

Abstract
Metal induced free radicals are important mediators of neurotoxicity in several neurodegenerative conditions such as Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease. Similar evidence is now emerging for prion diseases, a group of neurodegenerative disorders of humans and animals. The main pathogenic agent in all prion disorders is PrP-scrapie (PrPSc), a β-sheet rich isoform of a normal cell surface glycoprotein known as the prion protein (PrPC). Deposits of PrPSc in the brain parenchyma are believed to induce neurotoxicity through poorly understood mechanisms. Recent reports suggest that imbalance of brain metal homeostasis is a significant cause of PrPSc-associated neurotoxicity, though the underlying mechanisms are difficult to explain based on existing information. Proposed hypotheses include a functional role for PrPC in metal metabolism, and loss of this function due to aggregation to the disease associated PrPSc form as the cause of brain metal imbalance. Other views suggest gain of toxic function by PrPSc due to sequestration of PrPC-associated metals within the aggregates, resulting in the generation of redox-active PrPSc complexes. The physiological implications of some PrPC-metal interactions are known, while others are still unclear. The pathological implications of PrPC-metal interaction include metal-induced oxidative damage, and in some instances conversion of PrPC to a PrPSc-like form. Despite its significance, only limited information is available on PrP-metal interaction and its implications on prion disease pathogenesis. In this review, we summarize the physiological significance and pathological implications of PrP-metal interaction on prion disease pathogenesis.
Introduction
The current challenge facing prion research is the lack of an effective therapeutic strategy for prion disorders, a group of invariably fatal neurodegenerative conditions of humans and animals. Research in this area has been hampered due to incomplete understanding of the underlying mechanisms of neurotoxicity in these devastating diseases. The most favored hypothesis supported by numerous studies suggests that neurotoxicity in all prion disorders is mediated by PrP-scrapie (PrPSc), a β-sheet rich conformation of a cell surface glycoprotein known as the prion protein (PrPC). The change in conformation of PrPC to PrPSc confers certain biochemical and biophysical properties to PrPSc, which, unlike its counterpart PrPC, becomes insoluble in non-ionic detergents and resists limited digestion by proteinase-K (PK). Deposits of PrPSc in the brain parenchyma are considered the principal cause of neurotoxicity, though the pathways involved in this process are poorly understood (Prusiner, 1998; Aguzzi and Polymenidou, 2004, Caughey and Baron, 2006). Intense research in this area has clarified several aspects of this process. It is now clear that PrPSc in the extracellular space does not induce toxicity in the absence of PrPC expression on the neuronal plasma membrane, implicating PrPC in mediating the toxic signal (Malluci et al., 2003; Chesebro et al., 2005). On the other hand, accumulation of PrPSc only on astrocytes induces neurotoxicity, suggesting indirect pathways of toxicity by PrPSc (Jeffrey et al., 2004). Suggested pathways include secretion of toxic chemokines and factors by microglia in response to PrPSc, physical damage to the membrane structure by PrPSc aggregates, interference with synaptic transmission, and toxicity through a labile by-product of the PrPC to PrPSc conversion reaction (reviewed in Harris and True, 2006; Caughey and Baron, 2006). Evidence from cell model studies of familial prion disorders indicates abnormal processing and metabolism of mutant PrP as a possible cause of cytotoxicity, though the mechanism varies with specific mutations (Jin et al., 2000; Mishra et al., 2002; Gu et al., 2003). Other mechanisms such as activation of cell death pathways due to intracellular accumulation of PrPSc (Yadavalli et al., 2004; Kristiansen et al., 2005), cross-linking of neuronal PrPC on the plasma membrane by PrPSc (Solforosi et al., 2004), and toxicity due to C-transmembrane and cytosolic forms of PrPC through unspecified pathways have also been suggested, but the details of underlying mechanisms remain unclear (Hegde et al., 1998; 1999; Ma et al., 2002; Ma and Lindquist, 2002; Heller et al., 2003; Wang et al., 2006).
An overlooked but equally important cause of neurotoxicity is the loss of normal function of PrPC due to conversion to the PrPSc form. Information on this subject is limited since the normal function of PrPC remains ambiguous despite its ubiquitous presence and conservation through evolution. Transgenic mice lacking PrPC expression (PrP−/−) do not develop a specific phenotype unless a prion-like protein Doppel is up regulated, making it difficult to ascertain the functional role of PrPC (Bueler et al., 1992). Other experimental models suggest several unrelated functions, including a role in signal transduction, cell adhesion, copper uptake and transport, as an anti-oxidant and anti-apoptotic protein, and others (Bounhar et al., 2001; Roucou et al., 2004; 2005; Roucou and LeBlanc, 2005; Harris and True, 2006). The diverse functions attributed to PrPC probably reflect its involvement in an essential biochemical pathway that affects several cellular processes. Loss of this activity by the change in conformation of PrPC to PrPSc is likely to induce toxicity due to loss of an essential function, together with gain of certain toxic properties by PrPSc.
One such pathway may involve the functional role of PrPC in copper and iron metabolism. Since these metals are redox-active, abnormal metabolism of either of these metals is likely to induce neurotoxicity due to the generation of free radicals. Such a mechanism has been observed in neurodegenerative conditions associated with protein aggregation such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) (Moos and Morgan, 2004; Gerlach et al., 1994; Hayashi et al., 2006; Gaasch et al., 2007; Smith et al., 1997; 2007; Barnham and Bush, 2008). Recent reports indicate the presence of a similar phenomenon in prion disorders (Petersen et al., 2005; Singh et al., 2009a). Markers of oxidative stress and imbalance of metal homeostasis have been reported in prion disease affected brains, lending support to this assumption (Kim et al., 2000; Rossi et al 2004; Petersen et al., 2005; Pamplona et al., 2008; Singh et al., 2009a). In this review, we summarize evidence related to the interaction of PrPC with various metals, followed by the physiological and pathological implications of PrP-metal interaction. In particular, the role of PrPC in copper and iron metabolism is reviewed since these metals are essential for several metabolic processes, and are also toxic if mismanaged due to their redox-active nature.
PrP-metal interactions
Several studies have reported the interaction of PrPC with metals using in vitro and in vivo models. The in vitro studies have been more revealing due to the simplicity and accuracy of the readout compared to cell and animal models where metal metabolism is complex and the interaction of individual proteins with metals is often missed due to low affinity or their transient nature. Observations from in vitro studies using re-folded, full-length recombinant PrP or its fragments have lead to important findings regarding PrP-metal interaction and its significance to prion disease pathogenesis. It is now clear that recombinant PrP binds several divalent cations, including copper, iron, zinc, manganese and nickel (Pan et al., 1992; Hornshaw et al., 1995a; 1995b; Brown et al., 2000; Jackson et al., 2001; Jones et al., 2004; 2005; Basu et al., 2007). The highly conserved octa-peptide repeat region of PrP is the principal metal binding site, and its affinity for metals is highest for copper, followed by nickel, zinc, and manganese (Jackson et al. 2001). A diagrammatic representation of the interaction of PrPC with copper and iron is shown in Figure 1. The interaction of PrPC with metals is important to understand because of the physiological and pathological implications of this association. For example, PrPC is believed to mediate the uptake of copper and iron, suggesting a role in cellular copper and iron metabolism (Brown and Harris, 2003; Singh et al., 2009b,c). On the other hand, interaction of recombinant and cell associated PrPC with certain metals induces a change in its conformation to the disease associated PrPSc form, suggesting their role in prion disease pathogenesis (Deleaut et al., 2007; Basu et al., 2007). The conversion of PrPC to the PrPSc form increases its affinity for nickel while decreasing the binding of zinc and manganese, indicating specific interaction of different metals with the normal and the disease associated PrPSc form (Jackson et al., 2001).
Figure 1.
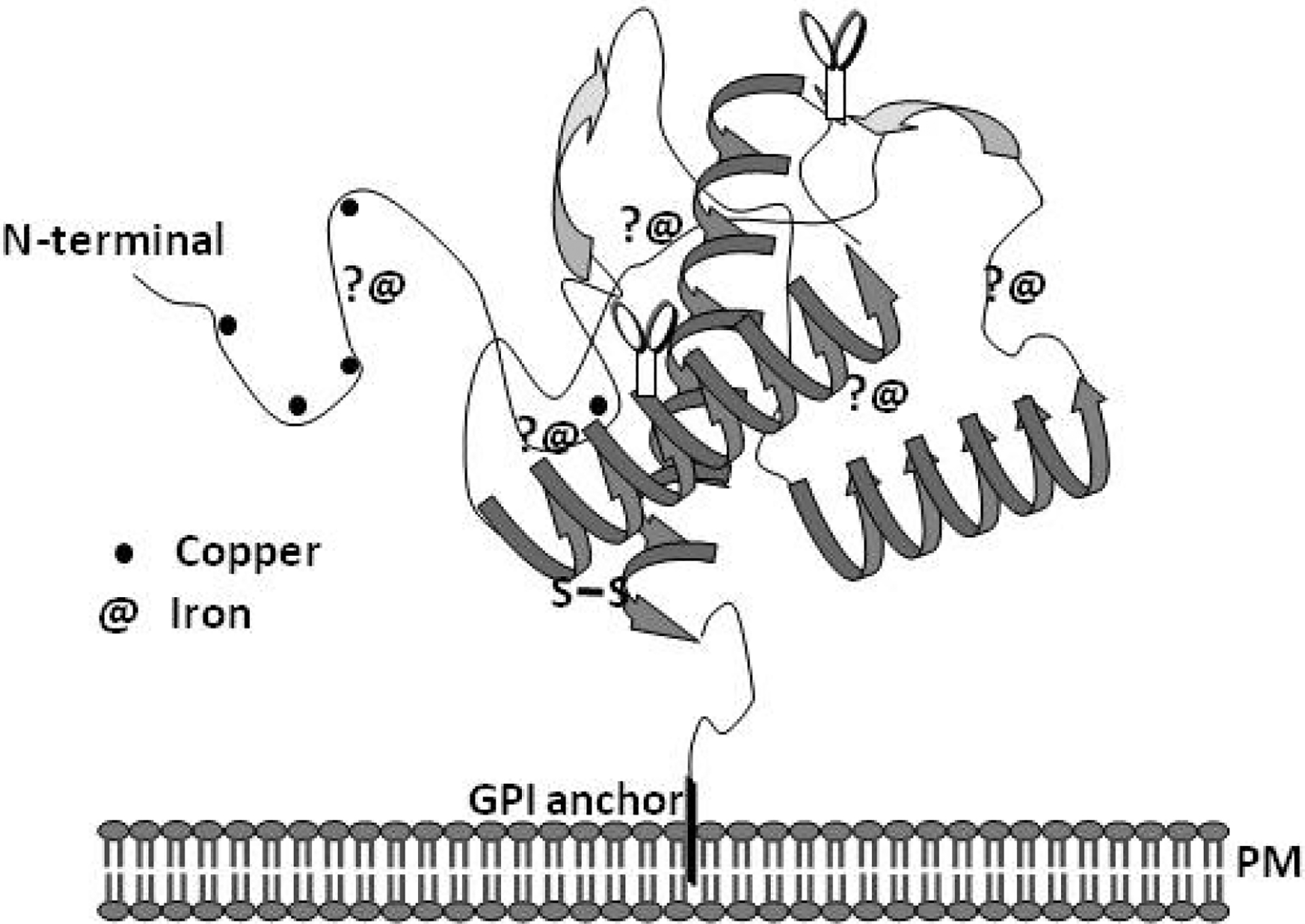
A model of PrPC demonstrating the known copper binding sites in the octa-peptide region and histidine residues 96 and 111. The association of PrPC with iron is based on unpublished data from recombinant full length PrPC and its fragments.
The association of PrPC with copper is better characterized than its interaction with other metals. Four copper binding sites have been identified within the octa-peptide repeat region of PrPC with additional sites on histidine residues 96 and 111 (Burns et al., 2003). Purified PrPC from mouse and human brains also binds copper, substantiating the in vitro observations (Brown et al., 2001; Wong et al., 2001b). The affinity of PrPC for copper is higher compared to zinc, and even a large excess of zinc cannot displace copper from the octa-peptide repeat region of full-length PrPC under in vitro conditions (Jackson et al., 2001; Walter et al., 2007). However, physiologically relevant levels of zinc in cell models alter the distribution of PrPC bound copper with relative ease, indicating the influence of other catalytic factors in determining the metal ion occupancy of PrPC (Watt and Hooper, 2003). These observations suggest that the interaction of PrPC with zinc may be more significant given the relative abundance of this metal in the brain (Qin et al., 2002; Watt and Hooper, 2003; Walter at al., 2007; Kenward et al., 2007). PrPC also binds manganese, probably in the C-terminal region between residues 91-230 or overlapping with the copper binding site at His-96 (Treiber et al., 2007; Brazier et al., 2008). Although these observations are useful, the data need to be interpreted with caution since in vitro reactions do not always represent the complex milieu of PrPC in cells or in the brain.
The interaction of PrPC with some of these metals alters its structure such that it simulates PrPSc in certain biochemical properties including detergent insolubility and resistance to limited digestion by proteinase-K. Other metals induce this change in PrPC by their absence such as in the presence of a specific chelator, suggesting that modification of metal ion occupancy of PrPC is equally significant in bringing about this change (Deleaut et al., 2007). Thus, addition of manganese promotes a change in the conformation of recombinant PrPC to PrPSc, while zinc is believed to inhibit fibril formation and promote inter-molecular reactions (Brown et al., 2000; Giese et al., 2004; Tsenkova et al., 2004; Bocharova et al., 2005; Kim et al., 2005; Abdelraheim et al, 2006; Treiber et al., 2006; Kenward et al., 2007). Addition of zinc and copper to the toxic peptide of PrP, a model often used for studying prion-mediated toxicity, increases its aggregation and toxicity to cells (Jobling et al., 1999; 2001). A similar change in the conformation of PrPC to a PrPSc-like form is noted when PrP-expressing yeast cells are grown in medium supplemented with copper or manganese (Treiber et al., 2006). This reaction is reversed by adding specific chelators such as bathocuproinedisulfonic acid and clioquinol to the medium, reinforcing the idea that only specific metals induce this reaction. Despite this information and additional data emphasizing the significance of PrPC and PrPSc-metal interaction, neither the physiological nor the pathological significance of this interaction is clearly understood.
Physiological significance of PrP-metal interaction
Observations from neuroblastoma cells suggest that PrPC binds extracellular copper ions and delivers to endocytic compartments, functioning as a copper uptake protein (Pauly and Harris, 1998; Brown and Harris, 2003). The octa-peptide repeats of PrPC are essential for this process since deletion of this region inhibits copper uptake (Perera and Hooper, 2001). The octa-peptide repeat region is also believed to function as a reductase, reducing Cu (II) ions before transport across the endosomal membrane to the cytosol (Miura et al., 2005). Although these observations suggest that PrPC may be a major copper delivery protein, surprisingly, there is minimal difference in the copper content of brains from wild type and transgenic mice lacking PrP expression (PrP−/−) (Giese et al., 2005). However, contradictory studies claim a significant difference in copper levels between wild type, PrP−/−, and scrapie infected mouse brains, suggesting a prominent role for PrPC in maintaining copper homeostasis in the brain (Brown et al., 1997, 1998; Wong et al., 2001c; Thackray et al., 2002). Likewise, conflicting results regarding brain copper levels have been reported in PrPC over-expressing mice (Herms et al., 1999; Kretzschmar et al., 2000; Waggoner et al., 2000; Stuermer and Plattner, 2005), leaving the matter unsettled.
The interaction of PrPC with iron is a relatively recent finding, and the physiological significance of this association is becoming increasingly clear from studies indicating a functional role for PrPC in cellular iron uptake and transport (Basu et al., 2007; Singh et al., 2009b; 2009c). Unlike most other divalent cations, iron is an essential component of enzymes and proteins and is required for optimal neuronal growth and function (Beard and Connor, 2003). On the other hand, iron is also considered a toxin due to its ability to exist in two oxidation states (ferric Fe3+ and ferrous Fe2+) (Thompson et al., 2001; Kaplan, 2002). Due to this reason, iron transport into and out of the cells is tightly regulated. It is surprising that PrPC influences the cellular iron pool within this tightly regulated mechanism of iron uptake, transport, and utilization (Singh et al., 2009b; 2009c). These observations have significant bearing on the pathogenesis of prion disorders since aggregation of PrPC is likely to disturb cellular iron homeostasis, resulting in neurotoxicity.
PrPC has been demonstrated to influence iron metabolism in cells expressing normal and mutant PrP forms and in PrP−/− transgenic mouse models (Singh et al., 2009b, 2009c). Over-expression of PrPC in cultured neuroblastoma cells increases the cellular labile iron pool (LIP) and iron saturation of ferritin, suggesting a role for PrPC in iron uptake. It is interesting to note that cells expressing pathogenic and non-pathogenic mutations of PrP alter the cellular LIP and iron saturation of ferritin differentially. This difference in the cellular iron content is maintained even when cells are cultured in the presence of excess extracellular iron, indicating a dominant role in iron uptake (Singh et al., 2009b). Furthermore, stimulation of endocytosis by a PrP-specific antibody increases intracellular iron stores, suggesting that PrPC mediates uptake of iron from the extracellular milieu. Unlike ceruloplasmin, PrPC does not mediate the efflux of excess iron from cells, confirming its role as an iron uptake protein (Jeong and David, 2003; Singh et al., 2009b). Presently it is unclear whether PrPC mediates iron uptake using a novel pathway or by interacting with the conventional pathway of iron uptake and transport. It has been hypothesized that PrPC may influence iron uptake by interacting with the transferrin/transferrin receptor pathway as described for HFE (Waheed et al., 2002), or function as a ferric reductase to facilitate the transport of ferric iron from endosomes to cytosolic ferritin (Singh et al., 2009b).
Similar observations suggesting a positive effect of PrPC on systemic iron levels are noted when wild type mice are compared with PrP−/− mouse models. Deletion of PrPC in PrP−/− mice induces iron deficiency in the latter by decreasing the efficiency of iron transport from the intestinal lumen to the blood stream, and uptake of iron from the blood by parenchymal cells and cells of the hematopoietic lineage. Re-expression of PrPC corrects the iron deficiency in these mice, confirming the functional role for PrP in iron uptake (Singh et al., 2009c). Considering that PrP−/− mice are only mildly iron deficient, it is likely that PrPC modulates the function of other iron uptake proteins or is involved in a pathway that compensates for its absence.
An important question that remains unanswered is the binding site and the affinity of PrPC for iron. In vitro experiments using recombinant PrPC and its fragments indicate that the octapeptide repeat region of PrPC is not essential for iron binding, and the iron and copper binding regions of PrPC do not overlap (unpublished observations). Limited data using different denaturing conditions suggest that the interaction of PrPC with iron depends on the conformation of PrPC rather than a specific amino acid sequence. These studies are limited to recombinant PrPC since it is difficult to identify iron associated cellular PrPC, suggesting that either the interaction of PrPC with iron is transient, or the affinity of PrPC for iron is not high enough for easy identification. A diagrammatic representation of possible mechanisms of iron uptake by PrPC is depicted in Figure 2. It is likely that after binding iron, PrPC moves from lipid rich domains in the plasma membrane to the vicinity of TfR, where it might influence the binding of Tf to the TfR, internalization of PrPC/TfR/Tf complex, or endocytosis of Tf/TfR complex. It is also possible that PrPC influences iron transport from the endosomes to the cytosol by functioning as a ferric-reductase as demonstrated for copper (Miura et al., 2005).
Figure 2.
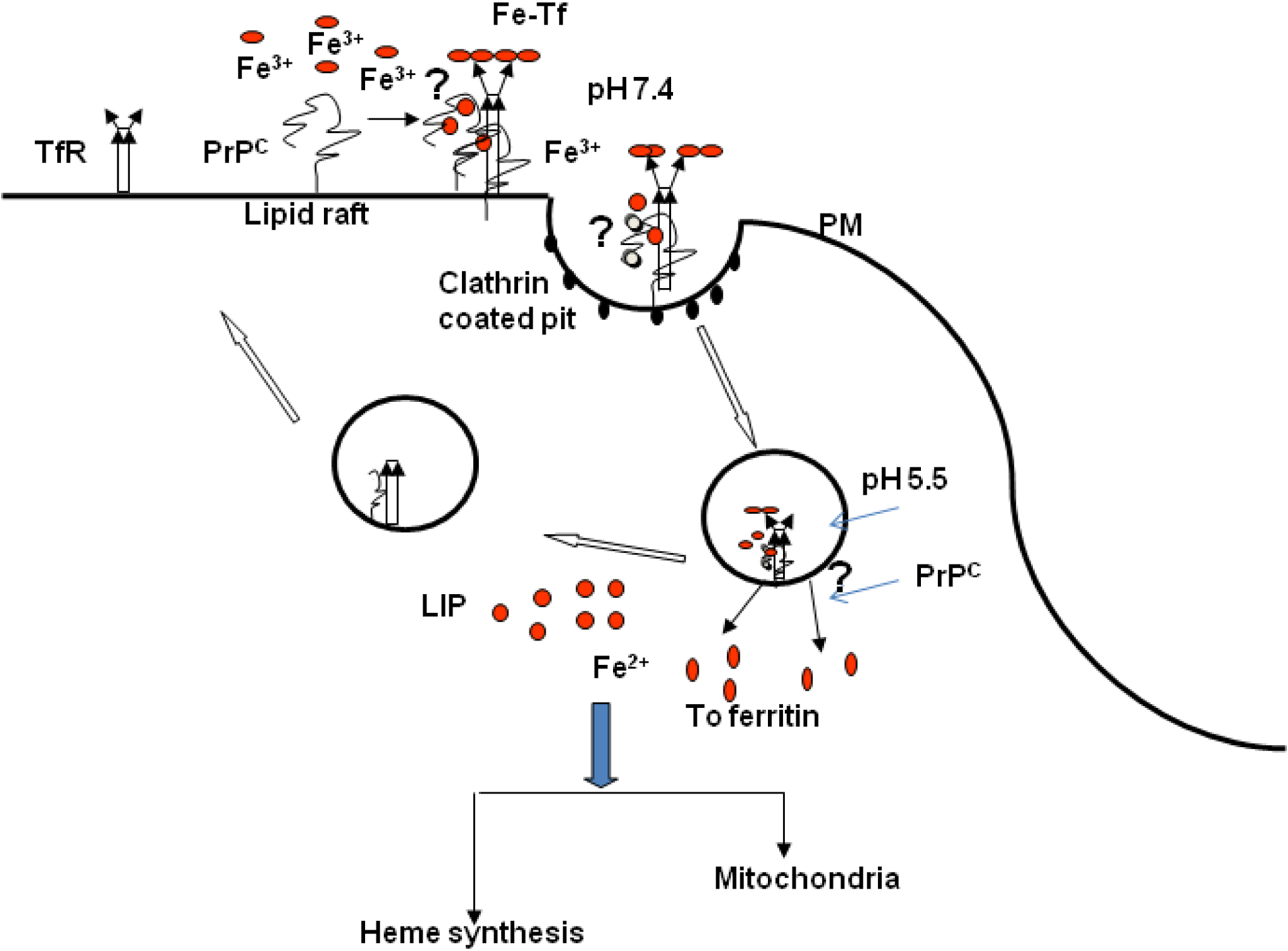
A representation of possible mechanisms by which PrPC is likely to mediate cellular iron uptake. At the plasma membrane, PrPC may bind iron directly for transport to the cytosol through endocytosis. It is also likely that PrPC influences iron uptake indirectly by modulating the endocytosis association or endocytosis of the Tf/TfR complex. Alternately, PrPC may facilitate the transport of iron from the endosomal compartment to the cytosol by functioning as a ferric-reductase. In the cytosol, iron is used for metabolic processes, or stored in ferritin in a relatively inert form.
Pathological implications of PrP-metal interaction
Copper and iron have significant pathological implications due to their redox-active nature. For example, both copper and iron induce the conversion of PrPC to the PrPSc form (Kim et al., 2005). Addition of copper increases the protease resistance and infectivity of denatured PrPSc in vitro, while copper chelation in vivo delays the onset of disease in prion infected mice (Pauly and Harris, 1998; Quaglio et al., 2001; Sigurdsson et al., 2003; Kuczius et al., 2004). Paradoxically, copper treatment inhibits PrPSc formation in cells infected with mouse prions and delays the onset of disease in scrapie infected hamsters, demonstrating a protective role, perhaps by augmenting the function of PrPC as a Cu/Zn superoxide dismutase (SOD) (Vassallo and Herms, 2003; Hijazi et al., 2003; Kiachopoulos et al., 2004; Orem et al., 2006). These conflicting observations can perhaps be explained from in vitro studies where copper inhibits the amplification of PrPSc from purified brain-derived PrPC and recombinant PrPC by stabilizing its α-helical structure, while it enhances the β-sheet structure of preformed PrP fibrils, thereby increasing their PrPSc content (Liu et al., 2007). Copper could therefore delay or augment disease progression based on the time when it is introduced to the animal.
An important consideration in evaluating the pathogenic consequences of PrPC-metal interaction is the cellular compartment in which metal bound PrPC is exposed to free radicals. It has been observed that exposure of PrPC expressing cells to a source of redox-active metal such as ferrous chloride induces the generation of PrPSc-like aggregates on the plasma membrane that accumulate within lysosomes in association with ferritin (Basu et al., 2007). These aggregates are also redox-active and initiate the generation of additional PrP-ferritin aggregates, propagating the PrPSc-like conformation within cells (Basu et al., 2007). Likewise, exposure of PrPC expressing cells to hemin, an iron containing compound, also results in the aggregation and degradation of PrPC (Lee et al., 2007), implicating redox-iron in the generation and propagation of PrPSc. Similar aggregation of α-synuclein is noted in response to iron and copper (Paik et al., 2000; Ostrerova-Golts et al., 2000; Golts et al., 2002; Takahashi et al., 2007), suggesting that protein aggregation by redox-active metals is not specific to PrPC. However, the generation of additional PrPSc aggregates by redox-active PrPSc appears to be unique to PrP, explaining in part the propagation of PrPSc once initiated. Surprisingly, chelation of iron from diseased brain homogenates decreases the total amount of PK-resistant PrPSc, suggesting that iron is also involved in the stability of PrPSc (Basu et al., 2007). This observation is of immense prophylactic value since iron chelation can be used as a means to decrease the infectivity of prion contaminated material.
Observations from mouse models of prion disease demonstrate an increase in the levels of iron regulatory proteins 1 and 2 and iron storage protein ferritin in the hippocampus and cerebral cortex of diseased brains, indicating the presence of brain iron imbalance (Kim et al., 2007). A similar imbalance of cellular iron homeostasis is observed in scrapie infected mouse neuroblastoma cells (ScN2a) that also show increased susceptibility to iron induced oxidative stress (Fernaeus et al., 2005; Fernaeus and Land, 2005). Imbalance of iron homeostasis is also observed in prion disease affected human, mouse, and hamster brains that demonstrate a phenotype of iron deficiency in the presence of increased total and redox-active Fe2+ iron (Singh et al., 2009a). The iron deficiency in scrapie infected hamster brains is noted ~6 weeks after inoculation of the infectious material and correlates with PrPSc levels, perhaps due to sequestration of iron in PrPSc-ferritin complexes (Singh et al., 2009a). The redox-active nature of PrPSc-ferritin aggregates is likely to induce the aggregation of additional PrPC, creating an ongoing state of iron imbalance and associated neurotoxicity in the diseased brain (Mishra et al., 2004; Basu et al., 2007). The associated iron deficiency caused by PrPSc-ferritin aggregates is equally harmful especially since iron is required for several vital metabolic processes (Figure 3).
Figure 3.
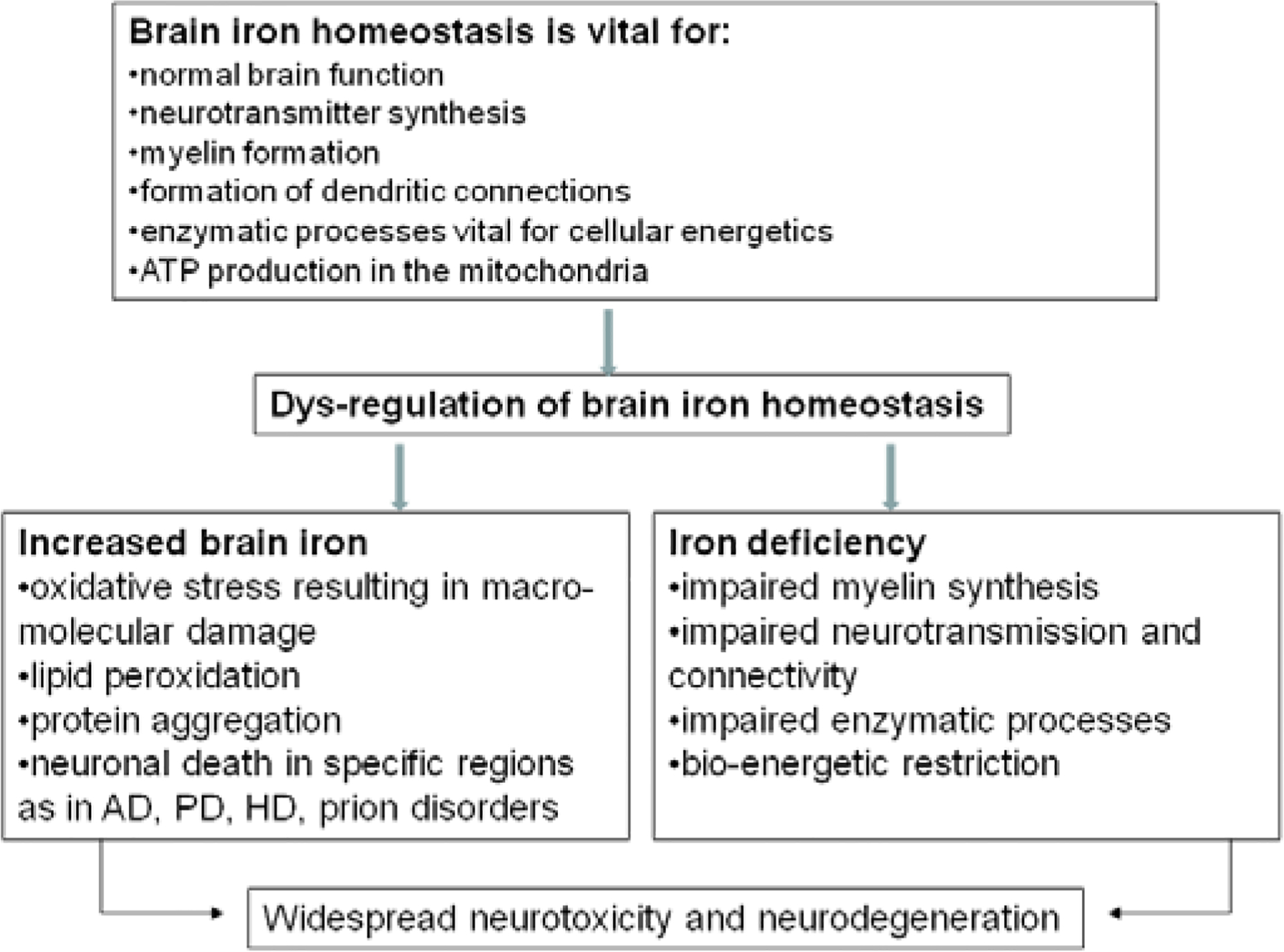
A model demonstrating the vital role of iron in brain function such as neurotransmitter synthesis, myelin formation, maintenance of dendritic connections, and other metabolic processes. Dys-regulation of brain iron homeostasis can induce neurotoxicity both by iron excess, and by iron deficiency. The former is a consequence of the redox-active nature of iron leading to oxidative stress, and the latter due to impairment of essential metabolic processes.
In addition to copper and iron, alteration in the homeostasis of manganese has also been reported in prion disease affected human and animal brains. In scrapie infected mice, levels of manganese are elevated in the peripheral blood, while in diseased cattle and sheep increased levels are noted in both the blood and brain tissue (Wong et al., 2001a, 2001b; Thackray et al., 2002; Hesketh et al. 2007; 2008). Surprisingly, altered levels of manganese have also been reported in scrapie resistant sheep that do not develop clinical disease, making it unlikely that the change is due to the disease process per se. However, limited studies make it difficult to explain the generality or the underlying cause of this abnormality. Other diseases that show elevated levels of manganese include hemochromatosis and diseases of the blood-brain barrier, information that may help in understanding the basis of manganese imbalance in prion disorders. Other reports indicate a decrease in the levels of copper and zinc in the brains of scrapie infected mice, although blood levels of zinc increase during disease progression as noted for manganese (Wong et al. 2001a, 2001b; Thackray et al., 2002). In human brains affected with sCJD, PrPC-associated copper is replaced by zinc, resulting in the loss of SOD-like activity of PrPC, a possible cause of increased neurotoxicity due to oxidative stress. Further studies are necessary to understand this phenomenon fully.
Summary and Perspective
Despite overwhelming evidence implicating PrPSc in the pathogenesis of prion disorders, the mechanistic details underlying the neurotoxicity associated with these disorders remain unclear. The association of PrPC with redox-active metals such as copper and iron provides new insight into the role of metal-induced oxidative stress in these disorders. Observations from cell and mouse models suggest that PrPC is involved in copper and iron uptake. Although PrPC is not a major iron or copper modulating protein, compromised levels of these metals due to aggregation of PrPC to the PrPSc form are likely to affect cell health due to their redox-active nature. At the same time, PrPSc assumes a redox-active nature due to sequestration of iron in PrPSc-ferritin aggregates, providing a logical explanation for imbalance of iron homeostasis in prion disease affected brains. This phenotype is likely to induce oxidative stress and neuronal damage, and could contribute significantly to prion disease associated neurotoxicity. Although the role of redox-active metals in prion disease pathogenesis is still at its formative stage, sufficient information exists to promt future investigations on this subject that may lead to the development of anti-oxidants and metal chelators as useful therapeutic agents.
Abbreviations
- Fe2+
Ferrous iron
- Fe3+
Ferric iron
- AD
Alzheimer’s disease
- CJD
Creutzfeldt-Jakob disease
- HFE
hemochromatosis protein
- HD
Huntington’s disease
- LIP
labile iron pool
- N2a
mouse neuroblastoma cells
- PD
Parkinson’s disease
- PK
proteinase-K
- PrP−/−
mice lacking PrP expression
- PrPC
prion protein
- PrPSc
PrP-scrapie
- ScN2a
scrapie infected mouse neuroblastoma cells
- SOD
Cu/Zn superoxide dismutase
- Tf
transferrin, TfR: transferrin receptor
References
- Aguzzi A, and Polymenidou M (2004). Mammalian prion biology: one century of evolving concepts. Cell. 116, 313–327. [DOI] [PubMed] [Google Scholar]
- Abdelraheim SR, Královicová S, and Brown DR (2006). Hydrogen peroxide cleavage of the prion protein generates a fragment able to initiate polymerisation of full length prion protein. Int J Biochem Cell Biol. 38,1429–1440. [DOI] [PubMed] [Google Scholar]
- Barnham KJ, and Bush AI (2008). Metals in Alzheimer’s and Parkinson’s diseases. Curr. Opin. Chem. Biol 12, 222–228. [DOI] [PubMed] [Google Scholar]
- Basu S, Mohan ML, Luo X, Kundu B, Kong Q, and Singh N (2007). Modulation of PK-resistant PrPSc in cells and infectious brain homogenate by redox-iron: Implications for prion replication and disease pathogenesis. Mol. Biol. Cell 18, 3302–3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard JL, and Connor JR (2003). Iron status and neural functioning. Ann. Rev. Nutr 23, 41–58. [DOI] [PubMed] [Google Scholar]
- Bocharova OV, Breydo L, Salnikov VV, and Baskakov IV (2005) Copper(II) inhibits in vitro conversion of prion protein into amyloid fibrils. Biochem. 44, 6776–6787. [DOI] [PubMed] [Google Scholar]
- Bounhar Y, Zhang Y, Goodyer CG,, and LeBlanc A (2001). Prion protein protects human neurons against Bax-mediated apoptosis. J. Biol. Chem 276, 39145–39149. [DOI] [PubMed] [Google Scholar]
- Brazier MW, Davies P, Player E, Marken F, Viles JH, and Brown DR (2008). Manganese binding to the prion protein. J. Biol. Chem 283, 12831–12839. [DOI] [PubMed] [Google Scholar]
- Brown DR, Qin K, Herms JW, Madlung A, Manson J, Strome R, Fraser PE, Kruck T, Von Bohlen A, Schulz-Schaeffer W, Giese A, Westaway D, and Kretzschmar H (1997). The cellular prion protein binds copper in vivo. Nature. 390, 684–687. [DOI] [PubMed] [Google Scholar]
- Brown DR, Hafiz F, Glasssmith LL, Wong BS, Jones IM, Clive C, and Haswell SJ (2000). Consequences of manganese replacement of copper for prion protein function and proteinase resistance, EMBO J. 19, 1180–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DR, Clive C, and Haswell SJ (2001). Antioxidant activity related to copper binding of native prion protein. J. Neurochem 76, 69–76. [DOI] [PubMed] [Google Scholar]
- Brown DR, Schmidt B, and Kretzschmar HA (1998). Effects of copper on survival of prion protein knockout neurons and glia. J. Neurochem 70, 1686–1693. [DOI] [PubMed] [Google Scholar]
- Brown LR, and Harris DA (2003). Copper and zinc cause delivery of the prion protein from the plasma membrane to a subset of early endosomes and the Golgi. J. Neurochem 87, 353–363. [DOI] [PubMed] [Google Scholar]
- Burns CS, Aronoff-Spencer E, Legname G, Prusiner SB, Antholine WE, Gerfen GJ, Peisach J, and Millhauser GL (2003). Copper coordination in the full-length, recombinant prion protein. Biochem. 42, 6794–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büeler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, Prusiner SB, Aguet M, and Weissmann C (1992). Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature. 356, 577–582. [DOI] [PubMed] [Google Scholar]
- Caughey B, and Baron GS (2006). Prions and their partners in crime. Nature 443, 803–810. [DOI] [PubMed] [Google Scholar]
- Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, Caughey B, Masliah E, and Oldstone M (2005)Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 308, 1435–1439. [DOI] [PubMed] [Google Scholar]
- Deleault NR, Harris BT, Rees JR, and Supattapone S (2007). Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. USA 104, 9741–9746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernaeus S, and Land T (2005). Increased iron-induced oxidative stress and toxicity in scrapie-infected neuroblastoma cells. Neurosci. Lett 382, 133–136. [DOI] [PubMed] [Google Scholar]
- Fernaeus S, Halldin J, Bedecs K, and Land T (2005). Changed iron regulation in scrapie-infected neuroblastoma cells. Brain Res. Mol. Brain Res 133, 266–273. [DOI] [PubMed] [Google Scholar]
- Gaasch JA, Lockman PR, Geldenhuys WJ, Allen DD, and Van der Schyf CJ (2007). Brain iron toxicity: differential responses of astrocytes, neurons, and endothelial cells. Neurochem Res. 32, 1196–1208. [DOI] [PubMed] [Google Scholar]
- Gerlach M, Ben-Shachar D, Riederer P, and Youdim MB (1994). Altered brain metabolism of iron as a cause of neurodegenerative diseases J Neurochem. 63, 793–807. [DOI] [PubMed] [Google Scholar]
- Giese A, Buchholz M, Herms J, and Kretzschmar HA (2005). Mouse brain synaptosomes accumulate copper-67 efficiently by two distinct processes independent of cellular prion protein. J. Mol. Neurosci 27, 347–54. [DOI] [PubMed] [Google Scholar]
- Giese A, Levin J, Bertsch U, and Kretzschmar HA (2004). Effect of metal ions on de novo aggregation of full-length prion protein. Biochem. Biophys. Res. Commun 320, 1240–1246. [DOI] [PubMed] [Google Scholar]
- Golts N, Snyder H, Frasier M, Theisler C, Choi P, and Wolozin B (2002). Magnesium Inhibits Spontaneous and Iron-induced Aggregation of α-Synuclein. J. Biol. Chem 277, 16116–16123. [DOI] [PubMed] [Google Scholar]
- Gu Y, Verghese S, Mishra RS, Xu X, Shi Y, and Singh N (2003). Mutant prion protein-mediated aggregation of normal prion protein in the endoplasmic reticulum: implications for prion propagation and neurotoxicity. J. Neurochem 84, 10–22. [DOI] [PubMed] [Google Scholar]
- Harris DA, and True HL (2006). New insights into prion structure and toxicity. Neuron. 50, 353–357. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Shoji M, and Abe K (2006). Molecular mechanisms of ischemic neuronal cell death--with relevance to Alzheimer’s disease. Curr Alzheimer Res. 3, 351–358. [DOI] [PubMed] [Google Scholar]
- Hegde RS, Mastrianni JA, Scott MR, DeFea KA, Tremblay P, Torchia M, DeArmond SJ, Prusiner SB, and Lingappa VR (1998). A transmembrane form of the prion protein in neurodegenerative disease. Science. 279, 827–834. [DOI] [PubMed] [Google Scholar]
- Hegde RS, Tremblay P, Groth D, DeArmond SJ, Prusiner SB, and Lingappa VR (1999). Transmissible and genetic prion diseases share a common pathway of neurodegeneration. Nature. 402, 822–826. [DOI] [PubMed] [Google Scholar]
- Heller U, Winklhofer KF, Heske J, Reintjes A, and Tatzelt J (2003). Post-translational import of the prion protein into the endoplasmic reticulum interferes with cell viability: a critical role for the putative transmembrane domain. J. Biol. Chem 278, 36139–36147. [DOI] [PubMed] [Google Scholar]
- Herms J, Tings T, Gall S, Madlung A, Giese A, Siebert H, Schürmann P, Windl O, Brose N, and Kretzschmar H (1999). Evidence of presynaptic location and function of the prion protein. J. Neurosci 19, 8866–8875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesketh S, Sassoon J, Knight R, and Brown DR (2008). Elevated manganese levels in blood and CNS in human prion disease. Mol. Cell Neurosci 37, 590–598. [DOI] [PubMed] [Google Scholar]
- Hesketh S, Sassoon J, Knight R, Hopkins J, and Brown DR (2007). Elevated manganese levels in blood and central nervous system occur before onset of clinical signs in scrapie and bovine spongiform encephalopathy. J. Anim. Sci 85, 1596–1609. [DOI] [PubMed] [Google Scholar]
- Hijazi N, Shaked Y, Rosenmann H, Ben-Hur T, and Gabizon R (2003). Copper binding to PrPC may inhibit prion disease propagation. Brain Res. 12, 192–200. [DOI] [PubMed] [Google Scholar]
- Hornshaw MP, McDermott JR, and Candy JM (1995a). Copper binding to the N-terminal tandem repeat regions of mammalian and avian prion protein. Biochem. Biophys. Res. Commun 207, 621–629. [DOI] [PubMed] [Google Scholar]
- Hornshaw MP, McDermott JR, Candy JM, and Lakey JH (1995b). Copper binding to the N-terminal tandem repeat region of mammalian and avian prion protein: structural studies using synthetic peptides, Biochem. Biophys. Res. Commun 214, 993–999. [DOI] [PubMed] [Google Scholar]
- Jackson GS, Murray I, Hosszu LL, Gibbs N, Waltho JP, Clarke AR, and Collinge J (2001). Location and properties of metal-binding sites on the human prion protein, Proc. Natl. Acad. Sci. U. S. A 98, 8531–8535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey M, Goodsir CM, Race RE, and Chesebro B (2004). Scrapie-specific neuronal lesions are independent of neuronal PrP expression. Ann. Neurol 55, 781–792. [DOI] [PubMed] [Google Scholar]
- Jeong SY, and David S (2003). Glycosylphosphatidyl-inositol-anchored ceruloplasmin is required for iron efflux from cells in the central nervous system. J. Biol. Chem 278, 27144–27148. [DOI] [PubMed] [Google Scholar]
- Jin T, Gu Y, Zanusso G, Sy M, Kumar A, Cohen M, Gambetti P, and Singh N (2000). The chaperone protein BiP binds to a mutant prion protein and mediates its degradation by the proteasome. J. Biol. Chem 275, 38699–38704. [DOI] [PubMed] [Google Scholar]
- Jobling MF, Huang X, Stewart LR, Barnham KJ, Curtain C, Volitakis I, Perugini M, White AR, Cherny RA, Masters CL, Barrow CJ, Collins SJ, Bush AI, and Cappai R (2001). Copper and zinc binding modulates the aggregation and neurotoxic properties of the prion peptide PrP106–126. Biochem. 40, 8073–84. [DOI] [PubMed] [Google Scholar]
- Jobling MF, Stewart LR, White AR, McLean C, Friedhuber A, Maher F, Beyreuther K, Masters CL, Barrow CJ, Collins SJ, and Cappai R (1999). The hydrophobic core sequence modulates the neurotoxic and secondary structure properties of the prion peptide 106–126. J. Neurochem 73, 1557–1565. [DOI] [PubMed] [Google Scholar]
- Jones CE, Abdelraheim SR, Brown DR, and Viles JH (2004). Preferential Cu2+ coordination by His96 and His111 induces beta-sheet formation in the unstructured amyloidogenic region of the prion protein. J. Biol. Chem 279, 32018–32027. [DOI] [PubMed] [Google Scholar]
- Jones CE, Klewpatinond M, Abdelraheim SR, Brown DR, and Viles JH (2005). Probing copper2+ binding to the prion protein using diamagnetic nickel2+ and 1H NMR: the unstructured N terminus facilitates the coordination of six copper2+ ions at physiological concentrations, J. Mol. Biol 346, 1393–1407. [DOI] [PubMed] [Google Scholar]
- Kaplan J (2002). Mechanisms of cellular iron acquisition: another iron in the fire. Cell. 111, 603–606. [DOI] [PubMed] [Google Scholar]
- Kenward AG, Bartolotti LJ, Burns CS (2007). Copper and zinc promote interactions between membrane-anchored peptides of the metal binding domain of the prion protein. Biochem. 46, 4261–4271. [DOI] [PubMed] [Google Scholar]
- Kiachopoulos S, Heske J, Tatzelt J, and Winklhofer KF (2004). Misfolding of the prion protein at the plasma membrane induces endocytosis, intracellular retention and degradation. Traffic. 5, 426–436. [DOI] [PubMed] [Google Scholar]
- Kim BH, Jun YC, Jin JK, Kim JI, Kim NH, Leibold EA, Connor JR, Choi EK, Carp RI, and Kim YS (2007). Alteration of iron regulatory proteins (IRP1 and IRP2) and ferritin in the brains of scrapie-infected mice. Neurosci. Lett 422, 158–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NH, Choi JK, Jeong BH, Kim JI, Kwon MS, Carp RI, and Kim YS (2005). Effect of transition metals (Mn, Cu, Fe) and deoxycholic acid (DA) on the conversion of PrPC to PrPres. FASEB J. 19, 783–785. [DOI] [PubMed] [Google Scholar]
- Kim NH, Park SJ, Jin JK, Kwon MS, Choi EK, Karp RI, and Kim YS (2000). Increased ferric iron content and iron-induced oxidative stress in the brains of scrapie-infected mice. Brain Res. 884, 98–103. [DOI] [PubMed] [Google Scholar]
- Kretzschmar HA, Tings T, Madlung A, Giese A, Herms J (2000). Function of PrP(C) as a copper-binding protein at the synapse. Arch. Virol. Suppl 16, 239–49. [DOI] [PubMed] [Google Scholar]
- Kristiansen M, Messenger MJ, Klohn PC, Brandner S, Wadsworth JD, Collinge J, and Tabrizi SJ (2005). Disease-related prion protein forms aggresomes in neuronal cells leading to caspase-activation and apoptosis. J. Biol. Chem 280, 38851–38861. [DOI] [PubMed] [Google Scholar]
- Kuczius T, Buschmann A, Zhang W, Karch H, Becker K, Peters G, and Groschup MH (2004). Cellular prion protein acquires resistance to proteolytic degradation following copper ion binding. Biol. Chem 385, 739–747. [DOI] [PubMed] [Google Scholar]
- Lee KS, Raymond LD, Schoen B, Raymond GJ, Kett L, Moore RA, Johnson LM, Taubner L, Speare JO, Onwubiko HA, Baron GS, Caughey WS, and Caughey B (2007). Hemin interactions and alterations of the subcellular localization of prion protein. J. Biol. Chem 282, 36525–36533. [DOI] [PubMed] [Google Scholar]
- Liu M, Yu S, Yang J, Yin X, and Zhao D (2007). RNA and CuCl2 induced conformational changes of the recombinant ovine prion protein. Mol. Cell Biochem 294, 197–203. [DOI] [PubMed] [Google Scholar]
- Ma J, and Lindquist S (2002). Conversion of PrP to a self-perpetuating PrPSc-like conformation in the cytosol. Science. 298, 1785–1788. [DOI] [PubMed] [Google Scholar]
- Ma J, Wollmann R, and Lindquist S (2002). Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science. 298, 1781–1785. [DOI] [PubMed] [Google Scholar]
- Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, and Collinge J (2003). Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science. 302, 871–874. [DOI] [PubMed] [Google Scholar]
- Mishra RS, Basu S, Gu Y, Luo X, Zou WQ, Mishra R, Li R, Chen SG, Gambetti P, Fujioka H, and Singh N (2004). Protease-resistant human prion protein and ferritin are cotransported across Caco-2 epithelial cells: implications for species barrier in prion uptake from the intestine. J. Neurosci 24, 11280–11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra RS, Gu Y, Bose S, Verghese S, Kalepu S, Singh N (2002). Cell surface accumulation of a truncated transmembrane prion protein in Gerstmann-Straussler-Scheinker disease P102L. J. Biol. Chem 277, 24554–24561. [DOI] [PubMed] [Google Scholar]
- Miura T, Sasaki S, Toyama A, and Takeuchi H (2005). Copper reduction by the octapeptide repeat region of prion protein: pH dependence and implications in cellular copper uptake. Biochem. 44, 8712–8720. [DOI] [PubMed] [Google Scholar]
- Moos T, and Morgan EH (2004). The metabolism of neuronal iron and its pathogenic role in neurological disease. Ann. N. Y. Acad. Sci 1012, 14–26. [DOI] [PubMed] [Google Scholar]
- Orem NR, Geoghegan JC, Deleault NR, Kascsak R, and Supattapone S (2006). Copper (II) ions potently inhibit purified PrPres amplification. J. Neurochem 96, 1409–1415. [DOI] [PubMed] [Google Scholar]
- Ostrerova-Golts N, Petrucelli L, Hardy J, Lee JM, Farer M, and Wolozin B (2000). The A53T alpha-synuclein mutation increases iron-dependent aggregation and toxicity. J. Neurosci 20, 6048–6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik SR, Shin HJ, and Lee JH (2000). Metal-catalyzed oxidation of alpha-synuclein in the presence of Copper (II) and hydrogen peroxide. Arch. Biochem. Biophys 378, 269–277. [DOI] [PubMed] [Google Scholar]
- Pamplona R, Naudí A, Gavín R, Pastrana MA, Sajnani G, Ilieva EV, Del Río JA, Portero-Otín M, Ferrer I, and Requena JR (2008). Increased oxidation, glycoxidation, and lipoxidation of brain proteins in prion disease. Free Radic Biol Med. 45, 1159–1166. [DOI] [PubMed] [Google Scholar]
- Pan KM, Stahl N, and Prusiner SB (1992). Purification and properties of the cellular prion protein from Syrian hamster brain, Protein Sci. 1, 1343–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauly PC, and Harris DA (1998). Copper stimulates endocytosis of the prion protein. J. Biol. Chem 273, 33107–33110. [DOI] [PubMed] [Google Scholar]
- Perera WSS, and Hooper NM (2001). Ablation of the metal ion-induced endocytosis of the prion protein by disease-associated mutation of the octarepeat region. Curr Biol. 11, 519–523. [DOI] [PubMed] [Google Scholar]
- Petersen RB, Siedlak SL, Lee HG, Kim YS, Nunomura A, Tagliavini F, Ghetti B, Cras P, Moreira PI, Castellani RJ, Guentchev M, Budka H, Ironside JW, Gambetti P, Smith MA, and Perry G (2005). Redox metals and oxidative abnormalities in human prion diseases. Acta Neuropathol. (Berl) 110, 232–238. [DOI] [PubMed] [Google Scholar]
- Prusiner SB (1998). Prions. Proc. Natl. Acad. Sci. U. S. A 95, 3363–3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin K, Yang Y, Mastrangelo P, and Westaway D (2002) Mapping Cu(II) binding sites in prion proteins by diethyl pyrocarbonate modification and matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometric footprinting. J. Biol. Chem 277, 1981–1990. [DOI] [PubMed] [Google Scholar]
- Quaglio E, Chiesa R, and Harris DA (2001). Copper converts the cellular prion protein into a protease-resistant species that is distinct from the scrapie isoform. J. Biol. Chem 276, 11432–11438. [DOI] [PubMed] [Google Scholar]
- Rossi L, Lombardo MF, Ciriolo MR, and Rotilio G (2004). Mitochondrial dysfunction in neurodegenerative diseases associated with copper imbalance. Neurochem Res. 29, 493–504. [DOI] [PubMed] [Google Scholar]
- Roucou X, and LeBlanc AC (2005). Cellular prion protein neuroprotective function: implications in prion diseases. J. Mol. Med 83, 3–11. [DOI] [PubMed] [Google Scholar]
- Roucou X, Gains M, and LeBlanc AC (2004). Neuroprotective functions of prion protein. J. Neurosci. Res 75, 153–161.14705136 [Google Scholar]
- Roucou X, Giannopoulos PN, Zhang Y, Jodoin J, Goodyer CG, LeBlanc A (2005). Cellular prion protein inhibits proapoptotic Bax conformational change in human neurons and in breast carcinoma MCF-7 cells. Cell Death Differ. 12, 783–795. [DOI] [PubMed] [Google Scholar]
- Sigurdsson EM, Brown DR, Alim MA, Scholtzova H, Carp R, Meeker HC, Prelli F, Frangione B, and Wisniewski T (2003). Copper chelation delays the onset of prion disease. J. Biol. Chem 278, 46199–46202. [DOI] [PubMed] [Google Scholar]
- Singh A, Isaac AO, Luo X, Mohan ML, Cohen ML, Chen F, Kong Q, Bartz J, and Singh N (2009a). Abnormal brain iron homeostasis in human and animal prion disorders. Plos Pathog. 5, e1000336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Mohan ML, Isaac AO, Luo X, and Singh N (2009b). Prion protein modulates cellular iron metabolism: Implications for prion disease pathogenesis. PlosOne. 4, e4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Kong Q, Luo X, Petersen RB, Meyerson H, and Singh N (2009c). Prion protein knock-out mice are iron deficient: A functional role for PrP in iron metabolism. Plos One. 4, e6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Harris PL, Sayre LM, and Perry G (1997). Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc. Natl. Acad. Sci. U. S. A 94, 9866–9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DG, Cappai R, and Barnham KJ (2007). The redox chemistry of the Alzheimer’s disease amyloid beta peptide. Biochim Biophys Acta.1768,1976–1990. [DOI] [PubMed] [Google Scholar]
- Solforosi L, Criado JR, McGavern DB, Wirz S, Sánchez-Alavez M, Sugama S, DeGiorgio LA, Volpe BT, Wiseman E, Abalos G, Masliah E, Gilden D, Oldstone MB, Conti B, and Williamson RA (2004). Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science. 303, 1514–1516. [DOI] [PubMed] [Google Scholar]
- Stuermer CA, and Plattner H (2005). The ‘lipid raft’ microdomain proteins reggie-1 and reggie-2 (flotillins) are scaffolds for protein interaction and signalling. Biochem. Soc. Symp 72, 109–118. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Ko LW, Kulathingal J, Jiang P, Sevlever D, and Yen SH (2007). Oxidative stress-induced phosphorylation, degradation and aggregation of alpha-synuclein are linked to upregulated CK2 and cathepsin D. Eur. J. Neurosci 26, 863–874. [DOI] [PubMed] [Google Scholar]
- Thackray AM, Knight R, Haswell SJ, Bujdoso R, and Brown DR (2002). Metal imbalance and compromised antioxidant function are early changes in prion disease. Biochem. J 362, 253–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson KJ, Shoham S, and Connor JR (2001). Iron and neurodegenerative disorders. Brain Res. Bull 55, 155–164. [DOI] [PubMed] [Google Scholar]
- Treiber C, Simons A, and Multhaup G (2006). Effect of copper and manganese on the de novo generation of protease-resistant prion protein in yeast cells. Biochem. 45, 6674–6680. [DOI] [PubMed] [Google Scholar]
- Treiber C, Thompsett AR, Pipkorn R, Brown DR, Multhaup G (2007). Real-time kinetics of discontinuous and highly conformational metal-ion binding sites of prion protein. J. Biol. Inorg. Chem 12, 711–720. [DOI] [PubMed] [Google Scholar]
- Tsenkova RN, Iordanova IK, Toyoda K, and Brown DR (2004). Prion protein fate governed by metal binding. Biochem. Biophys. Res. Commun 325, 1005–1012. [DOI] [PubMed] [Google Scholar]
- Vassallo N, and Herms J (2003). Cellular prion protein function in copper homeostasis and redox signalling at the synapse. J. Neurochem 86, 538–544. [DOI] [PubMed] [Google Scholar]
- Waggoner DJ, Drisaldi B, Bartnikas TB, Casareno RL, Prohaska JR, Gitlin JD, and Harris DA (2000). Brain copper content and cuproenzyme activity do not vary with prion protein expression level. J. Biol. Chem 275, 7455–7458. [DOI] [PubMed] [Google Scholar]
- Waheed A, Grubb JH, Zhou XY, Tomatsu S, Fleming RE, Costaldi ME, Britton RS, Bacon BR, and Sly WS (2002). Regulation of transferrin-mediated iron uptake by HFE, the protein defective in hereditary hemochromatosis. Proc. Natl. Acad. Sci. U. S. A 99, 3117–3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter ED, Stevens DJ, Visconte MP, and Millhauser GL (2007). The prion protein is a combined zinc and copper binding protein: Zn2+ alters the distribution of Cu2+ coordination modes. J. Am. Chem. Soc 129, 15440–15441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang F, Arterburn L, Wollmann R, and Ma J (2006). The interaction between cytoplasmic prion protein and the hydrophobic lipid core of membrane correlates with neurotoxicity. J. Biol. Chem 281, 13559–13565. [DOI] [PubMed] [Google Scholar]
- Watt NT, and Hooper NM (2003). The prion protein and neuronal zinc homeostasis. Trends Biochem. Sci 28, 406–410. [DOI] [PubMed] [Google Scholar]
- Wong BS, Brown DR, Pan T, Whiteman M, Liu T, Gambetti P, Olesik J, Rubenstein R, and Sy MS (2001a). Oxidative impairment in scrapie-infected mice is associated with brain metals. perturbations and altered antioxidant activities. J. Neurochem 79, 689–698. [DOI] [PubMed] [Google Scholar]
- Wong BS, Chen SG, Colucci M, Xie Z, Pan T, Liu T, Li R, Gambetti P, Sy MS, and Brown DR (2001b). Aberrant metal binding by prion protein in human prion disease. J. Neurochem 78, 1400–1408. [DOI] [PubMed] [Google Scholar]
- Wong BS, Liu T, Li R, Pan T, Petersen RB, Smith MA, Gambetti P, Perry G, Manson JC, Brown DR, and Sy M-S (2001c). Increased levels of oxidative stress markers detected in the brains of mice devoid of prion protein, J. Neurochem 76, 565–572. [DOI] [PubMed] [Google Scholar]
- Yadavalli R, Guttmann RP, Seward T, Centers AP, Williamson RA, and Telling GC (2004). Calpain-dependent endoproteolytic cleavage of PrPSc modulates scrapie prion propagation. J. Biol. Chem 279, 21948–21956. [DOI] [PubMed] [Google Scholar]