Intestinal lymphangiectasia in Foxc2lecKO mice. Green, LYVE1; blue, DAPI. Scale bars, 50 μm. LV area normalized to tissue area. *P < 0.01; n = 9 WT, n = 9 WT + Abx, n = 5 Foxc2lecKO, n = 7 Foxc2lecKO + Abx. (E) Microbiota depletion abrogates omental FALC enlargement of Foxc2lecKO mice. Green, CD45; red, LYVE1. Scale bars, 100 μm. FALC size and number normalized to tissue area in WT and Foxc2lecKO mice. *P < 0.001 WT versus Foxc2lecKO; *P < 0.01 Foxc2lecKO versus Foxc2lecKO + Abx, n = 8 WT; n = 8 WT + Abx, n = 4 Foxc2lecKO; n = 7 Foxc2lecKO + Abx. (F) Microbiota depletion abrogates enlargement of peripheral Foxc2lecKO LNs. *P < 0.01 inguinal LN (ingLN) in WT versus Foxc2lecKO; *P < 0.05, popliteal LN (popLN) in WT versus Foxc2lecKO, n = 12 WT, n = 14 WT + Abx, n = 7 Foxc2lecKO, n = 10 Foxc2lecKO + Abx. Data are means ± SD.
We also analyzed whether the cell-intrinsic effects of Foxc2 depletion on lymphatic vasculature were recovered upon antibiotic treatment. Because defective valves and rounded lymphangion LECs were still observed in antibiotic-treated Foxc2lecKO mice (fig. S12, E and F), this indicates that the beneficial effects of antibiotic treatment must be indirect and are due to either reduced infection or inflammatory status. Loss of Foxc2 did not lead to direct lung bacterial transfer and infection (fig. S13, A and B). We next analyzed the levels of inflammatory cytokines linked to the development of vascular leakage and lung edema, such as interleukin 6 (IL6), interferon γ (IFNγ), and tumor necrosis factor α (TNFα) (37) in antibiotic-treated and control WT and Foxc2lecKO mice. Although there was no difference in the levels of these cytokines in the blood of control WT and Foxc2lecKO mice, we found much lower levels of IL6, IFNγ, and TNFα in the blood of antibiotic-treated WT and Foxc2lecKO mice (fig. S13C). To answer how the same levels of inflammatory cytokines can be detrimental in Foxc2lecKO, but not WT, mice, we analyzed the circulating levels of angiopoietin-2 (ANGPT2) in WT and Foxc2lecKO animals. We focused on ANGPT2 because it is one of the most strongly induced transcripts in LECs from Foxc2lecKO mice in our single-cell RNA sequencing (scRNA-seq) data (see below). In addition, previous studies identified ANGPT2 as a critical factor sensitizing blood endothelial cell (BEC) responses to inflammatory stimuli such as TNFα and lung edema (37, 38). Loss of Foxc2 resulted in significantly increased circulating levels of ANGPT2 (fig. S13D) that were decreased upon antibiotic treatment. On the basis of these results, we propose that antibiotic treatment promotes the survival of Foxc2lecKO mice by decreasing the combined effects of circulating ANGPT2 and inflammatory cytokines on systemic inflammation.
Collectively, these data establish the microbiome as being a major contributor to the pathophysiological phenotype of adult Foxc2lecKO mice and highlight the importance of environmental factors in the regulation of organ-specific lymphatic and immunological functions.
Single-cell transcriptome analysis defines molecular features of adult LEC functional subsets
To understand the role of Foxc2 at the molecular level, we analyzed mesenteric LECs using scRNA-seq 3 weeks after Foxc2 inactivation and before development of overt lymphatic phenotypes. We sorted CD45− Pdpn+ CD31+ LECs with low stringency gating to avoid loss of a potentially altered LEC population upon Foxc2 depletion (Fig. 7A and fig. S14A). Uniform manifold approximation and projection (UMAP) visualization and clustering revealed the presence of LECs, BECs, fibroblasts, SMC/pericytes, and mesothelial cells (fig. S14, B and E). Analysis of differentially expressed genes (DEGs) between Foxc2lecKO and WT cells confirmed that the transcriptional changes were highest in LECs, which also demonstrated significant reduction in Foxc2 expression levels (fig. S14, C and D).
Fig. 7. Single-cell transcriptome profiling defines molecular and functional subsets of adult LECs.
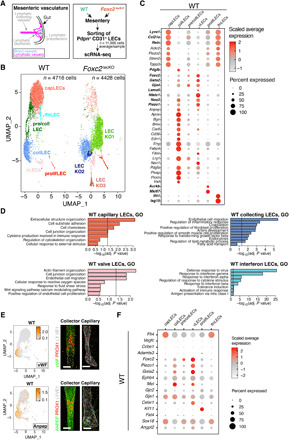
(A) Workflow of the scRNA-seq experiment. Cells were sorted from mesenteries of 6-week-old WT and Foxc2lecKO mice 3 weeks after Foxc2 depletion. n = 3 per genotype. (B) UMAP plot of WT and Foxc2lecKO LECs. Six major clusters of LECs in WT mice and nine major clusters in Foxc2lecKO mice were identified. (C) Dot plot of known markers (in bold) and selected new genes for WT LEC clusters: capillary (capLECs), collecting (collLECs), precollecting and collecting (pre/collLECs), valve (vLECs), proliferative (prolifLECs), and IFN (IfnLECs). The color code indicates scaled average expression level in each cluster, and the dot size indicates the percent of cells in each cluster expressing the given gene. (D) Bar plots of a subset of GO gene sets that were overrepresented among genes up-regulated in capLEC, collLEC, vLEC, and IfnLEC clusters of WT mice. (E) Expression level per cell (ln[normalized counts + 1]) of the new WT LEC markers vWF and Anpep overlaid on the UMAP and staining of adult mesenteric lymphatic collLVs and capillaries showing expression of vWF or ANPEP (green) and PROX1 (red). Scale bars, 50 μm. (F) Dot plot of expression of hereditary lymphedema–related genes in adult LEC subsets of WT mice. The color code indicates scaled average expression level in each cluster, and the dot size indicates the percent of cells in each cluster expressing the given gene.
We next analyzed in more detail WT and KO LEC populations expressing transcripts for CD31, Flt4, and Prox1 (Fig. 7B). On the basis of previously described markers (4, 5), we identified four major clusters of WT LECs: (i) capillary LEC cluster (Lyve1+, Ccl21a+, Reln+, Foxc2neg capLECs), (ii) collecting LEC cluster (Lyve1neg, Ccl21aneg, Relnneg, Foxc2low collLECs), (iii) valve LEC cluster (Lama5+, Foxc2+, Gata2high, Nfatc1high, Gja4high, Ccl21aneg, Lyve1neg vLECs), and (iv) pre-collecting LEC/small collecting LEC cluster (divided into LECs that are Ccl21a+, Lyve1+, Foxc2neg, and LECs that are Foxc2+, Ccl21aneg, Lyve1neg, pre/collLECs). In addition, we observed a small cluster of proliferating Aurkb+, Mki67+ LECs (prolifLECs) and a capLEC cluster expressing IFN-induced transcripts, such as Ifit1 and Isg15 (IfnLECs; Fig. 7C and table S3).
A high proportion of genes with subset-specific expression were not previously characterized as markers of the respective LEC subpopulations. For example, capLECs exposed to low-magnitude transmural flow expressed the light-touch mechanosensitive ion channel Piezo2, whereas the related Piezo1, which senses fluid shear stress, was enriched in collLECs and vLECs, which experience higher shear stress in vivo (Fig. 7C) (39). CapLECs expressed high levels of regulators of microtubule dynamics Stmn2 and Tppp3 and the decoy receptor for inflammatory chemokines Ackr2, highlighting the importance of microtubule cytoskeleton and regulated chemokine signaling in initial lymphatics (Fig. 7C).
CollLECs were enriched in transcripts for secreted growth factors regulating SMC/pericyte and fibroblast proliferation and recruitment (Tgfa, Pdgfb, Pdgfa, Bmp4, and Wnt2), transforming growth factor–β (TGF-β) co-receptors and TGF-β target genes (Lrg1, Eng, Bgn, and Fn1), transcripts encoding regulators of vascular tone (Edn1 and eNOS), and peptide processing (Anpep) (Fig. 7C and table S3). Unexpectedly, collLECs expressed BEC markers such as the plasmalemma vesicle–associated protein Plvap and Bmx and transcripts associated with the regulation of blood coagulation such as Thbd, Procr, Vwf, and Plat (Fig. 7C and table S3). Transcripts related to ion homeostasis (Car8, Clca3a1, and Slco2a1) and to lipid metabolism and fatty acid transport (ApoE, Ccdc3, Cd36, and Fabp4) were also enriched, pointing to additional new functions of collLECs (Fig. 7C and table S3).
Analysis of GO biological processes revealed enrichment in specific and meaningful biological functions associated with each subset, such as regulation of SMC proliferation and TGF-β signaling for collLECs or IFN signaling for some capLECs (Fig. 7D and table S4). To validate these scRNA-seq results, we performed staining for selected new markers, such as vWF and ANPEP, which confirmed their expression in collLECs (Fig. 7E).
To evaluate the clinical relevance of our scRNA-seq atlas, we examined the expression of genes, mutated in different subtypes of human primary lymphedemas (Fig. 7F) (16). High expression of Flt4 in capLECs is consistent with lymphatic capillary hypoplasia in Vegfr3-haploinsufficient mice and in Milroy disease patients (16). Selective expression of Met in collLECs and vLECs predicts defects of collLVs in patients with mutations in MET and its ligand hepatocyte growth factor (HGF) (40). Collectively, our single-cell transcriptomic analysis identified at least five adult LEC subsets and revealed their distinct and biologically relevant molecular profiles.
Foxc2 loss modifies the molecular identity of the entire lymphatic vascular network
We next compared Foxc2lecKO and WT mesenteric LECs at the single-cell transcriptome level to determine whether Foxc2 depletion altered cellular identity. Intriguingly, the Foxc2high valve and precollLECs clusters were almost absent in Foxc2lecKO samples, underscoring the essential role of Foxc2 in their maintenance (Fig. 8A). This finding agrees with our previous observations, demonstrating rapid selective clearance of Foxc2-deficient valve cells in early postnatal LVs (5). Unexpectedly, only few cells of the capLEC cluster remained, while a new KO LEC cluster termed “KO1” emerged located between capLECs and collLECs (Fig. 7B). The collecting and remaining KO vLEC clusters were also shifted within their respective transcriptional space (Fig. 7B). Persistence of the vLEC cluster agrees with partial conservation of the valve architecture in adult Foxc2lecKO mesenteric LVs (Fig. 3B). Staining for PROX1, which is highly expressed in all vLECs (8, 11), revealed rings of PROX1high cells in Foxc2lecKO lymphangions (fig. S1D), reminiscent of early stages of valve formation (11).
Fig. 8. Foxc2 loss modifies the molecular identity of the entire lymphatic vascular network.
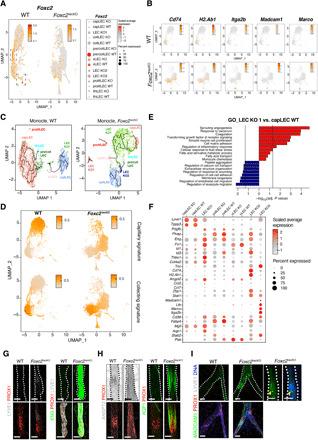
(A) Foxc2 inactivation eliminates Foxc2high LECs. Foxc2 expression per cell (ln[normalized counts + 1]) overlaid on UMAP plot, split by genotype. Square area: Foxc2 expression in WT LECs. Color code: Scaled average expression level in each cluster; the dot size denotes the percent of Foxc2+ cells in each cluster. (B) New Foxc2lecKO immune-related LEC subsets. Expression level per cell (ln[normalized counts + 1]) of the indicated transcripts overlaid on WT or Foxc2lecKO UMAP plots. (C) Trajectory analysis of WT and Foxc2lecKO LECs. UMAP was generated independently for each genotype using Monocle3. (D) capLEC and collLEC phenotypes converge upon loss of Foxc2. Module score per cell overlaid on UMAP plot, split by genotype. Scores calculated using WT collLECs versus capLECs up- or down-regulated transcripts. Higher score indicates that more signature genes are expressed in a cell. (E) GO terms overrepresented among up-regulated (red bars) or down-regulated (blue bars) LEC cluster KO1 transcripts versus WT capLECs. Vertical lines: Adjusted P value = 0.05. (F) Dot plot of markers for the indicated LEC subsets in WT and Foxc2lecKO LECs. Color code: Scaled average expression level in each cluster; the dot size denotes the percent of cells in each cluster expressing the given gene. (G) LYVE1 and ENG are reexpressed in Foxc2lecKO collLVs (collecting LVs) or capLVs (capillary LVs). Scale bars, 50 μm. (H) Increased ANGPT2 (white) and AQP1 (green) in PROX1+ (red) Foxc2lecKO LVs. Scale bars, 50 μm. (I) LN LEC cells are located in Foxc2lecKO collLVs. Green, MADCAM1; red, PROX1; white, LYVE1; blue, DNA. Right: Accumulating DAPI+ (blue) cells inside the MADCAM1+ (green) LVs. Scale bars, 50 μm.
We also observed the emergence of two additional new clusters with immune-related phenotypes: LEC cluster “KO2,” characterized by expression of antigen presentation–related transcripts such as Cd74 and H2Ab1, and LEC cluster “KO3” with LN LEC–specific transcripts such as Itga2b, Madcam1, Ltb, and Marco (Fig. 8B and table S5) (12).
Foxc2 loss induces LECs toward an inflammatory, profibrotic, and metabolically active phenotype
To model the relationships between different WT and KO LEC populations, we analyzed their transcriptional trajectories using Monocle 3 (41, 42). The analysis of WT LECs indicated that two major populations, collLECs and capLECs, formed discrete networks, with little interconversion between the two phenotypes (Fig. 8C). In contrast, valve and collLECs were connected by a linear trajectory, consistent with their physical proximity (Fig. 8C).
Trajectory analysis of Foxc2lecKO LECs revealed a transcriptional proximity and direct link of the remaining capLEC cluster and the new cluster LEC KO1, supporting the notion that KO1 LECs originate from capLECs. Analyses of collLEC and capLEC signatures derived from the WT transcriptomes demonstrated that KO collLECs acquired some capLEC traits (Fig. 8D). Unexpectedly, capillary-derived KO1 LECs gained the expression of multiple collLEC markers, such as Pdgfb, Plvap, and Eng (Fig. 8F). Staining for lymphatic capillary marker LYVE1 and ENG confirmed these observations (Fig. 8G). Together, these results indicate that Foxc2 inactivation leads to emergence of convergent cell populations with mixed capLEC and collLEC identities.
Analysis of GO biological processes enriched in Foxc2lecKO versus WT LECs revealed activation of pathways related to inflammation, TGF-β signaling, and lipid metabolism (Fig. 8E and fig. S15A), indicating the acquisition of inflammatory, pro-fibrotic, and metabolically active phenotypes. Of note, the GO term “response to bacterium” was among the top enriched pathways, consistent with the bacterial translocation we observed in Foxc2lecKO animals (Fig. 8E and fig. S15A).
Relevant transcriptional changes were observed in all Foxc2lecKO subsets (table S6). Overall, in comparison to WT capLECs, the KO1 LEC cluster was characterized by higher expression of transcripts for extracellular matrix components (Fn1, Col4a2, and Tnc), TGF-β co-receptors and target genes (Eng, Fn1, Id1, Id3, and Thbs1), SMC chemotactic growth factor Pdgfb, coagulation cascade (Plat and Procr), fatty acid transport and metabolism regulators (Cd36, Fabp4, and Mgll), and antagonistic ligand of Tie receptors Angpt2 (Fig. 8, F to H, fig. S15B) (43). The increased expression of Angpt2 in KO LECs validates our finding of increased levels of this molecule in the blood of Foxc2lecKO mice (fig. S13D) and its potential role in provoking blood vessel permeability, therefore contributing to animal demise. KO1 LECs, but not WT LECs, expressed transcripts encoding monocyte chemoattractants Ccl2 and Ccl7 (Fig. 8F). There was also a higher proportion of prolifLECs (1.3% of Foxc2lecKO versus 0.3% of WT LECs; fig. S16A), and a subset of Foxc2-deficient LECs expressed YAP/TAZ target genes Ctgf and Ankrd1, thus confirming previous observations of increased TAZ signaling and proliferation in FOXC2-deficient LECs (5).
Foxc2 inactivation resulted in the emergence of two new immune-related clusters KO2 and KO3 (Fig. 7B). KO2 LECs expressed high levels of antigen presentation–related Cd74 and H2ab1, the transcriptional regulator of IFN response Stat1, and the cytosolic viral sensor Z-DNA–binding protein 1 (Zbp1) (Fig. 8, B and F). KO3 LECs expressed transcripts characteristic of LN LECs (Fig. 8, B and F). LN-like LECs could originate from mLN contamination during sample collection; therefore, we investigated their spatial localization by staining for MADCAM1. We observed well-demarcated areas of MADCAM1high LECs within the collLVs of Foxc2lecKO mice (Fig. 8I). Unexpectedly, these areas were associated with intra-lymphatic immune cell aggregates, suggesting cross-talk of collLECs with stagnating lymphocytes in the generation of LN LEC identity.
To establish whether the transcriptional changes observed in Foxc2-deficient LECs were recapitulated in another model that displays defective lymphatic valves and flow, we analyzed Cx37−/− mice (11). Although both models shared similar primary defects (defective LV valves and lymph flow), Cx37−/− mice displayed normal life span and did not develop pleural effusion or peritoneal ascites and had normal body weight, contrary to Foxc2lecKO mice (fig. S17, A to C). We studied whether transcriptional changes observed in LECs upon Foxc2 depletion were also induced in mesenteric LECs of Cx37−/− mice. Representative genes strongly up-regulated upon Foxc2 loss were not up-regulated in Cx37−/− LECs compared to WT LECs (fig. S17, D to F).
In summary, single-cell transcriptome profiling of Foxc2lecKO LECs demonstrated that continuous Foxc2 expression is crucial for maintaining their specialized phenotypes and revealed an intriguing inflammatory and pro-fibrotic phenotype together with the emergence of new subsets of cells with LN and immune response traits (fig. S18).
DISCUSSION
Major progress has been made in understanding LEC developmental origins and embryonic lymphangiogenesis (4). However, less attention has been paid to the importance of the maintenance mechanisms of adult LVs. An important current challenge is to understand such mechanisms and the role of LVs in coordinating the interactions between different organs to sustain the organism’s physiological health. Here, we analyzed a mouse model with LEC-specific inactivation of Foxc2 presenting multiple systemic alterations, and we report a broad analysis of the role of collLV functions in adult homeostasis.
Organ-specific lymphatic vascular defects by loss of Foxc2 in adulthood
FOXC2 plays a crucial role in specialization of capillaries and collLVs during embryonic and postnatal development (5, 8, 11, 15). Our current results extend these observations by showing that continuous expression of FOXC2 is necessary for the maintenance of lymphatic valves and directional lymph flow in all vascular beds throughout life. We also report markedly organ-specific physiological repercussions of systemic LEC-specific Foxc2 deficiency, namely, in the peritoneal cavity and intestine followed by pleural effusion and animal demise (fig. S19), while the cutaneous lymphatic vascular network was less affected. In particular, Foxc2lecKO mice develop expansion of FALCs and selected peripheral LNs, as a result of ongoing peritoneal inflammation and rerouting of intestinal lymph. LDS patients develop peritoneal lymphoid aggregates (17) and have enlarged inguinal LNs (ingLNs) (44), indicating that loss of intestinal lymph compartmentalization is also a part of human pathology.
Our observations highlight the distinct needs and impact of the microenvironment on the maintenance of adult organ-specific lymphatic vasculatures. Maintenance of adult intestinal lacteals, but not dermal capillaries, requires continuous vascular endothelial growth factor receptor 3 (VEGFR3) stimulation (45, 46). In addition, lack of the adherens junction protein VE-cadherin in adult LECs leads to fragmentation of intestinal lacteals and mesenteric, but not dermal, collLVs (47). Higher lability of intestinal/peritoneal LVs could be due to their distinct developmental origin, which potentially programs lower expression of junctional LEC components (47), higher microenvironmental levels of lymphangiogenic factors, and/or exposure to organ-specific immune cells or extracellular matrix components. Also, intestinal lymph transports dietary fat and associated bacterial lipopolysaccharides (LPS) and fat-derived free fatty acids have been shown to increase lymphatic vascular permeability (48). Thus, developmentally programmed features of organ-specific lymphatics and microenvironmental cues must have combinatorial impact on the different outcome of the lack of lymphatic genes in different body compartments.
Role of collLVs in organ-specific lymph compartmentalization and organ-to-organ communication
On the organ level, lymphatic Foxc2 deficiency led to a compromised intestinal epithelial barrier, dysbiosis, and expansion of colitogenic bacteria in the gut, increased translocation of gut microbiota to peripheral LNs, and changes in systemic blood metabolites. Moreover, the circulating levels of the antagonistic Tie2 ligand ANGPT2 were increased upon Foxc2 loss. The absence of microbiota rendered Foxc2lecKO mice less susceptible to the loss of FOXC2. Depletion of gut microbiota rescued both local symptoms of intestinal and peritoneal cavity LV dysfunction and development of pleural effusion, resulting in improved overall survival. Antibiotics strongly reduced the levels of inflammatory cytokines IL6, IFNγ, and TNFα (fig. S13C), which were previously shown to synergize with ANGPT2 in the induction of blood vessel inflammation. We therefore envision that a combination of defective lymphatics and inflammatory status leads to lung dysfunction and, finally, animal demise, which can be rescued by antibiotics administration.
Loss of intestinal lymphatic capillary function in Dll4- or Vegfr3-deficient mice reduced dietary lipid absorption but did not impair intestinal health or result in pulmonary dysfunction (45, 46). These results highlight the central and active role of FOXC2 in intestinal and peritoneal collLVs and, in particular valves, in the maintenance of physiological health of the entire organism, notably through compartmentalization of intestinal lymph and communication with other organs, such as lungs.
Dysfunctional collLVs affect microbiota and blood metabolites
A pioneering study showed that acute intestinal infection with Yersinia pseudotuberculosis has long-term effects on the gut, inducing inflammation that leads to leaky mesenteric lymphatics. This prevents dendritic cell trafficking to mLNs, leading to compromised immune mucosal tolerance (26). Our data show that dysfunctional collecting lymphatics provoke immune dysregulation, which may facilitate the alteration and translocation of microbiota, and therefore initiate a feedback loop, further reinforcing lymphatic vascular dysfunction upon Foxc2 loss.
Intestinal infection has been suggested to be a driver of Crohn’s disease pathogenesis, and similar postinfection-like symptoms are observed in such patients (49). Long-term intestinal collLV dysfunction could therefore be a driver of intestinal bowel disease. Moreover, the mesentery of Crohn’s disease patients, where the flow of lymph is known to be altered, has been shown to harbor tertiary lymphoid organs (50).
Intestinal inflammation has been shown previously to disrupt the intestinal barrier and generate dysbiosis through a plethora of effects, such as increased secretion of cytokines, chemokines, and lipid-derived mediators (e.g., prostaglandins and leukotrienes) that attract and activate macrophages and neutrophils and alter the gut oxidative and metabolic environment affecting commensal growth (51). Although we do not know what causes the increased abundance of H. hepaticus in Foxc2-deficient mice, based on our data, we propose that chylomicron-rich lymph stagnation may represent an initial event leading to focal intestinal barrier damage. This damage would lead to increased intestinal inflammation and generate a further vicious cycle that could promote bacterial dysbiosis. A reverse transport of interstitial protein- and lipid-rich fluid to the gut lumen, which may alter the microbiota composition, is another potential contributing factor. Moreover, future studies will be needed to decipher whether an impaired antigen presentation response could be implicated in the increased abundance of H. hepaticus.
Changes in composition and function of the gut microbiota are closely related to the progression of metabolic and chronic diseases (52). Here, lymphatic loss of Foxc2 led to alteration of purine metabolism, as we detected elevated levels of the final product of purine degradation xanthine in the blood of Foxc2lecKO mice. This rise was prevented upon depletion of microbiota. The source and pathophysiological significance of higher blood xanthine in our model needs to be investigated in the future. Of interest, pathological stress–induced CD4+ cells are a major source of plasma xanthine in mice (53). Thus, lymphatic Foxc2 deficiency could potentially affect peripheral CD4+ cells to produce high levels of xanthine. Another potential driver of increased purine metabolites in the blood of Foxc2lecKO mice could be gut dysbiosis. H. hepaticus infection in immunodeficient mice has been previously related to changes in numerous serum metabolome pathways, including purine metabolites (54). A recent paper has shown that specific gut microbiota species potentiate checkpoint blockade immunotherapy through production of inosine and activation of antitumor T cells (55). Increased xanthine production in Foxc2lecKO mice may therefore have a systemic effect on immune cells. Our strategy of analyzing blood metabolome in a model of lymphedema-distichiasis may offer a new angle to investigate circulatory biomarkers of the disease as diagnostic tools.
Molecular LEC subtypes and their relevance for human diseases
Single-cell transcriptome profiling uncovered molecular features for various adult LEC subtypes. CapLECs express genes for environmental sensing, microtubule dynamics, and chemokine signaling, highlighting that LECs directly communicate with the interstitial space for immune cell trafficking. A cluster of capLECs with high IFN signaling may represent cells that closely interact with or recently encountered transmigrating IFN-producing immune cells.
In line with their role in lymph transport, collLECs produced higher levels of transcripts encoding basement membrane components and proteins regulating SMC differentiation, recruitment, and contractility. Their unexpected expression of multiple BEC markers underscores the plasticity of adult endothelial cell populations and suggests the potential for LEC-to-BEC transdifferentiation. Adult LVs do not transport platelets; however, collLECs express multiple coagulation cascade proteins, therefore suggesting their potential platelet-independent function. Adult collLVs are enveloped in white adipose tissue, and we found them to express multiple fatty acid transport–related genes. Lipid products generated by collLECs may thus regulate lymphatic SMCs and/or nearby adipocytes or used for their own metabolism.
Fifteen genes are already known to be causative for various forms of primary lymphedema; however, many new causative mutations remain to be identified (16). Our survey of LEC subsets and associated stromal subpopulations (Fig. 7 and fig. S14), together with previously published scRNA-seq analyses of endothelial cells, will be useful to make informed predictions about the mechanism of action of lymphedema-associated genes. As one example, collLECs exclusively express the receptor tyrosine kinase MET, mutated in late-onset primary lymphedema (40), suggesting a novel role of HGF/MET signaling in the development and/or function of collLVs.
Foxc2 maintains the identity of the whole lymphatic network
Foxc2 depletion led to disappearance of Foxc2high populations (Fig. 8A), and we observed corresponding partial regression of lymphatic valves and precollLVs in vivo (Fig. 3, A to C), highlighting the importance of Foxc2 for their maintenance. Analysis of valve sites revealed ring-like and focal PROX1+ structures, reminiscent of embryonic valve initiation sites [fig. S1D and (11)]. Previous in vitro studies showed that FOXC2 depletion increases migration of LECs under flow (5). Thus, in the absence of Foxc2, at least some valve cells may migrate back to lymphangion regions, recapitulating early steps of valve formation. Increased motility of LECs in small precollLVs, where Foxc2 is also expressed in lymphangion cells, and in omental capLECs would explain their thinning and disruption.
Emergence of a major new KO1 LEC cluster derived from capLECs is counterintuitive, given low-to-absent expression of Foxc2 in this compartment (Fig. 8A). However, widespread changes were documented in the embryonic lymphatic vasculature of Foxc2-deficient mice (15). Single-cell transcriptome analyses established that Foxc2 inactivation leads to a major loss of capillary and, to a lesser extent, collecting endothelial cell identities and to their convergence toward a mixed capillary/collecting phenotype (Fig. 8D). Regression of lymphatic capillaries upon VEGFR3/VEGF-C blockade has little impact on the specialization of collLVs (56); thus, our results reveal an unexpected role of collLVs in the maintenance of lymphatic capillary phenotype and function.
Foxc2-deficient LECs acquired an inflamed and pro-fibrotic phenotype. The latter is likely driven by TGF-β signaling, which promotes fibrosis in multiple other contexts. We observed heightened expression of TGF-β co-receptor endoglin and multiple targets and effectors of TGF-β pathways, such as Fn1, Thbs1, Id1, and Id3 (57, 58), as well as growth factors inducing mural cell recruitment and proliferation (Pdgfb and Wnt2) (9, 59). FOXC2 knockdown in cultured LECs does not induce TGF-β signaling (5). Thus, TGF-β activation in vivo likely reflects cross-talk of Foxc2-deficient LECs with other cell types, such as stagnating immune cells and nearby fibroblasts. Our data suggest that TGF-β–dependent tissue fibrosis is an early event in the pathology of LDS and that therapeutic targeting of TGF-β activation may be of interest for LDS treatment.
The transcriptional changes in Foxc2-deficient LECs were not recapitulated in another model with defective lymphatic valves and flow, Cx37−/− mice (fig. S17). Such difference may be a result of distinct collecting vessel defects of these models. Notably, loss of Cx37 prevents development of lymphatic valves without affecting collecting vessel LECs (11), whereas Foxc2lecKO animals develop degeneration of lymphatic valves and thinning and disruption of vessels, a process that we have previously shown to be associated with increased perivalvular and valvular LEC proliferation and apoptosis and loss of junctional integrity in collecting vessels (5).
Role of collecting LECs in generation of LN LEC identity
Two new single-cell clusters with immune-related phenotypes were observed in Foxc2lecKO LECs: KO2, with high expression of antigen presentation– and inflammation-related transcripts, and KO3, expressing LN LEC–specific transcripts (Fig. 8B and table S5). The expansion of LN-like LECs within the collLVs of Foxc2lecKO mice was unexpected. A small number of cells with LN LEC phenotype already exist in the WT LEC population (Fig. 8B), suggesting that collLECs are primed for generation of LECs with LN identity. Areas of LN-like LVs contained intralymphatic aggregates of lymphocytes (Fig. 8I). Thus, interaction of collLECs with lymphocytes may generate a feed-forward loop to promote expansion of LECs with LN identity.
In addition to intravascular B cell aggregates, we observed perivascular T cells surrounding defective valves in Foxc2lecKO collLVs (Fig. 3, F and G). Lymph that is prone to leak from vulnerable valve sinuses may be triggering signals for FALC formation or LVs themselves secrete factors, such as chemokines that may attract T cells.
In summary, our study sheds light on the fundamental molecular basis of adult lymphatic vascular organization in which FOXC2 plays a central role. FOXC2 is needed to maintain the directional transport of lymph, the connection between different lymphatic compartments, and the identity of the whole lymphatic network. Our results also emphasize the role of a correct LV network wiring in preventing systemic pathogen spread. As both local and systemic effects of defective lymph flow can be rescued upon gut microbiota depletion, our results also highlight the role of LVs in ensuring compartmentalized inter-organ communication.
MATERIALS AND METHODS
Study design
This study was designed to identify the mechanisms of lymphatic collecting vessel maintenance and the biological consequences of their impaired function beyond defective tissue drainage to identify potential therapeutic strategies. For this purpose, we used Foxc2fl/fl;Prox1-CreERT2 (5) mice. First, we investigated the outcome of adult mice lacking Foxc2 and their capacity to transport lymph (Fig. 1), we analyzed peritoneal and pleural compartments and the status of LNs (Fig. 2), and we characterized the status of collecting, precollecting, vessels and lymphatic capillaries in different vascular beds (Figs. 3 and 4). Second, we focused not only on the organ-specific (Fig. 4) but also on the systemic consequences of the loss of Foxc2 (Fig. 5). Third, we tested the hypothesis that commensal microbiota has an impact on the phenotype observed and that, by its depletion, the phenotype can be restored (Fig. 6). In the last section, we made use of single-cell transcriptomic analysis to decipher the molecular consequences of Foxc2 inactivation in adult LECs (Figs. 7 and 8).
Mouse procedures
Animal model
All experiments were approved by the Animal Ethics Committee of Vaud, Switzerland. Foxc2fl/fl;Prox1-CreERT2 (5) and Cx37−/− mice (60) were bred to at least the tenth generation on C57BL6/J background. For conditional gene inactivation, Foxc2fl/fl (WT) and Foxc2fl/fl;Prox1-CreERT2 (KO) adult mice received three intraperitoneal injections of tamoxifen (50 μg/g body weight) in Kolliphor EL (Sigma-Aldrich) every other day. We used sex-matched littermates that were either Foxc2fl/fl (WT) or Foxc2fl/fl Prox1-CreERT2 (KO). For the analysis of survival, mice were treated with tamoxifen at different ages. In all other experiments, gene inactivation was done in 5-week-old mice. WT and Foxc2lecKO mice were analyzed from 5 weeks (short-term depletion) to 16 to 17 weeks after the last tamoxifen injection (long-term deletion). All mice used in our experiments were littermates and optimized for sample size and sex matching.
Computed tomography
Thoracic cavity computed tomography (CT) scan images were acquired using an X-RAD 320ix irradiator (Precision X-Ray) to assess pleural effusion status in Foxc2lecKO and WT mice. Mice were anesthetized with 2% isoflurane, positioned supine in the animal holder, aligning its longitudinal axis transverse to the image plane. A two-dimensional (2D) prescan image was taken to confirm the animal position and define the boundaries of the region of interest. Tube voltage was 40 kVp (kilovolt peak), and tube current was 3 mA. The scan was operated on three windows that displayed axial, sagittal, and 25 coronal slices. The lung and thoracic cavity volumes were calculated using OsiriX viewer (61). Signal levels were adjusted (WL115, WW300), the edges of the lungs were first defined in the stack images taken, and the volume of the lungs was calculated with OsiriX software. The same method was used to calculate the volume of the whole thoracic cavity, taking as edges the first fifth ribs of the thoracic cavity from the appearance of neck vertebra until the appearance of the spine vertebra. The 3D volume was reconstructed, the region of interest was analyzed to provide its volume, and the ratio of the volume of the lungs and the thoracic cavity was calculated.
Ascites measurements
For peritoneal liquid measurements, a wick method was used, in which a previously weighted 1.5 cm × 1.5 cm Whatman filter paper was inserted in the peritoneal cavity until all the liquid was absorbed. The paper was then weighted, and the difference between the final and initial weight was normalized versus the body weight of the mouse.
Lymphatic function assays
For the analysis of dermal lymph transport, mice were terminally anesthetized and 5 μl of DyLight 594 Lycopersicon esculentum (tomato) lectin (1 mg/ml; Vector Laboratories) was injected into the surgically exposed ingLN. For lymph transport from the peritoneal cavity, 50 μl of 2000-kDa FITC-dextran in phosphate-buffered saline (PBS) (10 mg/ml; Sigma-Aldrich) was injected intraperitoneally. Mice were allowed to move freely in the cage for 30 min before euthanasia. For the evaluation of the intestinal barrier integrity and lymph leakage, mice were gavaged with 150 μl of 4-kDa FITC-dextran (80 mg/ml; Sigma-Aldrich) and euthanized 1 hour later. Images were acquired using a Leica microscope DFC 3000 G camera equipped with LAS X software (Leica Microsystems) and quantified using Photoshop CC v2015.1.2 (Adobe) and ImageJ Fiji software. For the evaluation of lung vascular permeability, mice were injected intravenously with 100 μl of Evans blue (10 mg/ml) 20 min before dissection. After right ventricle intracardiac perfusion with PBS, lungs were collected and weighed and Evans blue was extracted with formamide.
Antibiotic treatment
WT and Foxc2lecKO mice were co-housed since weaning age. Eight weeks after tamoxifen administration, mice received enrofloxacin (2.5 mg/ml; Baytril 10%, Bayer) for 2 weeks in drinking water, followed by 6 weeks of amoxicillin (0.8 mg/ml) and clavulanic acid (0.114 mg/ml; Co-Amoxi-Mepha, Mepha) in drinking water.
Staining procedures and image acquisition
Whole mount
Tissues were fixed with 4% paraformaldehyde (PFA), washed with PBS, permeabilized with 0.5% Triton X-100 (AppliChem), and incubated with blocking solution containing 5% donkey serum, bovine serum albumin (BSA; AppliChem), and 0.5% Triton X-100. Tissues were incubated with primary antibodies, followed by secondary antibodies (Thermo Fisher Scientific) for 3 days on rotating platform at 4°C. Tissues were cleared for minimum 3 days at 70 rpm at 4°C in 1.62 M Histodenz (Sigma-Aldrich) and 0.1% Tween 20 (Sigma-Aldrich) and mounted in Histodenz medium supplemented with Hoechst (0.2 μg/ml; Thermo Fisher Scientific). Confocal images represent maximum intensity projection of Z-stacks of single tile scan images.
Cryosection
Tissues were fixed in 4% PFA for 12 hours, equilibrated in 30% sucrose for 48 hours, and embedded in optimal cutting temperature (OCT) compound (Tissue-Tek, Sakura). Cryosections were cut at 8-μm thickness and fixed with 4% PFA, washed with PBS, permeabilized with 0.5% Triton X-100, and incubated with blocking solution followed by antibody staining. Sections were mounted in Fluoromount-G mounting medium supplemented with 4′,6-diamidino-2-phenylindole (DAPI; eBioscience).
Paraffin sections
Sections were cut at 5-μm thickness, deparaffinized, and subjected to heat-induced epitope retrieval using low- or high-pH retrieval solutions (DAKO), incubated with antibodies, and mounted in Fluoromount-G mounting medium supplemented with DAPI (eBioscience).
Images were taken using confocal Zeiss Zen v11.0.0.0 (Carl Zeiss) and analyzed using Imaris 8 (v.8.0.2) (Bitplane) and Photoshop CC v2015.1.2 (Adobe) software. Large image tiles (omental tissue pictures) were acquired using a motorized Zeiss time-lapse Axio Observer.Z1 with Zeiss Axiovision v4.8.2.0 software (Carl Zeiss). Bright-field microscope images were taken using a Leica MZ16 microscope with a Leica Microsystems DFC 295 camera and LAS v4.2 software (Leica Microsystems). The antibodies used for immunostaining are listed in table S7.
Quantifications from histological images
Lymphatic valves were quantified manually under a confocal Zeiss Zen v11.0.0.0 (Carl Zeiss). For skin collLV draining, quantification of total collLV length and Lycopersicon esculentum (tomato)–lectin–filled collLV length from ingLN to the axillary LN was quantified using Photoshop CC v2015.1.2 (Adobe) and ImageJ Fiji software. For histological analyses, at least three pictures per each sample were taken. LEC’s width versus length ratio was quantified in collLVs using ImageJ Fiji software. To measure lymphatic capillary diameter in ear skin, whole-mount pictures of LYVE-1 staining were taken using 10× objective with a Zeiss Zen v11.0.0.0 confocal microscope. Quantifications were performed with ImageJ Fiji software. Pictures of lymphatic vessels in lungs were acquired using an upright Zeiss Axio Imager Z1 using 20× objective. LV area in the lungs was quantified as the VEGFR3+ area normalized to the total tissue area using ImageJ Fiji software. For the quantification of LV area in the ileum, images were acquired using Zeiss Axio Imager Z1 and 20× objective. By means of ImageJ Fiji software, the images were transformed into 8-bit and a threshold was set up in the LYVE1 channel to visualize the LVs. The same threshold was kept for all the pictures. The area of LVs was measured and normalized to the corresponding tissue area. For low-magnification pictures and to quantify the area and number of FALCs of the entire omentum and mesenteric branches, an upright Zeiss Axio Imager Z1 using the tile scan and stitching function and the 10× objective was used. FALC quantification was performed with the ImageJ Fiji software, a threshold was fixed for the CD45 channel to visualize immune cell aggregates, and the area of FALCs versus the total area of the tissue was used as output. For the quantification of B220+ cells within LVs, ×20 magnification confocal pictures of whole-mount stainings were used. The number of B220+ cells was calculated per vessel area using Fiji software; IMARIS software was used to confirm the internal position of the cells in the vessels.
Immune cell analyses
Peritoneal and pleural wash flow cytometry
Washes were obtained by injecting 3% fetal calf serum in PBS, 150 and 70 μl/g of mouse weight in the peritoneal and thoracic cavities, respectively. Red and white blood cells including lymphocytes, monocytes, and granulocytes were quantified using a Mythic 18 Vet hematology analyzer (Woodly). For fluorescence-activated cell sorting (FACS), peritoneal and pleural washes were centrifuged at 1200 rpm at 4°C for 10 min, resuspended in red blood cell (RBC) lysis buffer (eBioscience) for 5 min at room temperature, and washed with FACS buffer (2 mM EDTA and 2% BSA in PBS). Cells were incubated with Fc block (antibody to CD16/32) for 20 min on ice and stained with conjugated antibodies in FACS buffer for 30 min. Biotin-conjugated primary antibodies were detected with fluorochrome-coupled streptavidin. The cell suspension was passed through a 40-μm strainer, and dead cells were excluded using a Zombie Aqua kit (BioLegend). B cells were identified as CD45+CD19+B220+, and T cells were identified as CD45+CD19−CD90+cells. For plasma cells, following CD19+ selection, a second positive selection for CD138 was performed. For detailed analysis of peritoneal immune cells, the following strategy was performed: B cells were identified as CD45+B220+CD19+, B2 cells as CD45+CD19+B220high, B1 cells as CD45+CD19+B220low, B1a cells as CD45+CD19+B220low CD5+, B1b cells as CD45+CD19+B220low CD5−, plasma cells as CD45+B220+CD19+CD138+IgM+, T cells as CD45+CD19−CD90+, neutrophils as CD45+CD19−CD3−Cd11b+Gr1+, eosinophils as CD45+CD19−CD3−Gr1−Cd11b+SiglecF+SSCAhigh, macrophages as CD45+CD19−CD3−Gr1−SiglecF−Cd11b+ F4/80+, and monocytes as CD45+CD19−CD3−Gr1−SiglecF−Cd11b+F4/80− Ly6C+. Samples were acquired on an LSR-II (BD Biosciences) with compensation using single-cell staining and analyzed with FlowJo 10.4.2 software (Tree Star Inc.). For flow cytometry analysis, the frequency of each cell type was calculated as percentage among viable CD45+ cells. Gating strategies are presented in fig. S2.
LNs, spleen, and bone marrow flow cytometry
The spleen, bone marrow from femurs, and popliteal and inguinal LNs were harvested in PBS with 2% serum and mechanically dissociated using 40-μm cell strainers before staining. Briefly, cells were negatively selected for CD11b and CD11c and positively selected for CD19 for B cells, GL7 for germinal center (GC) B cells, and CD3 for T cells. For plasma cells, following CD19+ selection, a second positive selection for CD138 was performed. Data were acquired on a FACSCanto (BD Biosciences) with compensations based on single stainings and analyzed with FlowJo 10.4.2 software (Tree Star Inc.). For flow cytometry analysis, the frequency of each cell type was calculated as percentage among viable CD45+ cells. This number was then used along with the cellularity to calculate the total cell number for each cell type. Gating strategies are presented in fig. S8.
Intestinal lamina propria flow cytometry
The second half of jejunum and the complete ileum were dissected and flushed with cold PBS. Peyer’s patches and mesenteric fat were removed, and the remaining small intestine was cut longitudinally and then washed with cold PBS. Tissue pieces (0.5 to 1 cm) were incubated for 20 min in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco) containing 10 mM EDTA with gentle stirring at 37°C. The tubes were vigorously shaken every 10 min to remove epithelial cells, intraepithelial lymphocytes, and mucus. The remaining intestinal tissues were incubated for 20 min at 37°C in digestion buffer containing DMEM, 2% serum, Liberase TL (0.2 mg/ml; Roche), deoxyribonuclease (DNase) I (1 U/ml; Invitrogen), and 1% gentamicin with gentle stirring. To favor tissue dissociation, the pieces of intestine were mixed by pipetting up/down several times after 10 and 20 min. Isolated cells in the supernatant were harvested and kept on ice with a 1:1 volume of complete DMEM until the end of the digestion process. In total, three identical digestions were performed for the complete dissociation of the intestine. The cell suspension was passed through a 40-μm strainer, centrifuged for 6 min at 150g at 4°C, and resuspended in FACS buffer (PBS, 2% serum, and 2 mM EDTA). Cells were incubated with Fc block (antibody to CD16/32) for 20 min on ice and stained with conjugated antibodies in FACS buffer for 30 min. Biotin-conjugated primary antibodies were detected with fluorochrome-coupled streptavidin. IgA+ plasma cells were selected as CD45+F4/80−CD11b−CD11c−FSC-Ahi IgA+. Data were acquired on an LSRII (BD Biosciences) flow cytometer with compensations based on single stainings and analyzed with FlowJo 10.4.2 software. The frequency of each cell type was calculated out of all viable CD45+ cells found in each sample. Gating strategy is presented in fig. S11.
LN LEC sorting
Inguinal, axillary, brachial, and popliteal LNs from 8-week-old mice deleted for 2 weeks (one injection each week) with tamoxifen (50 μg/g body weight) were collected in complete medium (phenol red–free DMEM and 5% serum). LNs were minced with scissors and digested with collagenase A (1 mg/ml; Roche), DNase (0.1 mg/ml), and dispase II (0.8 mg/ml) in 0.1% BSA in PBS with shaking at 330 rpm for 30 min at 37°C. EDTA (0.5 M) was added in 1:50 volume for 5 min, and the suspension was filtered through 40-μm strainer and washed twice in complete medium. Cells were blocked with Fc block (antibody to CD16/32) for 10 min and stained for 30 min with CD45, CD31, and podoplanin (PDPN) antibodies coupled with different fluorophores. Single-stain and unstained samples were used as controls. Following exclusion of multiplets and dead cells using DAPI, single CD45− CD31+ PDPN+ cells were gated and sorted in RLT lysis buffer containing β-mercaptoethanol (RNeasy Micro Kit, Qiagen).
Mesenteric LEC sorting
Whole mesenteries (mLNs were removed) from three WT and three Foxc2lecKO mice were collected 3 weeks after the first tamoxifen injection. Samples were digested for 30 min in digestion buffer containing Hanks’ balanced salt solution (calcium-free, Gibco), 2% BSA, 1% penicillin-streptomycin, Liberase TL (0.2 mg/ml; Roche), and DNase I (1 U/ml; Invitrogen) at 37°C with gentle stirring. Samples were then washed in complete medium [phenol red–free DMEM (Gibco) and 5% serum] and incubated for 5 min at room temperature with RBC lysis buffer (eBioscience). Cells were then washed again, blocked with anti-CD16/32 antibodies, and then incubated with conjugated antibodies specific for CD45, CD31, and PDPN. Following exclusion of dead cells using DAPI and/or RedDot1 Far-Red Nuclear Stain reagent (Biotium) and gating on single cells, single CD45− CD31+ PDPN+ cells were gated and sorted directly in lysis buffer RLT containing β-mercaptoethanol (RNeasy Micro Kit, Qiagen). Gating strategies are presented in figs. S1 and S14.
Cells were sorted on a FACSAria II (SORP) v8.0.1 cell sorter with FACSDiva software (BD Biosciences). All antibodies used for flow cytometry are listed in table S7.
Biochemical analyses
LPS concentration was determined using a Limulus amoebocyte lysate assay kit (LAL QCL, Lonza). Tissue extracts in radioimmunoprecipitation assay (RIPA) buffer and serum were diluted in LAL reagent water (1:40) and heated at 75°C for 10 min. An internal LPS control from the kit was included. Serum cytokines IL6, IFNγ, and TNFα were determined by using a LEGENDplex mouse inflammation panel kit (BioLegend) using CytoFlex S1 machine (Beckman Coulter) and analyzed using the BioLegend software provided with the kit. A mouse/rat ANGPT2 enzyme-linked immunosorbent assay (ELISA) (R&D Systems) was used for measurement of ANGPT2 levels. Total IgM levels in blood were determined using an ELISA mouse IgM kit (Thermo Fisher Scientific).
Lymph sampling and in vitro studies
Collection of intestinal lymph from the mesenteric lymphatic duct was performed according to previously described methods (62, 63), with some modifications. Briefly, to facilitate visualization of the lymphatic duct, 8 μl of olive oil (Sigma-Aldrich) per gram of body weight was administered by oral gavage to mice 1 hour before general anesthesia using 2% isoflurane. The abdomen was opened by midline incision, and the intestine and colon were retracted to the left side of the animal to identify and locate the mesenteric lymphatic duct. Under a microsurgical microscope (OPMI MD with Universal S3 Stand; Carl Zeiss), a 30-gauge needle linked to 0.5-ml syringe (BD Biosciences) was inserted to the lymphatic duct and advanced for 2 to 3 mm. About 10 to 20 μl of intestinal lymph were obtained from each mouse. Five percent intestinal lymph in complete DMEM was incubated with confluent transformed mouse intestinal epithelial cells (31) for 16 hours. Cell-cell junctions were analyzed by staining for E-cadherin and ZO-1. Five percent DSS was used as positive control for epithelial cell-cell junction disruption.
Tissue nucleic acid isolation and RT-qPCR
RNA isolation
Tissue samples were washed twice in ice-cold PBS, incubated for 30 min in RNA later solution (Invitrogen), transferred into RLT lysis buffer with β-mercaptoethanol (Qiagen), and snap-frozen on dry ice. Following homogenization with a FastPrep-24 disruptor (MP Biomedicals), the RNA was isolated using the Qiagen RNeasy Plus Mini Kit (Qiagen). RNA concentration and quality were analyzed using 2100 Bioanalyzer (Agilent). In the case of LNs, RNA was amplified using Ovation Pico WTA System V2 (Nugen).
DNA isolation
DNA extraction from tissues was performed by incubating the samples for 2 hours in proteinase K and heated for 15 min at 95°C. This DNA was used as a template for PCR amplification. DNA concentration and quality were assessed using NanoDrop 1000 (Thermo Fisher Scientific).
Reverse transcription quantitative polymerase chain reaction
We used reverse transcriptor First Strand cDNA Synthesis Kit (Roche Diagnostics), StepOnePlus Real-Time PCR instrument (Applied Biosystems), and SYBR Green PCR Master Mix (Thermo Fisher Scientific) for qPCR analyses. Data were analyzed using the comparative Ct (ΔΔCt) method as described by the manufacturer. Gene expression was normalized to the housekeeping gene 18S. RT-qPCR results are shown as fold change over controls. The nucleotide sequences of the real-time qPCR primers used in this study are described in table S7.
Tissue bulk RNA-seq
Tissue preparation and sequencing procedure for bulk RNA-seq
For lungs, the left lobule of lungs was dissected. For mesentery, the mesentery part from ileum and jejunum was entirely dissected and mLN was removed. For both tissues, RNA was isolated as described above (see the “RNA isolation” section). RNA quality check, library preparations, and sequencing reactions were conducted at Lausanne Genomic Technologies Facility (https://wp.unil.ch/gtf/). Total RNA samples were quantified using Qubit 2.0 Fluorometer (Thermo Fisher Scientific), and RNA integrity was checked with 4200 TapeStation (Agilent Technologies). All RNAs had an RNA quality number above 7.5. RNA-seq libraries were prepared from 500 ng of total RNA. RNA-seq library preparation used the TruSeq Stranded RNA Library Prep Kit for Illumina by following the manufacturer’s recommendations (Illumina). Briefly, enriched RNAs were fragmented for 15 min at 94°C. First- and second-strand complementary DNAs (cDNAs) were subsequently synthesized. cDNA fragments were end-repaired and adenylated at 3′ ends, and universal adapter was ligated to cDNA fragments, followed by index addition and library enrichment with limited PCR cycles. Sequencing libraries were validated on the Agilent TapeStation (Agilent Technologies) and quantified by using Qubit 2.0 Fluorometer (Invitrogen) as well as by quantitative PCR (Applied Biosystems). After clustering, the flowcell was loaded on the Illumina HiSeq instrument according to the manufacturer’s instructions. The samples were single-end sequenced (150 nucleotides) using HiSeq 4000 SR, each sample being sequenced on three separate lanes to increase coverage per sample. Image analysis and base calling were conducted by the HiSeq Control Software. Raw sequence data (.bcl files) generated from Illumina HiSeq were converted into fastq files and demultiplexed using Illumina’s bcl2fastq 2.17 software (Illumina). One mismatch was allowed for index sequence identification.
Bulk RNA-seq data analysis
Raw sequencing read alignment and count summarization were performed using the bcbio-nextgen pipeline (v.19.03, https://github.com/bcbio/bcbio-nextgen). Quality encoding of sequencing reads was converted to Sanger format using seqtk (https://github.com/lh3/seqtk). Sequencing reads obtained on the three lanes per sample were concatenated and aligned to the Mus musculus genome (mm10) using HISAT2 (v.2.1.0) (64). Sequence alignment map files were converted to binary alignment map files and indexed using SAMtools (v.1.9) (65). Read count summarization at the gene level was performed using the featureCounts method (66) of the Subread package (v.1.6.3) using the mm10 genome annotation.
Subsequent statistical analyses were performed using R (v.3.5.3 and v.3.6.0). Each tissue was analyzed independently from the other tissues. First, counts were filtered to remove undetected genes. Normalization factors were calculated using the trimmed mean of M values method (67) implemented in the edgeR package (v.3.28.1) for R. The CQN (68) (v.1.33.0) package for R was used to calculate generalized linear model (GLM) offset values, by providing GC content and longest transcript length of each gene to the cqn function. Percentage of GC and longest transcript length were obtained using the biomaRt (69) (v.2.38.0) package for R. Differential gene expression analysis between KO and WT mice for each tissue was performed by fitting gene-wise negative binomial generalized linear model using the glmFit function of the edgeR package, followed by likelihood ratio tests using the glmLRT function. Genes were considered as significantly differentially expressed at a Benjamini-Hochberg (BH) (70)–adjusted P value of <0.05.
Gene set enrichment analysis (GSEA) was performed following the method described by Subramanian et al. (18). We tested enrichment against the GO (71, 72) gene sets downloaded from the MSigDB (18) website (v.7.1). This list of gene sets was filtered to remove the ones with less than 40 genes, and log2(fold change) values were provided as the gene statistic. A normalized enrichment score and associated P value were calculated for each gene set by randomizing gene labels 1000 times. A gene set was considered as statistically significant at BH-adjusted P value of <0.05.
Single-cell RNA-seq
Cell sorting and sequencing procedure for single-cell RNA-seq
Mesentery LECs were isolated from three WT and three Foxc2lecKO samples following the protocol described above (see the “Mesenteric LEC sorting” section). Cells were sorted at 4°C on a Beckman Coulter MoFlo Astrios EQ. The instrument was configured with a 70-μm nozzle at a pressure setting of 60 psi. Viability was evaluated on the sorter and was, on average, 80%. Viable cells were identified as being redDot positive and DAPI negative. The staining was performed for 5 min at room temperature at a 1× redDot concentration and a DAPI concentration of 0.1 μg/ml. Cells were gated as shown in fig. S14.
LECs were recovered in Hank’s Balanced Salt solution (HBSS) with 40% serum. A total of 21,000 LECs were sorted, on average, per sample. Of those, an average of 11,000 cells were loaded on a Chromium Next GEM Chip B (10x Genomics). Droplets encapsulating single cells were generated using the 10x Genomics technology (Single Cell 3′ v3 chemistry). Briefly, an emulsion encapsulating single cells, reverse transcription reagents, and cell barcoding oligonucleotides was generated. After the actual reverse transcription step, the emulsion was broken and double-stranded cDNA was generated and amplified in a bulk reaction for 12 PCR cycles. This cDNA was fragmented, a P7 sequencing adaptor was ligated, and a 3′ gene expression library was generated by a 14-cycle PCR amplification.
The sequencing libraries were prepared according to the manufacturer’s recommendations (Chromium Single Cell 3′ GEM Kit v3). Libraries were quantified by a fluorimetric method (QubIT, Thermo Fisher Scientific), and their quality was assessed on a Fragment Analyzer system (Agilent Technologies). Cluster generation was performed with 1.4 nM of an equimolar pool from the resulting libraries, including 10% PhiX spike, using the Illumina HiSeq 3000/4000 PE Cluster Kit reagents (Illumina). Sequencing was performed on two runs in the Illumina HiSeq 4000 using HiSeq 3000/4000 SBS Kit reagents (Illumina) according to Illumina 10x Genomics recommendations at the Lausanne Genomic Technologies Facility (https://wp.unil.ch/gtf/). Sequencing data were demultiplexed using the bcl2fastq2 Conversion Software (version 2.20, Illumina) and aligned to the mouse reference genome (mm10) using the 10x Genomics Cell Ranger pipeline (version 3.1.0). A minimum of 33,000 reads/cell were obtained, and 18,271 genes, on average, were detected.
scRNA-seq data analysis
The aggr function of the Cell Ranger pipeline was used to generate a single count matrix including all six WT and KO samples. The filtered feature × barcode matrix was imported into R (v.3.6.0) and subsequently analyzed using the Seurat package (73) for R (v.3.1.5). We filtered the count matrix to retain cells that had between 500 and 5800 genes detected, less than 20% of mitochondrial gene counts, and less than 6% of dissociation-related genes (74) (n = 21,962 cells retained). Genes not detected in at least three cells were removed (n = 19,054 genes retained). The counts were log-normalized (scale factor = 10,000) and scaled using the NormalizeData and ScaleData functions implemented in the Seurat package. Most variable gene detection and principal component (PC) reduction were performed using the FindVariableFeatures and RunPCA functions using default parameters. Cell clustering of all WT and KO cells was performed using the shared nearest neighbor modularity optimization–based algorithm implemented in the FindNeighbors and FindClusters functions, using 19 PCs and a 0.4 resolution. UMAP dimensionality reduction was performed using the RunUMAP function of the Seurat package with parameters reduction = “pca” and 20 PCs.
Annotation of cell types was performed using known and marker genes of each cluster (fig. S14, B and E) (75, 76). Genes differentially expressed between each cluster (i.e., marker genes) and all other cells were determined using the FindAllMarkers function with parameters log2 (fold change) threshold = 0.15, test.use = “wilcox”. Genes significant with a false discovery rate (FDR)–adjusted P value below 0.05 were retained. Within each cluster, genes differentially expressed between WT and KO cells were determined using the FindMarkers function of the Seurat package with default parameters.
The raw counts of cells belonging to clusters 0 (LECs, capillaries), 1 (LECs, lymphangion), 4 (LECs in KO), 11 (proliferating ECs), 12 (LECs, LN-like), and 13 (BECs/LECs) (fig. S14, B and E) were extracted and normalized anew using the same functions as described for all cells using the following modifications: The percentage of dissociation-related genes was regressed out during scaling; the clustering resolution was set to 0.5; the UMAP dimensionality reduction was generated using 19 PCs.
Subsequent analyses were performed only on LECs defined as Cd31+, Prox1+, Vegfr3+, Flt1− cells. Marker genes of each LEC cluster were determined for WT and KO separately, using the FindAllMarkers function with default parameters. Last, genes differentially expressed between WT and KO within each cluster were determined using the FindMarkers function using default parameters.
After determining which genes were specifically differentially expressed between WT capLEC and WT collLEC, we generated a signature score for each cell by using the addModuleScore (77) function implemented in the Seurat package. Two scores were generated for each cell: one for the up-regulated genes and one for the down-regulated genes in WT capLEC cluster versus WT collLEC cluster.
Across the WT LEC subsets, 1947 of 19,054 genes (10.22%) were differentially expressed. Among these DEGs, we identified, on average, 220 LEC subset–specific transcripts, defined as genes with a log2 fold change value higher than 0.25 in each cluster compared to the other cells and FDR-adjusted P value below 0.05 (table S3). Collectively, 887 of 19,054 genes (4.65%) were differentially expressed between WT and Foxc2 KO LECs. Overrepresentation analysis of GO gene sets for any list of DEGs was performed using the enricher function implemented in the clusterProfiler (v. 3.14.13) package (78) for R, with P values adjusted using the BH adjustment method (70). Up-regulated or down-regulated genes were tested separately against the GO biological process gene sets (71, 72) downloaded from the MSigDB database (v. 7.1) (18).
Single-cell trajectory analysis was performed using the Monocle method implemented in the monocle3 (v.0.2.2) (42) package for R. The WT and KO LEC clusters were split, and a new UMAP was regenerated independently for each genotype using the reduce_dimension function of monocle3. We provided the new UMAP coordinates and the cluster label of each cell generated by Seurat to the learn_graph function, otherwise using default parameters.
16S rRNA gene sequencing
Bacterial DNA isolation
One fecal pellet from each mouse was collected into sterile 1.5-ml Biopure tube (Eppendorf) and immediately kept on dry ice and stored at −80°C until further processing. Total bacterial DNA was isolated using the QiaAMP Fast DNA Stool Mini Kit (QIAGEN) according to the manufacturer’s instructions. DNA was eluted with 100 μl of AE elution buffer (provided with the kit). DNA was stored at 4°C until being used for the PCR.
16S rRNA gene library preparation and sequencing
DNA was extracted from fecal contents as described above, and the V1-V2 hypervariable regions of 16S rRNA gene were amplified using 27F and 338R universal barcoded primers. The nucleotide sequences were as follows: 27F-5′-AATGATACGGCGACCACCGAGATCTACACTATGGTAATTCCAGMGTTYGATYMTGGCTCAG-3′ and 338R-5′-CAAGCAGAAGACGGCATACGAGATNNNNNN NNNNNNAGTCAGTCAGAAGCTGCCTCCCGTAGGAGT-3′ (bold: Illumina adaptor sequences; italic: linkers, NNNNNNNNNNNN sample-specific MID tag barcodes). PCRs were performed in duplicates in a volume of 20 μl each using an AccuPrime Taq DNA polymerase high-fidelity kit (Invitrogen), 4 μl of template DNA, and 0.44 μl each primer (stock at 10 μM). PCR program was as follows: 3 min at 94°C (initial denaturation), followed by 30 cycles of 30 s at 94°C (denaturation), 30 s at 56°C (annealing), 1.5 min at 72°C (extension), and 5 min at 72°C (final extension). Duplicates were pooled, and amplicon quantity and size were determined with LabChip GX capillary electrophoresis (Perkin Elmer). PCR products were pooled in equimolar amounts and purified using Agencourt AMPure XP magnetic beads (Beckman Coulter). Sequencing was performed on an Illumina MiSeq platform with MiSeq reagent kit V2-500 (pair-end, 2 × 250) as previously described (79) following the basic protocol for stool samples.
16S rRNA gene sequencing data analysis
A table of amplicon sequence variants (ASVs) was derived from the sequencing reads using the DADA2 pipeline (R package dada2) with the Silva 16S database version 132. ASV filtering, normalization, ordination, and diversity analyses were performed using Genocrunch platform (https://genocrunch.epfl.ch). Only samples with >1000 ASVs were considered for downstream analyses. The number of observations per samples was equalized using the rarefaction method with a sampling depth of 21,149 (random sampling without replacement) (R package vegan). Values were converted into per-million per column, and a log transformation was applied [log2(x + 1)]. ASVs were aggregated at each taxonomic level (sum of ASVs with the same taxonomic classification). Abundances were compared between experimental groups at each taxonomic level using fold change and analysis of variance (ANOVA). Rarefaction curves based on species richness were used to compare diversity between experimental groups, on which an ANOVA was performed at a rarefaction depth of 21,140 counts per sample. To compare the relative abundance of Epsilonbacteraeota at the phylum level, a two-tailed t test was performed on ASV proportions (%) using GraphPad Prism 8 software (GraphPad).
Metabolomics
Mandibular blood plasma samples were collected at different time points after Foxc2 depletion. WT, Foxc2lecKO, and antibiotic-treated mice were included. Polar metabolites were extracted from 20 μl of plasma with 180 μl of room temperature 80% methanol. Untargeted metabolomics of extracts was done by flow injection analysis–time-of-flight mass spectrometry on an Agilent 6550 Q-TOF instrument as previously described (80). Mass spectra were recorded from a mass/charge ratio of 50 to 1000 in high-resolution negative ionization mode. Ions were annotated by matching their measured mass with reference compounds derived from the Human Metabolome Database (HMDB 4.0), allowing a tolerance of 1 mDa. Significance was calculated using Student’s t test and corrected for multiple hypothesis testing with the BH method (70). Ion intensities data are provided in table S2.
Statistical analysis
Data analyses were performed using GraphPad Prism 8 for MacOS (GraphPad). The number of mice analyzed is indicated for each image in the figure legends. For in vivo experiments comparing WT and Foxc2lecKO animals, normality tests and two-tailed unpaired Student’s t test were performed to determine statistical significance between two means. For in vivo experiments comparing control and antibiotic-treated WT and Foxc2lecKO animals, we used one-way ANOVA with Tukey post hoc test. Scattered dot plot data are shown as means ± SD, where each single dot represents an individual mouse. P value is stated in each figure. Differences were considered statistically significant at P < 0.05.
Acknowledgments
We thank N. Miura for providing the Foxc2 fl/fl mice and the rat anti-Foxc2 antibody. T. Makinen (Uppsala University, Sweden) for sharing Prox1-CreERT2 mice; L. Sorokin (University of Münster, Münster, Germany) for laminin-α5 antibody; G. Y. Koh (KAIST, Daejeon, South Korea) for angiopoietin-2 antibody; C. Beauverd for mouse genotyping, colony maintenance, and immunostainings; J. Laube for participation in FALC and initial intestinal permeability analyses; and B. Petit and M.-C. Vozenin for help with hematology analyzer and CT acquisition and analysis. We thank J. Bernier-Latmani for reading the manuscript and useful discussions. Mouse Pathology, Flow Cytometry, Genomic Technologies Facility (GTF), Animal, and Cellular Imaging Facilities of the University of Lausanne are gratefully acknowledged. Funding: This work was supported by the Swiss National Science Foundation (CRSII5_177191 to T.V.P. and N.Z.; 31ER30_160674, 31003A-156266, and CRSK-3_190200 to T.V.P.; and 310030_185226/1 to S.A.L.), People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme FP7/2007-2013/ under REA grant agreement 317250 (to T.V.P.), interdisciplinary grant of the Faculty of Biology and Medicine of UNIL (to T.V.P. and D.V.), and two postdoctoral fellowships from the Human Frontier Science Program Organization (LT000074/2019-L to J.K. and LT000633/2020-L to S.A.-M.). Work in the laboratory of G.G. was supported by the Swiss National Science Foundation (310030_185185), the Fondazione San Salvatore, Lugano, and the Novartis Foundation. Author contributions: A.G.-L. and T.V.P. planned the study, and T.V.P. supervised the research. A.G.-L. prepared the mouse models, performed the mouse studies, and analyzed the data related to Figs. 1 (A to G), 2, 3 (B to G), 4 (B to H), and 5 to 8 and figs. S1 (A to F), S2, S3 to S7, and S9 to S19; E.B. contributed to Figs. 1 (B, G, and H), 3A, and 4A and figs. S1 (B, F, and G), S5A, and S8 and independently reproduced similar data to Fig. 2F; J.K. contributed to Figs. 4G and 6 and fig. S10; T.W.L. contributed to Figs. 7 and 8 and figs. S14 to S16 and tables S1 and S3 to S6; A.S. contributed to Fig. 1B and figure preparation; F.R. contributed to Fig. 2B and figs. S2A and S11; S.A.-M. contributed to fig. S2 (E and F); A.R. and T.P.W. contributed to Fig. 5 (A and B) and fig. S12 (B and C); G.R. contributed to Fig. 4A and fig. S8; S.D. contributed to Fig. 5C, fig. S12D, and table S2; and A.S., N.Z., D.V., B.M., G.G., M.D., and S.A.L. provided reagents, discussions, and advice on the study, and A.G.-L. and T.V.P. wrote the paper. All authors read, commented on, and approved the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The scRNA-seq and bulk RNA-seq data generated for this publication have been deposited in NCBI’s Gene Expression Omnibus (GEO) and are accessible through GEO Series accession number GSE156320 (https://ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE156320). 16S raw sequence data have been deposited in the European Nucleotide Archive (ENA) database with the accession number PRJEB39832. This study did not generate new unique reagents. All other data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/29/eabf4335/DC1
REFERENCES AND NOTES
- 1.Petrova T. V., Koh G. Y., Biological functions of lymphatic vessels. Science 369, eaax4063 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Oliver G., Kipnis J., Randolph G. J., Harvey N. L., The lymphatic vasculature in the 21st century: Novel functional roles in homeostasis and disease. Cell 182, 270–296 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.González-Loyola A., Petrova T. V., Development and aging of the lymphatic vascular system. Adv. Drug Deliv. Rev. 169, 63–78 (2021). [DOI] [PubMed] [Google Scholar]
- 4.Ulvmar M. H., Mäkinen T., Heterogeneity in the lymphatic vascular system and its origin. Cardiovasc. Res. 111, 310–321 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sabine A., Bovay E., Demir C. S., Kimura W., Jaquet M., Agalarov Y., Zangger N., Scallan J. P., Graber W., Gulpinar E., Kwak B. R., Makinen T., Martinez-Corral I., Ortega S., Delorenzi M., Kiefer F., Davis M. J., Djonov V., Miura N., Petrova T. V., FOXC2 and fluid shear stress stabilize postnatal lymphatic vasculature. J. Clin. Invest. 125, 3861–3877 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Randolph G. J., Ivanov S., Zinselmeyer B. H., Scallan J. P., The lymphatic system: Integral roles in immunity. Annu. Rev. Immunol. 35, 31–52 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jackson D. G., Leucocyte trafficking via the lymphatic vasculature—Mechanisms and consequences. Front. Immunol. 10, 471 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Norrmen C., Ivanov K. I., Cheng J., Zangger N., Delorenzi M., Jaquet M., Miura N., Puolakkainen P., Horsley V., Hu J., Augustin H. G., Yla-Herttuala S., Alitalo K., Petrova T. V., FOXC2 controls formation and maturation of lymphatic collecting vessels through cooperation with NFATc1. J. Cell Biol. 185, 439–457 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y., Jin Y., Mäe M. A., Zhang Y., Ortsäter H., Betsholtz C., Mäkinen T., Jakobsson L., Smooth muscle cell recruitment to lymphatic vessels requires PDGFB and impacts vessel size but not identity. Development 144, 3590–3601 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kazenwadel J., Betterman K. L., Chong C. E., Stokes P. H., Lee Y. K., Secker G. A., Agalarov Y., Demir C. S., Lawrence D. M., Sutton D. L., Tabruyn S. P., Miura N., Salminen M., Petrova T. V., Matthews J. M., Hahn C. N., Scott H. S., Harvey N. L., GATA2 is required for lymphatic vessel valve development and maintenance. J. Clin. Invest. 125, 2979–2994 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sabine A., Agalarov Y., Maby-El Hajjami H., Jaquet M., Hagerling R., Pollmann C., Bebber D., Pfenniger A., Miura N., Dormond O., Calmes J. M., Adams R. H., Makinen T., Kiefer F., Kwak B. R., Petrova T. V., Mechanotransduction, PROX1, and FOXC2 cooperate to control connexin37 and calcineurin during lymphatic-valve formation. Dev. Cell 22, 430–445 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Jalkanen S., Salmi M., Lymphatic endothelial cells of the lymph node. Nat. Rev. Immunol. 20, 566–578 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Avraham T., Daluvoy S., Zampell J., Yan A., Haviv Y. S., Rockson S. G., Mehrara B. J., Blockade of transforming growth factor-β1 accelerates lymphatic regeneration during wound repair. Am. J. Pathol. 177, 3202–3214 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huggenberger R., Siddiqui S. S., Brander D., Ullmann S., Zimmermann K., Antsiferova M., Werner S., Alitalo K., Detmar M., An important role of lymphatic vessel activation in limiting acute inflammation. Blood 117, 4667–4678 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petrova T. V., Karpanen T., Norrmen C., Mellor R., Tamakoshi T., Finegold D., Ferrell R., Kerjaschki D., Mortimer P., Yla-Herttuala S., Miura N., Alitalo K., Defective valves and abnormal mural cell recruitment underlie lymphatic vascular failure in lymphedema distichiasis. Nat. Med. 10, 974–981 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Martin-Almedina S., Mortimer P., Ostergaard P., Development and physiological functions of the lymphatic system—Insights from genetic studies of lymphedema. Physiol. Rev. , (2021). [DOI] [PubMed] [Google Scholar]
- 17.Brice G., Mansour S., Bell R., Collin J. R., Child A. H., Brady A. F., Sarfarazi M., Burnand K. G., Jeffery S., Mortimer P., Murday V. A., Analysis of the phenotypic abnormalities in lymphoedema-distichiasis syndrome in 74 patients with FOXC2 mutations or linkage to 16q24. J. Med. Genet. 39, 478–483 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., Mesirov J. P., Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benezech C., Luu N. T., Walker J. A., Kruglov A. A., Loo Y., Nakamura K., Zhang Y., Nayar S., Jones L. H., Flores-Langarica A., McIntosh A., Marshall J., Barone F., Besra G., Miles K., Allen J. E., Gray M., Kollias G., Cunningham A. F., Withers D. R., Toellner K. M., Jones N. D., Veldhoen M., Nedospasov S. A., McKenzie A. N. J., Caamano J. H., Inflammation-induced formation of fat-associated lymphoid clusters. Nat. Immunol. 16, 819–828 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meza-Perez S., Randall T. D., Immunological functions of the omentum. Trends Immunol. 38, 526–536 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reed H. O., Wang L., Sonett J., Chen M., Yang J., Li L., Aradi P., Jakus Z., D’Armiento J., Hancock W. W., Kahn M. L., Lymphatic impairment leads to pulmonary tertiary lymphoid organ formation and alveolar damage. J. Clin. Invest. 129, 2514–2526 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas S. N., Rutkowski J. M., Pasquier M., Kuan E. L., Alitalo K., Randolph G. J., Swartz M. A., Impaired humoral immunity and tolerance in K14-VEGFR-3-Ig mice that lack dermal lymphatic drainage. J. Immunol. 189, 2181–2190 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bovay E., Sabine A., Prat-Luri B., Kim S., Son K., Willrodt A. H., Olsson C., Halin C., Kiefer F., Betsholtz C., Jeon N. L., Luther S. A., Petrova T. V., Multiple roles of lymphatic vessels in peripheral lymph node development. J. Exp. Med. 215, 2760–2777 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shimoda H., Bernas M. J., Witte M. H., Dysmorphogenesis of lymph nodes in Foxc2 haploinsufficient mice. Histochem. Cell Biol. 135, 603–613 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Mowat A. M., Agace W. W., Regional specialization within the intestinal immune system. Nat. Rev. Immunol. 14, 667–685 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Fonseca D. M., Hand T. W., Han S. J., Gerner M. Y., Glatman Zaretsky A., Byrd A. L., Harrison O. J., Ortiz A. M., Quinones M., Trinchieri G., Brenchley J. M., Brodsky I. E., Germain R. N., Randolph G. J., Belkaid Y., Microbiota-dependent sequelae of acute infection compromise tissue-specific immunity. Cell 163, 354–366 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bazigou E., Xie S., Chen C., Weston A., Miura N., Sorokin L., Adams R., Muro A. F., Sheppard D., Makinen T., Integrin-α9 is required for fibronectin matrix assembly during lymphatic valve morphogenesis. Dev. Cell 17, 175–186 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bernier-Latmani J., Petrova T. V., Intestinal lymphatic vasculature: Structure, mechanisms and functions. Nat. Rev. Gastroenterol. Hepatol. 14, 510–526 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Blikslager A. T., Moeser A. J., Gookin J. L., Jones S. L., Odle J., Restoration of barrier function in injured intestinal mucosa. Physiol. Rev. 87, 545–564 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Cash H. L., Whitham C. V., Behrendt C. L., Hooper L. V., Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science 313, 1126–1130 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin E. S., Belmont P. J., Sinnamon M. J., Richard L. G., Yuan J., Coffee E. M., Roper J., Lee L., Heidari P., Lunt S. Y., Goel G., Ji X., Xie Z., Xie T., Lamb J., Weinrich S. L., VanArsdale T., Bronson R. T., Xavier R. J., Vander Heiden M. G., Kan J. L., Mahmood U., Hung K. E., Development of a colon cancer GEMM-derived orthotopic transplant model for drug discovery and validation. Clin. Cancer Res. 19, 2929–2940 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cooper H. S., Murthy S. N., Shah R. S., Sedergran D. J., Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab. Investig. 69, 238–249 (1993). [PubMed] [Google Scholar]
- 33.Fox J. G., Ge Z., Whary M. T., Erdman S. E., Horwitz B. H., Helicobacter hepaticus infection in mice: Models for understanding lower bowel inflammation and cancer. Mucosal Immunol. 4, 22–30 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Le Roy T., Lécuyer E., Chassaing B., Rhimi M., Lhomme M., Boudebbouze S., Ichou F., Haro Barceló J., Huby T., Guerin M., Giral P., Maguin E., Kapel N., Gérard P., Clément K., Lesnik P., The intestinal microbiota regulates host cholesterol homeostasis. BMC Biol. 17, 94 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang S., Huang M., You X., Zhao J., Chen L., Wang L., Luo Y., Chen Y., Gut microbiota mediates the anti-obesity effect of calorie restriction in mice. Sci. Rep. 8, 13037 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson E. L., Heaver S. L., Waters J. L., Kim B. I., Bretin A., Goodman A. L., Gewirtz A. T., Worgall T. S., Ley R. E., Sphingolipids produced by gut bacteria enter host metabolic pathways impacting ceramide levels. Nat. Commun. 11, 2471 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Millar F. R., Summers C., Griffiths M. J., Toshner M. R., Proudfoot A. G., The pulmonary endothelium in acute respiratory distress syndrome: Insights and therapeutic opportunities. Thorax 71, 462–473 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Fiedler U., Reiss Y., Scharpfenecker M., Grunow V., Koidl S., Thurston G., Gale N. W., Witzenrath M., Rosseau S., Suttorp N., Sobke A., Herrmann M., Preissner K. T., Vajkoczy P., Augustin H. G., Angiopoietin-2 sensitizes endothelial cells to TNF-α and has a crucial role in the induction of inflammation. Nat. Med. 12, 235–239 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Wu J., Lewis A. H., Grandl J., Touch, tension, and transduction—The function and regulation of piezo ion channels. Trends Biochem. Sci. 42, 57–71 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Finegold D. N., Schacht V., Kimak M. A., Lawrence E. C., Foeldi E., Karlsson J. M., Baty C. J., Ferrell R. E., HGF and MET mutations in primary and secondary lymphedema. Lymphat. Res. Biol. 6, 65–68 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trapnell C., Cacchiarelli D., Grimsby J., Pokharel P., Li S., Morse M., Lennon N. J., Livak K. J., Mikkelsen T. S., Rinn J. L., The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cao J., Spielmann M., Qiu X., Huang X., Ibrahim D. M., Hill A. J., Zhang F., Mundlos S., Christiansen L., Steemers F. J., Trapnell C., Shendure J., The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eklund L., Kangas J., Saharinen P., Angiopoietin-Tie signalling in the cardiovascular and lymphatic systems. Clin. Sci. 131, 87–103 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kinmonth J. B., Wolfe J. H., Fibrosis in the lymph nodes in primary lymphoedema. Histological and clinical studies in 74 patients with lower-limb oedema. Ann. R. Coll. Surg. Engl. 62, 344–354 (1980). [PMC free article] [PubMed] [Google Scholar]
- 45.Nurmi H., Saharinen P., Zarkada G., Zheng W., Robciuc M. R., Alitalo K., VEGF-C is required for intestinal lymphatic vessel maintenance and lipid absorption. EMBO Mol. Med. 7, 1418–1425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bernier-Latmani J., Cisarovsky C., Demir C. S., Bruand M., Jaquet M., Davanture S., Ragusa S., Siegert S., Dormond O., Benedito R., Radtke F., Luther S. A., Petrova T. V., DLL4 promotes continuous adult intestinal lacteal regeneration and dietary fat transport. J. Clin. Invest. 125, 4572–4586 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hägerling R., Hoppe E., Dierkes C., Stehling M., Makinen T., Butz S., Vestweber D., Kiefer F., Distinct roles of VE-cadherin for development and maintenance of specific lymph vessel beds. EMBO J. 37, e98271 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sawane M., Kajiya K., Kidoya H., Takagi M., Muramatsu F., Takakura N., Apelin inhibits diet-induced obesity by enhancing lymphatic and blood vessel integrity. Diabetes 62, 1970–1980 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peyrin-Biroulet L., Gonzalez F., Dubuquoy L., Rousseaux C., Dubuquoy C., Decourcelle C., Saudemont A., Tachon M., Béclin E., Odou M. F., Neut C., Colombel J. F., Desreumaux P., Mesenteric fat as a source of C reactive protein and as a target for bacterial translocation in Crohn’s disease. Gut 61, 78–85 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Randolph G. J., Bala S., Rahier J. F., Johnson M. W., Wang P. L., Nalbantoglu I., Dubuquoy L., Chau A., Pariente B., Kartheuser A., Zinselmeyer B. H., Colombel J. F., Lymphoid aggregates remodel lymphatic collecting vessels that serve mesenteric lymph nodes in Crohn disease. Am. J. Pathol. 186, 3066–3073 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Allaire J. M., Crowley S. M., Law H. T., Chang S. Y., Ko H. J., Vallance B. A., The intestinal epithelium: Central coordinator of mucosal immunity. Trends Immunol. 39, 677–696 (2018). [DOI] [PubMed] [Google Scholar]
- 52.Durack J., Lynch S. V., The gut microbiome: Relationships with disease and opportunities for therapy. J. Exp. Med. 216, 20–40 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fan K. Q., Li Y. Y., Wang H. L., Mao X. T., Guo J. X., Wang F., Huang L. J., Li Y. N., Ma X. Y., Gao Z. J., Chen W., Qian D. D., Xue W. J., Cao Q., Zhang L., Shen L., Tong C., Zhong J. Y., Lu W., Lu L., Ren K. M., Zhong G., Wang Y., Tang M., Feng X. H., Chai R. J., Jin J., Stress-induced metabolic disorder in peripheral CD4+ T cells leads to anxiety-like behavior. Cell 179, 864–879.e19 (2019). [DOI] [PubMed] [Google Scholar]
- 54.Lu K., Knutson C. G., Wishnok J. S., Fox J. G., Tannenbaum S. R., Serum metabolomics in a Helicobacter hepaticus mouse model of inflammatory bowel disease reveal important changes in the microbiome, serum peptides, and intermediary metabolism. J. Proteome Res. 11, 4916–4926 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mager L. F., Burkhard R., Pett N., Cooke N. C. A., Brown K., Ramay H., Paik S., Stagg J., Groves R. A., Gallo M., Lewis I. A., Geuking M. B., McCoy K. D., Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 369, 1481–1489 (2020). [DOI] [PubMed] [Google Scholar]
- 56.Zarkada G., Heinolainen K., Makinen T., Kubota Y., Alitalo K., VEGFR3 does not sustain retinal angiogenesis without VEGFR2. Proc. Natl. Acad. Sci. U.S.A. 112, 761–766 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goumans M. J., Liu Z., ten Dijke P., TGF-β signaling in vascular biology and dysfunction. Cell Res. 19, 116–127 (2009). [DOI] [PubMed] [Google Scholar]
- 58.Derynck R., Budi E. H., Specificity, versatility, and control of TGF-β family signaling. Sci. Signal. 12, eaav5183 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mill C., George S. J., Wnt signalling in smooth muscle cells and its role in cardiovascular disorders. Cardiovasc. Res. 95, 233–240 (2012). [DOI] [PubMed] [Google Scholar]
- 60.Simon A. M., Goodenough D. A., Li E., Paul D. L., Female infertility in mice lacking connexin 37. Nature 385, 525–529 (1997). [DOI] [PubMed] [Google Scholar]
- 61.Ariani A., Carotti M., Gutierrez M., Bichisecchi E., Grassi W., Giuseppetti G. M., Salaffi F., Utility of an open-source DICOM viewer software (OsiriX) to assess pulmonary fibrosis in systemic sclerosis: Preliminary results. Rheumatol. Int. 34, 511–516 (2014). [DOI] [PubMed] [Google Scholar]
- 62.Nagahashi M., Yamada A., Aoyagi T., Allegood J., Wakai T., Spiegel S., Takabe K., Sphingosine-1-phosphate in the lymphatic fluid determined by novel methods. Heliyon 2, e00219 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Banan B., Wei Y., Simo O., Tso P., Abumrad N. N., Flynn C. R., Sundaresan S., Albaugh V. L., Intestinal lymph collection via cannulation of the mesenteric lymphatic duct in mice. J. Surg. Res. 260, 399–408 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim D., Langmead B., Salzberg S. L., HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; 1000 Genome Project Data Processing Subgroup , The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liao Y., Smyth G. K., Shi W., featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 67.Robinson M. D., Oshlack A., A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hansen K. D., Irizarry R. A., Wu Z., Removing technical variability in RNA-seq data using conditional quantile normalization. Biostatistics 13, 204–216 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Durinck S., Spellman P. T., Birney E., Huber W., Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 4, 1184–1191 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Benjamini Y., Hochberg Y., Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B. Methodol. 57, 289–300 (1995). [Google Scholar]
- 71.Ashburner M., Ball C. A., Blake J. A., Botstein D., Butler H., Cherry J. M., Davis A. P., Dolinski K., Dwight S. S., Eppig J. T., Harris M. A., Hill D. P., Issel-Tarver L., Kasarskis A., Lewis S., Matese J. C., Richardson J. E., Ringwald M., Rubin G. M., Sherlock G., Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat. Genet. 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.The Gene Ontology Consortium , The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 47, D330–D338 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W. M., Hao Y., Stoeckius M., Smibert P., Satija R., Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.van den Brink S. C., Sage F., Vértesy Á., Spanjaard B., Peterson-Maduro J., Baron C. S., Robin C., van Oudenaarden A., Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat. Methods 14, 935–936 (2017). [DOI] [PubMed] [Google Scholar]
- 75.Vanlandewijck M., He L., Mäe M. A., Andrae J., Ando K., Del Gaudio F., Nahar K., Lebouvier T., Laviña B., Gouveia L., Sun Y., Raschperger E., Räsänen M., Zarb Y., Mochizuki N., Keller A., Lendahl U., Betsholtz C., A molecular atlas of cell types and zonation in the brain vasculature. Nature 554, 475–480 (2018). [DOI] [PubMed] [Google Scholar]
- 76.Muhl L., Genové G., Leptidis S., Liu J., He L., Mocci G., Sun Y., Gustafsson S., Buyandelger B., Chivukula I. V., Segerstolpe Å., Raschperger E., Hansson E. M., Björkegren J. L. M., Peng X. R., Vanlandewijck M., Lendahl U., Betsholtz C., Single-cell analysis uncovers fibroblast heterogeneity and criteria for fibroblast and mural cell identification and discrimination. Nat. Commun. 11, 3953 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tirosh I., Izar B., Prakadan S. M., Wadsworth M. H., Treacy D., Trombetta J. J., Rotem A., Rodman C., Lian C., Murphy G., Fallahi-Sichani M., Dutton-Regester K., Lin J. R., Cohen O., Shah P., Lu D., Genshaft A. S., Hughes T. K., Ziegler C. G., Kazer S. W., Gaillard A., Kolb K. E., Villani A. C., Johannessen C. M., Andreev A. Y., Van Allen E. M., Bertagnolli M., Sorger P. K., Sullivan R. J., Flaherty K. T., Frederick D. T., Jané-Valbuena J., Yoon C. H., Rozenblatt-Rosen O., Shalek A. K., Regev A., Garraway L. A., Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yu G., Wang L. G., Han Y., He Q. Y., clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rapin A., Pattaroni C., Marsland B. J., Harris N. L., Microbiota analysis using an illumina MiSeq platform to sequence 16S rRNA genes. Curr. Protoc. Mouse Biol. 7, 100–129 (2017). [DOI] [PubMed] [Google Scholar]
- 80.Fuhrer T., Heer D., Begemann B., Zamboni N., High-throughput, accurate mass metabolome profiling of cellular extracts by flow injection-time-of-flight mass spectrometry. Anal. Chem. 83, 7074–7080 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/29/eabf4335/DC1