

Abstract
Highly tractable 1-aryl-1-propynes, which are readily accessible via Sonogashira coupling, serve as chiral allylmetal pronucleophiles in ruthenium-JOSIPHOS-catalyzed anti-diastereo- and enantioselective aldehyde (α-aryl)allylations with primary aliphatic or benzylic alcohol proelectrophiles. This method enables convergent construction of homoallylic sec-phenethyl alcohols bearing tertiary benzylic stereocenters. Both steric and electronic features of aryl sulfonic acid additives were shown to contribute to the efficiency with which a more selective and productive iodide-bound ruthenium catalyst is formed. As corroborated by isotopic labelling studies, a dual catalytic process is operative in which alkyne-to-allene isomerization is followed by allene-carbonyl reductive coupling via hydrogen auto-transfer. Crossover of ruthenium hydrides emanating from these two discrete catalytic events is observed. The utility of this method is illustrated by conversion of selected reaction products to the corresponding phenethylamines and the first total syntheses of the neolignan natural products (−)-crataegusanoids A-D.
Graphical Abstract

Introduction
Carbonyl addition is the Proteus of metal-mediated C-C couplings.1 Recent analysis of >9 million patents from pharmaceutical industry shows that carbonyl addition (alongside the Suzuki coupling) persists as one of the most widely utilized methods for C-C bond formation in process R&D.2 Despite its importance, the majority of methods for carbonyl addition require preformed carbanions, which can be hazardous and are often generated using multiple sacrificial reagents, for example through halogenation-metalation-transmetalation sequences. Metal-catalyzed carbonyl reductive coupling of unsaturated pronucleophiles has emerged as an alternative to stoichiometric carbanions, but many reductants used in such processes are not ideal for chemical manufacture on scale (e.g. Mn, Zn, Et3B, Et2Zn, SiR3).3,4 We have advanced a broad, new family of metal-catalyzed carbonyl reductive couplings that exploit feedstock pronucleophiles in combination with feedstock reductants (H2, 2-PrOH, HCO2H), as well as related hydrogen autotransfer processes wherein alcohols serve dually as reductants and carbonyl proelectrophiles.4 These efforts include processes that exploit alkynes as allylmetal pronucleophiles.5,6,7
Given the tractability of 1-aryl-1-propynes and their wide availability via Sonogashira coupling (eq. 1), we sought to
 |
(eq. 1) |
develop catalytic enantioselective carbonyl (α-aryl)allylations via transfer hydrogenative couplings of 1-aryl-1-propynes with primary alcohols (Figure 1). Despite decades of work on asymmetric carbonyl allylation,7 enantioselective carbonyl (α-aryl)allylations are largely limited to isolated examples that embody moderate levels of asymmetric induction and deliver simple aryl fragments. These methods fall into two categories: (a) those involving chiral auxiliaries8 and (b) catalytic enantioselective protocols.9,10 In the former category, one systematic study involving allylbenzene pronucleophiles was reported by Gong,8g but this method is restricted to aryl aldehydes (Figure 1). In the latter category, systematic studies are limited to activated aldehydes (glyoxamides9e formaldehyde,10a fluoral and difluoroacetaldehyde10b) and a Nozaki-Hiyama-Kishi (α-aryl)allylation to form quaternary benzylic stereocenters.9i Catalytic enantioselective carbonyl (α-aryl)allylations applicable to both aliphatic and aromatic aldehydes are unknown, and would enable convergent construction of tertiary benzylic stereocenters and stereogenic C-O bonds, which are ubiquitous among natural products and FDA approved drugs. Here, using a ruthenium catalyst modified by the JOSIPHOS ligand SL-J009-1, we report that diverse 1-aryl-1-propynes engage in C-C coupling with primary aliphatic or benzylic alcohols to furnish products of (α-aryl)allylation bearing relatively complex aryl moieties in good yield with complete anti-diastereoselectivity and high levels of enantioselectivity.
Figure 1.

Convergent construction of sec-phenethyl alcohols bearing tertiary benzylic stereocenters via enantioselective carbonyl anti-(α-aryl)allylation of unactivated aldehydes.
Results and Discussion
In prior work from our laboratory,6c it was found that protonation of H2Ru(CO)(PPh3)3 by the aryl sulfonic acid 2,4,6-(iPr)3PhSO3H delivers a cationic ruthenium(II) complex that exists in equilibrium with a ruthenium(0) complex. This ruthenium(0) species promotes two discrete catalytic events: (a) alkyne-to-allene isomerization, and (b) transfer hydrogenative allene-carbonyl reductive coupling by way of a transient oxaruthenacycle. This process converts alkyl-substituted alkynes and primary alcohols to linear secondary (Z)-homoallylic alcohols (Scheme 1, eq. 2).6c Remarkably, in the presence of iodide and a chelating phosphine ligand, an alternate dual catalytic process becomes operative in which alkyne-to-allene isomerization is followed by hydrometalation of the transient allene to form an allylruthenium(II) species. This process converts alkyl-substituted alkynes and primary alcohols to branched secondary homoallylic alcohols with excellent control of regio-, diastereo- and enantioselectivity (Scheme 1, eq. 3).6e As corroborated by deuterium labelling studies, both processes involve alkyne-to-allene isomerization. The fate of the resulting allene largely depends on the intervention of cationic vs neutral ruthenium complexes, which partition entry into catalytic cycles involving either allene-carbonyl oxidative coupling or allene hydrometalation, respectively.6c,e,11
Scheme 1.
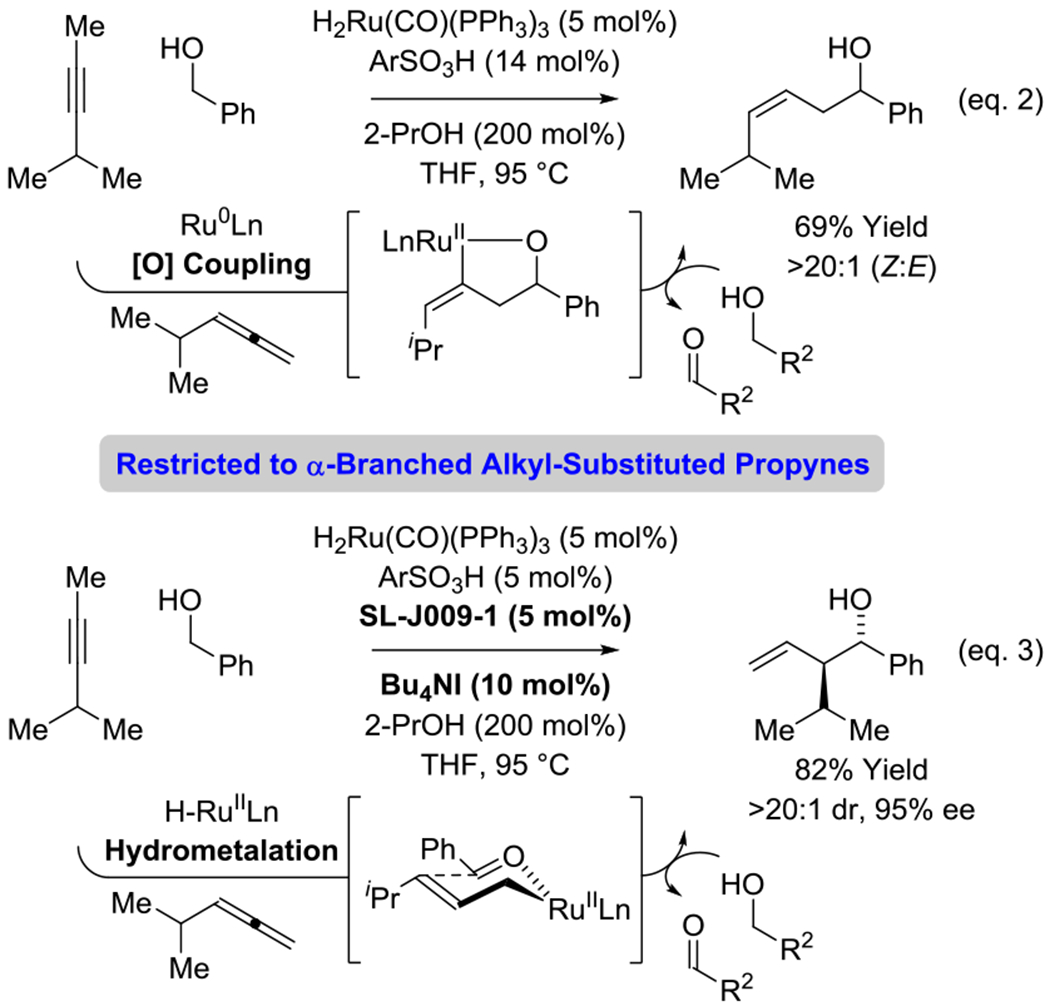
Alkynes as latent allenes in alcohol-mediated hydrohydroxyalkylation to form linear or branched homoallylic alcohols (Ar = 2,4,6-triisopropylphenyl).
Both catalytic processes are largely restricted to α-branched alkyl-substituted propynes, such as 4-methyl-2-pentyne. We speculate that α-branched alkyl groups at the acetylenic position may facilitate alkyne-to-allene isomerization by favorably influencing the regioselectivity of alkyne hydrometalation. Initial attempts to exploit 1-aryl-1-propynes as pronucleophiles for asymmetric carbonyl (α-aryl)allylation were inefficient, and especially low isolated yields were observed for 1-aryl-1-propynes bearing electron deficient aromatic rings. To overcome this limitation, efforts to optimize the carbonyl (α-aryl)allylation of 1-(4-CF3-phenyl)-1-propyne 1a and primary aliphatic alcohol 2a were undertaken (Scheme 2). Under conditions effective for couplings of 4-methyl-2-pentyne (but without 2-PrOH, which is used to reduce uncoupled aldehyde so it can reenter the catalytic cycle),6d the product of carbonyl (α-aryl)allylation 3a was obtained in 17% yield with only modest levels of enantiomeric enrichment (Scheme 1, entry 1). When catalyst loading was doubled, a proportionate increase in the yield of 3a was observed (Scheme 1, entry 2). Replacing 2,4,6-(iPr)3PhSO3H with 4-MePhSO3H and 4-NO2PhSO3H led to successive improvements (Scheme 1, entries 3 and 4, respectively). These data are significant, as they reveal both steric and electronic features of the catalyst impact efficiency and enantioselectivity. Using 2,4,6-(iPr)3PhSO3H in DME, slightly higher enantioselectivity was observed (Scheme 1, entries 2 vs 5), but lower temperatures limited conversion (Scheme 1, entry 6). The iodide-bound catalyst is generated through the acid-base reaction of the ruthenium dihydride with the arylsulfonic acid followed by substitution by iodide (eq. 4).12 Notably, the more
 |
(eq. 4) |
acidic, less hindered arylsulfonic acid 4-NO2PhSO3H appears to enhance the efficiency of this process, allowing temperature to be reduced, augmenting enantioselectivity without diminishing conversion (Scheme 1, entry 7). Under these conditions, moving from THF to DME solvent, 3a could be obtained in 85% yield with >20:1 anti-diastereoselectivity and high enantioselectivity (88% ee) (Scheme 1, entry 8). Omission of Bu4NI led to a significant decrease in yield and selectivity that could not be fully restored through use of Bu4NCl or Bu4NBr (Scheme 1, entries 9-11). Racemic product was obtained using the achiral ligand DIPPF in combination with (+)-camphor sulfonic acid (Scheme 1, entry 12). The collective data suggest both conversion and enantioselectivity depends on the efficiency with which the iodide-bound catalyst is formed.13
Scheme 2.

Influence of arylsulfonic acid in the reaction of 1-(4-CF3-phenyl)-1-propyne 1a and alcohol 2a to form the product of carbonyl (α-aryl)allylation 3a.a
aYields of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary phase HPLC analysis. Diastereoselectivities were determined via 1H NMR analysis of crude reaction mixtures. bConditions described in Scheme 1, eq. 3 were applied using H2Ru(CO)(PPh3)3 (5 mol%), SL-J009-1 (5 mol%), Bu4NI (10 mol%). cDIPPF (10 mol%) was used as ligand.
Under these conditions, the ruthenium-catalyzed coupling of diverse 1-aryl-1-propynes 1a-1ff with primary alcohols 2a-2ff to form enantiomerically enriched phenethyl alcohols 3a-3ff was explored (Table 1). As illustrated by the formation of phenethyl alcohols 3c, 3x and iso-3x, ortho-substituted aryl propynes are competent partners for C-C coupling. Heteroaryl-substituted propynes are converted to adducts 3d-3h, 3p and 3s, establishing compatibility with Lewis basic sulfur (3d) and nitrogen (3e-3h, 3p, 3s) functional groups. Additionally, as demonstrated by the formation of adducts 3g-3o, primary alcohols bearing a tethered N-Boc amine (3g) or pyrazole (3h), 2- aminopyridine (3i), oxazole (3j), furan (3k), thiophene (3l), and indole (3m-3o) moieties are competent partners for arylpropyne-mediated asymmetric (α-aryl)allylation. Notably, adducts 3j and 3n derive from the FDA approved therapeutic agents oxaprozin and indomethacin, respectively, highlighting the potential applicability of this method to drug discovery. Adducts derived from primary alcohols that incorporate strained saturated ring systems, including cyclopropanes (3t), difluorocyclobutanes (3u), azetidines (3r, 3v) and oxetanes (3w) were well tolerated. Whereas low conversion was associated with the use of acyclic α-stereogenic primary alcohols, the corresponding β-stereogenic primary alcohols were converted to products of (α-aryl)allylation (3x, iso-3x, 3y, iso-3y) with high levels of catalyst-directed diastereoselectivity. Finally, primary benzylic alcohols undergo (α-aryl)allylation as shown by the formation of adducts 3aa-3ff. Here, compatibility of pinacolboronate functional groups, as demonstrated by formation of 3bb and 3ff, is significant. In certain cases, minor decreases or increases in reaction temperature were made to improve enantioselectivity or increase conversion, respectively. The assignment of absolute stereochemistry for adducts 3a-3ff is made in analogy to that determined for compound 3r by single crystal X-ray diffraction analysis. Attempted coupling of the more highly substituted aryl alkyne, 1-phenyl-1-butyne, results in internal redox-isomerization to form the terminal π-allyl, delivering products of carbonyl anti-(α-benzyl)allylation in low yield.
Table 1.
Ruthenium-catalyzed coupling of 1-aryl-1-propynes 1a-1ff with primary alcohols 2a-2ff to form enantiomerically enriched phenethyl alcohols 3a-3ff.a
 |
Yields of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary phase HPLC analysis. Diastereoselectivities were determined via 1H NMR analysis of crude reaction mixtures. For standard conditions see Scheme 2, entry 7, 0.2 mmol scale, 48 h. See Supporting Information for further experimental details.
75 °C.
90 °C.
SL-J009-2.
65 °C.
Phenethylamines represent a broad class of psychoactive substances.14 To further illustrate the potential utility of this method to discovery efforts in pharmaceutical research, representative adducts 3d, 3j and 3r were transformed to the corresponding N-Boc-protected phenethylamines (Scheme 3). The phenethyl alcohols 3d, 3j and 3r were exposed to diphenylphosphoryl azide in the presence of diisopropyl azodicarboxylate and triphenylphosphine to furnish the azides 4d, 4j and 4r with complete inversion of stereochemistry and only trace quantities of competing elimination to form the conjugated dienes.15 One-pot Staudinger reduction16 of 4d, 4j and 4r followed by treatment with di-tert-butyl dicarbonate provided phenethylamines 5d, 5j and 5r, respectively, in good yield.
Scheme 3.
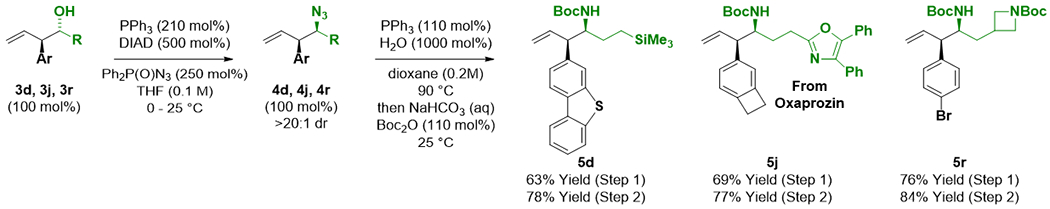
Conversion of phenethyl alcohols 3d, 3j and 3r to phenethylamines 5d, 5j and 5r.a
aYields of material isolated by silica gel chromatography. See Supporting Information for further experimental details.
(−)-Crataegusanoids A-D17 were recently isolated from the fruit of the Chinese mountain hawthorn tree, Crataegus pinnatifida, which are used to make “haw flakes,” a traditional candy from northern China. In an in vitro evaluation against two human hepatocellular carcinoma cell lines, HepG2 and Hep3B, (−)-crataegusanoids A and B displayed moderate cytotoxicity. To further illustrate the utility of the present method for asymmetric alkyne-mediated carbonyl (α-aryl)allylation, total syntheses of neolignan natural products (−)-crataegusanoids A-D were undertaken (Scheme 4). To this end, phenethyl alcohol ent-3aa was subjected to ozonolysis followed by treatment with NaBH4 to provide a 1,3-diol. Acetal or ketal formation followed by concomitant cleavage of the TIPS silyl ether and phenolic tosylate moieties delivered (−)-crataegusanoids A-D.
Scheme 4.

Total syntheses of neolignan natural products (−)-crataegusanoids A-D.a
aYields of material isolated by silica gel chromatography. See Supporting Information for further experimental details.
A series of experiments were performed to probe the reaction mechanism (Scheme 5). Under standard reaction conditions, allene iso-1r is converted to the product of carbonyl (α-aryl)allylation 3r in 45% yield (eq. 5). This experiment demonstrates that allenes are competent partners for carbonyl (α-aryl)allylation, corroborating their role as reactive intermediates. Notably, the yield of 3r obtained from allene iso-1r is significantly lower than the yield of 3r obtained from the corresponding 1-aryl-1-propyne 1r (eq. 5). These data highlight the value of utilizing tractable 1-aryl-1-propynes as reservoirs for less stable and less abundant aryl-substituted allenes. Exposure of deuterio-1ee to furfuryl alcohol 2ee under standard conditions delivers deuterio-3ee-I (eq. 6). Deuterium is transferred to the internal vinylic position (50% 2H at Hc) and allylic positions (20% 2H at Hd). These data corroborate alkyne isomerization through successive, reversible alkyne hydrometalation-β-hydride elimination, and that the ruthenium hydrides initiating hydrometalation and arising via β-hydride elimination can emanate from either the alkyne or the alcohol. Additionally, small but significant loss of deuterium is observed at the olefinic terminus (93.5% 2H at Ha,b), indicating allene hydrometalation occurs with reversibly incomplete regioselectivity. In alignment with this interpretation, the reaction of alkyne 1ee with the deuterated furfuryl alcohol deuterio-2ee (eq. 7) also results in transfer of deuterium to the internal vinylic position (50% 2H at Hc), and hydrogen-deuterium exchange occurs at the carbinol position of the primary alcohol in both reactions (7% 2H at He, eq. 6; 90% 2H at He, eq. 7) likely via reversible alcohol dehydrogenation. In equation 6, deuterium content is not completely conserved, which may be due to exchange with adventitious water.
Scheme 5.

Experiments corroborating intervention of allenes as reactive intermediates.a
aYields of material isolated by silica gel chromatography. See Supporting Information for further experimental details.
The collective data are consistent with the indicated catalytic cycle (Scheme 6). Ruthenium-catalyzed alkyne-to-allene isomerization is followed by allene hydrometalation to form fluxional σ-allyl- and π-allylruthenium complexes I.19 Aldehyde coordination followed by stereospecific carbonyl addition by way of the (E)-σ-allyliridium through the chair-like transition structure II delivers a homoallylic ruthenium alkoxide III, which upon exchange with the primary alcohol releases the product of carbonyl (α-aryl)allylation and forms the ruthenium alkoxide IV. β-Hydride elimination from ruthenium alkoxide IV delivers the ruthenium hydride V along with aldehyde to close the catalytic cycle. The π-bound alkene in III prevents β-hydride elimination at this stage by occupying the adjacent coordination site. Notably, two discrete catalytic events are operative: (a) alkyne-to-allene isomerization and (b) transfer hydrogenative carbonyl addition. Yet, as demonstrated by deuterium labelling studies (eq. 5 and 6), crossover of ruthenium hydrides that arise in these two catalytic processes is observed.
Scheme 6.
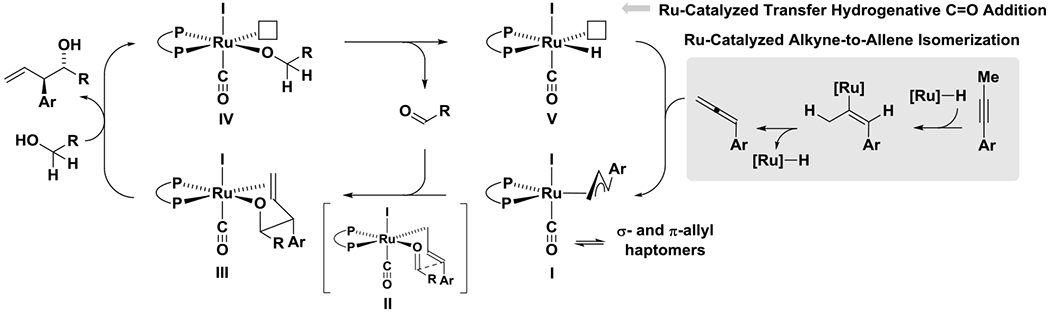
Proposed catalytic cycle for ruthenium-catalyzed C-C coupling of 1-arylpropynes with primary alcohols to form products of carbonyl (α-aryl)allylation.
Conclusions
In summary, we report that abundant, tractable 1-aryl-1-propynes serve as chiral allylmetal pronucleophiles in reactions with primary alcohol proelectrophiles to form products of carbonyl (α-aryl)allylation. These hydrogen auto-transfer processes enable access to homoallylic phenethyl alcohols with excellent control of diastereo- and enantioselectivity. This method was successfully applied to the synthesis of psychoactive phenethyl amines, as well as the neolignan natural products (−)-crataegusanoids A-D. Both steric and electronic features of the aryl sulfonic acid additive were shown contribute to the efficiency with which a more productive and selective iodide-bound ruthenium catalyst is formed. As established by deuterium labelling studies, the present processes contribute to a growing class of enantioselective metal-catalyzed C-C and C-X coupling reactions in which alkynes serve as reservoirs for less abundant and less stable allenes.5,6 Future work will focus on the development of related catalytic C-C couplings of π-unsaturated feedstocks that occur in the absence of stoichiometric organometallic reagents.4f,20,21
Supplementary Material
ACKNOWLEDGMENT
The Robert A. Welch Foundation (F-0038) and the NIH-NIGMS (RO1-GM069445) are acknowledged for partial support of this research. Mr. Shisuke Goto is acknowledged for skillful technical assistance.
Footnotes
Supporting Information. Experimental procedures and spectroscopic data for all new compounds (1H NMR, 13C NMR, IR, HRMS), including images of NMR spectra and HPLC traces for racemic and enantiomerically enriched compounds. Single crystal X-ray diffraction data for compound 3r.
The authors declare no competing financial interest.
REFERENCES
- (1).For selected reviews on carbonyl addition chemistry, see:; (a) Noyori R; Kitamura M Enantioselective Addition of Organometallic Reagents to Carbonyl Compounds: Chirality Transfer, Multiplication and Amplification. Angew. Chem. Int. Ed 1991, 30, 49–69. [Google Scholar]; (b) Soai K; Shibata T Alkylation of Carbonyl Groups. In Comprehensive Asymmetric Catalysis I-III; Jacobsen EN, Pfaltz A, H. Yamamoto, Eds.; Springer-Verlag Berlin Heidelberg: Germany, 1999; Vol. 2, pp 911–922. [Google Scholar]; (c) Pu L; Yu H-B Catalytic Asymmetric Organozinc Additions to Carbonyl Compounds. Chem. Rev 2001, 101, 757–824. [DOI] [PubMed] [Google Scholar]; (d) Catalytic Enantioselective Addition of Allylic Organometallic Reagents to Aldehydes and Ketones. Chem. Rev 2003, 103, 2763–2793. [DOI] [PubMed] [Google Scholar]; (e) Trost BM; Weiss AH The Enantioselective Addition of Alkyne Nucleophiles to Carbonyl Groups. Adv. Synth. Catal 2009, 351, 963–983. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Comprehensive Organic Synthesis, 2nd ed.; Knochel P; Molander GA, Eds.; Elsevier: Oxford, 2014; Vols. 1 and 2. [Google Scholar]
- (2).Schneider N; Lowe DM; Sayle RA; Tarselli MA; Landrum GA Big Data from Pharmaceutical Patents: A Computational Analysis of Medicinal Chemists’ Bread and Butter. J. Med. Chem 2016, 59, 4385. [DOI] [PubMed] [Google Scholar]
- (3).For selected reviews on metal-catalyzed carbonyl reductive coupling, see:; (a) Montgomery J Nickel-Catalyzed Reductive Cyclizations and Couplings. Angew. Chem. Int. Ed 2004, 43, 3890–3908. [DOI] [PubMed] [Google Scholar]; (b) Kimura M; Tamaru Y Nickel-Catalyzed Reductive Coupling of Dienes and Carbonyl Compounds. Top. Curr. Chem 2007, 279, 173–207. [Google Scholar]; (c) Jeganmohan M; Cheng C-H Cobalt- and Nickel-Catalyzed Regio-and Stereoselective Reductive Coupling of Alkynes, Allenes, and Alkenes with Alkenes. Chem. Eur. J 2008, 14, 10876–10886. [DOI] [PubMed] [Google Scholar]; (d) Moragas T; Correa A; Martin R Metal-Catalyzed Reductive Coupling Reactions of Organic Halides with Carbonyl-Type Compounds. Chem. Eur. J 2014, 20, 8242–8258. [DOI] [PubMed] [Google Scholar]; (e) Nguyen KD; Park BY; Luong T; Sato H; Garza VJ; Krische MJ Metal Catalyzed Reductive Coupling of Olefin-Derived Nucleophiles: Reinventing Carbonyl Addition. Science 2016, 354, No. aah5133. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Holmes M; Schwartz LA; Krische MJ Intermolecular Metal-Catalyzed Reductive Coupling of Dienes, Allenes, and Enynes with Carbonyl Compounds and Imines. Chem. Rev 2018, 118, 6026–6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).For a recent reviews on metal-catalyzed carbonyl reductive coupling mediated by H2, 2-PrOH, or through hydrogen auto-transfer, see:; Doerksen RS; Meyer CC; Krische MJ Feedstock Reagents in Metal-Catalyzed Carbonyl Reductive Coupling: Minimizing Preactivation for Efficiency in Target Oriented Synthesis. Angew. Chem., Int. Ed 2019, 58, 14055–14064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).For a review on the use of alkynes as π-allyl precursors in catalysis, see:; Haydl AM; Breit B; Liang T; Krische MJ Alkynes as Electrophilic or Nucleophilic Allylmetal Precursors in Transition Metal Catalysis. Angew. Chem. Int. Ed 2017, 56, 11312–11325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).For alkynes as latent nucleophilic allylmetal equivalents in metal-catalyzed carbonyl reductive coupling and related redox-neutral processes, see:; (a) Obora Y; Hatanaka S; Ishii Y Iridium-Catalyzed Coupling Reaction of Primary Alcohols with 1-Aryl-1-propynes Leading to Secondary Homoallylic Alcohols. Org. Lett 2009, 11, 3510–3513. [DOI] [PubMed] [Google Scholar]; (b) Obora Y; Sawaguchi T; Tsubakimoto K; Yoshida H; Ogawa S; Hatanaka S Iridium-Catalyzed Synthesis of ω-Hydroxy Homoallylic Alcohols. Synthesis 2013, 2115–2119. [Google Scholar]; (c) Park BY; Nguyen KD; Chaulagain MR; Komanduri V; Krische MJ Alkynes as Allylmetal Equivalents in Redox-Triggered C-C Couplings to Primary Alcohols: (Z)-Homoallylic Alcohols via Ruthenium Catalyzed Propargyl C-H Activation. J. Am. Chem. Soc 2014, 136, 11902. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chen Q-A; Cruz FA; Dong VM Alkyne Hydroacylation: Switching Regioselectivity by Tandem Ruthenium Catalysis. J. Am. Chem. Soc 2015, 137, 3157–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Liang T; Nguyen KD; Zhang W; Krische MJ Enantioselective Ruthenium Catalyzed Carbonyl Allylation via Alkyne-Alcohol C-C Bond Forming Transfer Hydrogenation: Allene Hydrometallation vs. Oxidative Coupling. J. Am. Chem. Soc 2015, 137, 3161–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Liang T; Zhang W; Chen T-Y; Nguyen KD; Krische MJ Ruthenium Catalyzed Diastereo- and Enantioselective Coupling of Propargyl Ethers with Alcohols: Siloxy-Crotylation via Hydride Shift Enabled Conversion of Alkynes to π-Allyls. J. Am. Chem. Soc 2015, 137, 13066–13071. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Liang T; Zhang W; Krische MJ Iridium Catalyzed C-C Coupling of a Simple Propargyl Ether with Primary Alcohols: Enantioselective Homoaldol Addition via Redox-Triggered (Z)-Siloxyallylation. J. Am. Chem. Soc 2015, 137, 16024–16027. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Zhang W Chen W; Xiao H; Krische MJ Carbonyl anti-(α-Amino)allylation via Ruthenium Catalyzed Hydrogen Auto-Transfer: Use of an Acetylenic Pyrrole as an Allylmetal Pronucleophile. Org. Lett 2017, 19, 4876–4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).For selected reviews on enantioselective carbonyl allylation, see:; (a) Denmark SE; Fu J Catalytic Enantioselective Addition of Allylic Organometallic Reagents to Aldehydes and Ketones. Chem. Rev 2003, 103, 2763–2794. [DOI] [PubMed] [Google Scholar]; (b) Hall DG Lewis and Brønsted Acid Catalyzed Allylboration of Carbonyl Compounds: From Discovery to Mechanism and Applications. Synlett 2007, 11, 1644–1655. [Google Scholar]; (c) Hargaden GC; Guiry PJ The Development of the Asymmetric Nozaki—Hiyama—Kishi Reaction. Adv. Synth. Catal 2007, 349, 2407–2424. [Google Scholar]; (d) Yus M; González-Gómez JC; Foubelo F Catalytic Enantioselective Allylation of Carbonyl Compounds and Imines. Chem. Rev 2011, 111, 7774–7854. [DOI] [PubMed] [Google Scholar]; (e) Huo H-X; Duvall JR; Huang M-Y; Hong R Catalytic Asymmetric Allylation of Carbonyl Compounds and Imines with Allylic Boronates. Org. Chem. Front 2014, 1, 303–320. [Google Scholar]; (f) Spielmann K; Niel G; de Figueiredo RM; Campagne J-M Catalytic Nucleophilic ‘Umpoled’ π-Allyl Reagents. Chem. Soc. Rev 2018, 47, 1159–1173. [DOI] [PubMed] [Google Scholar]
- (8).(a) Riediker M; Duthaler RO Enantioselective Allylation of Carbonyl Compounds with Titanium-Carbohydrate Complexes. Angew. Chem., Int. Ed. Engl 1989, 28, 494–495. [Google Scholar]; (b) Hafner A; Duthaler RO; Marti R; Rihs G; Rothe-Streit P; Schwarzenbach F Enantioselective syntheses with titanium carbohydrate complexes. Part 7. Enantioselective Allyltitanation of Aldehydes with Cyclopentadienyldialkoxyallyltitanium Complexes. J. Am. Chem. Soc 1992,114, 2321–2336. [Google Scholar]; (c) Sebelius S; Szabó KJ Allylation of Aldehyde and Imine Substrates with In Situ Generated Allylboronates – A Simple Route to Enatioenriched Homoallyl Alcohols. Eur. J. Org. Chem 2005, 2539–2547. [Google Scholar]; (d) Vogt M; Ceylan S; Kirschning A Stereocontrolled Palladium-Catalysed Umpolung Allylation of Aldehydes with Allyl Acetates. Tetrahedron 2010, 66, 6450–6456. [Google Scholar]; (e) Haddad TD; Hirayama LC; Singaram B Indium-Mediated Asymmetric Barbier-Type Allylations: Additions to Aldehydes and Ketones and Mechanistic Investigation of the Organoindium Reagents. J. Org. Chem 2010, 75, 642–649. [DOI] [PubMed] [Google Scholar]; (f) De Sio V; Massa A; Scettri A Chiral Sulfoxides as Activators of Allyl Trichlorosilanes in the Stereoselective Allylation of Aldehydes. Org. Biomol. Chem 2010, 8, 3055–3059. [DOI] [PubMed] [Google Scholar]; (g) Li L-L; Tao Z-L; Han Z-Y; Gong L-Z Double Chiral Induction Enables a Stereoselective Carbonyl Allylation with Simple Alkenes under the Sequential Catalysis of Palladium Complex and Chiral Phosphoric Acid. Org. Lett 2017, 19, 102–105. [DOI] [PubMed] [Google Scholar]; (h) Tekle-Smith MA; Williamson KS; Hughes IF; Leighton JL Direct, Mild, and General n-Bu4NBr-Catalyzed Aldehyde Allylsilylation with Allyl Chlorides. Org. Lett 2017, 19, 6024–6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).With the exception of reference (e) and (i), only isolated examples of catalytic enantioselective carbonyl (α-aryl)allylations have been described:; (a) Nakajima M; Saito M; Hashimoto S One-Pot Enantioselective Synthesis of Optically Active Homoallylic Alcohols from Allyl Halides. Chem. Pharm. Bull 2000, 48, 306–307. [DOI] [PubMed] [Google Scholar]; (b) Zanoni G; Gladiali S; Marchetti A; Piccinini P; Tredici I; Vidari G Enantioselective Catalytic Allylation of Carbonyl Groups by Umpolung of π-Allyl Palladium Complexes. Angew. Chem. Int. Ed 2004, 43, 846–849. [DOI] [PubMed] [Google Scholar]; (c) Zhu S-F; Yang Y; Wang L-X; Liu B; Zhou Q-L Synthesis and Application of Chiral Spiro Phospholane Ligand in Pd-Catalyzed Asymmetric Allylation of Aldehydes with Allylic Alcohols. Org. Lett 2005, 7, 2333–2335. [DOI] [PubMed] [Google Scholar]; (d) Hemelaere R; Carreaux F; Carboni B; Cross-Metathesis / Isomerization / Allylboration Sequence for a Diastereoselective Synthesis of Anti-Homoallylic Alcohols from Allylbenzene Derivatives and Aldehydes. Chem. Eur. J 2014, 20, 14518–14523. [DOI] [PubMed] [Google Scholar]; (e) Evans DA; Aye Y; Wu J Asymmetric, anti-Selective Scandium-Catalyzed Sakurai Additions to Glyoxyamide. Applications to The Syntheses of N-Boc d-Alloisoleucine and d-Isoleucine. Org. Lett 2006, 8, 2071–2073. [DOI] [PubMed] [Google Scholar]; (f) Bai B; Zhu H-J; Pan W Structure Influence of Chiral 1,1′-Biscarboline-N,N′-dioxide on The Enantioselective Allylation of Aldehydes with Allyltrichlorosilanes. Tetrahedron 2012, 68, 6829–6836. [Google Scholar]; (g) Miura T; Nishida Y; Morimoto M; Murakami M Enantioselective Synthesis of Anti Homoallylic Alcohols from Terminal Alkynes and Aldehydes Based on Concomitant Use of a Cationic Iridium Complex and a Chiral Phosphoric Acid. J. Am. Chem. Soc 2013, 135, 11497–11500. [DOI] [PubMed] [Google Scholar]; (h) Tan Z; Wan X; Zang Z; Qian Q; Deng W; Gong H Ni-Catalyzed Asymmetric Reductive Allylation of Aldehydes with Allylic Carbonates. Chem. Commun 2014, 50, 3827–3830. [DOI] [PubMed] [Google Scholar]; (i) Xiong Y; Zhang G Enantioselective Synthesis of Quaternary Stereocenters via Chromium Catalysis. Org. Lett 2016, 18, 5094–5097. [DOI] [PubMed] [Google Scholar]
- (10) (a).Garza VJ; Krische MJ Hydroxymethylation beyond Carbonylation: Enantioselective Iridium Catalyzed Reductive Coupling of Formaldehyde with Allylic Acetates via Enantiotopic π-Facial Discrimination. J. Am. Chem. Soc 2016, 138, 3655–3658. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cabrera JM; Tauber J; Zhang W; Xiang M; Krische MJ Selection between Diastereomeric Kinetic vs Thermodynamic Carbonyl Binding Modes Enables Enantioselective Iridium-Catalyzed anti-(α-Aryl)allylation of Aqueous Fluoral Hydrate and Difluoroacetaldehyde Ethyl Hemiacetal. J. Am. Chem. Soc 2018, 140, 9392–9395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).For an exhaustive summary of ruthenium-catalyzed transfer hydrogenative carbonyl reductive couplings of allene pronucleophiles, see reference 3f.
- (12).For acid-base reactions of ruthenium dihydrides with Brønsted acids, see:; (a) Dobson A; Robinson SR;Uttley MF Complexes of the Platinum Metals. V. Perfluorocarboxylato Derivatives J. Chem. Soc., Dalton Trans 1974, 370–377. [Google Scholar]; (b) McInturff EL; Yamaguchi E; Krische MJ Chiral Anion Dependent Inversion of Diastereo- and Enantioselectivity in Carbonyl Crotylation via Ruthenium Catalyzed Butadiene Hydrohydroxyalkylation. J. Am. Chem. Soc 2012, 134, 20628–20631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Maitlis PM; Haynes A; James BR; Catellani M; Chiusoli GP Iodide Effects in Transition Metal Catalyzed Reactions. Dalton Trans 2004, 3409–3419 [DOI] [PubMed] [Google Scholar]
- (14).For selected reviews on phenethylamines, see:; (a) Aghajanian GK; Marek GJ Serotonin and Hallucinogens. Neuropsychopharm 1999, 21(2s), 16S–23S. [DOI] [PubMed] [Google Scholar]; (b) Trachsel D Fluorine in Psychedelic Phenethylamines. Drug Test. Anal 2012, 4, 577–590. [DOI] [PubMed] [Google Scholar]; (c) King LA New Phenethylamines in Europe. Drug Test. Anal 2013, 6, 808–818. [DOI] [PubMed] [Google Scholar]; (d) Inan F; Brunt, Tibor M; Contrucci RR; Hondebrink L; Franssen, Eric JF. Novel Phenethylamines and Their Potential Interactions With Prescription Drugs: A Systematic Critical Review. Ther. Drug Monit 2020, 42, 271–281. [DOI] [PubMed] [Google Scholar]
- (15).Lal B; Pramanik BN; Manhas MS; Bose AK Diphenylphosphoryl Azide: A Novel Reagent for The Stereospecific Synthesis of Azides from Alcohols. Tetrahedron Lett. 1977, 1977–1980. [Google Scholar]
- (16).For a review on the Staudinger reduction, see:; Gololobov YG; Zhmurova IN; Kasukhin LF Sixty Years of Staudinger Reaction. Tetrahedron 1981, 37, 437–472. [Google Scholar]
- (17).Guo R; Shang X-Y; Lv T-M; Yao G-D; Lin B; Wang X-B; Huang XX; Song S-J Phenylpropanoid Derivatives from The Fruit of Crataegus pinnatifida Bunge and Their Distinctive Effects on Human Hepatoma Cells. Phytochemistry 2019, 164, 252–261. [DOI] [PubMed] [Google Scholar]
- (18).Tse SKS; Xue P; Lin Z; Jia G Hydrogen/Deuterium Exchange Reactions of Olefins with Deuterium Oxide Mediated by the Carbonylchlorohydrido- tris(triphenylphosphine)ruthenium(II) Complex. Adv. Synth. Catal 2010, 352, 1512–1522. [Google Scholar]
- (19).For the reaction of HXRu(CO)(PR3)3 (X = Cl, Br) with allenes and dienes to form π-allylruthenium species, see:; (a) Hiraki K; Ochi N; Sasada Y; Hayashida H; Fuchita Y; Yamanaka S Organoruthenium(II) Complexes Formed by Insertion Reactions of Some Vinyl Compounds and Conjugated Dienes into a Hydrido—Ruthenium Bond. J. Chem. Soc., Dalton Trans 1985, 873–877. [Google Scholar]; (b) Hill AF; Ho CT; Wilton-Ely IDET The Coupling of Methylene and Vinyl Ligands at a Ruthenium(II) Centre. Chem. Commun 1997, 2207–2208. [Google Scholar]; (c) Xue P; Bi S; Sung HHY; Williams ID; Lin Z; Jia G Isomerism of [Ru(η3-allyl)Cl(CO)(PPh3)2] Organometallics 2004, 23, 4735–4743. [Google Scholar]
- (20).In an exciting advance, unactivated olefins were used as allylmetal pronucleophiles in branch-selective asymmetric carbonyl additions. These processes exploit a ternary hybrid catalyst system comprising a photoredox catalyst, a hydrogen-atom-transfer catalyst, and a chromium complex. A single example of carbonyl (α-aryl)allylation was reported (>20:1 dr, 72% ee). For this and related photoredox-mediated carbonyl allylations, see:; Tanabe S; Mitsunubu H Kanai M Catalytic Allylation of Aldehydes Using Unactivated Alkenes. J. Am. Chem. Soc 2020, 142, 12374–12381 [DOI] [PubMed] [Google Scholar]; and references cited therein. [Google Scholar]
- (21).A referee of the present manuscript brought to our attention an “archived preprint” (not yet peer-reviewed) on enantioselective cobalt-BPE-catalyzed carbonyl (α-aryl)allylation employing allylbenzenes as allylmetal pronucleophiles. Sacrificial AlMe3 (150 mol%) is required to mediate π-allyl formation. Relatively modest yields and stereoselectivities are observed (typically 50-60% yield, 90:10 dr, 80% ee):; Zhang H; Huang J; Meng F Cobalt-Catalyzed Diastereo- and Enantioselective Allyl Addition to Aldehydes and α-Ketoesters through Allylic C–H Functionalization. DOI: 10.21203/rs.3.rs-58188/v1. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.