

Abstract
The heavy metal Cadmium (Cd), a widespread environmental contaminant, poses serious hazards to human health and is considered a metallohormone and carcinogen. In women with uterine fibroids, there is a significant association between blood Cd levels and increased fibroid tumor size. The aim of this study was to determine if benign human uterine leiomyoma (fibroid) cells could be malignantly transformed in vitro by continuous Cd exposure and, if so, explore a molecular mechanism by which this could occur. We found when fibroid cells were exposed to 10 µM CdCl2 for 8 weeks, a robust and fast‐growing Cd‐Resistant Leiomyoma (CR‐LM) cell culture was established. The CR‐LM cells formed viable colonies in soft agar and had increased cytoplasmic glycogen aggregates, enhanced cell motility, a higher percentage of cells in G2/M phase, and increased expression of the proliferation marker Ki‐67. NanoString analysis showed downregulation of genes encoding for extracellular matrix (ECM) components, such as collagens, fibronectins, laminins, and SLRP family proteins, whereas genes involved in ECM degradation (MMP1, MMP3, and MMP10) were significantly upregulated. A volcano plot showed that the top differentially genes favored cancer progression. Functional analysis by ingenuity pathway analysis predicted a significant inhibition of TGFB1 signaling, leading to enhanced proliferation and attenuated fibrosis. Prolonged Cd exposure altered phenotypic characteristics and dysregulated genes in fibroid cells predicative of progression towards a cancer phenotype. Therefore, continuous Cd exposure alters the benign characteristics of fibroid cells in vitro, and Cd exposure could possibly pose a health hazard for women with uterine fibroids.
Keywords: cadmium, extracellular matrix, metallohormone, NanoString, proliferation, TGFB1 signaling, uterine fibroid cells, uterine leiomyoma
Abbreviations
- AIG
anchorage‐independent growth
- BGN
biglycan
- CR‐LM
cadmium resistant leiomyoma
- DCN
decorin
- DEG
differentially expressed genes
- ECM
extracellular matrix
- FMOD
fibromodulin
- Ht‐UtLM
human uterine leiomyoma cell line immortalized via retroviral transfection of telomerase
- IPA
ingenuity pathway analysis
- Ki‐67
antigen Ki‐67 identified by monoclonal antibody Ki‐67
- LUM
lumican
- MMP1
matrix metalloproteinase 1
- PTGS2
prostaglandin‐endoperoxide synthase 2
- SLRP
small leucine‐rich proteoglycans
- SMAD3
SMAD family member 3
- SPP1
secreted phosphoprotein 1
- SULF1
sulfatase 1
- TGFB1
transforming growth factor beta 1
- THBS1
thrombospondin 1
- VEGFA
vascular endothelial growth factor A
1. INTRODUCTION
Uterine fibroids (leiomyomas) are common benign smooth muscle neoplasms of the uterus. Fibroids have both smooth muscle and extracellular matrix (ECM) components that contribute to a substantial amount of fibrous matrix in many tumors, hence the colloquial term, “fibroid.” Uterine fibroids cause symptoms such as uterine bleeding and pelvic pain that can be severe resulting in infertility or major surgery.1 Fibroid risk is associated with race, with Black women having a higher incidence of developing fibroids earlier in life.2 Fibroids do not generally occur before menarche, and they shrink dramatically after menopause. Because fibroids are hormonally regulated, it has been suggested that exposure to exogenous estrogens and metallohormones, such as Cd, may contribute to fibroid growth.1 In occupational settings, workers may be exposed to Cd in activities such as smelting, zinc refining, electroplating, and welding. In nonoccupational settings, Cd exposure typically results from tobacco consumption and ingestion of contaminated foods and drinking water. Chronic exposure to Cd, such as cigarette smoke, is thought to contribute to cancer development, and epidemiological studies show causal associations between Cd exposure and the risk of prostate, breast, and lung cancer.3 In human tissues, Cd concentrations have been reported to range from 0.8 µM to up to 1700 µM depending on age, duration of exposure, environmental exposure concentration, occupation, sex, and smoking status.4, 5, 6 Additionally, Cd bioaccumulates in many tissues with a half‐life of approximately 10‐30 years.7, 8
It is thought that Cd may mimic the effects of estrogen and contribute to endocrine disruption of the reproductive systems of wildlife and to the high incidence of hormonally related cancers and diseases observed in human populations.9 Previous studies have shown that Cd acts like steroidal estrogens in vitro in breast cancer cells due to its ability to form a high‐affinity complex with the hormone binding domain of the classical estrogen receptor alpha (ESR1).10, 11, 12 We have found that Cd can act as a metalloestrogen by nongenomic mechanisms upregulating membrane‐associated G protein‐coupled estrogen receptor 1 (GPER1) and epidermal growth factor receptor (EGFR), and stimulating proliferation of hormonally responsive human uterine leiomyoma cells.13, 14 We have also shown that in human uterine leiomyoma cells exposed to Cd at concentrations of 10−4 to 200 µM, significant proliferative effects were observed at doses of 0.1 to 20 µM at 24, 48, and 72 hours.13
An evolving body of evidence supports that Cd may be associated with multiple adverse outcomes in women's reproductive health.15 The correlation between uterine fibroid volume and blood heavy metal concentrations was investigated, and it was found that blood Cd concentrations correlated with fibroid volume.1 In this study, we examined the effects of chronic Cd exposure on fibroid cells to determine if Cd could induce phenotypic and signaling pathway alterations that would suggest possible cancer progression. Our results indicate that prolonged Cd exposure may pose a carcinogenic hazard for women with uterine fibroids and may have important clinical implications.
2. MATERIALS AND METHODS
2.1. Cell cultures
A human uterine leiomyoma cell line immortalized via retroviral transfection of telomerase (ht‐UtLM) was generated by our laboratory and maintained in medium as previously described.16 Human fibroblast and vulvar leiomyosarcoma (SK‐LMS‐1) cell lines were cultured in DMEM medium supplemented with 10% FBS. All cultures were kept in a standard tissue culture incubator at 37°C with 5% CO2.
2.2. Cadmium treatment
For 1‐week Cd exposure, ht‐UtLM cultures were maintained in the culture medium16 supplemented with 10 μM CdCl2 for 1 week with fresh medium replacement on day 3. For 8‐week Cd‐exposure, the ht‐UtLM cultures were maintained continuously in ht‐UtLM culture medium supplemented with 10 μM CdCl2 for 8 weeks. The culture medium was replaced with fresh medium every 3 days. Passage‐matched control ht‐UtLM cultures were run concurrently with Cd exposed cells. All data assessments were done following 8 weeks of continuous Cd exposure using the Cd exposure protocol described above.
2.3. RNA isolation and NanoString transcriptional profiling
Total RNA was isolated from three RNA samples from three independent experiments for control and Cd‐exposed leiomyoma cells by extracting with Trizol Reagent (ThermoFisher Cat# 15596026) and subsequently purifying with RNeasy Mini kit (Qiagen Cat#74104). The mRNA expression was examined using the NanoString platform with Cancer Progression (NS_Cancerprog_C4972) and Cancer Pathways (NS_CancerPath_C2535) panels. For both codesets, RNA expression was quantified on the nCounter Digital Analyzer, and the counts were generated with nSolver (v4.0) software. Data were adjusted utilizing the manufacturer's positive and negative control probes, as well as internal house‐keeping genes which had the lowest variability across all samples. The nSolver adjusted data were then imported into Partek (v7.0), log2 transformed and quantile normalized.
2.4. Ki‐67 antibody staining and quantification
Approximately 3.0 mL of ht‐UtLM and CR‐LM cell cultures (about 75 000 cells) were inoculated into chamber slides (ThermoFisher Scientific, Lab‐Tek Flaskette Chamber Slide system, Cat# 177453) and grown to 80% confluency in Cd‐free culture medium. The slides were fixed in 4% paraformaldehyde in PBS for 10 minutes, rinsed with 1× automation buffer (Biocare Medical Cat# TWB945M), permeabilized in 0.2% Triton X‐100, and washed in 1× automation buffer. Rabbit polyclonal anti‐Ki67 (Biocare Medical Cat#CRM325) antibody was applied to the slides at a dilution of 1:100 for 30 minutes. The slides were then incubated with anti‐Rabbit IgG HRP Polymer (BioCare Medical #MP‐7401) for 30 minutes. The slides were then treated with 3,3‐diaminobenzidine to develop chromogenic reaction. Slides were counterstained with hematoxylin and scanned with Leica Aperio AT2 Digital Whole Slide Scanning system. Three slides for each treatment group were prepared and quantified.
2.5. Immunofluorescence staining
The ht‐UtLM and CR‐LM cells were stained with COL1A1 (E6A8E) Rabbit mAb (Cell Signaling Cat# 39952) and Fibronectin (EP5) mAb (Sana Cruz Biotechnology Cat# sc‐8422). The procedure for immunofluorescence staining and confocal microscopy observations was done as described previously.17 Three slides were stained and evaluated per study group.
2.6. Transmission electron microscopy
The sample cells were detached with 0.25% trypsin solution, resuspended in 1× PBS and centrifuged at 1000 rpm. The pellets were fixed in 4F:1G fixative18 and submitted to NIEHS EM core for processing, and the detailed procedure for sample processing was described previously.19 Digital images were captured with a Gatan Orius SC1000/SC600 attached to a FEICO Tecnai T12 transmission electron microscope. Three independent blocks were prepared and evaluated by electron microscopy for each group of control and Cd‐treated cells.
2.7. Cell migration tracking
The CR‐LM and control cells were grown in glass bottom microwell dishes (MatTek Corporation, Ashland, MA, USA, Part No# P35G‐1.5‐14‐C) for 72 hours to reach about 80% confluency in the Cd‐free culture medium and stained with 1:10 000 dilution of Hoechst 33342 for 2 hours. The time‐lapse images in 3 × 3 tiles were recorded with Zeiss LSM 880 confocal microscope (objective Zeiss Plan‐Apochromate 20X/0.8 M27). The time interval was set at 5 minutes, and a total of 100 frames were recorded. The tracking lengths for individual nuclei were calculated by autoregressive motion algorithm by IMARIS. The track length histograms were produced by GraphPad PRISM 8, and the probability density distribution was produced by RStudio.
2.8. Cell cycle analysis
The sample cells were fixed with cold 70% ethanol overnight. Prior to analysis with BD LSRFortessa flow cytometer, the samples were stained with 1.0 mL Propidium Iodide solution (20 μg PI/mL, 10 units/mL RNase ONE Ribonuclease in 1× PBS) for 30 minutes in dark. To ensure that the samples were free from cell aggregates that may block the cytometer, the stained samples were filtered in a Falcon tube (5 mL Polystyrene Round‐bottom Tube with Cell‐Strainer Cap, Falcon Cat# 352235), collected at least 10 000 cells in a LO flow rate, and the data were analyzed by ModFit LT. Three independent samples were prepared for control and CR‐LM groups under conditions of normal (20% serum), starvation (cultured in serum‐free medium for 48 hours), and resupplemented (added 20% serum to the culture medium for 24 hours).
2.9. Soft agar assay
Tissue culture 6‐well plates (BECTON DICKINSON Cat# 353046) were used for soft agar assays. In each well, 1.5 mL 0.5% noble agar containing 1× MEM and 10% FBS was used to serve as the bed agar. The sample cells to be tested in the amount of 5 × 104 cells per well were seeded into 1.5 mL 0.3% noble agar containing MEM and 10% FBS on top of the bed agar. Three weeks later, the plates were stained overnight with 0.05% vital stain (P‐Iodonitrotetrazolium Violet, Sigma Cat# I‐8377) and imaged with inverted epifluorescence microscope. Six wells were setup for each cell type evaluated.
2.10. nSolver analysis
We imported RCC and RLF files to nSolver 4.0 for each panel and built a MultiRLF experiment that combined the data from the two panels and normalized the data with overlapping probes. The Box Plot graphs were exported from MultiRLF experiment. The volcano plot was generated by differential expression module with Bejamini–Hochberg correction and modified with RStudio for better readability.
2.11. Ingenuity pathway analysis
The NanoString dataset containing gene identifiers and corresponding expression values was uploaded into ingenuity pathway analysis (IPA). A fold‐change cutoff of 2.0 was set to identify molecules whose expression was significantly differentially expressed. Right‐tailed Fisher's exact test was used to calculate a p‐value determining the probability that each biological function assigned to that network due to chance alone (Krämer A., et. al., 2014).
2.12. Matrix metalloproteinases protein array
The cells were lysed with cold lysis buffer (150 mM NaCl, 0.5% Triton X‐100, 0.05% Na deoxycholate, 4% glycerol, 1 mM DTT, 10 mM Tris‐HCl, pH 7.4) supplemented with protease inhibitor cocktail (Roche Life Science, Indianapolis, IN, Cat# 11836153001). The proteomic profile of the matrix metalloproteinases (MMP) was produced with RayBio C‐Series Human Matrix Metalloproteinase Antibody Array C1 (RayBiotech Cat# AAH‐MMP‐1‐8).
2.13. TGFB phospho antibody array
TGFB phospho antibody array (Full Moon Biosystems, Cat# PTG176), which includes 176 antibodies with six replicates for each antibody, was used to analyze the Cd treated and control samples. The proteins were extracted with Antibody Assay Kit (Full Moon Biosystems, Cat# KAS02). The lysates were purified and labeled by biotin, and then combined with antibodies in the array. The slides were scanned by Agilent Technologies Microarray scanner.
2.14. Statistical analysis
The statistical significance of the differences was determined by t‐test (one‐tailed distribution, two‐sample unequal variance). Statistical significance was defined as P < .05.
3. RESULTS
3.1. Cd exposure altered cell morphology and ultrastructure
The control ht‐UtLM cells were typically elongated (Figure 1A); however, upon exposure to 10 μM Cd for 7 days, many of the cells had lost their typical elongated morphology. Beneath masses of floating spherical cells, there were a few elongated cells that remained tightly attached (Figure 1B). Interestingly, by 8 weeks, Cd exposure at 10 μM resulted in the formation of robust Cd‐Resistant ht‐UtLM (CR‐LM) cultures with focal cellular aggregates (Figure 1C). As shown in Figure 1D, the nuclei of control cells were intact and oval‐shaped, with a relatively smooth nuclear surface. However, the nuclei of 7‐day Cd‐treated cells had pitted surfaces, some with multiple lobulations, indicating significant cellular and nuclear damage (Figure 1E). However, at 8 weeks, Cd‐exposed cells had smooth nuclear surfaces with no evidence of cellular damage (Figure 1F). Ultrastructurally, CR‐LM showed large deposits of glycogen granules at the periphery of the cells compared with ht‐UtLM cells (Figure 1G‐J).
FIGURE 1.
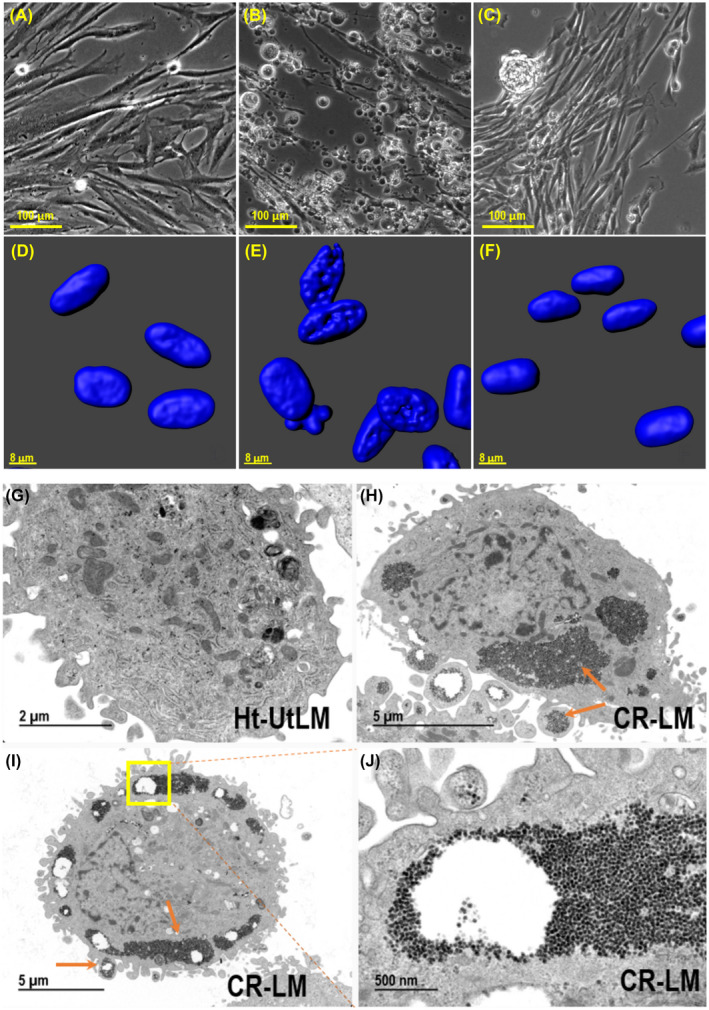
The effects of 10 μM Cd exposure on ht‐UtLM cells and ultrastructural characteristics of ht‐UtLM and 8‐week Cd‐exposed ht‐UtLM cells (designated CR‐LM). A, No Cd exposure; (B) Cd exposure for 1 week. (C) Cd exposure for 8 weeks. Bottom row: The Z‐stack images acquired with Zeiss LSM 880 confocal microscope, DAPI signal only. D, No Cd exposure; (E) Cd exposure for 1 week; (F) Cd exposure for 8 weeks. Ultrastructural images: (G) Ht‐UtLM cell; (H) CR‐LM cell; note the peripheral cytoplasmic accumulations of glycogen; (I) CR‐LM cell; (J) Higher magnification of region in box shown in panel (I). A–C (scale bar = 100 µm); D–F (scale bar = 8 µm); G (scale bar = 2 µm); H and I (scale bar = 5 µm); J (scale bar = 500 nm)
3.2. Cd exposure increased Ki‐67 expression and promoted cell cycle progression
To evaluate cell proliferation, cell cycle progression and expression of the proliferation marker, Ki‐67, were assessed. As shown in Figure 2, there was a higher percentage of Ki‐67 negative cells (with blue nuclei) in the control (Figure 2A,B), whereas CR‐LM cells had fewer Ki‐67 negative nuclei (Figure 2C,D). Further automated counting of the nuclei with Imaris showed that in the control population, the average percentage of Ki‐67 negative cells (G0 quiescent phase) was 37.9%. In contrast, the average percentage of Ki‐67 negative cells in CR‐LM was 4.8% (Figure 2E). Therefore, prolonged Cd exposure effectively decreased the percentage of quiescent cells by 7.9‐fold (37.9%/4.8%; P < .001) and rendered CR‐LM cells to be mostly in active phases of the cell cycle. To determine DNA synthesis dynamics, we conducted cell cycle analysis with flow cytometry. The CR‐LM culture had a significantly higher (P < .001) percentage of cells in G2/M phases (8.8%) versus controls (3.4%) when cultured in normal medium (Figure 2F). Under serum starvation conditions for 48 hours, the percentage of G2/M cells in CR‐LM were 3.4‐fold greater (P < .001) than the ht‐UtLM controls (Figure 2F). Specifically, after serum resupplementation for 24 hours, the percentage of cells in S phase from CR‐LM (51.5%) was significantly greater (P < .01) than the control (35.3%) (Figure 2F).
FIGURE 2.
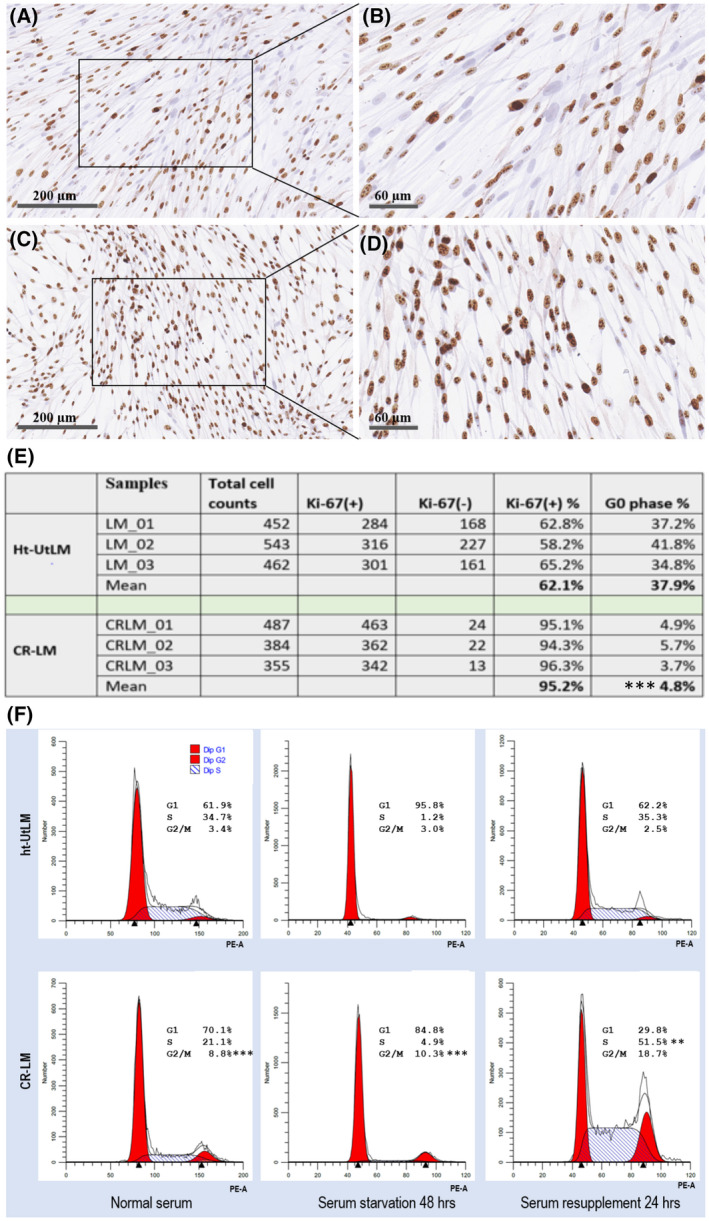
Prolonged Cd exposure stimulated Ki‐67 expression and cell cycle progression. A and B, Ht‐UtLM cells. C and D, CR‐LM. E, Quantification of Ki‐67 positive and negative cells. The difference in Ki‐67 positive cells between the groups was statistically significant (***P < .001). F, Prolonged Cd exposure promoted higher percentages of CR‐LM cells in the G2/M phase of the cell cycle compared with ht‐UtLM cells as analyzed by flow cytometry under normal and serum starvation conditions (***P < .001). After serum resupplementation for 24 hours, the percentage of cells in S phase from CR‐LM was significantly greater (**P < .01) than the control.
3.3. Cd exposure promoted anchorage‐independent growth and cell migration
Soft agar assays were conducted to evaluate whether Cd exposure would promote anchorage‐independent growth. Fibroblast (negative control), ht‐UtLM, CR‐LM, and SK‐LMS‐1 (human vulvar leiomyosarcoma, positive control) cells were tested for their ability to grow in soft agar (Figure 3). After growing in soft agar for 3 weeks, neither fibroblast or ht‐UtLM cells developed viable colonies (Figure 3A,B). Multiple viable small colonies were detected in CR‐LM (Figure 3C). As expected, SK‐LMS‐1 cells developed multiple viable larger colonies (Figure 3D). These findings suggest that CR‐LM has acquired the ability to grow independently of being anchored.
FIGURE 3.

Soft agar assays to assess anchorage‐independent growth. A, Fibroblast cells served as negative controls. B, Ht‐UtLM cells. C, CR‐LM, the viable colonies are indicated by arrows. D, SK‐LMS‐1 cells served as positive controls. Images acquired by epifluorescence microscopy in phase contrast mode. A‐D, (scale bar = 1 mm)
As illustrated by full track visualization, 8‐week Cd exposure accelerated CR‐LM cell motility (Figure 4A,B). Quantitative cell migration analysis was done by tracking individual nuclei with Imaris. As shown in the histograms of track length distributions in Figure 4C,D, there were 319 and 747 ht‐UtLM and CR‐LM cells, respectively, which passed the threshold of the tracking parameters. In ht‐UtLM, the top 3 fast‐moving cells fell into the bin at 175 μm, and the peak was located at the bin at 45 μm (Figure 4C). Whereas in CR‐LM, the fastest moving cell fell into the bin at 285 μm, and the peak was positioned at 65 μm (Figure 4D). The average migration distance (track length) for the control was 54.15 μm with SD 31.06 μm, whereas the average migration distance for CR‐LM was 88.25 μm with SD 43.70 μm (Figure 4C,D ). To show normalized distributions, an R density function was used to plot the probability density for both populations (Figure 4E). The one‐tailed t‐test with a two‐sample unequal variance returned a P‐value of 1.34E‐42, indicating that CR‐LM cells had significantly greater motility than ht‐UtLM controls.
FIGURE 4.
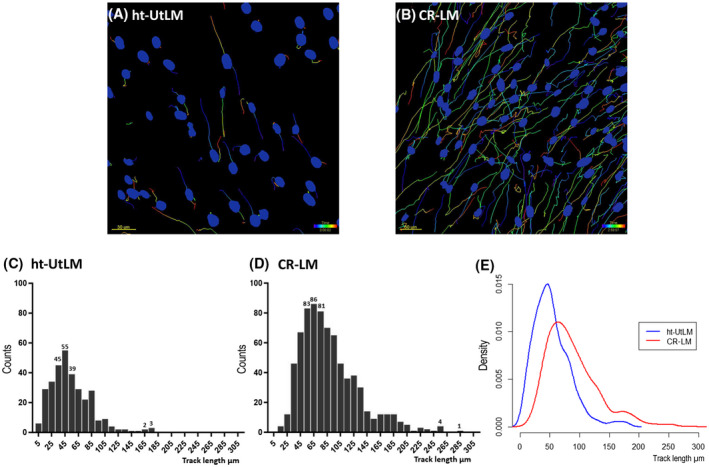
Prolonged Cd exposure stimulated cell motility in CR‐LM cells. A and B, The full track visualization of time‐lapse experiments for ht‐UtLM (A) and CR‐LM (B) cells, respectively. C and D, Frequency distribution of the track length values for ht‐UtLM (C) and CR‐LM (D) cells as shown in the histograms. E, The superimposed track length distributions of ht‐UtLM and CR‐LM cells by probability density plotted by density function with RStudio.
3.4. Differential gene expression induced by Cd exposure
There were 580 and 600 codesets for PanCancer Pathways and Progression Panels, respectively, with 132 overlapping codesets. The data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus20 and are accessible through GEO Series accession number GSE161456 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE161456 ). For CR‐LM and control cells, after applying a fold‐change filter (cutoff at 2.0), there were 180 DEGs (Dataset S1). The RCC (Recorder Code Counts) files derived from the two panels were imported into nSolver to display overall gene expression profile in volcano plot (Figure 5A). The top upregulated DEG in CR‐LM was SPP1, and the top downregulated DEG was SULF1 (Figure 5A). Interestingly, both SPP1 and SULF1 genes encode matricellular proteins that exist in ECM and serve as non‐structural regulatory proteins. The matricellular proteins modulate cell function by interacting with cell‐surface receptors, proteases, hormones, and other bioeffector molecules, as well as with structural matrix proteins. In CR‐LM, many genes encoding for ECM constitutive components were significantly downregulated. The genes encoding for collagens such as COL1A1, COL3A1, COL5A2, COL5A1, COL4A1, COL1A2, COL6A1, COL4A2, COL11A1, and COL18A1 were all reduced ranging from approximately 2‐ to nearly 10‐fold (Table 1). Additionally, genes encoding for fibronectin and laminins, also primary high‐molecular weight glycoproteins of ECM, were downregulated ranging from 3.55‐ to 4.49‐fold. The members of SLRP family, such as LUM, BGN, DCN, and FMOD, were downregulated 22.22‐, 9.06‐, 2.39‐, and 2.58‐fold, respectively. Also, THBS1, a gene encoding an adhesive glycoprotein in ECM that can bind to collagen, laminin, and fibronectin and mediates cell‐matrix interactions, was downregulated more than 40‐fold (Table 1). However, genes encoding for MMPs were significantly upregulated. For example, the transcript of MMP1, MMP3, and MMP10 were upregulated by 26.35‐, 6.71‐, and 4.22‐fold, respectively.
FIGURE 5.
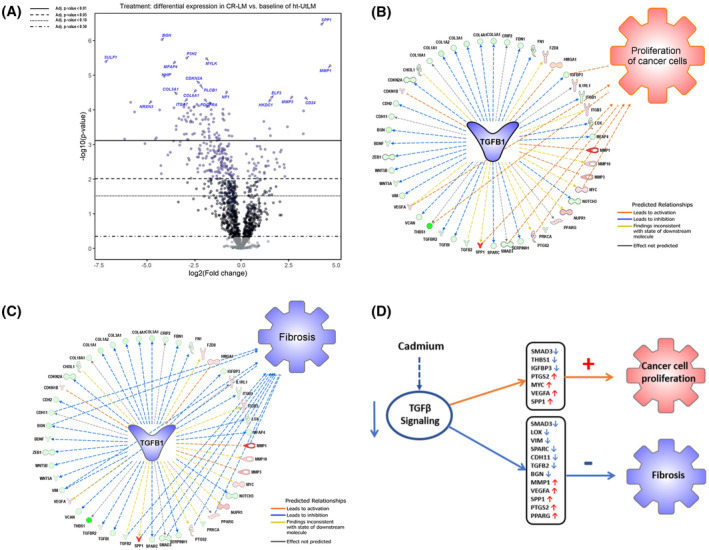
Cd exposure induced differentially expressed genes and signaling pathway changes. A, Volcano plot of altered gene expression patterns in Cd‐exposed CR‐LM cells. The volcano plot shows each gene's −log10(P‐value) and log2 fold change with the chosen covariate. Highly significant genes fall at the top of the plot and those genes highly and differentially expressed fall to either side. The horizontal lines indicate different false discovery rate (FDR) thresholds. Genes are colored if the resulting P‐value is below the given FDR threshold. The 20 most statistically significant genes are labeled. B, IPA predicted that the inhibited TGFB1 signaling favored cancer cell proliferation. Among the 47 nodes (molecules) within the TGFB1 network, seven relationship lines were identified to be associated with activation of cancer cell proliferation. The nodes implicated as having proliferative stimulatory effects (orange lines) were SPP1, MYC, VEGFA, PTGS2, IGFBP3, SMAD3, and THBS1. C, IPA predicted that the inhibited TGFB1 signaling in CR‐LM suppressed fibrosis. Among the 47 nodes within the TGFB1 network, 12 relationship lines were identified to be associated with the suppression of fibrosis. The nodes implicated as having an inhibitory effect (blue lines) were MMP1, LOX, CDH1, BGN, VIM, VEGFA, TGFB2, SPP1, SPARC, SMAD3, PTGS2 and PPARG. D, IPA analysis predicted that Cd exposure inhibits TGFB signaling, promotes cell proliferation, and inhibits fibrosis
TABLE 1.
Differential expression of extracellular and metalloproteinase genes in CR‐LM and ht‐UtLM cells
| Symbol | Entrez gene name | Expr FDR (q‐value) | Expr fold change | Location |
|---|---|---|---|---|
| MMP1 | Matrix metallopeptidase 1 | 2.03E−06 | 26.35 | Extracellular space |
| MMP3 | Matrix metallopeptidase 3 | 1.07E−05 | 6.71 | Extracellular space |
| MMP10 | Matrix metallopeptidase 10 | 1.48E−02 | 4.22 | Extracellular space |
| COL1A1 | Collagen type I alpha 1 chain | 5.41E−04 | −9.94 | Extracellular space |
| COL3A1 | Collagen type III alpha 1 chain | 7.52E−06 | −8.55 | Extracellular space |
| COL5A2 | Collagen type V alpha 2 chain | 1.04E−04 | −8.51 | Extracellular space |
| COL5A1 | Collagen type V alpha 1 chain | 2.66E−04 | −8.49 | Extracellular space |
| COL4A1 | Collagen type IV alpha 1 chain | 7.05E−05 | −5.33 | Extracellular space |
| COL1A2 | Collagen type I alpha 2 chain | 5.96E−04 | −4.37 | Extracellular space |
| COL6A1 | Collagen type VI alpha 1 chain | 8.02E−04 | −3.40 | Extracellular space |
| COL4A2 | Collagen type IV alpha 2 chain | 2.44E−04 | −2.77 | Extracellular space |
| COL11A1 | Collagen type XI alpha 1 chain | 1.64E−04 | −2.68 | Extracellular space |
| COL18A1 | Collagen type XVIII alpha 1 chain | 1.99E−04 | −2.46 | Extracellular space |
| FN1 | Fibronectin 1 | 2.50E−02 | −3.55 | Extracellular space |
| LAMA3 | Laminin subunit alpha 3 | 1.39E−03 | −3.80 | Extracellular space |
| LAMA4 | Laminin subunit alpha 4 | 7.05E−05 | −4.49 | Extracellular space |
| LUM | Lumican | 4.96E−07 | −22.22 | Extracellular space |
| BGN | Biglycan | 1.07E−05 | −9.06 | Extracellular space |
| FMOD | Fibromodulin | 2.85E−07 | −2.58 | Extracellular space |
| DCN | Decorin | 1.66E−03 | −2.39 | Extracellular space |
| THBS1 | Thrombospondin 1 | 1.85E−04 | −41.99 | Extracellular space |
This table was derived from IPA Canonical Pathway analysis on combined Cancer Progression and Cancer Pathways panels.
Pink = Upregulated gene expression fold changes; Green = Downregulated gene expression fold changes.
3.5. Cd exposure resulted in TGFB signaling inhibition
Based on IPA analysis of NanoString datasets, TGFB1 was identified to be the top upstream regulator being significantly inhibited with an activation z‐score of −2.75 and P‐value of 4.87E−29 (Dataset S2). TGFB genes encode secreted ligands of TGFB superfamily. Ligands of this family bind to various TGFB receptors leading to recruitment and activation of SMAD family transcription factors. In our IPA dataset, TGFB2 and TGFBR2 molecules involved in early events of TGFB signaling were downregulated by 6.85‐ and 2.18‐fold (Dataset S2). SMAD3, a regulator in TGFB signaling and downstream of TGFB receptor, was downregulated 2.50‐fold. Notably, THBS1, a major TGFB ligand activator, was downregulated by 41.99‐fold (Dataset S2), which may have contributed to the inhibition of TGFB signaling. To directly visualized barcode counts for critical genes in TGFB signaling, we generated boxplots based on RCC files. As shown in Figure S1, the barcode counts for SMAD3, TGFB2, TGFB3, and TGFBR2 were all suppressed in CR‐LM.
3.6. The inhibition of TGFB signaling promotes cancer cell proliferation and inhibits fibrosis
IPA revealed that the inhibited TGFB pathway was associated with enhanced cancer cell proliferation (Figure 5B). In TGFB1 network, 14 relationship lines were found to be associated with cancer cell proliferation. Among the 14 relationship lines, there were seven orange lines (leading to activation), five yellow (inconsistent prediction), two black (effect not predicted), and zero blue (leading to inhibition). For the sake of clarity, the yellow, black, and blue lines are not shown in Figure 5B. The seven nodes associated with enhanced proliferation were: 1. SPP1; 2. VEGFA; 3. PTGS2; 4. MYC (MYC proto‐oncogene bHLH transcription factor); 5. IGFBP3 (insulin‐like growth factor binding protein 3); 6. SMAD3; and 7. THBS1. The Cd exposure mediated expression of the seven genes (orange lines) aligned with the expression direction of the same molecules in Ingenuity Knowledge Base for cancer cell proliferation. It was therefore strongly suggested that long‐term Cd exposure would render fibroid cells to be more proliferative (Figure 5B, Dataset S2). IPA also revealed that the inhibited TGFB1 signaling was responsible for decreased fibrosis (Dataset S2, Figure 5C). In TGFB1 network, 21 relationship lines were found to be associated with fibrosis. There were zero orange, four yellow, five gray, and 12 blue lines. For the sake of clarity, only the blue lines were displayed. The 12 nodes directly associated with the suppressed fibrosis were 1. CDH11 (cadherin 11); 2. BGN; 3. VIM (vimentin); 4. VEGFA; 5. TGFB2 (transforming growth factor β2); 6. SPP1; 7. SPARC (secreted protein acidic and cysteine rich); 8. SMAD3; 9. PTGS2; 10. PPARG (peroxisome proliferator activated receptor γ); 11. MMP1; and 12. LOX (lysyl oxidase). IPA analysis predicted that Cd exposure inhibits TGFB signaling, promotes cell proliferation, and decreases fibrosis (Figure 5D).
3.7. Confirmation of NanoString data with protein array and confocal microscopy
We stained the fibroid cells with collagen 1 and fibronectin antibodies to visualize expression of these major ECM proteins. As illustrated in Figure 6A‐D, both collagen and fibronectin protein expression were reduced in CR‐LM cells; however, COL1A1 was mostly localized in the cytoplasm, and fibronectin was mostly expressed along the cell surface of ht‐UtLM cells. Additionally, a human MMP antibody protein array was used to quantify MMP protein levels in cell extracts. As shown in Figure 6E‐H, the levels of MMP1, MMP3, and MMP10 in protein array blots were all increased in CR‐LM. Further densitometric quantification showed that the expression of MMP1, MMP3, and MMP10 was upregulated by 32.4‐, 4.8‐, and 4.5‐fold, respectively (P‐value < .0001). The strong correlation between NanoString results and the protein expression data suggests that the NanoString platform is robust and reliable.
FIGURE 6.
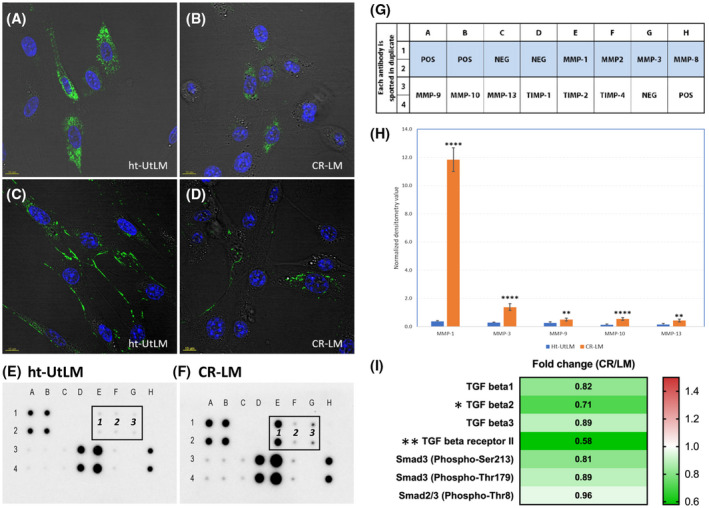
Prolonged Cd exposure reduced the protein expression of collagen 1 and fibronectin and increased MMPs. A and B, COL1A1 expression in ht‐UtLM and CR‐LM cells. C and D, Fibronectin expression in ht‐UtLM and CR‐LM cells. E, MMPs and TIMPs protein expression profile of ht‐UtLM. The positions of MMP1, MMP2, and MMP3 are denoted as 1, 2, and 3. F, CR‐LM expression profile. G, The array layout. H, Histogram of normalized signal intensity for significantly expressed MMPs. The protein expression profiles were acquired with RayBio C‐Series Human Matrix Metalloproteinase Antibody Array C1. The signal intensity for each protein was normalized with Image J. I, Decreased expression of TGFB signaling proteins following continuous Cd exposure detected by TGFB phospho‐antibody array. *P < .05; **P < .01; ****P < .0001
We investigated the protein expression and phosphorylation levels of critical proteins in TGFB signaling pathway using a TGFB phospho antibody array. In accordance with our NanoString data, the expression of TGFB1, TGFB2, TGFB3, and TGFB receptor II was all decreased in CR‐LM cells (Figure 6I). Additionally, the phosphorylation levels of SMAD3 (phospho‐Ser213 and phospho‐Thr179) and SMAD2/3(Phospho‐Thr8) were also decreased. However, only fold changes for TGFB2 (P < .05) and TGFB receptor 2 (P < .01) were statistically significant. Overall, the direction of the expression changes in these critical proteins in TGFB signaling detected by the TGFB phospho antibody array reinforces that prolonged Cd exposure inhibits TGFB signaling.
4. DISCUSSION
The effects of prolonged Cd exposure on phenotypic and signaling pathway changes in human uterine fibroid cells were investigated in this study. Our results showed that Cd exposure altered fibroid cell morphology, increased cell proliferation, induced anchorage‐independent growth, and enhanced cell motility. When fibroid cells were maintained in Cd‐containing medium for 8 weeks, a subset of the once severely stressed cells developed into fast‐growing Cadmium‐Resistant Leiomyoma (CR‐LM) cultures. Previous studies indicated that Cd and Cd‐containing compounds have carcinogenic potential in experimental animals and humans.21 It is therefore no surprise that Cd exposure could induce morphological changes and the formation of focal aggregates showing a proclivity towards cancer progression.
Long‐term Cd exposure significantly upregulated Ki‐67 expression and enhanced cell proliferation. We reported earlier that ht‐UtLM cells are highly proliferative following short‐term Cd exposures.13, 14 Ki‐67 is a proliferation marker, expressed in all active phases of the cell cycle (G1, S, G2, and mitosis), but absent in quiescent phase. Cd exposure significantly altered the cell cycle, with the percentage of cells in G2/M phase being increased in CR‐LM cells. The higher percentage of cells in G2/M correlated well with the increased Ki‐67 expression in CR‐LM cells. Moreover, the abundance of peripheral deposits of cytoplasmic glycogen granules suggested increased glycogen reserves for possible increased cell motility and proliferation.22 Cancer cells under aerobic conditions consume more glucose than required for ATP generation via oxidative phosphorylation, a phenomenon known as the Warburg effect.22, 23
AIG (Anchorage‐independent growth) is the ability of transformed cells to grow independently of a solid surface and is one of the hallmarks of cancer cells. The acquisition of AIG is thought to be a crucial step for malignancy to allow cells to metastasize, colonize in other tissues, and grow at distant sites.24 Previously, we demonstrated that ht‐UtLM cells do not show AIG and fail to produce viable colonies in soft agar.16 In this study, CR‐LM developed multiple viable colonies in soft agar, which would suggest that CR‐LM cells have acquired AIG characteristics and are progressing towards a cancer phenotype. Overexpression of ECM component, lumican, in melanoma cells has been reported to inhibit AIG in agarose gel and the capacity to invade the extracellular matrix.25 In our dataset, lumican expression was inhibited by 22‐fold (Table 1), suggesting that lumican downregulation may have been a contributor to AIG in CR‐LM. Also, prolonged Cd exposure significantly increased CR‐LM cell motility. Metastatic cancer cells undergo dramatic remodeling of ECM and cytoskeletal network and display enhanced migration.26 The significant upregulation of multiple matrix metalloproteinases and the significant downregulation of ECM components such as lumican, fibronectin, and collagen may all contribute to the enhanced motility.
In the NanoString dataset, genes encoding for ECM components (collagens, fibronectin, laminin,s and proteoglycans) were inhibited significantly. In the volcano plot, the top differentially expressed genes (such as SULF1, LUM, BGN, SPP1, and MMP1) were all regulators or components of the ECM and thought to be important in cancer progression and metastasis. Downregulation of SULF 27 and LUM gene expression in breast cancer is associated with rapid cancer progression and poor survival.25 The multiple structural components of the ECM have been reported to prevent invasion of cancer cells and possesses an intrinsic mechanism to downregulate signaling processes that are required for cancer cell proliferation. Small Leucine Rich Proteoglycans (SLRPs) such as LUM, BGN, DCN, and FMOD are ubiquitous ECM components involved in matrix structural organization and as such can potentially regulate cancer cell multiplication, angiogenesis and migration.28 In breast cancer, it was shown that the presence of laminins in ECM is critical for healthy breast cells to form organized clusters in vitro that resemble breast glands. In the absence of laminins, however, the communication between the breast cells and ECM is lost, and the healthy breast cells become disorganized and start to form clumps that resemble tumors, which highlights the importance of laminins and other ECM components in maintaining the differentiated state.29 It was shown that LUM has a direct role in regulating cancer progression. For example, reduced LUM expression in breast cancer is associated with rapid cancer progression and poor survival. On the other hand, high levels of LUM significantly suppressed subcutaneous tumor formation in a syngeneic mouse model.25 Both DCN and LUM may be considered anti‐tumor factors due to their roles in negative regulation of tumor cell proliferation. Therefore, in CR‐LM top differentially expressed genes identified in a volcano plot suggested a direction towards cancer progression. Interestingly, MMP genes, which are responsible for degrading ECM components, were highly overexpressed in CR‐LM. It is worth noting that the predominant role of MMPs is to mobilize growth factors and cytokines by dissociating them from the ECM.30
There are more than 560 putative proteases in the human degradome that are responsible for the irreversible post‐translational modifications, with MMPs as key nodal proteases. The MMPs directly regulated a host of signaling molecules, including most of the 54 chemokines in human.31, 32 Elevated expression of MMP1 is closely associated with lymph node metastasis in patients with advanced cervical cancer.33 Interestingly, Cd exposure not only significantly increased MMP1 transcript levels by as much as 26.35‐fold but also boosted MMP1 protein expression level by 32.4‐fold in CR‐LM cells. Additionally, the Cd exposure dramatically suppressed transcript and protein expression levels of ECM structural components such as fibronectin and collagens. The differential alterations in the MMP and ECM expression might greatly contribute to the Cd‐mediated cellular proliferation and migration stimulation.
The node TGFB1 was identified as the top upstream regulator being significantly inhibited in CR‐LM cells. The biological and physiological functions of TGFB ligands and receptors are of critical importance in human diseases. TGFB signaling has been implicated as one of the major players in uterine fibroid pathophysiology that mediates cellular migration, fibroid growth, and metabolism. The upregulation of TGFB signaling in uterine fibroids results in excessive extracellular matrix production and accumulation.34 This excessive overexpression of TGFB may be responsible for clinically symptomatic uterine fibroids.34 Importantly, genetic studies have consistently demonstrated that TGFB signaling serves as a tumor suppressor and plays a critical role in preventing carcinogenesis in normal epithelial cells.35 The tumor suppressive effects of TGFB in normal epithelial cells include cell cycle arrest, apoptosis, and prevention of cell immortalization. In CR‐LM, the major TGFB ligand activator THBS1 was downregulated more than 40‐fold (Dataset S1, Table 1), which could contribute to the inhibition of TGFB signaling. The modulation of THBS1 activity may represent a novel approach to selectively regulate TGFB signaling in fibrotic disease.36 The prolonged Cd exposure significantly attenuated TGFB signaling, which might compromise the tumor suppressive activities of TGFB and thereby favor pro‐cancer events.
Based on the direction of the gene expression changes of downstream nodes (molecules), IPA predicted significant inhibition of the TGFB1 pathway. Our TGFB protein array data confirmed decreased expression of TGFB1, TGFB2, TGFB3, and TGFBRII, all major TGFB signaling proteins in CR‐LM cells exposed to Cd for 2 months. However, it should be noted that in the protein phosphorylation array analysis, all the fold‐change values were marginal although TGFB2 and TGFBRII were significant. The TGFB ligand is a multifunctional cytokine that signals to nucleus through cell surface transmembrane receptors with serine/threonine kinase activity and cytoplasmic effectors, including SMAD proteins. Many members of the SLRP family such as decorin, biglycan, asporin, and fibromodulin are able to bind to and modulate TGFB pathways. Moreover, decorin modulates the TGFB pathway and regulates matrix organization and mechanical characteristics of three‐dimensional collagen matrices.37 Multiple lines of evidence suggest that both lumican and decorin inhibit cancer proliferation and invasion and are considered as anti‐tumor factors. Decorin has been implicated in the negative regulation of tumor cell proliferation via interaction with TGFB signaling network.25 In our dataset, the significant downregulation of SLRP transcripts aligned well with the predicted inhibition of TGFB signaling.
In summary, prolonged Cd exposure rendered fibroid cells morphologically distinctive, anchorage‐independent, and highly proliferative. Our data strongly suggest that the Cd induced phenotypic characteristics and dysregulated gene expression patterns in fibroid cells are indicative of progression from a benign cell type producing excessive amounts of ECM components towards a proliferative cancerous phenotype. Further studies are required to assess whether long‐term environmental Cd exposures pose a health hazard for woman with benign uterine fibroids.
CONFLICT OF INTEREST
All authors declare that they have no competing interests.
AUTHOR CONTRIBUTIONS
All authors have read and approved the manuscript. Y. Yan participated in design of the research, performed experiments, analyzed data, and wrote manuscript; J. Liu, A. Lawrence, M.J. Dykstra, R. Fannin, K. Gerrish, C.J. Tucker, E. Scappini conducted experiments and analyzed data; D. Dixon designed research and wrote manuscript.
Supporting information
Fig S1
Dataset‐S1
Dataset‐S2
ACKNOWLEDGMENTS
The authors thank Dr Agnes Janoshazi, Fluorescence Microscopy and Imaging Center; Dr Carl Bortner and Ms Maria Sifre, Flow Cytometry Center; Ms Deloris Sutton, Electron Microscopy Core; Ms Natasha Clayton and Ms Heather Jensen, Immunohistochemistry Core, and Mr Trey Saddler, Office of Data Science for their excellent technical support. This research was supported by the Intramural Research Program of the NIEHS and DNTP (ES021196‐27).
Yan Y, Liu J, Lawrence A, et al. Prolonged cadmium exposure alters benign uterine fibroid cell behavior, extracellular matrix components, and TGFB signaling. The FASEB Journal. 2021;35:e21738. 10.1096/fj.202100354R
REFERENCES
- 1.Ye S, Chung HW, Jeong K, et al. Blood cadmium and volume of uterine fibroids in premenopausal women. Ann Occup Environ Med. 2017;29:22. 10.1186/s40557-017-0178-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baird DD, Dunson DB, Hill MC, Cousins D, Schectman JM. High cumulative incidence of uterine leiomyoma in black and white women: ultrasound evidence. Am J Obstet Gynecol. 2003;188:100‐107. [DOI] [PubMed] [Google Scholar]
- 3.Luevano J, Damodaran C. A review of molecular events of cadmium‐induced carcinogenesis. J Environ Pathol Toxicol Oncol. 2014;33:183‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nogawa K, Honda R, Yamada Y, et al. Critical concentration of cadmium in kidney cortex of humans exposed to environmental cadmium. Environ Res. 1986;40:251‐260. [DOI] [PubMed] [Google Scholar]
- 5.Satarug S. Dietary cadmium intake and its effects on kidneys. Toxics. 2018;6(1):15. 10.3390/toxics6010015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elinder C‐G. Normal values for cadmium in human tissues, blood, and urine in different countries. In: Friberg L, Elinder C‐G, Kjellstrom T, Nordberg GF, eds. Cadmium and Health: A Toxicological and Epidemiological Appraisal Volume 1: Exposure, Dose, and Metabolism. Vol. 1. Boca Raton, FL: CRC Press, Inc.; 1985:81‐102. [Google Scholar]
- 7.Rafati Rahimzadeh M, Rafati Rahimzadeh M, Kazemi S, Moghadamnia AA. Cadmium toxicity and treatment: an update. Caspian J Intern Med. 2017;8:135‐145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Järup Lars, Åkesson Agneta. Current status of cadmium as an environmental health problem. Toxicol. Appl. Pharmacol. 2009;238(3):201–208. 10.1016/j.taap.2009.04.020. [DOI] [PubMed] [Google Scholar]
- 9.Johnson MD, Kenney N, Stoica A, et al. Cadmium mimics the in vivo effects of estrogen in the uterus and mammary gland. Nat Med. 2003;9:1081‐1084. [DOI] [PubMed] [Google Scholar]
- 10.Stoica A, Katzenellenbogen BS, Martin MB. Activation of estrogen receptor‐alpha by the heavy metal cadmium. Mol Endocrinol. 2000;14:545‐553. [DOI] [PubMed] [Google Scholar]
- 11.Ali I, Penttinen‐Damdimopoulou PE, Mäkelä SI, et al. Estrogen‐like effects of cadmium in vivo do not appear to be mediated via the classical estrogen receptor transcriptional pathway. Environ Health Perspect. 2010;118:1389‐1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Z, Yu X, Shaikh ZA. Rapid activation of ERK1/2 and AKT in human breast cancer cells by cadmium. Toxicol Appl Pharmacol. 2008;228:286‐294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao X, Yu L, Moore AB, Kissling GE, Waalkes MP, Dixon D. Cadmium and proliferation in human uterine leiomyoma cells: evidence of a role for EGFR/MAPK pathways but not classical estrogen receptor pathways. Environ Health Perspect. 2015;123:331‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu J, Yu L, Castro L, et al. A nongenomic mechanism for “metalloestrogenic” effects of cadmium in human uterine leiomyoma cells through G protein‐coupled estrogen receptor. Arch Toxicol. 2019;93:2773‐2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pollack AZ, Ranasinghe S, Sjaarda LA, Mumford SL. Cadmium and reproductive health in women: a systematic review of the epidemiologic evidence. Curr Environ Health Rep. 2014;1:172‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carney SA, Tahara H, Swartz CD, et al. Immortalization of human uterine leiomyoma and myometrial cell lines after induction of telomerase activity: molecular and phenotypic characteristics. Lab Invest. 2002;82:719‐728. [DOI] [PubMed] [Google Scholar]
- 17.Yan Y, Yu L, Castro L, Dixon D. ERα36, a variant of estrogen receptor α, is predominantly localized in mitochondria of human uterine smooth muscle and leiomyoma cells. PLoS ONE. 2017;12:e0186078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dykstra MJ. Why 4F:1G fixative works for me. Microscopy Today. 2010;18:50‐53. [Google Scholar]
- 19.Castro L, Gao X, Moore AB, et al. A high concentration of genistein induces cell death in human uterine leiomyoma cells by autophagy. Expert Opin Environ Biol. 2016;5(Suppl 1). 10.4172/2325-9655.S1-003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waalkes MP, Rehm S. Chronic toxic and carcinogenic effects of cadmium chloride in male DBA/2NCr and NFS/NCr mice: strain‐dependent association with tumors of the hematopoietic system, injection site, liver, and lung. Fundam Appl Toxicol. 1994;23:21‐31. [DOI] [PubMed] [Google Scholar]
- 22.Zois CE, Harris AL. Glycogen metabolism has a key role in the cancer microenvironment and provides new targets for cancer therapy. J Mol Med. 2016;94:137‐154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu J. The Warburg metabolism fuels tumor metastasis. Cancer Metastasis Rev. 2019;38:157‐164. [DOI] [PubMed] [Google Scholar]
- 24.Guadamillas MC, Cerezo A, Del Pozo MA. Overcoming anoikis–pathways to anchorage‐independent growth in cancer. J Cell Sci. 2011;124:3189‐3197. [DOI] [PubMed] [Google Scholar]
- 25.Naito Z. Role of the small leucine‐rich proteoglycan (SLRP) family in pathological lesions and cancer cell growth. J Nippon Med Sch. 2005;72:137‐145. [DOI] [PubMed] [Google Scholar]
- 26.Yilmaz M, Christofori G. Mechanisms of motility in metastasizing cells. Mol Cancer Res. 2010;8:629‐642. [DOI] [PubMed] [Google Scholar]
- 27.Lai JP, Sandhu DS, Shire AM, Roberts LR. The tumor suppressor function of human sulfatase 1 (SULF1) in carcinogenesis. J Gastrointest Cancer. 2008;39:149‐158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Appunni S, Anand V, Khandelwal M, Gupta N, Rubens M, Sharma A. Small leucine rich proteoglycans (decorin, biglycan and lumican) in cancer. Clin Chim Acta. 2019;491:1‐7. [DOI] [PubMed] [Google Scholar]
- 29.Furuta S, Ren G, Mao JH, Bissell MJ. Laminin signals initiate the reciprocal loop that informs breast‐specific gene expression and homeostasis by activating NO, p53 and microRNAs. eLife. 2018;7:e26148. 10.7554/eLife.26148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pardo A, Selman M. MMP‐1: the elder of the family. Int J Biochem Cell Biol. 2005;37:283‐288. [DOI] [PubMed] [Google Scholar]
- 31.Puente XS, Sánchez LM, Overall CM, López‐Otín C. Human and mouse proteases: a comparative genomic approach. Nat Rev Genet. 2003;4:544‐558. [DOI] [PubMed] [Google Scholar]
- 32.Overall CM, Kleifeld O. Tumour microenvironment—opinion: validating matrix metalloproteinases as drug targets and anti‐targets for cancer therapy. Nat Rev Cancer. 2006;6:227‐239. [DOI] [PubMed] [Google Scholar]
- 33.Tian R, Li X, Gao Y, Li Y, Yang P, Wang K. Identification and validation of the role of matrix metalloproteinase‐1 in cervical cancer. Int J Oncol. 2018;52:1198‐1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ciebiera M, Włodarczyk M, Wrzosek M, et al. Role of transforming growth factor β in uterine fibroid biology. Int J Mol Sci. 2017;18(11):2435. 10.3390/ijms18112435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lebrun JJ. The dual role of TGFβ in human cancer: from tumor suppression to cancer metastasis. ISRN Mol Biol. 2012;2012:381428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murphy‐Ullrich JE, Suto MJ. Thrombospondin‐1 regulation of latent TGF‐β activation: a therapeutic target for fibrotic disease. Matrix Biol. 2018;68–69:28‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schaefer L, Iozzo RV. Biological functions of the small leucine‐rich proteoglycans: from genetics to signal transduction. J Biol Chem. 2008;283:21305‐21309. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Dataset‐S1
Dataset‐S2