

Abstract
Purpose:
VEGF is upregulated in glioblastoma and may contribute to immunosuppression. We performed a phase II study of pembrolizumab alone or with bevacizumab in recurrent glioblastoma.
Patients and Methods:
Eighty bevacizumab-naïve patients with recurrent glioblastoma were randomized to pembrolizumab with bevacizumab (cohort A, n = 50) or pembrolizumab monotherapy (cohort B, n = 30). The primary endpoint was 6-month progression-free survival (PFS-6). Assessed biomarkers included evaluation of tumor programmed death-ligand 1 expression, tumor-infiltrating lymphocyte density, immune activation gene expression signature, and plasma cytokines. The neurologic assessment in neuro-oncology (NANO) scale was used to prospectively assess neurologic function.
Results:
Pembrolizumab alone or with bevacizumab was well tolerated but of limited benefit. For cohort A, PFS-6 was 26.0% [95% confidence interval (CI), 16.3–41.5], median overall survival (OS) was 8.8 months (95% CI, 7.7–14.2), objective response rate (ORR) was 20%, and median duration of response was 48 weeks. For cohort B, PFS-6 was 6.7% (95% CI, 1.7–25.4), median OS was 10.3 months (95% CI, 8.5–12.5), and ORR was 0%. Tumor immune markers were not associated with OS, but worsened OS correlated with baseline dexamethasone use and increased posttherapy plasma VEGF (cohort A) and mutant IDH1, unmethylated MGMT, and increased baseline PlGF and sVEGFR1 levels (cohort B). The NANO scale contributed to overall outcome assessment.
Conclusions:
Pembrolizumab was ineffective as monotherapy and with bevacizumab for recurrent glioblastoma. The infrequent radiographic responses to combinatorial therapy were durable. Tumor immune biomarkers did not predict outcome. Baseline dexamethasone use and tumor MGMT warrant further study as potential biomarkers in glioblastoma immunotherapy trials.
Introduction
Immunotherapeutic agents have transformed treatment for several cancers, although in most settings only a subset of patients benefit. In contrast, phase III studies of immunotherapies including vaccines (1) and inhibitors of immune checkpoint molecules (2), have not improved survival for glioblastoma, the most common primary adult malignant brain tumor. Explanations for these disappointing results remain unclear, but multiple complementary, immunosuppressive factors in the tumor microenvironment likely contribute (3).
Glioblastoma is a highly angiogenic cancer characterized histopathologically by vascular proliferation induced by high VEGF production by tumor cells. VEGF inhibition with bevacizumab, a humanized VEGF blocking antibody, significantly prolongs progression-free survival (PFS) but not overall survival (OS) for recurrent (4) and newly diagnosed patients (5, 6). VEGF may also contribute to tumor immunosuppression through several mechanisms (7) and preclinical studies (8–11) as well as a growing number of clinical reports (12–18) show that VEGF inhibition can enhance immunotherapy benefit for a variety of cancers.
Pembrolizumab, a high-affinity, humanized IgG4-κ inhibitory mAb of the immunomodulatory receptor programmed-death 1 (PD-1), is currently approved for the treatment of multiple malignancies based on its safety profile and efficacy. Potential biomarkers of therapeutic benefit with PD-1 blockade in other tumor types include tumor programmed death-ligand 1 (PD-L1) expression, tumor-infiltrating lymphocyte (TIL) density, and immune activation gene expression profile (GEP; ref. 19). Although a retrospective study reported modestly prolonged PFS in a minority of patients with recurrent high-grade glioma (20), the activity of pembrolizumab for glioblastoma has not been prospectively assessed. In addition, a heavily pretreated, multifocal patient with recurrent glioblastoma with a hypermutated tumor due to a germline POLE mutation achieved a dramatic response (21).
Our investigator-initiated, multicenter, randomized phase II study evaluated the hypothesis that the addition of VEGF blockade could enhance the antitumor activity of anti-PD-1 therapy for recurrent glioblastoma, and we also evaluated the single-agent activity of pembrolizumab for this indication. The study was not designed to directly compare the arms but rather to evaluate the efficacy of each arm independently. Because bevacizumab is known to prolong PFS in recurrent glioblastoma, the addition of bevacizumab in our study offered the possibility to extend pembrolizumab exposure to enhance its potential for therapeutic benefit. Our study incorporated a comprehensive analysis of tumor immune biomarkers and detailed analysis of circulating cytokines as well as prospective integration of the neurologic assessment in neuro-oncology (NANO) scale (22) to monitor neurologic function.
Patients and Methods
Study design and patients
This phase II, multicenter, open-label, two-cohort study enrolled adults with histologically confirmed glioblastoma at first or second relapse and a Karnofsky performance status of ≥70 who were ≥28 days from prior surgery and ≥12 weeks from prior radiation. Patients requiring >4 mg/day of dexamethasone or who received PD-1/PD-L1 or VEGF/VEGFR inhibitors were excluded. Additional eligibility criteria are listed in the Supplementary Data.
The study (NCT02337491) was compliant with the Declaration of Helsinki and guidelines on Good Clinical Practice. Ethics approval was obtained at all participating centers and all patients provided informed consent. The study sponsor was Dana-Farber Cancer Institute (Boston, MA).
Study procedures
Initially a safety lead-in using a 3+3 design was performed to establish the MTD or recommended phase II dose (RP2D) of pembrolizumab combined with bevacizumab because these two agents had not been combined previously for patients with glioblastoma. The initial dose level for the safety lead-in evaluated the established dose of each agent when administered as monotherapy including 200 mg of pembrolizumab i.v. every 3 weeks and 10 mg/kg of bevacizumab i.v. biweekly. Subsequent planned dose levels included deescalation of pembrolizumab dosing if the MTD was exceeded. Thereafter, eligible patients were randomized (5:3) to include a total of 50 patients to receive pembrolizumab plus bevacizumab (cohort A) and 30 patients to receive pembrolizumab monotherapy (cohort B). Patients who were treated at the RP2D during the safety lead-in were included in the intent-to-treat (ITT) population for cohort A. Each cohort accrued using a single-stage design. Treatment continued until tumor progression, unacceptable toxicity, noncompliance, or withdrawal of consent. Additional information on study procedures is provided in the Supplementary Data.
Toxicity was graded using Common Terminology Criteria for Adverse Events version 4.0. Investigator assessed response occurred every 6 weeks using clinical examination and contrast-enhanced MRI according to the radiologic assessment in neuro-oncology (RANO) criteria (23). Clinically stable patients with radiologic progression were allowed to continue study therapy for up to 3 months pending progression confirmation as per the immunotherapy response assessment criteria in neuro-oncology criteria including backdating to date of initial progression for those patients who had confirmation of progression noted on follow-up imaging (24). Patient neurologic function was assessed at baseline and at MRI assessments using the NANO scale (22).
Biomarker analyses
Immunocorrelative studies including PD-L1 expression, TIL density, immune activation GEP by nanostring, as well as plasma biomarkers were performed as described previously (25–29) and are detailed in the Supplementary Data.
Outcomes
The primary endpoint for each cohort, PFS-6, and secondary endpoints of objective response rate (ORR), PFS, OS, and overall safety, were assessed on the ITT population which included patients who were randomized to receive study therapy. Time-to-event analyses used the Kaplan–Meier method. Exploratory endpoints included association of outcome with tumor PD-L1 expression, TIL density and immune activation GEP, levels of circulating cytokines, as well as changes in NANO scale (22).
Statistical analysis
Cohort A included a safety lead-in with a planned deescalation of pembrolizumab dosing if more than one dose-limiting toxicity (DLT) was observed among the initial 6 patients. Upon determination of the MTD/RP2D in the safety lead-in, patients were randomized 5:3 to the combination of pembrolizumab plus bevacizumab (cohort A) or single-agent pembrolizumab (cohort B). Although a contemporaneous control arm of standard therapy was not included, success for each study arm was defined by a noteworthy improvement in primary endpoint relative to established historical benchmarks. For cohort A, accrual of 50 patients provided 87% power with an overall type I error of 0.05 to detect a PFS-6 ≤40% versus >60%. A PFS-6 rate of 40% was chosen based on phase II data for bevacizumab in the same target population (30, 31). For cohort B, accrual of 30 patients provided 84% power with an overall type I error of 0.05 to detect a PFS-6 ≤10% versus >30%. A 10% PFS-6 rate was chosen based on meta-analysis data for the same target population treated with salvage therapy, excluding antiangiogenic therapy (32).
Additional statistical methods are described in the Supplementary Data.
Results
Patients and treatment
No DLTs were noted during the safety lead-in, which established the RP2D at 200 mg of pembrolizumab i.v. every 3 weeks plus 10 mg/kg of bevacizumab biweekly. Eighty-eight patients enrolled and 80 were deemed eligible between February 2015 and June 2016. Fifty patients were treated on cohort A (including the 6 patients enrolled to the safety lead-in) and 30 on cohort B (Fig. 1). Summary demographics and baseline patient characteristics were comparable for both cohorts (Table 1).
Figure 1.
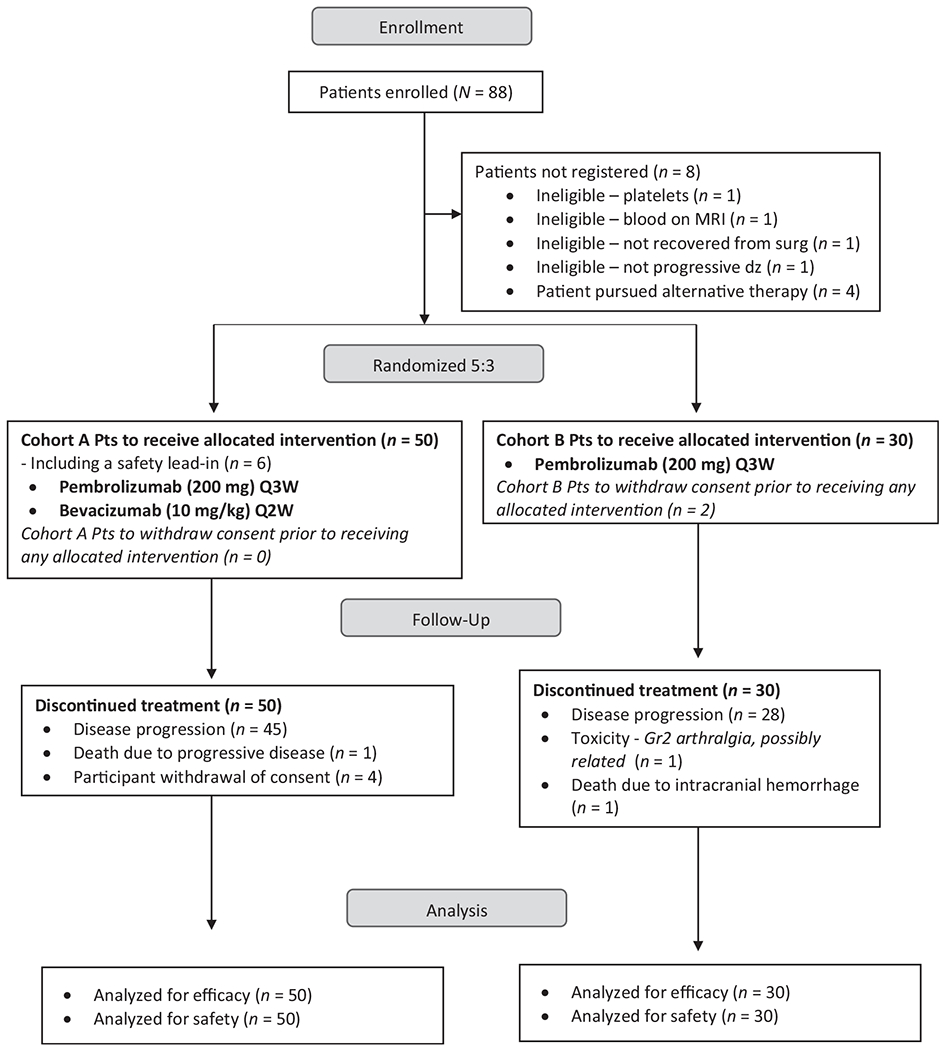
Study profile. CONSORT diagram showing the number of patients who were enrolled, treated with pembrolizumab plus bevacizumab (cohort A) or pembrolizumab (cohort B), discontinued treatment, and were analyzed for efficacy and safety.
Table 1.
Patient characteristics and study dispositiona.
| Characteristic | Cohort A: P+B (n = 50) | Cohort B: P alone (n = 30) | Total (n = 80) |
|---|---|---|---|
| Median age (years, IQR) | 52 (42–59) | 55 (42–62) | 53 (42–60) |
| <65 years (%) | 41 (82.0) | 26 (86.7) | 67 (83.8) |
| ≥65 years (%) | 9 (18.0) | 4 (13.3) | 13 (16.2) |
| Gender, male (%) | 35 (70.0) | 19 (63.3) | 54 (67.5) |
| Diagnosis at enrollment | |||
| GBM | 50 (100) | 30 (100) | 80 (100) |
| GS | 0 (0) | 0 (0) | 0 (0) |
| KPS (%) | |||
| 90–100 | 26 (52.0) | 13 (43.4) | 39 (48.8) |
| 80 | 17 (34.0) | 16 (53.3) | 33 (41.2) |
| 70 | 7 (14.0) | 1 (3.3) | 8 (10.0) |
| # prior PD | |||
| 1 | 35 (70.0) | 24 (80.0) | 59 (73.8) |
| 2 | 15 (30.0) | 6 (20.0) | 21 (26.2) |
| Resection prior to study | |||
| Gross total | 6 (12.0) | 7 (23.3) | 13 (16.2) |
| Subtotal | 2 (4.0) | 2 (6.7) | 4 (5.0) |
| Biopsy | 1 (2.0) | 5 (16.7) | 6 (7.5) |
| Not done | 41 (82.0) | 16 (53.3) | 57 (71.2) |
| Initial glioma diagnosis | |||
| Grade II glioma | 1 (2.0) | 1 (3.3) | 2 (2.5) |
| Grade III glioma | 2 (4.0) | 5 (16.7) | 7 (8.8) |
| Grade IV glioma | 47 (94.0) | 24 (80.0) | 71 (88.8) |
| Dexamethasone use | |||
| At study entry | 12 (24.0) | 7 (23.3) | 19 (23.8) |
| Required initiation after study start | 5/38 (13.2) | 5/23 (21.7) | 10/61 (16.4) |
| Required increase after study start | 0/12 (0) | 1/7 (14.3) | 1/19 (5.3) |
| Mean time from initial GBM diagnosis to enrollment in weeks (SD) | 75.9 (70.9) | 85.0 (78.2) | 79.3 (73.4) |
| MGMT status | |||
| Methylated | 20 (40.0) | 9 (30.0) | 29 (36.2) |
| Unmethylated | 17 (34.0) | 10 (33.3) | 27 (33.8) |
| Unknown | 13 (26.0) | 11 (36.7) | 24 (30.0) |
| IDH1 status | |||
| Mutant | 8 (16.0) | 4 (13.3) | 12 (15.0) |
| Wild-type | 35 (70.0) | 21 (70.0) | 56 (70.0) |
| Unknown | 7 (14.0) | 5 (16.7) | 12 (15.0) |
| Tumor source for immunocorrelatives | |||
| Archival | 31 (62.0) | 15 (50.0) | 46 (57.5) |
| Relapse | 15 (30.0) | 11 (36.7) | 26 (32.5) |
| None | 4 (8.0) | 4 (13.3) | 8 (10.0) |
| PD-L1 expression | |||
| Present | 22 (44.0) | 9 (30.0) | 31 (38.8) |
| Absent | 23 (46.0) | 16 (53.3) | 39 (48.8) |
| Not done | 5 (10.0) | 5 (16.7) | 10 (12.5) |
| TIL density | |||
| 0–1 | 25 (50.0) | 10 (33.3) | 35 (43.8) |
| 2–3 | 20 (40.0) | 15 (50.0) | 35 (43.8) |
| Not done | 5 (10.0) | 5 (16.7) | 10 (12.5) |
| Inflammatory gene expression signature | |||
| ≥ −0.3 | 4 (8.0) | 6 (20.0) | 10 (12.5) |
| > −0.3 | 36 (72.0) | 17 (56.7) | 53 (66.3) |
| Not done | 10 (20.0) | 7 (23.3) | 17 (21.3) |
| # study cycles completed | |||
| Median (IQR) | 3.0 (2.0–4.0) | 1.5 (1.0–3.0) | 2.0 (1.0–4.0) |
| Reason off study | |||
| PD | 45 (90.0) | 28 (93.3) | 73 (91.3) |
| Toxicity | 0 (0) | 1 (3.3) | 1 (1.2) |
| Consent withdrawal | 4 (8.0) | 0 (0) | 4 (4.9) |
| Death | 1 (2.0) | 1 (3.3) | 2 (2.4) |
| Current status | |||
| Dead | 45 (90.0) | 28 (93.3) | 73 (91.2) |
| Alive | 5 (10.0) | 2 (6.7) | 7 (8.8) |
Abbreviations: B, bevacizumab; GBM, glioblastoma; GS, gliosarcoma; IDH1, isocitrate dehydrogenase 1; IQR, interquartile range; KPS, Karnofsky performance score; MGMT, methylguanine methyltransferase; P, pembrolizumab; PD, progressive disease; PD-L1, programmed death-ligand 1; SD, standard deviation; TIL, tumor-infiltrating lymphocytes.
Percents are correct for denominators in each cell.
All patients have discontinued study therapy, including 91% due to progressive disease. Six percent electively discontinued study therapy after a median of 102.6 weeks (range, 54.0–114.0). The median number of completed cycles for cohorts A and B were 3.0 and 1.5, respectively. Seventy-three patients have died while 7 remain in active follow-up.
Efficacy
With a median follow-up for cohort A of 48.6 months [95% confidence interval (CI), 48.6–not reached], PFS-6 rate was 26% (95% CI, 16.3–41.5), while median PFS and OS were 4.1 months (95% CI, 2.8–5.5) and 8.8 months (95% CI, 7.7–14.2), respectively (Fig. 2). With a median follow-up for cohort B of 49.4 months (95% CI, 48.6–not reached), the PFS-6 rate was 6.7% (95% CI, 1.7–25.4), while the median PFS and OS were 1.43 months (95% CI, 1.4–2.7) and 10.3 months (95% CI, 8.5–12.5), respectively.
Figure 2.
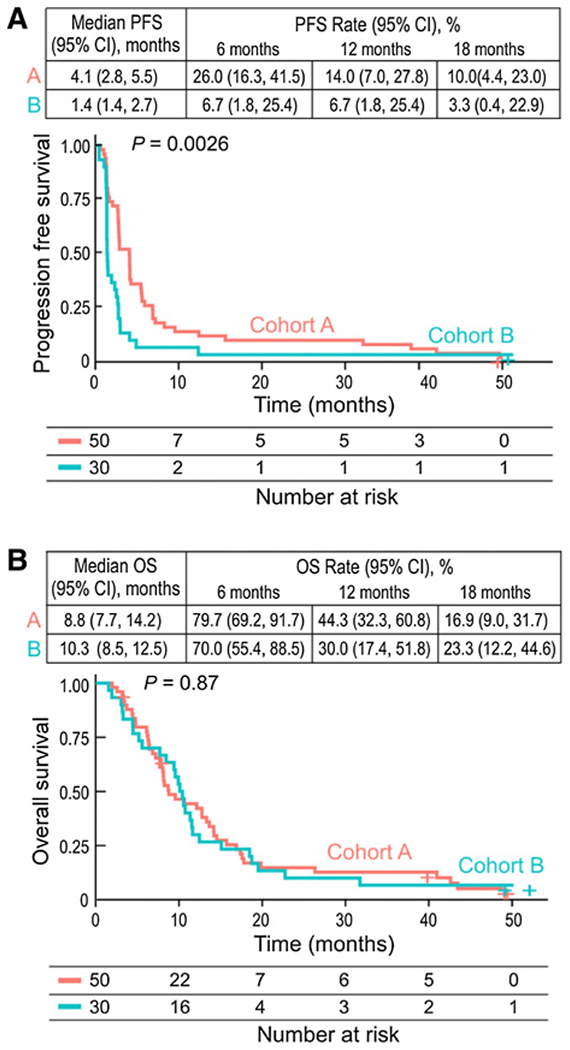
PFS and OS in all patients. A, The number of events; median PFS; PFS rates at 6, 12, and 18 months; and the Kaplan–Meier curve for PFS in all patients treated with nivolumab or nivolumab plus bevacizumab. B, The number of events; median OS; OS rates at 6, 12, and 18 months; and the Kaplan–Meier curve for OS per investigator assessment in patients treated with nivolumab or nivolumab plus bevacizumab. Symbols indicate censored observations. HRs and CIs were estimated using a Cox proportional hazards model. HR, hazard ratio; OS, overall survival; PFS, progression-free survival.
Maturity of follow-up allowed for detection of a possible “tail of the curve” for OS which was not observed for either study cohort. Ten patients, all on cohort A (20.0%), achieved a radiographic response by RANO criteria including nine partial responses and one complete response. Most responses occurred in patients not receiving dexamethasone (90%), were treated at first progression (80%), or had an IDH1 wild-type tumor (90%). Age, MGMT methylation, and tumor location were not associated with response. The median duration of radiographic response was 48 weeks (range, 10.9–174.4+). A summary of univariate factors and association with OS are summarized in Fig. 3. Poor survival was associated with baseline dexamethasone use (HR = 3.27; 95% CI, 1.6–6.7) for cohort A and IDH1 mutation (HR = 6.4; 95% CI, 1.7–23.3) or lack of MGMT promoter methylation (HR = 3.4; 95% CI, 1.1–10.5) for cohort B. Median OS for patients on baseline dexamethasone versus not for cohort A were 6.23 months (95% CI, 4.44–NA) and 12.8 (95% CI, 8.25–17.4; P = 0.0011), respectively. No difference in OS (P = 0.777) was observed among cohort B patients when stratified by baseline dexamethasone use. A trend for worse OS was also associated with enrollment at second relapse for cohort A but did not reach statistical significance (HR = 1.86; 95% CI, 0.97–3.59).
Figure 3.
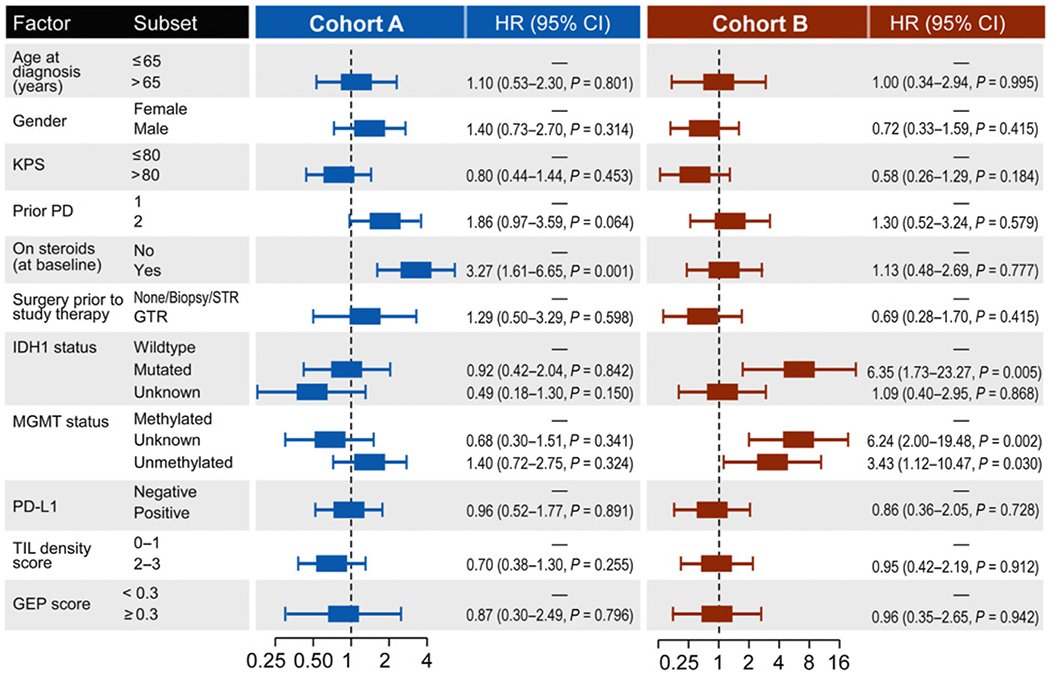
OS in patient subgroups. Forest plot of univariate HR for death of patient and tumor characteristics in the analysis of treatment effect in patient subgroups by cohort.
Safety
Most treatment-related adverse events (TRAE) were low grade (Table 2A), including immune-related adverse events (AE; Table 2B). No grade 5 TRAEs occurred. For cohort A, no grade 4 TRAEs occurred and hypertension was the most common grade 3 event (20%). For cohort B, a single grade 4 TRAE occurred and was cerebral edema that developed at tumor progression during a dexamethasone taper. Headache was the most common grade 3 event (10%). One patient who was treated on cohort B discontinued study therapy due to a TRAE (grade 2 arthralgia).
Table 2A.
Grade ≥2 adverse events at least possibly related to study therapy in ≥5% of patients by cohort.
| Cohort A: P+B (n = 50) |
Cohort B: P alone (n = 30) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Grade | 2 (%) | 3 (%) | 4 (%) | Total (%) | 2 (%) | 3 (%) | 4 (%) | Total (%) | |
| Event | |||||||||
| Hypertension | 15 (30) | 10 (20) | 0 | 25 (50) | 0 | 0 | 0 | 0 | |
| Headache | 4 (8) | 4 (8) | 0 | 8 (16) | 6 (20) | 3 (10) | 0 | 9 (30) | |
| Fatigue | 9 (18) | 0 | 0 | 9 (18) | 5 (17) | 0 | 0 | 5 (17) | |
| Infection | 6 (12) | 1 (2) | 0 | 7 (14) | 0 | 0 | 0 | 0 | |
| Proteinuria | 7 (14) | 0 | 0 | 7 (14) | 0 | 0 | 0 | 0 | |
| Arthralgia | 4 (8) | 0 | 0 | 4 (8) | 1 (3) | 0 | 0 | 1 (3) | |
| Diarrhea | 2 (4) | 1 (2) | 0 | 3 (6) | 1 (3) | 0 | 0 | 1 (3) | |
| Seizures | 3 (6) | 0 | 0 | 3 (6) | 2 (7) | 0 | 0 | 2 (7) | |
| Anorexia | 1 (2) | 0 | 0 | 1 (2) | 2 (7) | 0 | 0 | 2 (7) | |
| Hyperglycemia | 2 (4) | 0 | 0 | 2 (4) | 1 (3) | 1 (3) | 0 | 2 (7) | |
| Lymphopenia | 2 (4) | 0 | 0 | 2 (4) | 2 (7) | 0 | 0 | 2 (7) | |
Abbreviations: B, bevacizumab; n, number; P, pembrolizumab.
Table 2B.
Immune-related adverse events at least possibly related to study therapy.
| Event | Cohort Grade | A: P+B | B: P alone | A: P+B | B: P alone | A: P+B | B: P alone | A: P+B | B: P alone |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | ||||||
| Abdominal pain | 3 (6%) | 0 | 1 (2%) | 0 | 0 | 0 | 0 | 0 | |
| Alkaline phosphatase elevation | 2 (4%) | 1 (3%) | 0 | 0 | 0 | 0 | 0 | 0 | |
| ALT elevation | 7 (14%) | 1 (3%) | 0 | 0 | 1 (2%) | 0 | 0 | 0 | |
| AST elevation | 7 (14%) | 0 | 0 | 0 | 1 (2%) | 0 | 0 | 0 | |
| Arthralgia | 6 (12%) | 0 | 4 (8%) | 1 (3%) | 0 | 0 | 0 | 0 | |
| Bilirubin increase | 1 (2%) | 0 | 1 (2%) | 0 | 0 | 0 | 0 | 0 | |
| Cerebral edema | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (3%) | |
| Colitis | 0 | 0 | 1 (2%) | 0 | 0 | 0 | 0 | 0 | |
| Conjunctivitis | 1 (2%) | 1 (3%) | 0 | 0 | 0 | 0 | 0 | 0 | |
| Creatinine increase | 2 (4%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Diarrhea | 7 (14%) | 0 | 2 (4%) | 1 (3%) | 0 | 0 | 0 | 0 | |
| Dyspnea | 1 (2%) | 0 | 1 (2%) | 0 | 0 | 0 | 0 | 0 | |
| Hyperglycemia | 5 (10%) | 0 | 2 (4%) | 1 (3%) | 0 | 1 (3%) | 0 | 0 | |
| Hyperthyroidism | 4 (8%) | 0 | 1 (2%) | 0 | 0 | 0 | 0 | 0 | |
| Hypothyroidism | 3 (6%) | 1 (3%) | 2 (4%) | 1 (3%) | 0 | 0 | 0 | 0 | |
| Infusion reaction | 0 | 0 | 1 (2%) | 0 | 0 | 0 | 0 | 0 | |
| Myalgia | 6 (12%) | 0 | 1 (2%) | 0 | 0 | 0 | 0 | 0 | |
| Rash | 5 (10%) | 1 (3%) | 1 (2%) | 0 | 0 | 0 | 0 | 0 | |
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; B, bevacizumab; P, pembrolizumab.
Tumor immune biomarker analyses
Tumor was available from either original diagnosis (archival; n = 46; 58%) or relapse prior to study enrollment (n = 26; 33%) for 72 patients (90%; Table 1). There was no difference in OS for patients with archival versus relapsed tumor samples (P = 0.34). PD-L1 expression, detected in 31 tumors (39%), correlated with PFS for cohort B but not cohort A, and was not associated with OS for either cohort (Fig. 3). TIL density, which was low (IHC score 0–1) in 35 tumors (44%) and increased (IHC score 2–3) in 35 tumors (44%), was not associated with outcome for either cohort. Immune activation GEP was evaluable for 63 tumors (79%), including 40 (80%) from cohort A and 23 (77%) from cohort B. GEP scores below −0.3 predict rapid progression with pembrolizumab whereas scores above −0.3 associate with variable progression times including a higher likelihood of longer PFS (26, 28). Of note, 66% of our study patients had a GEP score below −0.3. The median GEP score was higher among relapsed compared with archival tumors but distributions were overlapping (Supplementary Fig. S1; P = 0.62). Similarly, median GEP score trended higher with increased PD-L1 expression (P = 0.11) and TIL score (P = 0.52) but wide, overlapping distributions without statistical significance were observed (Supplementary Fig. S2A and S2B). For both cohorts, GEP score was not associated with ORR, PFS, or OS.
Plasma biomarker analyses
Plasma samples were available from 32 cohort A (64%) and 19 cohort B patients (63%). Posttreatment (day 84), cohort A had decreased ANG-2 and VEGF levels and increased PlGF levels, whereas no significant changes were noted for cohort B (Supplementary Table S1). Baseline VEGF was higher for cohort A versus cohort B (P = 0.0052). When evaluated for correlation with outcome, elevated baseline PlGF and sVEGFR1 for cohort B and posttreatment VEGF for cohort A correlated with poor OS (Supplementary Table S2).
Integration of NANO
The overall NANO compliance rate for all visits was 94%. Seven patients (9%) lacked a baseline NANO evaluation and were excluded from NANO analyses. Fifteen patients (19%) lacked an end of treatment NANO evaluation. At baseline, 35 patients (43%) had a normal NANO examination. Two NANO domains (strength and language) accounted for most changes in neurologic function during study treatment. Eighteen patients (25%, 9 per cohort) met NANO criteria for progression, including 2 without radiographic progression. Three patients (4%, all in cohort A) had NANO response and SD by MRI; these patients survived between 13.0 and 49.6 months. NANO assessment before cycle 3 correlated with RANO response (P = 0.011) and change in either Karnofsky performance score (KPS; P = 0.002) or dexamethasone requirement (P = 0.007). Patients with NANO progression at this time point had poorer median survival (9.57 months) compared with those without NANO progression (10.65 months), but this trend did not achieve statistical significance (P = 0.2).
Discussion
Glioblastoma has emerged as a profoundly immunotherapy refractory tumor that likely reflects multiple, complementary mechanisms of tumor-induced immunosuppression (3, 33–36). VEGF, which is highly expressed in glioblastoma and drives angiogenesis, also contributes to tumor immunosuppression via multiple mechanisms (7, 37–43). Clinical benefit of bevacizumab to block VEGF for glioblastoma is limited to an improvement in PFS but not OS (4–6). Our study evaluated the hypothesis that concurrent VEGF blockade may enhance the antitumor activity of pembrolizumab for patients with recurrent glioblastoma. The rationale underlying our study hypothesis is based on extensive preclinical (8–11) and clinical (12–14) data in multiple cancer types that has led to the approval of five such combinations for extracranial malignancies in the past 18 months (15–18, 44).
Our study data demonstrate that standard dosing of bevacizumab and pembrolizumab in combination failed to improve both PFS or OS relative to bevacizumab monotherapy indicating that a definitive study randomizing patients to bevacizumab versus bevacizumab plus anti-PD-1 therapy is not indicated (2). Among patients who achieved a response, however, its duration was significantly longer than that associated with bevacizumab monotherapy. This observation aligns with recently reported data indicating that response to anti-PD-1 therapy with nivolumab, although noted in a small percentage of patients with recurrent glioblastoma, was of noteworthy duration (2). In addition, our study confirmed that single-agent anti-PD-1 therapy was ineffective for recurrent glioblastoma with most patients progressing rapidly, within two cycles of therapy (2).
The observed failure of bevacizumab to improve outcome when combined with pembrolizumab in our study likely involves a number of possible explanations. First, our study results argue that the potential complementary benefit of dual blockade of VEGF and PD(L)-1 is dependent on tumor context and not applicable to glioblastoma. Second, our trial used an established, and relatively high bevacizumab dose, which is feasible due to its safety profile. However, preclinical studies suggest that lower dosing of antiangiogenics may induce normalization of the tumor vasculature with improved antitumor immune responses and survival whereas higher doses do not (9). Relatively high doses of anti-VEGF drugs can augment preexisting hypoxia within the tumor microenvironment that could worsen immunosuppression (7, 45, 46) and promote an intratumoral influx of immunosuppressive cells (47, 48). Third, antiangiogenic agents may also decrease intratumoral penetration of therapeutic mAbs such as cetuximab or trastuzumab (49), although it is unknown whether benefit of anti-PD-1 blocking antibodies requires intratumoral distribution in glioblastoma tumors. It is also possible that timing of anti-PD-1 therapy may be relevant based on two recent reports demonstrating that neoadjuvant anti-PD-1 therapy can trigger infiltration and activation of TILs in the tumor microenvironment which may enhance efficacy (50, 51).
Immune biomarkers including PD-L1 expression, TIL analysis, and immune activation GEP have been shown to predict a likelihood of therapeutic benefit with immune checkpoint blockade for some malignancies (26, 28, 52), but have not been previously evaluated prospectively in a comprehensive manner for glioblastoma. We noted that PD-L1 expression and increased TIL infiltrate were present in nearly 50% of tumor samples in our study but did not correlate with OS. Immune activation GEP was markedly low in most glioblastoma tumors in our study and also did not correlate with outcome. Insufficient tumor material precluded evaluation of tumor mutational burden or microsatellite instability status as potential biomarkers; however, a recent retrospective study indicates that tumor hypermutation following temozolomide chemotherapy in patients with recurrent glioblastoma was not associated with improved benefit following immune checkpoint blockade (53). Our findings indicate that tumor PD-L1 expression, TIL infiltrate, and immune activation GEP may not be informative to predict therapeutic benefit of anti-PD-1 therapy for patients with recurrent glioblastoma. A limitation of these analyses is that some tumor samples were from original diagnosis; however, archival tumor samples or a mixture of archival and at-treatment samples, as was incorporated for our study, have been utilized effectively for immunocorrelative analyses in solid tumor studies (54, 55) including glioblastoma (56).
Our study also incorporated a detailed analysis of clinical and tumor-associated biomarkers. Baseline dexamethasone use (cohort A) as well as lack of tumor MGMT methylation and IDH1 mutation (cohort B) correlated with poorer OS. Baseline dexamethasone use and tumor MGMT methylation status were also associated with outcome among patients with recurrent glioblastoma treated with nivolumab on a randomized phase III study (2). The detrimental effect of dexamethasone observed in our study may have been due to its ability to quantitatively and qualitatively decrease effector immune cells in patients with glioblastoma (57) or it may have reflected other relevant confounding factors such as a larger tumor burden. The effect of IDH1 mutation status should be interpreted cautiously due the small number of patients with IDH1-mutant tumors enrolled on cohort B (n = 4) and that the median time from original glioblastoma diagnosis to study enrollment was much longer in the IDH1-mutant patients compared with wild-type (1,238 days vs. 783 days). Our analysis of plasma angiokines revealed that elevated baseline PlGF and sVEGFR1 (cohort B) and posttherapy VEGF (cohort A) levels correlated with poorer survival. These inducible factors may reflect increased tumor hypoxia at baseline (in the anti-PD-1 therapy alone group) and posttreatment (in the combination group), which can contribute to immunosuppression (45, 46). Overall our findings are limited by sample size, thus are hypothesis generating and may warrant further study.
The NANO scale was developed by a multidisciplinary panel of neuro-oncology experts as an objective, user-friendly measure of neurologic function that provides a broader neurologic assessment than measures of function traditionally utilized for patients with glioblastoma such as KPS or the mini–mental status exam (22). We demonstrated that NANO, which has not been previously evaluated in a neuro-oncology clinical trial, can be performed efficiently in a multicenter trial as indicated by an overall acceptable compliance rate (94%); however, the lack of baseline and end-of-study assessments in some patients precluded accurate assessment of whether neurologic changes correlated with outcome. Nonetheless, in general, we observed preservation of baseline neurologic status by NANO during treatment in nonradiographic progressors and neurologic decline among radiographic progressors although this was impacted by tumor anatomic location relative to functional cortex as expected.
Several limitations of this study exist. Although our study did not incorporate a standard-of-care control arm, failure to generate a signal of improved outcome relative to established, historical benchmarks, argues that further investigation of our study regimen relative to a contemporaneous control arm is not justified. We incorporated a comprehensive analysis of relevant immunocorrelative biomarkers and demonstrated that these were not informative among patients with recurrent glioblastoma; however, the sample size for these analyses was relatively small and included some archival tumor samples which may not have reflected the tumor microenvironment at the time of study therapy. Finally, missed key assessment time points including those at baseline and end of study likely diminished our ability to fully assess the utility of NANO and future studies incorporating NANO should strive to improve compliance especially at these critical points of investigational therapy.
Overall, our results do not support future studies of anti-PD-1 monotherapy or in combination with bevacizumab administered using the FDA-approved, established dosing schedule. PD-L1, TIL density, and GEP score do not appear to be useful biomarkers among patients with recurrent glioblastoma undergoing anti-PD-1 therapy. Ongoing clinical trials (NCT02336165 and NCT03452579) are assessing whether a reduced bevacizumab dosing schedule may improve outcome with anti–PD-1/PD-L1 therapy for patients with glioblastoma. Future efforts should consider targeting alternative potential modulators of immunosuppression in glioblastoma tumors.
Supplementary Material
Translational Relevance.
VEGF promotes angiogenesis in glioblastoma but there is accumulating evidence that also implicates VEGF as a mediator of tumor-induced immunosuppression. Preclinical and clinical data in some cancers support a hypothesis that dual VEGF and immune checkpoint blockade may enhance antitumor immune responses and has led to approval of five such combinations for extracranial malignancies. Our randomized phase II trial in patients with recurrent glioblastoma demonstrates that bevacizumab administered using an established dosing schedule fails to improve outcome when added to programmed-death 1 (PD-1) blockade, which is ineffective as monotherapy. In addition, we show that tumor programmed death-ligand 1 expression, tumor-infiltrating lymphocyte density, and immune gene activation score do not identify patients more likely to benefit from anti-PD-1 therapy. Finally, better assessment of quality of life including preservation of neurologic function is needed for patients with glioblastoma. We demonstrate the feasibility of integrating the neurologic assessment in neuro-oncology scale to prospectively assess neurologic function in a clinical trial.
Acknowledgments
We thank the Ben and Catherine Ivy Foundation and the Jennifer Oppenheimer Cancer Research Initiative for their support of this clinical trial, Anna Khachatryan for outstanding technical support with biomarker analyses, Kathleen Shedlock, ANP for substantial collaborative input, and Debbie Ferreira for administrative support. We gratefully acknowledge Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. for provision of pembrolizumab and financial support of the conduct of this study. We also thank the study patients, their families, and all the participating investigators and research staff. This research was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA008748. L. Nayak acknowledges support from the Leukemia and Lymphoma Society Scholar in Clinical Research Award (LLS Grant ID: 2322–19). R.K. Jain acknowledges support from the NIH (R35CA197742, R01CA208205, U01CA224173), the Ludwig Center at Harvard, the Jane’s Trust Foundation, National Foundation for Cancer Research, and the Advanced Medical Research Foundation.
We gratefully acknowledge the following organizations for funding support: The Ben and Catherine Ivy Foundation; The Jennifer Oppenheimer Cancer Research Initiative; Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.; and NIH/NCI Cancer Center Support Grant P30 CA008748.
Authors’ Disclosures
L. Nayak reports grants from The Leukemia and Lymphoma Society and NIH/NCI outside the submitted work. K. Peters reports grants from Merck during the conduct of the study. J.L. Clarke reports other from Agios, Merck, and Novartis outside the submitted work. J.T. Jordan reports personal fees from Navio Theragnostics Inc., CereXis Inc, Health2047 Inc., and Neurofibromatosis Network, and grants from The Burke Foundation outside the submitted work. J. de Groot reports other from Carthera, HiaHe Pharma, Taiho Pharma, Janssen Global Services, Prelude Therapeutics, Merck Sharp & Dohme Co., Kiyatec, Novartis, and Debiopharm Therapeutics Inc., as well as personal fees from ResTORbio Inc., Roche, Magnolia Innovation LLC, Insightec, Syneos Health, Bioasis Technologies Inc., GenomiCare, Merk & Co., Del Mar Pharmaceuticals, and Tocagen outside the submitted work. L. Nghiemphu reports grants from NIH and Novartis outside the submitted work. T. Kaley reports nonfinancial support from NIH during the conduct of the study. H. Colman reports grants from Merck during the conduct of the study, as well as personal fees from Best Doctors/Teladoc, Karyopharm Therapeutics, Private Health, Orbus, Bayer, Forma Therapeutics, and Adastra Pharmaceuticals outside the submitted work. S. Gaffey is employed by Foundation Medicine, Inc. J.H. Yearley reports a patent 10647771 issued to Merck Sharp & Dohme and a patent 0241115 issued to Merck Sharp & Dohme. D.G. Duda reports grants and personal fees from Bayer and BMS; grants from Exelixis; and personal fees from Surface Oncology and Simcere during the conduct of the study. R.K. Jain reports grants from NIH, National Foundation for Cancer Research, The Ludwig Center at Harvard, The Jane’s Trust Foundation, and The Advanced Medical Research Foundation during the conduct of the study, as well as personal fees and other from Amgen, Chugai, Ophthotech, Merck, SPARC, SynDevRx, XTuit, Enlight, Accurius Therapeutics, Tekla Healthcare Investors, Tekla Life Sciences Investors, Tekla Healthcare Opportunities Fund, and Tekla World Healthcare Fund outside the submitted work. P.Y. Wen reports personal fees and other from Agios, AstraZeneca/Medimmune, Vascular Biogenics, and VBI Vaccines; other from Beigene, Eli Lily, Genentech/Roche, Kazia, MedicaNova, Merck, Novartis, and Oncoceutics; personal fees from Bayer, Blue Earth Diagnostics, Immunomic Therapeutics, Karyopharm, Kiyatec, Puma, Taiho, Deciphera, Tocagen, Voyager, QED, Imvax, and Elevate io; and other from Prime Oncology, UpToDate, and Elsevier during the conduct of the study. D.A. Reardon reports other from Merck during the conduct of the study; Dr. Reardon also reports other from Acerta Pharmaceuticals, Incyte, Omnio, and Tragara; personal fees and other from Agenus, Celldex, EMD Serono, and Inovio; and personal fees from Abbvie, Advantagene, Amgen, Bayer, Bristol-Myers Squibb, DelMar, Genentech/Roche, Imvax, Medicenna, Merck, Merck KGaA, Monteris, Novocure, Oncours, Oxigene, Regeneron, Stemline, Sumitono Dainippon Pharma, Taiho Oncology Inc, and Boston Biomedical outside the submitted work. No disclosures were reported by the other authors.
Footnotes
Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Prior presentation: 2018 American Society of Clinical Oncology Annual Meeting.
References
- 1.Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol 2017;18:1373–85. [DOI] [PubMed] [Google Scholar]
- 2.Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A, et al. Effect of nivolumab vs. bevacizumab in patients with recurrent glioblastoma: the CheckMate 143 phase 3 randomized clinical trial. JAMA Oncol 2020;6:1003–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mangani D, Weller M, Roth P. The network of immunosuppressive pathways in glioblastoma. Biochem Pharmacol 2017;130:1–9. [DOI] [PubMed] [Google Scholar]
- 4.Wick W, Gorlia T, Bendszus M, Taphoorn M, Sahm F, Harting I, et al. Lomustine and bevacizumab in progressive glioblastoma. N Engl J Med 2017;377:1954–63. [DOI] [PubMed] [Google Scholar]
- 5.Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med 2014;370:709–22. [DOI] [PubMed] [Google Scholar]
- 6.Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med 2014;370:699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol 2018;15:325–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allen E, Jabouille A, Rivera LB, Lodewijckx I, Missiaen R, Steri V, et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci Transl Med 2017;9:eaak9679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci U S A 2012;109:17561–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res 2010;70:6171–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bocca P, Di Carlo E, Caruana I, Emionite L, Cilli M, De Angelis B, et al. Bevacizumab-mediated tumor vasculature remodelling improves tumor infiltration and antitumor efficacy of GD2-CAR T cells in a human neuroblastoma preclinical model. Oncoimmunology 2017;7:e1378843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hodi FS, Lawrence D, Lezcano C, Wu X, Zhou J, Sasada T, et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res 2014;2:632–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wallin JJ, Bendell JC, Funke R, Sznol M, Korski K, Jones S, et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun 2016;7:12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med 2018;378:2288–301. [DOI] [PubMed] [Google Scholar]
- 15.Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med 2020;382:1894–905. [DOI] [PubMed] [Google Scholar]
- 16.Rini BI, Plimack ER, Stus V, Gafanov R, Hawkins R, Nosov D, et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med 2019;380:1116–27. [DOI] [PubMed] [Google Scholar]
- 17.Makker V, Rasco D, Vogelzang NJ, Brose MS, Cohn AL, Mier J, et al. Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer: an interim analysis of a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol 2019;20:711–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reck M, Mok TSK, Nishio M, Jotte RM, Cappuzzo F, Orlandi F, et al. Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir Med 2019;7:387–401. [DOI] [PubMed] [Google Scholar]
- 19.Lu S, Stein JE, Rimm DL, Wang DW, Bell JM, Johnson DB, et al. Comparison of biomarker modalities for predicting response to PD-1/PD-L1 checkpoint blockade: a systematic review and meta-analysis. JAMA Oncol 2019;5:1195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reiss SN, Yerram P, Modelevsky L, Grommes C. Retrospective review of safety and efficacy of programmed cell death-1 inhibitors in refractory high grade gliomas. J Immunother Cancer 2017;5:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johanns TM, Miller CA, Dorward IG, Tsien C, Chang E, Perry A, et al. Immunogenomics of hypermutated glioblastoma: a patient with germline POLE deficiency treated with checkpoint blockade immunotherapy. Cancer Discov 2016;6:1230–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nayak L, DeAngelis LM, Brandes AA, Peereboom DM, Galanis E, Lin NU, et al. The Neurologic Assessment in Neuro-Oncology (NANO) scale: a tool to assess neurologic function for integration into the Response Assessment in Neuro-Oncology (RANO) criteria. Neuro Oncol 2017;19:625–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wen PY, Macdonald DR, Reardon DA, Cloughesy TF, Sorensen AG, Galanis E, et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol 2010;28:1963–72. [DOI] [PubMed] [Google Scholar]
- 24.Okada H, Weller M, Huang R, Finocchiaro G, Gilbert MR, Wick W, et al. Immunotherapy response assessment in neuro-oncology: a report of the RANO working group. Lancet Oncol 2015;16:e534–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dolled-Filhart M, Locke D, Murphy T, Lynch F, Yearley JH, Frisman D, et al. Development of a prototype immunohistochemistry assay to measure programmed death ligand-1 expression in tumor tissue. Arch Pathol Lab Med 2016;140:1259–66. [DOI] [PubMed] [Google Scholar]
- 26.Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 2017;127:2930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reardon DA, Lassman AB, Schiff D, Yunus SA, Gerstner ER, Cloughesy TF, et al. Phase 2 and biomarker study of trebananib, an angiopoietin-blocking peptibody, with and without bevacizumab for patients with recurrent glioblastoma. Cancer 2018;124:1438–48. [DOI] [PubMed] [Google Scholar]
- 28.Ott PA, Bang YJ, Piha-Paul SA, Razak ARA, Bennouna J, Soria JC, et al. T-cell-inflamed gene-expression profile, programmed death ligand 1 expression, and tumor mutational burden predict efficacy in patients treated with pembrolizumab across 20 cancers: KEYNOTE-028. J Clin Oncol 2019;37:318–27. [DOI] [PubMed] [Google Scholar]
- 29.Habra MA, Stephen B, Campbell M, Hess K, Tapia C, Xu M, et al. Phase II clinical trial of pembrolizumab efficacy and safety in advanced adrenocortical carcinoma. J Immunother Cancer 2019;7:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kreisl TN, Kim L, Moore K, Duic P, Royce C, Stroud I, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol 2009;27:740–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol 2009;27:4733–40. [DOI] [PubMed] [Google Scholar]
- 32.Wu W, Lamborn KR, Buckner JC, Novotny PJ, Chang SM, O’Fallon JR, et al. Joint NCCTG and NABTC prognostic factors analysis for high-grade recurrent glioma. Neuro Oncol 2010;12:164–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rutledge WC, Kong J, Gao J, Gutman DA, Cooper LA, Appin C, et al. Tumor-infiltrating lymphocytes in glioblastoma are associated with specific genomic alterations and related to transcriptional class. Clin Cancer Res 2013;19:4951–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, et al. T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin Cancer Res 2018;24:4175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woroniecka KI, Rhodin KE, Chongsathidkiet P, Keith KA, Fecci PE. T-cell dysfunction in glioblastoma: applying a new framework. Clin Cancer Res 2018;24:3792–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chongsathidkiet P, Jackson C, Koyama S, Loebel F, Cui X, Farber SH, et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat Med 2018;24:1459–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gabrilovich D, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S, et al. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood 1998;92:4150–66. [PubMed] [Google Scholar]
- 38.Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med 2014;20:607–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Terme M, Pernot S, Marcheteau E, Sandoval F, Benhamouda N, Colussi O, et al. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res 2013;73:539–49. [DOI] [PubMed] [Google Scholar]
- 40.Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med 2015;212:139–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sitkovsky MV, Kjaergaard J, Lukashev D, Ohta A. Hypoxia-adenosinergic immunosuppression: tumor protection by T regulatory cells and cancerous tissue hypoxia. Clin Cancer Res 2008;14:5947–52. [DOI] [PubMed] [Google Scholar]
- 42.Ohm JE, Gabrilovich DI, Sempowski GD, Kisseleva E, Parman KS, Nadaf S, et al. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood 2003;101:4878–86. [DOI] [PubMed] [Google Scholar]
- 43.Allen E, Missiaen R, Bergers G. Therapeutic induction of high endothelial venules (HEVs) to enhance T-cell infiltration in tumors. Oncotarget 2017;8:99207–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Motzer RJ, Penkov K, Haanen J, Rini B, Albiges L, Campbell MT, et al. Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med 2019;380:1103–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Palazon A, Aragones J, Morales-Kastresana A, de Landazuri MO, Melero I. Molecular pathways: hypoxia response in immune cells fighting or promoting cancer. Clin Cancer Res 2012;18:1207–13. [DOI] [PubMed] [Google Scholar]
- 46.Wei J, Wu A, Kong LY, Wang Y, Fuller G, Fokt I, et al. Hypoxia potentiates glioma-mediated immunosuppression. PLoS One 2011;6:e16195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu XD, Hoang A, Zhou L, Kalra S, Yetil A, Sun M, et al. Resistance to antiangiogenic therapy is associated with an immunosuppressive tumor microenvironment in metastatic renal cell carcinoma. Cancer Immunol Res 2015;3:1017–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu-Emerson C, Snuderl M, Kirkpatrick ND, Goveia J, Davidson C, Huang Y, et al. Increase in tumor-associated macrophages after antiangiogenic therapy is associated with poor survival among patients with recurrent glioblastoma. Neuro Oncol 2013;15:1079–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arjaans M, Oosting SF, Schroder CP, de Vries EG. Bevacizumab-induced vessel normalization hampers tumor uptake of antibodies–response. Cancer Res 2013;73:7147–8. [DOI] [PubMed] [Google Scholar]
- 50.Huang AC, Orlowski RJ, Xu X, Mick R, George SM, Yan PK, et al. A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat Med 2019;25:454–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med 2019;25:477–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014;515:563–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Touat M, Li YY, Boynton AN, Spurr LF, Iorgulescu JB, Bohrson CL, et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 2020;580:517–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Prat A, Navarro A, Pare L, Reguart N, Galvan P, Pascual T, et al. Immune-related gene expression profiling after PD-1 blockade in non-small cell lung carcinoma, head and neck squamous cell carcinoma, and melanoma. Cancer Res 2017;77:3540–50. [DOI] [PubMed] [Google Scholar]
- 55.Higgs BW, Morehouse CA, Streicher K, Brohawn PZ, Pilataxi F, Gupta A, et al. Interferon gamma messenger RNA signature in tumor biopsies predicts outcomes in patients with non-small cell lung carcinoma or urothelial cancer treated with durvalumab. Clin Cancer Res 2018;24:3857–66. [DOI] [PubMed] [Google Scholar]
- 56.Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med 2019;25:462–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gustafson MP, Lin Y, New KC, Bulur PA, O’Neill BP, Gastineau DA, et al. Systemic immune suppression in glioblastoma: the interplay between CD14+HLA-DRlo/neg monocytes, tumor factors, and dexamethasone. Neuro Oncol 2010;12:631–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.