

Summary
Toxin-antitoxin (TA) systems are widespread in bacteria, but their activation mechanisms and bona-fide targets remain largely unknown. Here, we characterize a type III TA system, toxIN, that protects E. coli against multiple bacteriophage, including T4. Using RNA-sequencing, we find that the endoribonuclease ToxN is activated following T4 infection and blocks phage development primarily by cleaving viral mRNAs and inhibiting their translation. ToxN activation arises from T4-induced shutoff of host transcription, specifically of toxIN, leading to loss of the intrinsically unstable toxI antitoxin. Transcriptional shutoff is necessary and sufficient for ToxN activation. Notably, toxIN does not strongly protect against another phage, T7, which incompletely blocks host transcription. Thus, our results reveal a critical trade-off in blocking host transcription: it helps phage commandeer host resources, but can activate potent defense systems. More generally, our results now reveal the native targets of an RNase toxin and activation mechanism of a phage-defensive TA system.
Keywords: toxin-antitoxin, phage defense, endoribonuclease, bacterial transcription, T4 phage, E. coli
Introduction
Bacteria must continually protect themselves against bacteriophage predation. The need to survive abundant and diverse phage predators has produced an equally diverse and sophisticated set of immunity mechanisms that can interfere with nearly every aspect of a phage’s life cycle. These mechanisms include cell surface modifications that directly block adsorption or genome injection, as well as CRISPR-Cas and restriction-modification systems that can cleave phage DNA or RNA (Hampton et al., 2020). A more indirect ‘last-resort’ immunity mechanism is abortive infection (Abi), which induces cell death after infection but before phage reproduction has completed, thereby protecting uninfected neighbors in a population (Lopatina et al., 2020). Unlike CRISPR-Cas and restriction-modification systems, which are essentially always on but able to distinguish self from non-self, Abi systems must remain off until phage infection occurs and then be rapidly activated upon infection. However, it remains unknown how Abi systems specifically sense and respond to phage infection. Additionally, the specific, direct targets of most Abi systems during phage infection are unknown.
One particularly poorly-understood class of abortive infection systems are toxin-antitoxin (TA) systems. These genetic modules are encoded in the chromosomes of nearly all bacteria and archaea, and are typically composed of a growth inhibitory toxin and a cognate antitoxin that are co-expressed and form a complex, which neutralizes the toxin (Harms et al., 2018). How, and under what conditions, TA systems are activated to liberate toxins remains poorly defined (Harms et al., 2018; Song and Wood, 2020). Plasmid-borne TA systems can promote plasmid stability, but most TA systems are chromosomally-encoded (Blower et al., 2012; Leplae et al., 2011). Many abiotic stresses can stimulate TA transcription, but there is no compelling evidence that it produces active toxin following most stresses (Fraikin et al., 2019; LeRoux et al., 2020). TA systems may contribute to the formation of persister cells, dormant cells that non-heritably tolerate antibiotics (Dörr et al., 2010; Helaine et al., 2014), but this function is controversial (Fraikin et al., 2019; Harms et al., 2017).
Several TA systems have been implicated in phage defense (Dy et al., 2014; Koga et al., 2011; Pecota and Wood, 1996) including type III TA systems (Fig. 1A), which were first identified in the plant pathogen Pectobacterium atrosepticum (Fineran et al., 2009). For type III systems, the antitoxin is an RNA encoded as an array of short tandem repeats followed by the protein coding toxin gene, with an intervening Rho-independent transcription terminator that likely controls the ratio of antitoxin to toxin (Blower et al., 2012; Fineran et al., 2009). Type III toxins are endoribonucleases that cleave the repetitive antitoxin precursor to yield repeat monomers that bind to toxins, thereby neutralizing further toxin activity (Blower et al., 2011; Short et al., 2013). Following phage infection, these endoribonuclease toxins are presumably liberated from their antitoxins, but the underlying mechanism is not known (Fig. 1A). More generally, the mechanistic basis of toxin activation for all TA systems is unknown, except for the plasmid-borne system ccdAB in which CcdB toxin accumulates after plasmid loss and the consequent inability of cells to replenish the proteolytically unstable CcdA antitoxin (Melderen et al., 1994).
Fig. 1. E. coli toxIN is a type III toxin-antitoxin system.
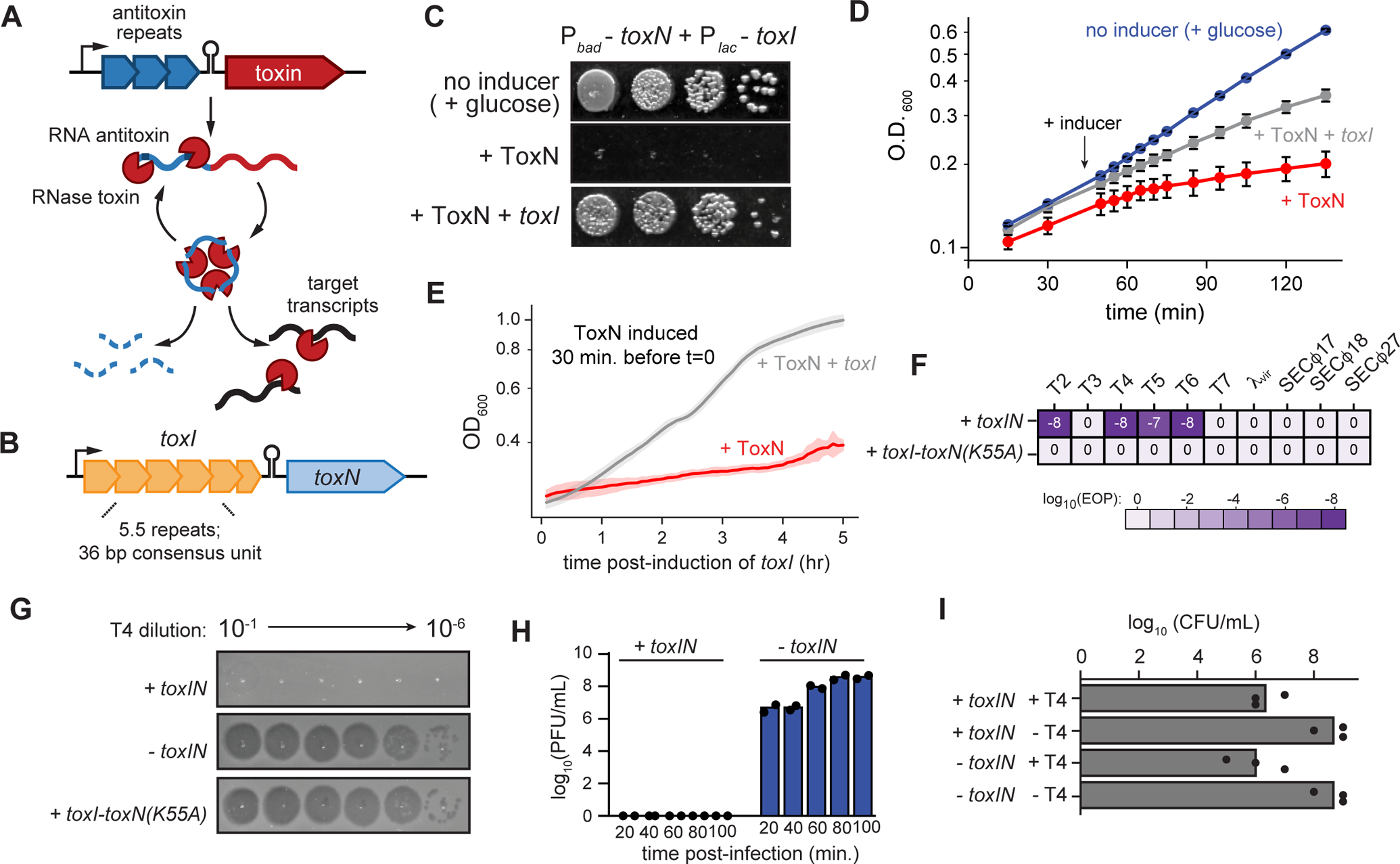
(A-B) Schematic summarizing key properties of type III TA systems (A) and E. coli toxIN (B).
(C) Representative plating assay showing toxI rescue of ToxN toxicity. Plasmids harboring toxN and toxI under arabinose- and IPTG-inducible promoters, respectively, were transformed into E. coli MG1655. ToxN, ToxN and toxI, or neither were induced as indicated; glucose represses transcription from both promoters.
(D) Growth curve showing toxI rescue of ToxN toxicity following simultaneous induction of both.
(E) Growth curve showing toxI rescue of ToxN toxicity following ToxN induction for 30 min (red curve in Fig. 1D). Data are the average of two technical replicates each of three biological replicates, with shaded areas indicating S.D.
(F) Efficiency of plaquing for +toxIN and +toxI toxN(K55A) cells infected with a panel of coliphages. Data are the mean of three biological replicates rounded to the nearest order of magnitude.
(G) Serial dilution plaque assays for T4 spotted on +toxIN, −toxIN, and +toxI toxN(K55A) cells.
(H) Plaque-forming units (PFU)/mL of T4 in +toxIN and −toxIN cells (MOI = 0.01) at 20, 40, 60, 80 and 100 min post-infection. Data are the mean of two biological replicates, with individual datapoints indicated.
(I) Colony-forming units (CFU)/mL of +toxIN and −toxIN cells infected with T4 (MOI = 5) compared to uninfected cells treated identically. Data are the mean of three biological replicates rounded to the nearest order of magnitude, with individual data points indicated.
Also see Fig. S1.
For type III TA systems, the precise targets of the endoribonuclease (RNase) toxins, other than precursor antitoxin RNA, remain unclear. Notably, RNase toxins are also common in type II TA systems, where the antitoxin is a protein rather than an RNA; these toxins have been shown to cleave a variety of mRNAs, tRNAs, and rRNAs (Culviner and Laub, 2018; Pedersen et al., 2003; Schifano et al., 2016). However, these toxins have been studied almost exclusively by artificial overexpression. Identifying the bona fide targets of RNase toxins ultimately requires knowledge of the conditions in which these toxins become active.
Here, we characterize a type III TA system, toxIN, from an environmental isolate of E. coli that confers robust defense against multiple phage, including T4. Using RNA sequencing (RNA-seq), we demonstrate that ToxN is a sequence-specific endoribonuclease that is activated relatively late in the T4 life cycle. ToxN prevents productive phage infection primarily by directly cleaving viral mRNAs, not host mRNAs. Thus, our work suggests that ToxN does not trigger cell death, as in canonical abortive infection systems, but instead blocks the production of mature virions, with cell death triggered by the phage. Importantly, we also elucidate the mechanistic basis of ToxN activation, finding that T4-induced shutoff of host transcription, including of toxIN, is both necessary and sufficient for activation. Further, we find that a different phage, T7, is partially resistant to toxIN-based immunity because it does not induce a complete transcription shutoff of toxIN. Collectively, this work reveals, for the first time to our knowledge, (i) the precise targets of an RNase toxin following its native activation rather than by toxin overexpression and (ii) the molecular mechanism behind an inducible, phage-defensive TA system.
Results
Identification of a type III toxin-antitoxin system in E. coli
Type III toxins have been classified into three families (toxIN, tenpIN, and cptIN) (Blower et al., 2012). We used JACKHMMER to find new homologs of each family of toxins in enterobacteria and then verified a subset of these hits as likely type III toxins by searching for upstream tandem repeats and Rho-independent transcription terminators. We found several examples of toxIN-like systems in environmental isolates of E. coli, including one from E. coli GCA_001012275 with 81% similarity to the previously-characterized P. atrosepticum toxIN (toxINPa) (Fineran et al., 2009) (Fig. 1B, Fig. S1A–S1B). Hereafter we refer to this locus as toxIN for simplicity or as toxINEc if necessary to distinguish it from toxINPa. For toxINEc, the toxI contains 5.5 repeats of a 36 bp consensus unit, followed by a predicted transcriptional terminator and then by toxN, which encodes a putative endoribonuclease. To verify that toxINEc is a functional toxin-antitoxin system, we cloned toxI and toxN into vectors that allow for separate and inducible expression of each, and co-transformed these plasmids into E. coli MG1655. As expected for a TA system, inducing ToxN inhibited the growth of E. coli, and co-induction of toxI rescued this toxicity, on both solid media and in culture (Fig. 1C–1D). We also established that ToxN is a bacteriostatic toxin, as inducing toxI 30 min after inducing ToxN could still rescue its toxicity (Fig. 1E).
To determine whether toxIN could protect E. coli MG1655 from phage infection, we cloned toxIN, flanked by its native promoter and the transcription terminator that follows toxN, into a low-copy number plasmid. We then infected cells harboring toxIN with a panel of diverse coliphage, including T2, T4, T5, T6, T7, λvir, SECϕ17, SECϕ18, and SECϕ27. We compared the number of plaques each phage formed on +toxIN cells to the number formed on a control strain bearing an empty vector (−toxIN) (Fig. 1F–G, S1C). Strikingly, toxIN strongly protected E. coli MG1655 from infection by T2, T4, T5 and T6, with an efficiency of plaque formation (EOP) relative to the control strain of less than 10−7. This protection was dependent on ToxN RNase activity, as expression of a toxN mutant (K55A) predicted to ablate endoribonuclease activity rendered cells fully susceptible to phage infection (Fig. 1F–G). In the presence of toxIN, the burst size of T4 following a single round of infection (80 min) decreased from ~75 to 0, indicating that T4 was unable to replicate in the presence of this TA system (Fig. 1H). Additionally, we found that after one round of T4 infection (at a multiplicity of infection, MOI, of 5 to ensure most cells were infected), the fraction of +toxIN cells that survived was equivalent to that of −toxIN cells, indicating that toxIN-mediated defense did not allow cells to recover following T4 infection (Fig. 1I). Taken together, these results indicate that toxIN is a bona fide type III toxin-antitoxin system that protects E. coli against infection by many phage, including T4, possibly via an abortive infection mechanism.
ToxN is a sequence-specific RNase that cleaves E. coli transcripts widely when overexpressed
To understand how ToxN RNase activity protects cells from phage infection, we first used RNA-sequencing (RNA-seq) to study the host-encoded targets of ToxN following its overexpression in uninfected E. coli MG1655 (Fig. 2A). We induced ToxN expression for 5–10 min., and then extracted RNA from these cells, as well as from a control strain harboring an empty vector that was treated identically. We generated and mapped RNA-seq libraries from both samples and counted the number of reads crossing each nucleotide in the genome. To identify regions that were cleaved by ToxN, we computed a ‘cleavage ratio’ (log2 of read counts +ToxN:empty vector) at each position (Culviner and Laub, 2018). By examining individual transcripts, we observed many regions with low cleavage ratios (grey bars in Fig. 2C–D), suggesting cleavage by ToxN (Fig. 2A). Across all 1,717 genes above our expression threshold (> 64 reads), 65% (or 1,118 genes) had a minimum cleavage ratio ≤ −1 following ToxN overexpression (Fig. 2B) indicating widespread RNA cleavage.
Fig. 2. ToxN is a sequence-specific endoribonuclease.
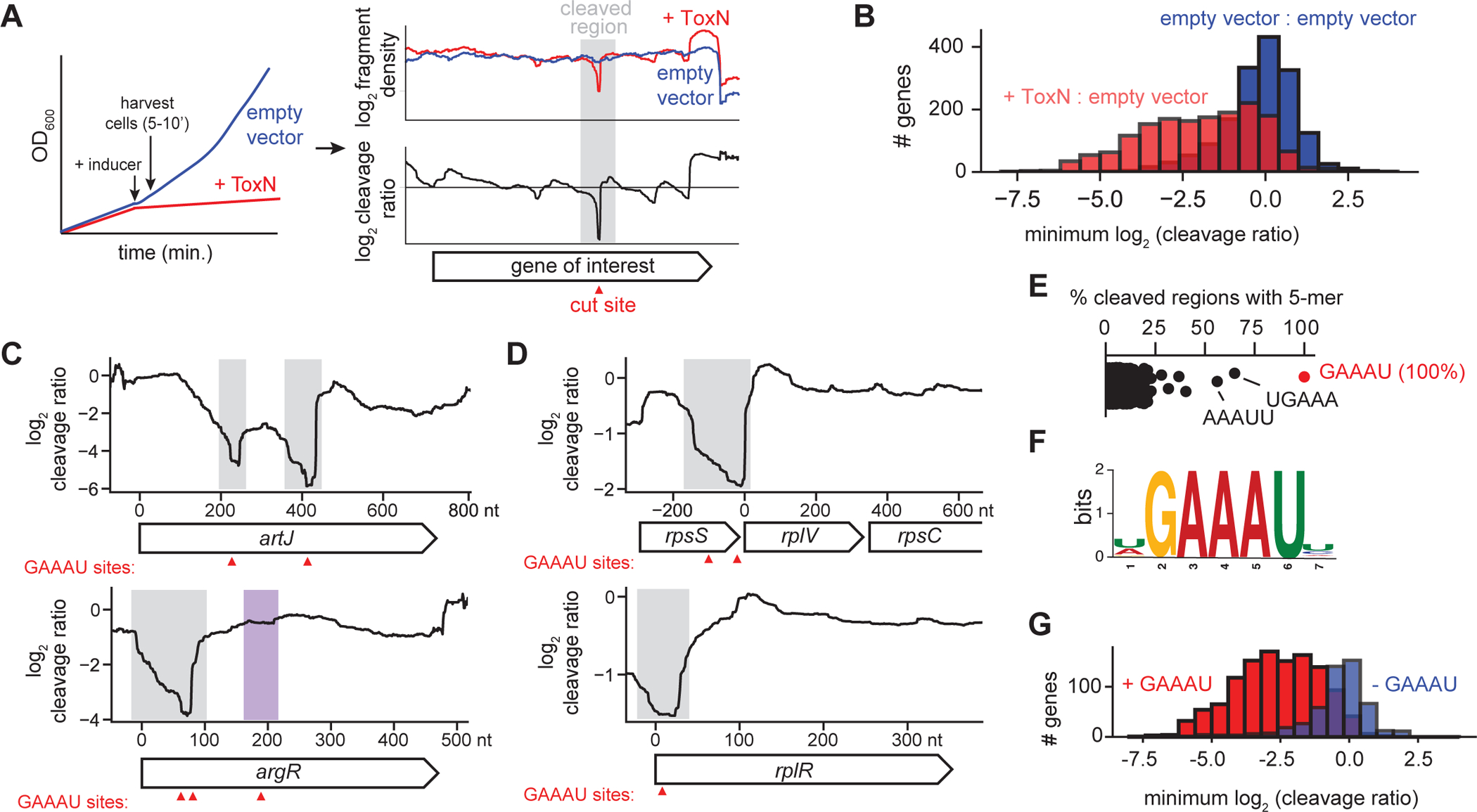
(A) Schematic overview of RNA-seq approach for determining ToxN cleavage sites.
(B) Histograms showing the distribution of the minimum cleavage ratios within well-expressed coding regions (n = 1,717) in E. coli when comparing cells overexpressing ToxN to those harboring an empty vector (red) or comparing two independent replicates harboring an empty vector (blue).
(C-D) Cleavage profiles for four well-cleaved E. coli transcripts. Sites of GAAAU motifs are indicated with red triangles. Well-cleaved instances of GAAAU are highlighted in gray, and a poorly cleaved region containing the motif is highlighted in purple.
(E) Percent of well-cleaved regions containing each possible 5-mer.
(F) Sequence logo for well-cleaved regions.
(G) Histograms showing the distribution of the minimum cleavage ratios within well-expressed coding regions in E. coli with (red, n = 1,195) and without (blue, n = 522) the ToxN motif.
Also see Fig. S2.
ToxN homologs are sequence-specific RNases; for example, ToxNPa cleaves AAAU sites in RNA (Short et al., 2013). To determine the sequence specificity of ToxNEc, we selected 141 deep, narrow valleys in the E. coli cleavage profiles (Fig. 2A) that represent clear, defined regions of ToxN-mediated cleavage. We found that the sequence GAAAU was present in 100% of these regions, with 5-mers containing part of this motif also highly enriched (Fig. 2E–F, S2A–F). All transcripts called as cleaved by ToxN contained at least one instance of GAAAU within their cleaved regions (Fig. 2C–D, 2F, S2G–H). Notably, GAAAU is also part of the toxI repeat sequence (Fig. S1B) and is likely cleaved within full-length toxI precursor by ToxN.
To confirm that the motif GAAAU was necessary for cleavage by ToxN, we divided all well-expressed E. coli transcripts into those with and without GAAAU and calculated the minimum cleavage ratio for all transcripts in each category (Fig. 2G). Transcripts containing the motif were generally much more cleaved than genes without it (median cleavage ratio of −2.62 and −0.24, respectively). Genes without the motif that had a low minimum cleavage ratio (<−1) were often co-operonic with well-cleaved genes containing the motif. Although 85% of transcripts containing a GAAAU motif were cleaved by ToxN (Fig. 2G), some GAAAU motifs were not detectably cleaved (see Fig. 2C, S2I), possibly because of RNA secondary structure or ribosome occupancy. Thus, although the GAAAU motif is necessary for cleavage by ToxN, it is not always sufficient, as is also true with E. coli MazF (Culviner and Laub, 2018).
ToxN primarily cleaves viral transcripts following T4 infection
The analyses above, like all global studies of endoribonuclease toxins to date, examined the cleavage of host-encoded transcripts following ToxN overexpression, which explains toxicity but may not accurately reflect the bona fide targets of ToxN. Our finding that toxIN protects E. coli from T4 infection offered an opportunity to examine the activity of an RNase toxin in its native context. To this end, we used the same RNA-seq pipeline as above to examine T4-infected cells harboring toxIN on a low-copy vector (+toxIN) or an empty vector control (−toxIN). We infected cells with T4 at an MOI of 5, and extracted RNA 2.5, 5, 10, 20, and 30 min. post-infection (−toxIN cells burst at ~60 min post-infection). We mapped the resulting libraries to both the E. coli and T4 genomes and then calculated the total mRNA reads coming from each genome at each timepoint, normalized to a spike-in control (Fig. 3A, S3A). T4 mRNAs were highly expressed relative to E. coli mRNAs in both +toxIN and −toxIN strains by 5 min post-infection, indicating that ToxN does not prevent expression of T4 mRNAs. Concomitant with the accumulation of T4 mRNAs was a decrease in E. coli mRNAs in both strains, consistent with the known ability of T4 to commandeer the host transcription machinery (Hinton, 2010). Because several T4 early gene products are responsible for blocking host transcription and degrading host mRNAs, we conclude that ToxN does not interfere with these early aspects of the T4 life cycle, but it may become active late in the T4 life cycle.
Fig. 3. ToxN aborts T4 infection via widespread cleavage of phage mRNAs.
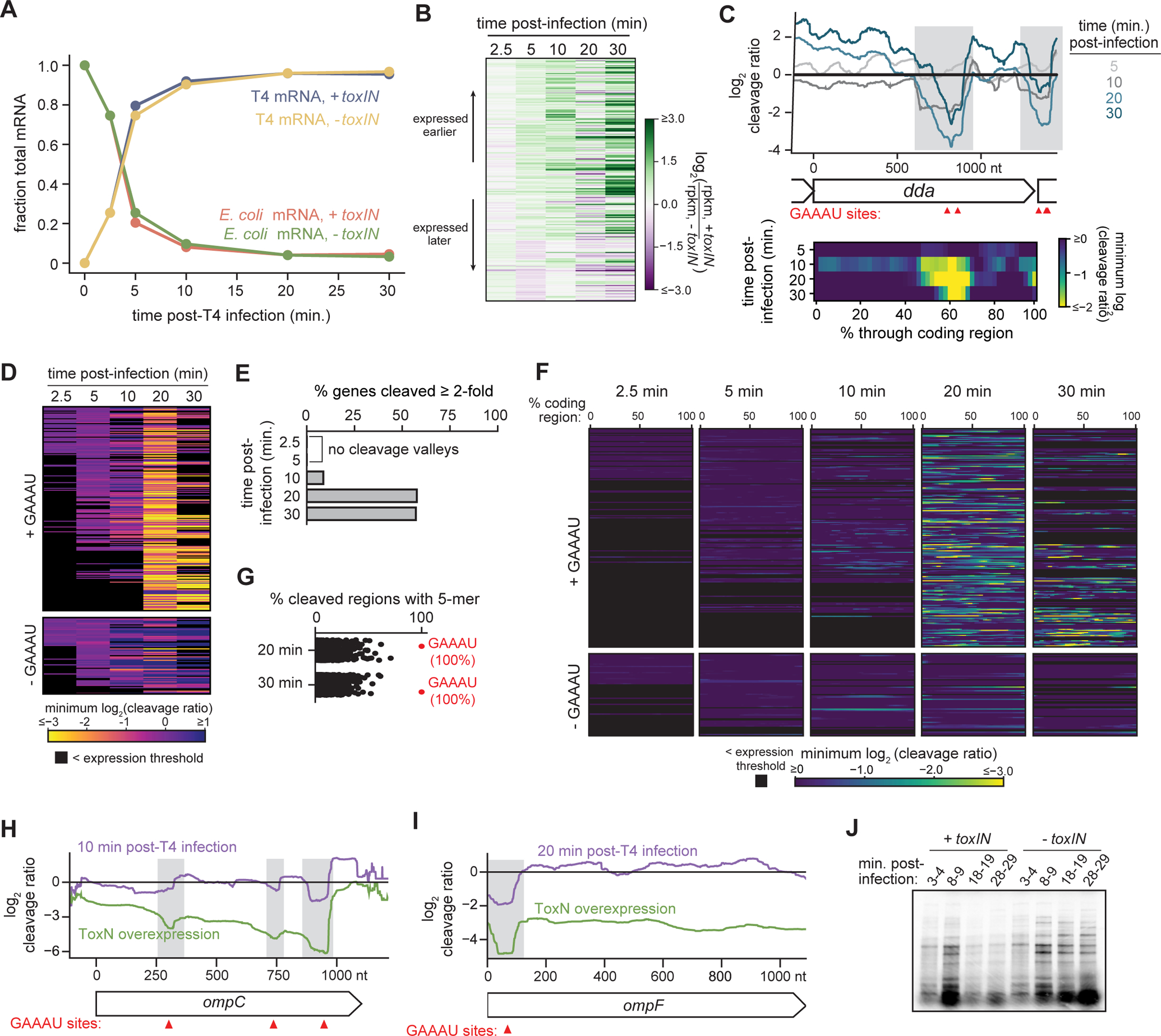
(A) Plot showing the fraction of total RNA-seq signal coming from host (E. coli) and phage (T4) mRNAs in +toxIN and −toxIN cells at the indicated times post-infection. Data are the mean of two biological replicates.
(B) Heatmap showing the ratio of log2(rpkm) for each T4 transcript at each timepoint post-infection in +toxIN cells to that in −toxIN cells. Each row corresponds to an individual gene, and genes are ordered based on their time of peak expression in the −toxIN dataset (see Fig. S3C).
(C) Top: Cleavage profile for the T4 gene dda at 5, 10, 20, and 30 min post-infection. GAAAU motifs are indicated with red triangles and cleaved regions with gray bars. Bottom: Heat map showing the minimum log2 (cleavage ratio) for 50 equally-sized regions in dda at 5, 10, 20, and 30 min post-infection.
(D) Heat map showing the minimum log2 (cleavage ratio) in every T4 transcript at 2.5, 5, 10, 20, and 30 min post-infection. Each row corresponds to an individual gene, and genes are ordered based on their time of peak expression during infection of −toxIN cells. Transcripts not well-expressed at a given timepoint are indicated in black.
(E) Bar graph quantifying the percent of well-expressed T4 transcripts cleaved ≥ 2-fold in both RNA-seq replicates at 2.5, 5, 10, 20 and 30 min post-infection. Note that at 2.5 and 5 min post-infection, no T4 genes were cleaved.
(F) Heat map, ordered as in (B), but showing the minimum log2 (cleavage ratio) for 50 equally-sized regions in every T4 transcript at 2.5, 5, 10, 20, and 30 min post-infection.
(G) Percent of well-cleaved regions in T4 transcripts 20 and 30 min post-infection that contain each possible 5-mer.
(H-I) Cleavage profiles for ompC (H) and ompF (I) following ToxN overexpression (green) and post-T4 infection (purple).
(J) Pulse-labeling of T4 proteins produced during the indicated times post-infection.
Also see Fig. S3.
To test whether toxIN interfered with later aspects of the T4 gene expression program, we calculated the log2(rpkm) for each T4 gene at each timepoint post-infection for both the +toxIN and −toxIN samples (Fig. S3B–I). In the absence of toxIN, the T4 gene expression program proceeded, as expected, with the sequential expression of early, delayed early, middle and late genes. In the presence of toxIN, the T4 gene expression program proceeded similarly over the first 10 min. However, by 20 min post-infection, early transcripts remained higher, and many late transcripts lower, relative to the −toxIN sample (Fig. 3B, S3B–E). We quantified this decrease using our spike-in control (Fig. S3F–I). T4 early, delayed early, and middle mRNAs were either comparable to or slightly more abundant in +toxIN cells than in −toxIN cells throughout the time course. T4 late transcripts were also comparable between the two strains over the first 10 min, but many were then ~50% lower in +toxIN cells at 20 and 30 min post-infection. These results suggest that ToxN activity likely accumulates during the first 20 min post-infection and then disrupts the expression or stability of T4 late genes.
To determine whether the defects in late gene expression arose directly due to ToxN cleavage activity, rather than from indirect effects of ToxN-dependent cleavage of a few key transcriptional regulators, we computed a cleavage ratio for each T4 transcript (log2 counts +toxIN : −toxIN), analogous to that done for ToxN overexpression (Fig. 2). For example, Fig. 3C shows the cleavage profiles spanning the early T4 gene dda at 5, 10, 20, and 30 min post-infection. For dda, two clear cleavage valleys emerged by 10 min post-infection (shaded grey regions in Fig. 3C), each of which contained two instances of the ToxN cleavage motif GAAAU. Similar patterns were seen for other T4 transcripts (Fig. S3J,L).
To systematically assess ToxN-mediated cleavage of all T4 transcripts, we computed the minimum cleavage ratio in each T4 transcript at each timepoint and plotted those values as a function of time (Fig. 3D). At early timepoints, very few T4 genes had low cleavage ratio minima, indicating little difference in their profiles +/− toxIN. However, for the majority of transcripts, their cleavage ratio minima progressively decreased over time, with most being ~4–8-fold lower by 20 min post-infection. Importantly, this effect was likely dependent on ToxN activity, as nearly all transcripts lacking the ToxN cleavage motif GAAAU maintained a minimum cleavage ratio close to 0 over the full 30 min time course, with a few exceptions that are likely co-operonic with transcripts harboring GAAAU sites. The fraction of T4 mRNAs with a minimum cleavage ratio ≤ −1 increased as a function of time (Fig. 3E), with 63% of all transcripts cleaved by 20 min post-infection.
To rule out the possibility that the downregulation of T4 transcripts was indirectly due to ToxN cleavage of a few key regulators, rather than to widespread degradation of the T4 transcriptome, we also looked for signatures of ToxN cleavage throughout each T4 transcript at each timepoint. We divided each T4 transcript into 50 equally-sized bins and then calculated the minimum cleavage ratio in each bin (Fig. 3C, 3F). Cleavage valleys were evident in a handful of transcripts by 10 min post-infection, and then became pervasive in most transcripts harboring GAAAU motifs by 20 and 30 min post-infection. Of the well-defined cleavage valleys at 20 and 30 min post-infection, 100% contained a GAAAU, indicating that cleavage throughout the transcriptome was a direct result of ToxN activity (Fig. 3G). Many transcripts had multiple GAAAU motifs and multiple cleavage valleys such that these RNAs had cleavage ratios < −1 across their entire lengths. Collectively, these results indicate that ToxN is active by ~10 min post-T4 infection and then directly cleaves the majority of T4 transcripts in a sequence-specific manner.
ToxN is not a canonical Abi system, but rather directly blocks the production of new virions
It has previously been proposed that type III TA systems trigger abortive infection through toxin-dependent cleavage of host transcripts, leading to host cell death and, consequently, an inability of phage to replicate (Short et al., 2018). However, because T4 blocks most host transcription and E. coli mRNAs generally have short half-lives, there were very few host transcripts above our read count threshold, particularly at the later timepoints when we observed robust ToxN-dependent cleavage of T4 transcripts (Fig. 3A, S3A). We did observe cleavage of a few highly-expressed host transcripts at 10 and 20 min. post-infection (Fig. 3H–3I, S3K), but by this time, host transcripts constituted only ~5% of mRNA reads in our RNA-seq data. Thus, we hypothesized that phage transcripts are the major targets of ToxN during infection, directly producing defects in phage particle production.
To test whether toxin cleavage disrupts T4 protein expression, we conducted pulse-labeling experiments in +toxIN and −toxIN cells (Fig. 3J). We radiolabeled newly synthesized proteins with [35S]methionine and [35S]cysteine for 2 minutes at various timepoints following T4 infection (MOI = 5) and analyzed total protein by SDS-PAGE. Consistent with our RNA-seq data, T4 early proteins (3–4 min.) were expressed at similar levels in +toxIN and −toxIN cells. At 8–9 min post-infection, the banding pattern was altered in +toxIN cells, and by 18–19 min post-infection new synthesis was virtually absent in +toxIN cells. Thus, the decrease in T4 protein synthesis generally coincides with when we detect cleavage by ToxN in our RNA-seq data. We conclude that ToxN activity in infected cells leads to widespread cleavage of phage transcripts, thereby disrupting T4 protein synthesis, particularly middle and late genes, which prevents the maturation of new T4 particles. More generally, these observations suggest that toxIN does not protect cells from infection by killing the host cell, as in canonical abortive infection systems, but rather by directly disrupting phage maturation. Cell death likely results from the action of early T4 proteins that disrupt essential host processes and are produced before ToxN is activated (Koerner and Snustad, 1979).
ToxN is activated by transcription shutoff of the toxIN locus
Importantly, in our experiments, toxIN was expressed from its native promoter, i.e. ToxN was not artificially overexpressed. How, then, does phage infection generate a pool of active ToxN? We envisioned three general models (Fig. 4A): (1) infection accelerates antitoxin degradation; (2) infection increases toxin synthesis relative to antitoxin expression; or (3) infection decreases toxIN transcription in conjunction with constitutively fast antitoxin degradation. To distinguish between these models, we sought to measure the levels and lifetimes of toxI and the levels of ToxN before and after T4 infection.
Fig. 4. ToxN is activated by transcription shutoff of toxIN.

(A) Schematic overview of three models for ToxN activation during T4 infection.
(B) Northern blot of toxI RNA using a probe complementary to a single toxI repeat (top) and Western blot of ToxN-His6 using an anti-His6 antibody (bottom) during T4 infection (MOI = 5, left) and rif treatment (300 μg/mL, right).
(C) Schematic overview of the rif growth recovery experiment in (D).
(D) Growth curves for +toxIN and −toxIN cells following rif treatment for 30 min. Data are the mean of two technical replicates each of three biological replicates, and shaded areas indicate the S.D. Estimated lag times are reported in parentheses.
(E) Cleavage profiles for two E. coli transcripts at the times indicated following addition of rif (300 μg/mL). GAAAU motifs are indicated with red triangles.
(F) Growth curves before and after CRISPRi-mediated shutoff of the toxIN promoter.
(G) Schematic overview of toxIN rescue experiments shown in (H).
(H) Serial dilution plaque assays for T4 on +toxIN cells co-transformed with plasmids encoding toxI under the control of the indicated T4 promoter.
Also see Fig. S4.
To assess toxI stability, we used Northern blotting to monitor toxI RNA (1) in uninfected cells following rifampicin (rif) treatment, which inhibits transcription, or (2) in T4-infected cells (Fig. 4B, top). To detect both processed and unprocessed (precursor) forms of toxI, we used a probe complementary to a single toxI repeat. We extracted total RNA from +toxIN cells immediately before and at several timepoints after treatment with rif or T4 infection, and then resolved each sample by 6% urea-PAGE. Full-length toxI levels decreased at the same approximate rate in both conditions, with a half-life of less than 2.5 min in each case. Notably, full-length toxI was undetectable by 10 min post-infection when we first detected ToxN cleavage activity by RNA-seq, suggesting this form of toxI is required for ToxN inhibition. In contrast to full-length antitoxin, the processed fragments of toxI were more stable in each condition, but again comparable in their turnover rates. These observations suggest that ToxN is not activated through an acceleration of toxI degradation, arguing against model 1 (Fig. 4A).
We next sought to determine whether ToxN protein levels changed concurrently with changes in toxI RNA levels during T4 infection and rif treatment. We tagged ToxN with a C-terminal His6 tag and, after verifying that this tag did not affect anti-phage activity (Fig. S4A), extracted protein at several timepoints following rif treatment and T4 infection. Using immunoblots, we found that ToxN protein levels did not change significantly in either condition (Fig. 4B, bottom; S4B), arguing against model 2.
Thus, our results favor model 3 in which T4-induced shutdown of host transcription coupled to the intrinsically fast turnover of toxI (Fig. 4A) leads to liberation of an existing pool of ToxN. Because the degradation kinetics of toxI following rif-mediated transcription shutoff were similar to that during T4 infection, model 3 predicts that ToxN should also be activated following rif treatment. To test this prediction, we treated +toxIN and −toxIN cells with rifampicin for 30 min, washed off the rif, transferred both strains to fresh media, and monitored growth (Fig. 4C). Notably, the +toxIN cells took ~1 hr longer to recover than the control strain following treatment with rif (50 or 300 μg/mL), but not in the absence of treatment (Fig. 4D). Importantly, chloramphenicol treatment, which inhibits protein synthesis instead of transcription, did not increase the lag time of +toxIN cells relative to the control, suggesting that the response observed was specific to a transcription shutoff (Fig. S4C).
To determine whether the growth delay in +toxIN cells treated with rif was a result of ToxN RNase activity, we performed RNA-seq on +toxIN and −toxIN cells following treatment with 300 μg/mL rif. We detected sequence-specific cleavage of ToxN targets, including rpsS and ompC, by 5 min post-treatment (Fig. 4E). By 10 min post-treatment, 89% of well-expressed E. coli genes were cleaved at least 2-fold in the presence of ToxN, and 100% of well-defined cleavage valleys contained the ToxN motif (Fig. S4D–E). Thus, ToxN is activated by rif treatment.
To further establish that the cleavage activity of ToxN following rif treatment results from transcription shutoff of the toxIN locus, and is not an indirect consequence of rif treatment, we used CRISPR interference to specifically block toxIN transcription. We used a plasmid-based system with constitutive expression of a guide RNA targeting the toxIN promoter and inducible expression of catalytically inactive Cas9 (dCas9), and we co-transformed this system into +toxIN and −toxIN cells. Within 15 minutes of inducing dCas9, the growth of +toxIN cells slowed relative to −toxIN cells (Fig. 4F). This growth defect results from ToxN cleavage activity, as cells expressing a ToxN active site mutant (K55A) grew at the same rate as the control strain following dCas9 induction. Taken all together, we conclude that transcription shutoff of toxIN is sufficient to rapidly activate ToxN.
Transcription shutoff of toxIN is necessary to activate ToxN during T4 infection
We next sought to determine whether transcription shutoff of toxIN is the mechanism by which ToxN is activated during T4 infection. We reasoned that if transcription shutoff of toxIN was necessary to liberate ToxN, then forcing synthesis of toxI RNA following T4 infection should prevent ToxN activation and therefore prevent ToxN-dependent defense. To test this idea, we cloned toxI under the control of T4 early, middle and late promoters, so that toxI would, in principle, be transcribed during T4 infection (Fig. 4G). We co-transformed cells with one plasmid harboring a native copy of toxIN or an empty vector and another plasmid expressing toxI from one of three different T4 promoters (PT4:toxI), and then infected these strains with T4 (Fig. 4H). In the presence of PT4 middle :toxI, we observed small plaques at lower T4 dilutions compared to cells harboring PT4 early:toxI or PT4 late:toxI or the control strain (Fig. 4H). These results indicated that the additional toxI expressed from a T4 middle promoter prevented toxIN from fully protecting cells. These results are also consistent with our observation that the disappearance of full-length toxI by 10 min post-T4 infection coincides with the emergence of ToxN activity; it is likely that the expression of toxI too early or too late in the T4 life cycle cannot sequester ToxN and bypass TA-mediated defense. We conclude that phage-induced transcription shutoff of the toxIN locus, and complete clearance of full-length toxI concurrent with middle gene expression, is necessary for the activation of ToxN and protection against T4 infection.
Complete transcription shutoff of toxI is crucial for TA-mediated defense
Our results suggest that the activation of ToxN during T4 infection is a result of phage-mediated shutoff of host transcription (Hinton, 2010). This is consistent with our observation that ToxN also protects against T2, T5, and T6 infection, all of which are known to shut off host transcription (Crawford, 1959; Koerner and Snustad, 1979). However, this property is common to other phage infections, including T3 and T7, raising the question of why toxIN did not protect against these phage (Mahadik et al., 1974; McAllister and Barrett, 1977) (Fig. 1F). To investigate this question, we focused on T7. We first measured the burst size of T7 in +toxIN and −toxIN cells. Interestingly, although T7 was able to replicate in +toxIN cells, its burst size decreased ~50% relative to the control strain (Fig. 5A). This result suggested that ToxN is also activated during T7 infection, but does not provide the magnitude of protection it does with T4.
Fig. 5. Complete transcription shutoff of toxIN is necessary for ToxN-mediated defense.
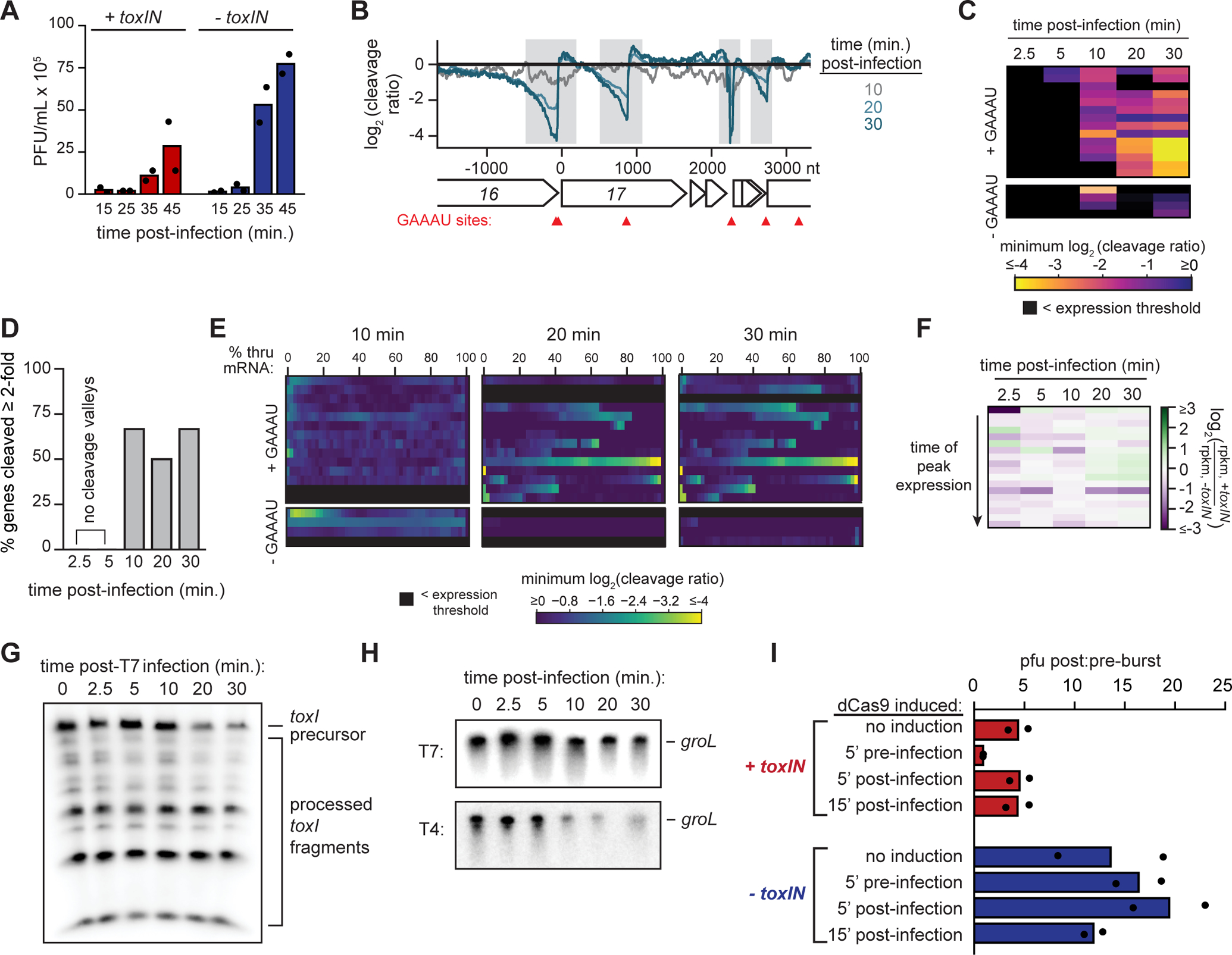
(A) Plaque-forming units (PFU)/mL of T7 in +toxIN and −toxIN cells (MOI = 0.01) at 15, 25, 35, and 45 min post-infection. Data are the mean of two biological replicates, with individual datapoints indicated.
(B) Cleavage profile for the region surrounding the T7 gene 17 at 10, 20 and 30 min post-infection.
(C) Heat map showing the minimum log2 (cleavage ratio) for every T7 mRNA at 2.5, 5, 10, 20, and 30 min post-infection. Each row corresponds to an individual mRNA, and mRNAs are ordered based on their time of peak expression during infection of −toxIN cells (see Fig. S5G).
(D) Bar graph quantifying the percent of well-expressed T4 mRNAs cleaved ≥ 2-fold in both RNA-seq replicates at 2.5, 5, 10, 20 and 30 min post-infection.
(E) Heat maps, ordered as in (C), showing the minimum log2 (cleavage ratio) for 50 equally-sized regions in every T7 mRNA at 2.5, 5, 10, 20, and 30 min post-infection.
(F) Heatmap showing the ratio of log2(rpkm) for each T7 mRNA at each timepoint post-infection in +toxIN cells to that in −toxIN cells. Each row corresponds to an individual mRNA, and mRNAs are ordered based on their time of peak expression in the −toxIN dataset (Fig. S5G).
(G) Northern blot of toxI RNA using a probe complementary to a single toxI repeat during T7 infection (MOI = 5).
(H) Northern blots of groL during T7 infection (top) and T4 infection (bottom) in −toxIN cells.
(I) Ratio of plaque-forming units (PFU)/mL of T7 in +toxIN and −toxIN cells (MOI = 0.01) 45 and 15 min post-infection, following induction of dCas9 targeting the toxIN promoter at the timepoints indicated. Data are the mean of two biological replicates, with individual datapoints indicated.
Also see Fig. S5.
To test whether ToxN was indeed active during T7 infection, we performed RNA-seq following an infection. As with T4, we saw clear cleavage of T7 transcripts by ToxN by 20 min post-infection, with > 60% of all T7 mRNAs cleaved at least two-fold in the presence of toxIN after 20 min, including 75% of T7 mRNAs with a GAAAU (Fig. 5B–E, S5A–C). As with T4, there were fewer total reads coming from phage RNAs in the +toxIN sample compared to the −toxIN sample (Fig. S5D), although we did not observe the same misregulation of T7 gene expression (Fig. 5F, S5E–G) seen with T4 (Fig. 3B, S3B–I), in particular for late transcripts. These data suggest that ToxN is activated and cleaves T7 mRNAs during infection, but that toxin activity is not sufficient to grossly interfere with the T7 life cycle.
Why is ToxN activity insufficient to completely block T7 replication? One possibility is that T7 is intrinsically less vulnerable to ToxN, either because it encodes an inhibitor of ToxN or because its transcriptome is less affected by ToxN cleavage; we ruled out the latter given that >75% of T7 transcripts have a GAAAU. Alternatively, T7 shutoff of host transcription may happen later or less completely than in T4 such that toxI is not degraded fully or rapidly enough to fully block T7 replication. To test the latter hypothesis, we used Northern blotting to measure toxI levels (Fig. 5G) and found that precursor levels decreased following T7 infection, but were not fully degraded, even after 30 min. This suggested that toxI was either (1) not completely shutoff transcriptionally or (2) was somehow stabilized during T7 infection. We favored the former as our RNA-seq data had already indicated that T7-mediated host transcription shutoff was incomplete compared to T4 infection (compare Fig. S3A to S5D). To further validate this model, we compared the stability of groL, another E. coli transcript, via Northern blot during T4 and T7 infection in −toxIN cells (Fig. 5H). As with toxI, groL transcript was far more stable during T7 infection than during T4 infection, suggesting that its transcription was not completely shut off.
Our results indicated that toxIN is not fully activated during T7 infection because enough toxI is transcribed to inactivate some ToxN. Thus, we hypothesized that forcing complete shutoff of toxIN transcription would be sufficient to protect cells from T7. To this end, we introduced into strains carrying either toxIN or an empty vector an inducible dCas9 and a constitutively expressed guide RNA that targets the toxIN promoter, as above (Fig. 4F). We then infected the strains with T7 at an MOI of 0.01 and measured the number of T7 particles present in each culture 15 min (pre-burst) and 45 min (post-burst) post-infection (Fig. 5I). Strikingly, cells harboring toxIN in which dCas9 was induced 5 min pre-infection showed a complete loss of T7 production, i.e. the pre-burst:post-burst ratio was 1, whereas −toxIN cells treated identically had a ratio of 16. A reduction in burst size was also seen for +toxIN cells without inducing dCas9, likely because of leaky dCas9 expression. We conclude that T7 is not intrinsically resistant to toxIN-mediated immunity. Instead, our results suggest that an incomplete transcription shutoff of toxIN during T7 infection results in less active ToxN and an incomplete block in T7 replication. More generally, these results highlight a fundamental trade-off associated with the extent of phage-driven shutoff of host transcription: it helps a phage commandeer host resources and maximize its reproduction, but it can activate potent anti-phage immune systems like toxIN.
Discussion
Mechanistic basis of ToxN activation during phage defense
Predation by phage represents a nearly constant evolutionary pressure to bacteria. Recent studies have revealed a remarkable diversity of systems that bacteria use to protect themselves from this threat (Doron et al., 2018; Gao et al., 2020). In some cases, such as restriction-modification, these systems are effectively in constant surveillance mode, using DNA modifications to distinguish host and foreign DNA. Others, including cyclic nucleotide-based systems and abortive infection systems, must get activated specifically in response to phage infection, but the underlying mechanisms are generally not known. We addressed this question in the context of toxin-antitoxin systems.
Based on our results, we propose the following model for type III TA-mediated phage defense (Fig. 6). toxI is normally produced at high rates and processed into monomers by active ToxN, but with little to no ToxN released to degrade other transcripts. Following phage infection, transcription of the toxIN locus is rapidly and completely shut off. Degradation of existing toxI precursors, via ToxN cleavage and other cellular RNases, leads to the rapid depletion of toxI antitoxin and liberation of active ToxN. By the time ToxN is liberated, most mRNAs in the cell are phage-derived. ToxN then cleaves the vast majority of these phage transcripts, thereby preventing the production of new phage particles and limiting spread of the phage to other cells.
Fig. 6. Model for type III TA-mediated phage defense.
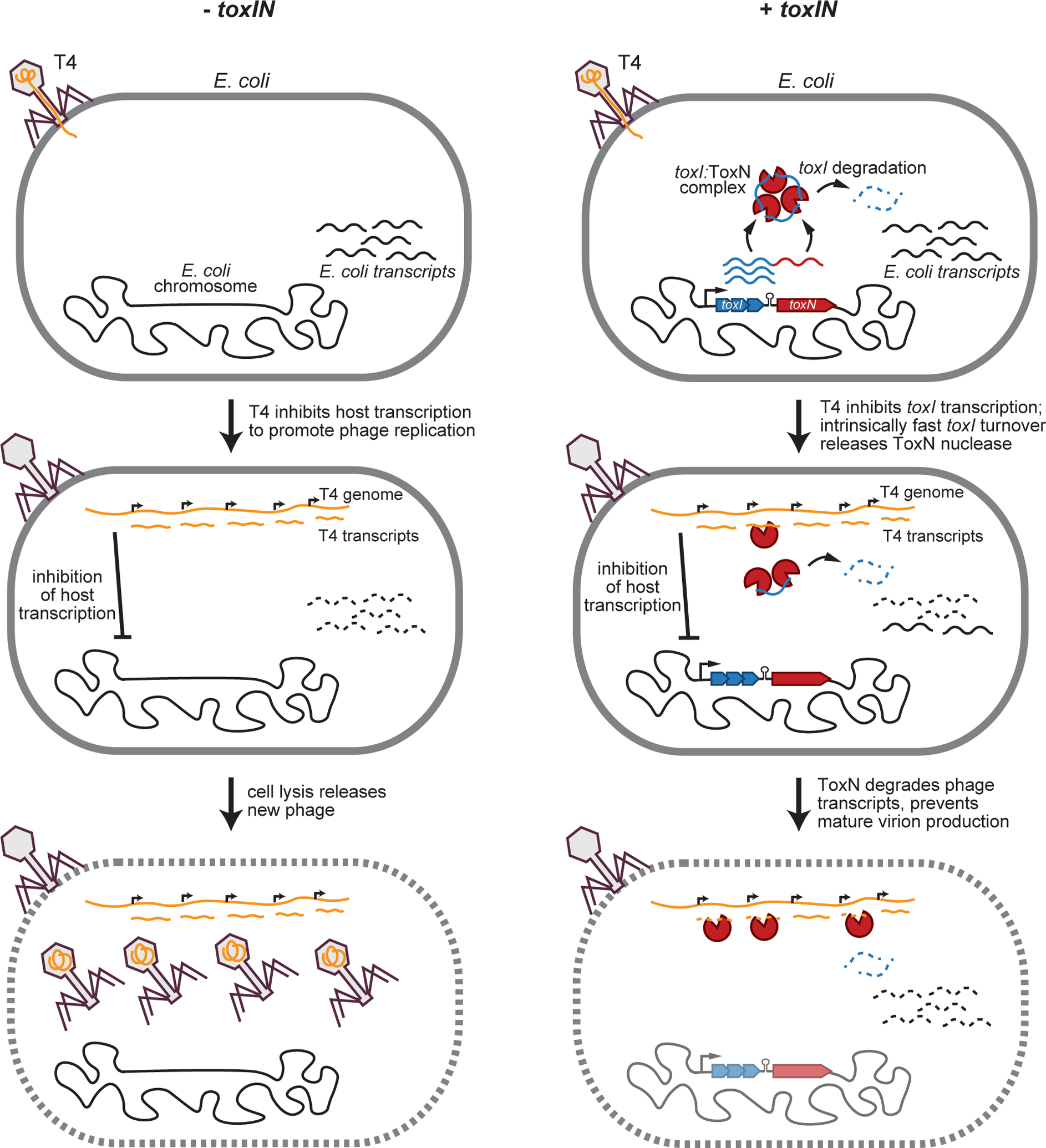
Model for T4 infection in −toxIN and +toxIN cells. (Left) Following infection of −toxIN cells, T4 inhibits host transcription to promote phage replication and the production of new phage particles that are released by eventual cell lysis. (Right) Following infection of +toxIN cells, T4 inhibits host transcription, including of toxIN. Because toxI is more labile than ToxN and toxI:ToxN complexes dissociate rapidly, free ToxN ribonuclease accumulates by ~10 min post-infection. ToxN then degrades phage transcripts, preventing the production of new T4 virions. Because T4 infection disrupts the host cell membrane and chromosome, the host cell does not survive but uninfected neighbors do.
Also see Fig. S6.
The notion that transcription shutoff activates TA systems has been proposed previously, but never experimentally demonstrated (Naka et al., 2017; Short et al., 2018; Song and Wood, 2020). Indeed, prior to this study, the mechanisms that activate TA systems were largely unknown, except for the plasmid-maintenance system ccdAB. For ccdAB, plasmid loss prevents cells from replenishing the unstable antitoxin CcdA, leading to an accumulation of free CcdB toxin, which kills cells (Melderen et al., 1994). The mechanism of action demonstrated here for toxIN is similar in that a block to synthesis leads to free, active ToxN. Prior work showed that P. atrosepticum ToxN is relatively stable whereas toxI, like CcdA, is unstable (Blower et al., 2017; Short et al., 2018), but it had not been established, until now, whether reduced synthesis was necessary or sufficient to activate ToxN during phage infection (Blower et al., 2017; Short et al., 2018)
Using phage-induced transcription shutoff to activate a defense system like ToxIN is a powerful way for host cells to detect infection without the risk of auto-immunity. A rapid and nearly complete cessation of host transcription is a hallmark of infection by many phage, particularly T4, which has several mechanisms for inhibiting host transcription as it repurposes E. coli RNA polymerase to produce phage RNA (Hinton, 2010). Relying on transcription shutoff, rather than on expression of a particular host or phage protein, might allow for wide host ranges and facile horizontal transfer of toxIN and related systems. Notably, the use of transcription shutoff as a triggering mechanism for toxIN confronts phage with an important trade-off: shutting down host transcription helps the phage shunt host resources into the production of new virions, but it runs the risk of activating a defense mechanism like that encoded by toxIN. Consistent with this idea, we found that toxIN did not provide complete protection against T7 and instead only reduced the burst size. Although T7 does inhibit the host RNA polymerase, its shutdown is not as complete as during T4 infection (Brunovskis and Summers, 1971; Snyder, 1972) (Fig. 5G–H).
ToxN endoribonuclease activity during T4 infection
To our knowledge, this is the first high-resolution, global study of toxin activity following native activation. Prior work on endoribonucleases has relied on toxin overexpression and only studied cleavage in uninfected cells. For TA systems that target phage like toxIN, such studies will have completely missed the primary targets. We found that ToxN becomes active relatively late (by ~10 min) during T4 infection; by this time, most E. coli mRNAs have been degraded. Thus, the vast majority of ToxN cleavage events we detected were in T4 transcripts. By 20 min post-infection, roughly 60% of all T4 transcripts were downregulated at least 2-fold in the presence of toxIN. Importantly, many of these genes are T4 late transcripts necessary for phage particle formation and cell lysis, and our pulse-labeling experiments indicated very little phage protein produced in the latest stages of T4 development.
Thus, our work indicates that infected cells die not because of ToxN activation, but because T4 triggers processes such as chromosome degradation and the disruption of membrane potential prior to ToxN liberation. Previous work proposed that type III TA systems act as traditional abortive infection modules by cleaving host transcripts (Short et al., 2018), thereby killing host cells to prevent phage infection from proceeding. However, neither the overexpression of ToxN nor the activation of ToxN via CRISPRi led to host cell death (Fig. 1D–E, 4F). Cells overexpressing ToxN simply ceased growing, and this growth arrest could be rescued by expressing toxI (Fig. 1E). Given these observations and that ToxN primarily cleaves phage mRNAs, we propose that toxIN does not trigger a canonical abortive infection. Instead, it is T4 phage infection that leads to host cell death with toxIN blocking the production of new mature virions to protect neighboring, uninfected cells in the population (Fig. S6).
Do other TA systems protect against phage infection?
There are now several examples of type III TA systems that function in phage defense, including the AbiQ system from Lactococci discovered in 1998, though it was not recognized as a type III TA system until later (Samson et al., 2013). Some type IV TA systems, including S. algalactiae abiEi/ii and Shewanella sp. sanaTA, are abortive infection systems, but how they are activated by phage is unclear (Dy et al., 2014; Sberro et al., 2013). A type I system, hok/sok, from the R1 plasmid protects against T4 if present on a high-copy plasmid (Pecota and Wood, 1996); but whether it functions in phage defense on the native R1 is not known, and a mechanism of activation has not been elucidated. The type II system mazEF was suggested to protect against P1 infection (Hazan and Engelberg-Kulka, 2004), but we could not replicate those results in spotting assays on E. coli MG1655 (Fig. S5H). The E. coli type II systems rnlAB and lsoAB do protect cells from dmd- T4 infection (Koga et al., 2011; Otsuka and Yonesaki, 2012), so other type II TA systems may also protect against phage. If they do, additional mechanisms for toxin activation likely remain to be discovered. Although the protein antitoxins of type II systems are typically more unstable than their cognate toxins, many are not degraded fast enough to liberate toxin through a phage-induced shutdown of transcription as shown here for toxIN (LeRoux et al., 2020).
Limitations
The functions of toxin-antitoxin systems remain poorly understood, and most studies have relied on toxin overexpression, as the native inducers were not known. Having identified a toxIN system that protects E. coli against T4 infection, we were able to elucidate the bona fide molecular targets of an RNase toxin and the molecular mechanism that naturally activates it during phage infection. We cannot, however, completely rule out toxIN plays other roles. We found that ToxN can be liberated by treatment with rifampicin, but whether toxIN promotes survival in rif is unclear. Some type III systems have been shown to stabilize plasmids, so the toxINEc examined here could function in both plasmid inheritance and phage defense. Finally, as noted above, the mechanism for toxin activation we elucidated may be common to phage-defensive TA systems featuring intrinsically labile antitoxins. For other systems, particularly type II systems, other mechanisms of activation are likely necessary. Future work is needed to identify these mechanisms for TA systems and, more broadly, many of the diverse bacterial immune systems being identified.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Questions about or requests for methods, strains, and resources generated in this study can be directed to the Lead Contact, Michael T. Laub (laub@mit.edu).
Materials Availability
Plasmids and strains generated in this study are available upon request to the Lead Contact.
Data and Code Availability
RNA-seq data is available at GEO – accession number GSE161756. Software used for RNA-seq analysis is available at https://github.com/peterculviner/genomearray. All images presented in the figures are available in Mendeley Data at doi:10.17632/d9dphh55cj.1.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
For all experiments in liquid media, Escherichia coli MG1655 strains were grown in M9 (10x stock made with 64 g/L Na2HPO4-7H2O, 15 g/L KH2PO4, 2.5 g/L NaCl, 5.0 g/L NH4Cl) medium supplemented with 0.1% casamino acids, 0.4% glycerol, 2 mM MgSO4, and 0.1 mM CaCl2 (M9-glycerol). Glucose was supplemented to 0.4% where stated (M9-glucose). For plasmid construction, E. coli DH5α cells were grown in Luria broth (LB) medium, supplemented with 0.4% glucose to suppress expression from the arabinose-inducible promoter PBAD when applicable. Antibiotics were used at the following concentrations (liquid ; plates): kanamycin (30 μg/mL ; 50 μg/mL), carbenicillin (50 μg/mL ; 100 μg/mL), chloramphenicol (20 μg/mL ; 30 μg/mL).
METHOD DETAILS
Plasmid construction
All primers, strains, and plasmids used in this study are listed in Tables S1, S2, and S3, respectively.
pBAD33-toxN and pEXT20-toxI: A truncated version of the toxIN locus from E. coli GCA_001012275 (containing 2.5 toxI repeats, the putative Rho-independent transcription terminator, and the toxN gene) was purchased as a gBlock (Integrated DNA Technologies). toxN was PCR amplified with primers CG-1 and CG-2 to add SacI and SalI restriction sites and cloned into the SacI and SalI sites of the arabinose-inducible pBAD33 vector (pBAD33-toxN). A truncated toxI locus containing 3.5 tandem repeats was constructed using annealing of CG-3 and CG-4, followed by PCR amplification with CG-5 and CG-6 to add sequences that overlapped with the IPTG-inducible pEXT20 vector. pEXT20 was digested with SacI and SalI, and then toxI was inserted into the digested plasmid using Gibson assembly (pEXT20-toxI).
pBR322–toxIN & pBR322 empty vector: To assemble the entire toxIN locus, (1) the regions 500 bp upstream of toxI and (2) toxN plus the 77 bp downstream of toxN were purchased as gBlocks (Integrated DNA Technologies). The entire toxI array of 5.5 repeats was assembled using overlap extension of CG-7 and CG-4, then PCR amplified with CG-8 and CG-9. To add sequences overlapping with the regions upstream and downstream of toxI, a second round of PCR with CG-10 and CG-9 was performed, followed by a third round with CG-11 and CG-9. The toxIN locus was then assembled by overlap extension of the two gBlocks and the PCR-amplified toxI region, followed by PCR amplification of the entire region with CG-12 and CG-13, which also contained regions of overlap with the backbone plasmid pBR322. pBR322 empty vector was constructed via round-the-horn PCR of the parental pBR322 plasmid with CG-14 and CG-15 to remove the tetracycline promoter and resistance cassette To construct pBR322–toxIN, the toxIN locus was inserted into the former site of the tetracycline promoter and resistance cassette in pBR322 empty vector using Gibson assembly. pBR322-toxI-toxN(K55A) and pBR322-toxI-toxN-His6 were constructed via round-the-horn PCR on pBR322–toxIN, using the primer pairs CG-16/CG-17 and CG-18/CG-19, respectively.
pACYC–toxIN & pACYC empty vector: The toxIN locus, along with its native promoter and the 77 bp downstream of toxN, were PCR-amplified from pBR322–toxIN with CG-20 and CG-21. A modified pACYC backbone constructed by PCR amplifying pACYCDuet-1 (Novagen) with CG-22 and CG-23 to remove lacI and the two multiple cloning sites. To construct pACYC–toxIN, the CG-21/22 PCR product was inserted into the pACYC PCR product using Gibson assembly. To construct pACYC empty vector, the pACYC PCR product was blunt-end ligated.
pBluescript-PT4-toxI: The T4 35.3 (PT4 early, the gene product 46 early promoter), rIIB2 (PT4 middle), and soc (PT4 late) promoters were amplified from T4 gDNA using the primer pairs CG-22/23, CG-24/25, and CG-26/27, respectively, which also contained homology to the multiple cloning site of pKVS45. These promoters were chosen based on information about their expression levels and timing reported in (Karam and Drake, 1994). pKVS45 was linearized via PCR with CG-28/29, and the promoter PCR products were inserted into this PCR product using overlap extension PCR with phosphorylated primers CG-28/23, CG-28/25, or CG-28/27, followed by blunt-end ligation, to generate three pKVS45-PT4 variants. The toxIN locus was then inserted into each pKVS45-PT4 backbone in two steps. First, toxIN, along with ~20 bp of the region upstream of toxI, was PCR amplified from pBR322–toxIN with CG-30/31. Each pKVS45-PT4 variant was linearized via PCR with CG-32/33, and toxIN was inserted into each backbone using Gibson assembly. Second, the overhang regions upstream of toxIN were removed via round-the-horn PCR using CG-34–37 to generate pKVS45-PT4–toxIN plasmids. Presumably because of leaky toxicity of PT4 early–toxIN, we were only able to isolate clones with an extra toxI repeat (6.5 instead of 5.5) for this construct. Additionally, there was a point mutation in the variable region of a single toxI repeat in PT4 late–toxIN, but it should not affect TA function. To generate pBluescript-PT4-toxI, PT4 early-toxI, PT4 middle-toxI, and PT4 late-toxI were amplified from pKVS45-PT4–toxIN variants using primer pairs CG-38/39, CG-40/39, and CG-41/39, respectively, which also contained regions overlapping the multiple cloning site in pBluescript II SK(+). pBluescript II SK(+) was linearized via PCR with CG-42/43. Each pBluescript-PT4-toxI variant was then assembled using Gibson assembly of PT4-toxI PCR products and the pBluescript PCR product.
pdCas9deg–toxIN gRNA: pdCas9deg3 contains a constitutively-expressed guide RNA and dCas9 with a degron tag under the control of an aTc-inducible promoter (Wiktor et al., 2016). A guide RNA sequence targeting the toxIN promoter was introduced into pdCas9deg3 using PCR with primers CG-44/45, which also contained a SpeI restriction site. The resulting PCR fragment was then digested with SpeI and ligated with T4 DNA ligase to generate the final plasmid.
toxN overexpression growth experiments
toxN and toxI overexpression experiments were conducted at 37 °C; all growth experiments were done at least three times independently with representative curves and plates shown in the figures. For liquid growth experiments, E. coli cells were grown with glucose until just prior to induction to prevent loss of toxicity. In addition, because pBAD33-toxN expression was leaky even in the absence of inducer, we performed all toxN overexpression experiments in a background containing pEXT20-toxI. For liquid growth curve experiments, single colonies were grown overnight in M9-glucose. Overnight cultures were back-diluted to OD600 = 0.01 and grown to OD600 ~ 0.4 in fresh M9-glucose. Cells were pelleted by centrifugation at 4 °C and 4000 g for 5 minutes. Pellets were washed twice in M9-glycerol and then resuspended into fresh M9-glycerol. All samples were back-diluted into fresh M9-glycerol at OD600 = 0.15, and cells were recovered for 45 mins before induction of toxin or antitoxin. toxN was induced with 0.2% arabinose, and toxI was induced with 100 μM IPTG. OD600 was monitored before and after induction using a UV-Vis Spectrophotometer. For serial dilution plating experiments, single colonies were grown to saturation in M9-glucose. Cells were pelleted by centrifugation at 4 °C and 4000 g for 5 minutes, washed twice in 1x phosphate-buffered saline (PBS), and resuspended to OD600 = 1.0 in 1x PBS. Cultures were serially diluted in 1x PBS and spotted onto M9L plates (M9-glycerol supplemented with 5% LB (v/v)) supplemented with 0.4% glucose, 0.2% arabinose, or 0.2% arabinose and 100 μM IPTG. Plates were then incubated at 37 °C for 24 h (0.4% glucose, toxin-repressing) or 48 h (0.2% arabinose, or 0.2% arabinose and 100 μM IPTG, toxin-inducing) before imaging.
RNA extraction for toxN overexpression studies
For RNA-seq experiments, toxN was overexpressed for 5–10 min before RNA was harvested. Cells were harvested by adding 1 mL of bacterial culture to 110 μL of stop solution (95% ethanol, 5% acid-buffered phenol) on ice and spinning at 21,000 g for 30 s. After removing the supernatant, cells were flash-frozen in liquid nitrogen and stored at −80 °C. To extract RNA, Trizol (Invitrogen) was pre-heated to 65 °C, and 400 μL of Trizol was added to each cell pellet. Resuspended pellets were shaken at 65 °C for 10 min at 2,000 rpm in a thermomixer and flash-frozen at −80 °C for at least 10 min. Samples were then thawed to room temperature and spun down at 4 °C and 14,000 g for 5 min. The supernatants were then mixed with 400 μL of 100% ethanol and spun through a DirectZol spin column (Zymo). Columns were washed with RNA prewash buffer (Zymo) twice and RNA wash buffer (Zymo) once, and RNA was eluted by spinning 90 μL diethyl pyrocarbonate (DEPC)-H2O through each column. To remove DNA, samples were then treated with 4 μL Turbo DNase I (Invitrogen) and 10 μL 10x Turbo DNase I buffer (Invitrogen) for 40 min at 37 °C.
Samples were then mixed with 96 μL DEPC H2O, extracted with 200 μL buffered acid phenol-chloroform, and precipitated for 4 h at −80 °C with 20 μL 3 M sodium acetate (NaOAc), 2 μL GlycoBlue (Invitrogen), and 600 μL ice-cold ethanol. RNA was pelleted via centrifugation at 4 °C and 21,000 g for 30 min. Pellets were washed twice with 500 μL of ice-cold 70% ethanol, air-dried, and resuspended in DEPC-H2O. RNA yield and integrity was verified by a NanoDrop spectrophotometer and a Novex 6% TBE-urea gel (Invitrogen), respectively.
Phage experiments in culture and efficiency of plaquing (EOP) titer determination
T4D was obtained from R. Young; T7, λvir, secΦ17, secΦ18, and secΦ27 from R. Sorek; T2, T3, T5 and T6 from the American Type Culture Collection (ATCC). P1vir was obtained from a lab stock used for transduction. Phage stocks were propagated in E. coli MG1655 at 37 °C in LB and were isolated from single plaques. For P1vir experiments, we grew cells in MMB media (LB supplemented with 0.1 mM MnCl2, 5 mM MgCl2, and 5 mM CaCl2). For PT4-toxI experiments, we used a derivative of T4D with a truncation in the T4 gene alc; this variant arose naturally during routine phage propagation and showed an increased ability to escape ToxN in the presence of PT4 middle-toxI. Phage stocks were titered by mixing them with E. coli MG1655 and LB + 0.5% agar, and plating this mixture on LB + 1.2% agar plates at 37 °C. To determine the efficiency of plaquing (EOP) of a particular phage, phage stocks were serially diluted in 1x FM buffer (20 mM Tris-HCl pH 7.4, 100 mM NaCl, 10 mM MgSO4), and 5 μL of each dilution was spotted on a lawn of bacteria seeded in an LB/agar/antibiotic plate. Plates were then grown at room temperature overnight and plaques quantified the following day. EOP was calculated by comparing the ability of the phage to form plaques on an experimental strain relative to the control strain and is reported as the mean of three independent replicates in all cases.
All phage experiments in liquid culture were conducted at 30 °C. Single colonies were grown overnight in M9-glucose. Overnight cultures were back-diluted to OD600 = 0.01 and grown to OD600 = 0.3 in fresh M9-glucose before addition of phage. For T4 experiments, L-tryptophan and thiamine were added at 15 μg/mL and 1 μg/mL, respectively.
One-step growth curves to measure burst size
One-step growth curve protocols were based on the protocol from (Patterson-West et al., 2018). Cells (900 μL) were mixed with phage (MOI ~ 0.01) and incubated at 30 °C for 10 min or 5 min for T4 or T7 infection, respectively, to allow for adsorption. To remove unadsorbed phage, cells were then spun down, washed twice in 500 μL M9-glucose, and resuspended in 1 mL fresh M9-glucose. Infected cells were then diluted 10,000-fold in 25 mL of M9-glucose at 30 °C in a shaking incubator (200 rpm). At each timepoint, 100 μL of infected cells were mixed with 100 μL of E. coli MG1655 in LB (OD ~ 0.4). The mixtures were then mixed with 4 mL of LB + 0.5% agar (heated to 50 °C) and spread on LB + 1.2% agar plates. Plates were then incubated overnight at 37 °C for T4 or room temperature for T7, and plaques were enumerated the following day. Burst size was calculated by dividing the average number of plaques for post-burst timepoints to that pre-burst.
CFU measurements pre- and post-T4 infection
Cell cultures were split into two 10 mL aliquots. One aliquot was mixed with T4 (MOI ~ 5) and and the other with an equal volume of LB (−T4 control). Both were grown in a shaking incubator (200 rpm) for 15 min at 30 °C. 1 mL of each culture was then spun down and resuspended in 100 μL 1x phosphate-buffered saline (PBS). Samples were then serially diluted 10-fold in 1x PBS and spotted on LB + 1.2% agar plates supplemented with carbenicillin. Plates were incubated for 24 hr at 37 °C before imaging. CFU was estimated by the 10-fold dilution at which each sample formed single plaques on the plate.
RNA extraction for RNA-seq and Northern blots following phage infection
RNA extraction protocols for RNA-seq and Northern blot experiments following phage infection were based on the protocol from (Luke et al., 2002). +toxIN and −toxIN cells were grown at 30 °C and infected with phage at MOI = 5. At every timepoint of interest, 500 μL cells were added to 500 μL boiling lysis buffer (SDS 2%, 4 mM EDTA pH = 8) (and 1 μL of a 1:2.5 dilution of the ERCC RNA Spike-In Mix (Invitrogen) when applicable), vortexed to mix, and incubated at 100 °C for 5 min. We included the spike-in control, which allows for assessments of absolute amounts of individual RNAs between +toxIN and −toxIN cells, for only one replicate of our T4 RNA-seq experiment and for both replicates of our T7 RNA-seq experiment. For Northern blot experiments, two 500 μL aliquots of cells were collected at every timepoint to ensure that enough RNA was harvested. Samples were then flash-frozen and stored at −80 °C until sample collection was complete. Samples were then thawed to room temperature. RNA was isolated from each sample with two rounds of extraction with 1 mL acid-buffered phenol heated to 67 °C and one round of extraction with 1 mL of acid-buffered phenol chloroform. RNA was then precipitated at −20 °C for at least 1 hour with 1x volume isopropanol, 1/10x volume 3 M NaOAc (pH = 5.5), and 1/100x volume GlycoBlue. RNA was pelleted via centrifugation at 4 °C and 21,000 g for 30 min. Pellets were washed twice with 500 μL of ice-cold 70% ethanol, air-dried, and resuspended in 90 μL DEPC-H2O. To remove DNA, samples were then treated with 4 μL Turbo DNase I (Invitrogen) and 10 μL 10x Turbo DNase I buffer (Invitrogen) for 40 min at 37 °C. Samples were then mixed with 96 μL DEPC H2O, extracted with 200 μL buffered acid phenol-chloroform, and precipitated for 4 h at −80 °C with 20 μL 3 M sodium acetate (NaOAc), 2 μL GlycoBlue (Invitrogen), and 600 μL ice-cold ethanol. RNA was pelleted via centrifugation at 4 °C and 21,000 g for 30 min. Pellets were washed twice with 500 μL of ice-cold 70% ethanol, air-dried, and resuspended in DEPC-H2O. RNA yield and integrity was verified by a NanoDrop spectrophotometer and a Novex 6% TBE-urea gel (Invitrogen), respectively.
Northern blot analysis following phage infection
1–4 μg DNase I-treated total RNA was diluted 1:1 in 2x sample buffer (Invitrogen), loaded onto Novex 6% TBE-Urea gels (Invitrogen), and run for 60–90 min at 100 V. RNA was then transferred onto Amersham Hybond-N+ nylon membrane (GE) with a Trans-Blot Turbo semi-dry transfer apparatus (BioRad) run for 90 minutes at 0.4 A. RNA was then crosslinked to the membrane using a UV Stratalinker 1800 (Stratagene) run on the autocrosslink setting. Oligonucleotide probes were radiolabeled by mixing 1 μL 10 μM oligo, 2.5 μL 10x T4 PNK buffer (NEB), 7.5 μL [γ−32P] ATP (PerkinElmer), 1 μL T4 PNK, and 13 μL DEPC H2O and incubating at 37 °C for 30 min. T4 PNK was then inactivated by incubating at 65 °C for 20 min and free ATP removed by spinning reactions through NucAway spin columns (Invitrogen). Membranes were pre-hybridized via incubation in a rotating hybridization oven with 10 mL ULTRAhyb-Oligo (Invitrogen) at 42 °C for 30 min. 5–10 μL radiolabeled probe was then added to each membrane, and membranes were incubated at 42 °C overnight to allow for probe hybridization. Membranes were then washed twice in 2x SSC (Invitrogen) / SDS 0.5%, wrapped in plastic wrap, and incubated at room temperature with a phosphor screen for 2–24 hr. Phosphor screens were then imaged with a Typhoon FLA 9500 (GE) instrument, and images were processed with ImageJ. All Northern blots shown are representative of two independent replicates.
Protein pulse-labeling
Pulse-labeling experiments were conducted at 30 °C. Overnight cultures of +toxIN and −toxIN cells were back-diluted to OD600 = 0.01 and grown to OD600 ~ 0.4 in 25 mL fresh M9-glucose. Cultures were then back-diluted to OD600 = 0.2 and grown at 30 °C for 30 min before infection with T4 at MOI = 5. At each labeling timepoint, 1 mL of culture were labeled with 25 μCi/mL [35S] EasyTag™ EXPRESS35S Protein Labeling Mix (Perkin Elmer) for 2 min, and then cells were pelleted and flash-frozen in liquid nitrogen. Pellets were then resuspended at OD600 = 10 (based on the OD600 of the culture prior to infection) in 2x Laemmi loading dye supplemented with 20 mM DTT. Samples were then analyzed by 4–20% SDS-PAGE, and the gel was dried and incubated at room temperature with a phosphor screen for 2–24 hr. Phosphor screens were then imaged with a Typhoon FLA 9500 (GE) instrument, and images were processed with ImageJ. Gel images shown are representative of two independent replicates.
Western blot analysis of ToxN
+toxI toxN-His6 cells were grown at 30 °C in M9-glucose to OD600 = 0.3 and infected with phage at MOI = 5. At every timepoint of interest, 1 mL of cells was pelleted and flash-frozen. Pellets were then resuspended at OD600 = 15 (based on the OD600 of the culture prior to infection) in 2x Laemmi loading dye supplemented with 20 mM DTT. As a loading control following T4 infection, His6-FolC was added to each resuspended cell pellet at a final concentration of 0.2 fM. Samples were then analyzed by 4–20% SDS-PAGE and transferred to a PVDF membrane. To visualize proteins, anti-His6 antibody (ThermoFisher) was used at a final concentration of 1:5000, and then SuperSignal West Femto Maximum Sensitivity Substrate (ThermoFisher) was used to develop the blots. Blots were then imaged with a FluorChem R Imager (ProteinSimple). As a loading control following rifampicin treatment, blots were then re-probed with anti-RpoA antibody at a final concentration of 1:5000. Blots shown are representative of two independent replicates.
One-step growth curve of T7 following CRISPRi knockdown of toxIN
+toxIN and −toxIN cells were grown at 30 °C in 25 mL cultures in M9-glucose. Cell aliquots (900 μL, one for each dCas9 induction condition) were mixed with T7 (MOI ~ 0.01) and incubated at 30 °C for 5 min to allow for adsorption. Infected cells were then diluted 10,000-fold in 25 mL of M9-glucose at 30 °C in a shaking incubator (200 rpm). To induce dCas9 expression 5 min pre-infection, 50 μL of 100 ng/μL anhydrous tetracycline (aTc) (final concentration = 200 ng/mL) was added to 25 mL cultures before aliquoting into 900 μL samples. To induce dCas9 5 min and 15 min post-infection, 50 μL of 100 ng/μL aTc (final concentration = 200 ng/mL) was added to 25 mL cultures following 10,000-fold dilution. To quantify pfu pre-and post-burst, 100 μL of infected cells were mixed with 100 μL of E. coli MG1655 in LB (OD ~ 0.4) 15 min post-infection (pre-burst) and 45 min post-infection (post-burst). The mixtures were then mixed with 4 mL of LB + 0.5% agar (heated to 50 °C) and spread on LB + 1.2% agar plates. Plates were incubated overnight at 37 °C, and plaques were enumerated the following day. Burst size was calculated by dividing the average number of plaques for the post-burst timepoint to that pre-burst.
Transcription shutoff-mediated ToxN activation
+toxIN and −toxIN cells were grown at 30 °C and treated with rifampicin at a final concentration of 300 μg/mL. Cells were then treated as described above for RNA extraction following phage infection. Northern blots were conducted as described above for Northern blots following phage infection. For Western blots, toxI toxN-His6+ cells were grown at 30 °C and treated with rifampicin at a final concentration of 300 μg/mL. Cells were then treated as above for Western blot analysis following T4 infection.
Growth recovery following rifampicin and chloramphenicol treatment
Rifampicin treatment experiments were conducted at 30 °C. Single colonies were grown overnight in M9-glucose. Overnight cultures were back-diluted to OD600 = 0.01 and grown to OD600 ~ 0.4 in 50 mL fresh M9-glucose. Cultures were then back-diluted to OD600 = 0.2, split into 3 × 25 mL aliquots, and grown at 30 °C for 30 min. 500 μL of 15 mg/mL rifampicin was added to samples to produce a final concentration of 300 μg/mL, 500 μL of 2.5 mg/mL rifampicin for 50 μg/mL, or 500 μL DMSO was added for untreated samples. Samples were then grown for an additional 30 min at 30 °C, after which 1 mL of each culture was spun down and washed twice in an equal volume of M9-glucose. Cells were then back-diluted to OD600 = 0.01, and growth was measured in 24-well plates at 5 min intervals with orbital shaking at 30 °C on a plate reader (Biotek). Data points reported are the mean of two technical replicates each for three independent growth curve experiments.
Chloramphenicol treatment experiments were conducted as described above for rifampicin treatment. 50 μL of 50 mg/mL and 12.5 mg/mL chloramphenicol was added to samples to yield a final concentration of 100 μg/mL and 25 μg/mL chloramphenicol, respectively, and an equal volume of 100% ethanol was added to untreated samples.
Growth following CRISPRi knockdown of toxIN
Growth experiments were conducted at 30 °C. Single colonies were grown overnight in M9-glucose. Overnight cultures were back-diluted to OD600 = 0.01 and grown to OD600 = 0.3 in 25 mL fresh M9-glucose. To induce dCas9 expression, 50 μL of 100 ng/μL aTc at a final concentration of 200 ng/mL was added to each culture. OD600 before and after induction was monitored using a UV-Vis Spectrophotometer.
Paired-end library preparation
Libraries were generated as described previously (Culviner et al., 2020). The library generation protocol was a modified version of the paired-end strand-specific dUTP method using random hexamer primers. For ToxN overexpression (both replicates) and one T4 infection library, rRNA was removed using a RiboZero kit (Illumina) using 2–3 μg total RNA as input. For the second T4 infection library, rRNA was removed using a MICROBExpress kit (ThermoFisher) using 3.3 μg total RNA as input. For rifampicin treatment and T7 infection libraries (two independent replicates of each), rRNA was removed using a recently-developed DIY E. coli rRNA depletion kit, using 2.5 μg total RNA as input (Culviner et al., 2020). Paired-end sequencing was performed on an Illumina NextSeq 5000 at the MIT BioMicroCenter.
QUANTIFICATION AND STATISTICAL ANALYSIS
Discovery of an E. coli type III toxin-antitoxin system
Our search for novel type III TA systems was modeled after that reported in (Blower et al., 2012). ToxN homologs were identified using two iterations of JACKHMMER (https://www.ebi.ac.uk/Tools/hmmer/search/jackhmmer). We restricted our search to the Ensembl Bacterial Genomes database and used Eubacterium rectale M104/1 ToxN as input. A list of E. coli ToxN homologs was then curated based on these results, with an E-value significance cutoff of 0.01. To verify that these toxins were part of a type III TA loci, we extracted the sequences 1 kb upstream of each putative toxin gene. We then analyzed these sequences for tandem repeats using Tandem Repeat Finder (tandem.bu.edu) using the default settings (match, mismatch, indels = 2,7,7; min score = 50). Finally, we analyzed the sequences between the tandem repeats and the toxin gene for an RNA hairpin followed by a poly-U tract using the mfold Web Server (http://unafold.rna.albany.edu/?q=mfold) using its default settings. The E. coli toxIN locus characterized in this study was derived from SuperContig CFSAN026791_contig0029 (positions 1,226–2,318) from E. coli strain GCA_001012275 in the Ensembl Bacteria database. To compare the toxI and ToxN sequences from E. coli and P. atrosepticum, we used the Clustal Omega Web Server (https://www.ebi.ac.uk/Tools/msa/clustalo/) using default settings.
RNA-sequencing read mapping and normalization
FASTQ files for each barcode were mapped to the MG1655 genome (NC_00913.2), the T4 genome (NC_000866), the T7 genome (V01146), and the sequences in the ERCC RNA Spike-In Mix (Invitrogen) as appropriate, using bowtie2 (version 2.1.0) with the following arguments: −D 20 −R 3 −N 0 −L 20 −i S,1,0.50 −p 6 −I 40 X 300 (Langmead and Salzberg, 2012). The samtools (version 0.1.19) suite (Li et al., 2009) was used via the pysam library (version 0.9.1.4) for interconversion of BAM and SAM file formats and conducting indexing. Gene names and coding region positions were extracted from NCBI annotations. For the T7 transcriptome, mRNA positions were extracted based on T7 promoter and terminator positions reported from NCBI annotations and in (Chen and Schneider, 2005). For analysis of fragment density across a given transcriptome, one count was added to all positions between and including the 5′ and 3′ ends of reads. To correct for variability in sequencing depth among samples, counts at each position were divided by a sample size factor. Briefly, for each sample, the total counts in each coding region was recorded and the geometric mean of these sums was calculated to yield a ‘reference sample’ for each library. For a given sample, the total counts in each coding region was then normalized by the reference sample, and the median of these ratios was the ‘size factor’ for that sample. Counts at every position were then scaled by this size factor. For analysis of counts in individual genes or gene classes, one count was added to the middle of each read. All reads mapping to a given coding region were then summed and normalized by reads per kilobase of transcript per million (RPKM). This normalized quantity was then used for all downstream analyses.
Identifying regions cleaved by ToxN
Cleavage valley identification was conducted as described in (Culviner and Laub, 2018), with a few modifications described below, using our lab’s custom genomearray Python package (https://github.com/peterculviner/genomearray). We looked for deep and narrow valleys in well-expressed coding regions (≥ 64 counts in all positions, except for Fig. 3B and 3D) in the cleavage ratio array that were likely to be a result of ToxN cleavage in our RNA-seq datasets. For heat maps of T4 transcripts during infection, we used an expression threshold of ≥ 16 counts to visualize as many cleaved transcripts as possible.
To find ToxN cleavage valleys following ToxN overexpression, we conducted Gaussian smoothing of the cleavage ratio array using σ = 40 nucleotides, truncating the filter at 4σ. We then searched for local minima in this smoothed cleavage ratio and searched for true minima in the region ± 25 nucleotides from these local minima in the unsmoothened cleavage ratio array. To ensure that the cleavage valley surrounding these local minima were narrow and well-defined, we looked at defined regions up and downstream of each local minimum and required that the cleavage ratio increased by some defined amount within that region (Fig. S3A–F); regions meeting those criteria in both replicates were defined as cleaved regions and, if not, they were removed from subsequent analyses. For data presented in Fig. 2E–F and S2G–I (toxN overexpression), we used the thresholds dx ≤ 50 nucleotides and dy ≥ 1.5 in the log2 cleavage ratio array on either side of the local minimum.
We identified ToxN cleavage valleys following T4 infection, rifampicin treatment, and T7 infection largely in the same way; however, because we noticed that cleavage profiles were wider and shallower than in our ToxN overexpression dataset, we used different dx and dy thresholds for identifying cleavage valleys. For T4 and T7 infection, we defined cleavage valleys as those regions with dx ≤ 100 and dy ≥ 1. For rifampicin treatment, we defined cleavage valleys as those regions with dx ≤ 100 upstream and dx ≤ 50 downstream of local minima and dy ≥ 1 on either side of local minima.
Identifying the ToxN cleavage motif
To determine the ToxN cleavage motif, we first analyzed cleaved regions defined by different dx and dy thresholds for enriched tri-, tetra-, penta-, and hexamer sequences using a custom script. To verify the 5-mer motif GAAAU and find any additional sequence specificity, we submitted the sequences of 141 cleaved regions with dx ≤ 50 and dy ≥ 1.5 to MEME to generate a sequence logo and position weight matrix. We then verified existence of the GAAAU 5-mer in our other RNA-seq datasets by again looking for enriched 5-mer sequences in cleaved regions.
Analysis of the T4 and T7 life cycles in +toxIN and −toxIN cells
The identities of T4 early, delayed early, middle, and late genes were taken from a microarray analysis of the T4 life cycle (Luke et al., 2002) and were used to generate Fig. S3D–I. For the heat maps in Fig. S3B–C, we determined the time of peak expression for every T4 gene by calculating the fraction of the total T4 transcriptome each gene occupied at every timepoint post-infection in the −toxIN background. The time of peak expression was defined as the timepoint at which an individual transcript occupied the largest fraction of the T4 transcriptome. Analysis of the progression of the T7 life cycle was conducted in largely the same way, except that we analyzed the peak expression of mRNA units instead of individual genes.
Analysis of growth curves
Growth curves shown in Fig. 4D and S4C were analyzed using the Curveball package (Ram et al., 2019). Briefly, growth curves were fit to the Logistic Lag2 model, and the lag time of each curve was estimated based on this fit.
Supplementary Material
Acknowledgments
We thank P. Culviner, M. Guo, and M. LeRoux for comments on the manuscript, S. Srikant for assistance with sequencing, and all members of the Laub lab for helpful discussions. This work was supported by an NSF predoctoral graduate fellowship to C.K.G. and an NIH grant to M.T.L. (R01GM082899), who is also an Investigator of the Howard Hughes Medical Institute.
Footnotes
Declaration of Interests
The authors declare no competing interests.
References
- Blower TR, Pei XY, Short FL, Fineran PC, Humphreys DP, Luisi BF, and Salmond GPC (2011). A processed noncoding RNA regulates an altruistic bacterial antiviral system. Nat Struct Mol Biol 18, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blower TR, Short FL, Rao F, Mizuguchi K, Pei XY, Fineran PC, Luisi BF, and Salmond GPC (2012). Identification and classification of bacterial Type III toxin–antitoxin systems encoded in chromosomal and plasmid genomes. Nucleic Acids Res 40, 6158–6173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blower TR, Chai R, Przybilski R, Chindhy S, Fang X, Kidman SE, Tan H, Luisi BF, Fineran PC, and Salmond GPC (2017). Evolution of Pectobacterium Bacteriophage ΦM1 To Escape Two Bifunctional Type III Toxin-Antitoxin and Abortive Infection Systems through Mutations in a Single Viral Gene. Appl Environ Microb 83, e03229–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunovskis I, and Summers WC (1971). The process of infection with coliphage T7 V. Shutoff of host RNA synthesis by an early phage function. Virology 45, 224–231. [DOI] [PubMed] [Google Scholar]
- Chen Z, and Schneider TD (2005). Information theory based T7-like promoter models: classification of bacteriophages and differential evolution of promoters and their polymerases. Nucleic Acids Res 33, 6172–6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford LV (1959). Nucleic acid metabolism in Escherichia coli infected with phage T5. Virology 7, 359–374. [DOI] [PubMed] [Google Scholar]
- Culviner PH, and Laub MT (2018). Global Analysis of the E. coli Toxin MazF Reveals Widespread Cleavage of mRNA and the Inhibition of rRNA Maturation and Ribosome Biogenesis. Mol Cell 70, 868–880.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culviner PH, Guegler CK, and Laub MT (2020). A Simple, Cost-Effective, and Robust Method for rRNA Depletion in RNA-Sequencing Studies. Mbio 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doron S, Melamed S, Ofir G, Leavitt A, Lopatina A, Keren M, Amitai G, and Sorek R (2018). Systematic discovery of antiphage defense systems in the microbial pangenome. Science 359, eaar4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dörr T, Vulić M, and Lewis K (2010). Ciprofloxacin Causes Persister Formation by Inducing the TisB toxin in Escherichia coli. Plos Biol 8, e1000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dy RL, Przybilski R, Semeijn K, Salmond GPC, and Fineran PC (2014). A widespread bacteriophage abortive infection system functions through a Type IV toxin–antitoxin mechanism. Nucleic Acids Res 42, 4590–4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fineran PC, Blower TR, Foulds IJ, Humphreys DP, Lilley KS, and Salmond GPC (2009). The phage abortive infection system, ToxIN, functions as a protein–RNA toxin–antitoxin pair. Proc National Acad Sci 106, 894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraikin N, Rousseau CJ, Goeders N, and Melderen LV (2019). Reassessing the Role of the Type II MqsRA Toxin-Antitoxin System in Stress Response and Biofilm Formation: mqsA Is Transcriptionally Uncoupled from mqsR. Mbio 10, e02678–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Altae-Tran H, Böhning F, Makarova KS, Segel M, Schmid-Burgk JL, Koob J, Wolf YI, Koonin EV, and Zhang F (2020). Diverse enzymatic activities mediate antiviral immunity in prokaryotes. Science 369, 1077–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton HG, Watson BNJ, and Fineran PC (2020). The arms race between bacteria and their phage foes. Nature 577, 327–336. [DOI] [PubMed] [Google Scholar]
- Harms A, Fino C, Sørensen MA, Semsey S, and Gerdes K (2017). Prophages and Growth Dynamics Confound Experimental Results with Antibiotic-Tolerant Persister Cells. Mbio 8, e01964–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms A, Brodersen DE, Mitarai N, and Gerdes K (2018). Toxins, Targets, and Triggers: An Overview of Toxin-Antitoxin Biology. Mol Cell 70, 768–784. [DOI] [PubMed] [Google Scholar]
- Hazan R, and Engelberg-Kulka H (2004). Escherichia coli mazEF-mediated cell death as a defense mechanism that inhibits the spread of phage P1. Mol Genet Genomics 272, 227–234. [DOI] [PubMed] [Google Scholar]
- Helaine S, Cheverton AM, Watson KG, Faure LM, Matthews SA, and Holden DW (2014). Internalization of Salmonella by Macrophages Induces Formation of Nonreplicating Persisters. Science 343, 204–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesselbach BA, and Nakada D (1977). “Host Shutoff” Function of Bacteriophage T7: Involvement of T7 Gene 2 and Gene 0.7 in the Inactivation of Escherichia coli RNA Polymerase. J Virol 24, 736–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton DM (2010). Transcriptional control in the prereplicative phase of T4 development. Virol J 7, 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karam JD, and Drake JW (1994). Molecular Biology of Bacteriophage T4 (Washington, DC: American Society for Microbiology; ). [Google Scholar]
- Koerner JF, and Snustad DP (1979). Shutoff of host macromolecular synthesis after T-even bacteriophage infection. Microbiol Rev 43, 199–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga M, Otsuka Y, Lemire S, and Yonesaki T (2011). Escherichia coli rnlA and rnlB Compose a Novel Toxin–Antitoxin System. Genetics 187, 123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leplae R, Geeraerts D, Hallez R, Guglielmini J, Drèze P, and Melderen LV (2011). Diversity of bacterial type II toxin–antitoxin systems: a comprehensive search and functional analysis of novel families. Nucleic Acids Res 39, 5513–5525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeRoux M, Culviner PH, Liu YJ, Littlehale ML, and Laub MT (2020). Stress Can Induce Transcription of Toxin-Antitoxin Systems without Activating Toxin. Mol Cell 79, 280–292.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Subgroup, 1000 Genome Project Data Processing (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopatina A, Tal N, and Sorek R (2020). Abortive Infection: Bacterial Suicide as an Antiviral Immune Strategy. Ann Rev Virol 7, 1–14. [DOI] [PubMed] [Google Scholar]
- Luke K, Radek A, Liu X, Campbell J, Uzan M, Haselkorn R, and Kogan Y (2002). Microarray Analysis of Gene Expression during Bacteriophage T4 Infection. Virology 299, 182–191. [DOI] [PubMed] [Google Scholar]
- Mahadik SP, Dharmgrongartama B, and Srinivasan PR (1974). Regulation of host ribonucleic acid synthesis in bacteriophage T3-infected cells. Properties of an inhibitory protein of Escherichia coli ribonucleic acid polymerase. J Biological Chem 249, 1787–1791. [PubMed] [Google Scholar]
- McAllister WT, and Barrett CL (1977). Roles of the Early Genes of Bacteriophage T7 in Shutoff of Host Macromolecular Synthesis. J Virol 23, 543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melderen L, Bernard P, and Couturier M (1994). Lon‐dependent proteolysis of CcdA is the key control for activation of CcdB in plasmid‐free segregant bacteria. Mol Microbiol 11, 1151–1157. [DOI] [PubMed] [Google Scholar]
- Naka K, Qi D, Yonesaki T, and Otsuka Y (2017). RnlB Antitoxin of the Escherichia coli RnlA-RnlB Toxin–Antitoxin Module Requires RNase HI for Inhibition of RnlA Toxin Activity. Toxins 9, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuka Y, and Yonesaki T (2012). Dmd of bacteriophage T4 functions as an antitoxin against Escherichia coli LsoA and RnlA toxins. Mol Microbiol 83, 669–681. [DOI] [PubMed] [Google Scholar]
- Patterson-West J, James TD, Fernández-Coll L, Iben JR, Moon K, Knipling L, Cashel M, and Hinton DM (2018). The E. coli Global Regulator DksA Reduces Transcription during T4 Infection. Viruses 10, 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecota DC, and Wood TK (1996). Exclusion of T4 phage by the hok/sok killer locus from plasmid R1. J Bacteriol 178, 2044–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen K, Zavialov AV, Pavlov M.Yu., Elf J, Gerdes K, and Ehrenberg M (2003). The Bacterial Toxin RelE Displays Codon-Specific Cleavage of mRNAs in the Ribosomal A Site. Cell 112, 131–140. [DOI] [PubMed] [Google Scholar]
- Ram Y, Dellus-Gur E, Bibi M, Karkare K, Obolski U, Feldman MW, Cooper TF, Berman J, and Hadany L (2019). Predicting microbial growth in a mixed culture from growth curve data. Proc National Acad Sci 116, 14698–14707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson JE, Spinelli S, Cambillau C, and Moineau S (2013). Structure and activity of AbiQ, a lactococcal endoribonuclease belonging to the type III toxin–antitoxin system. Mol Microbiol 87, 756–768. [DOI] [PubMed] [Google Scholar]
- Sberro H, Leavitt A, Kiro R, Koh E, Peleg Y, Qimron U, and Sorek R (2013). Discovery of Functional Toxin/Antitoxin Systems in Bacteria by Shotgun Cloning. Mol Cell 50, 136–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schifano JM, Cruz JW, Vvedenskaya IO, Edifor R, Ouyang M, Husson RN, Nickels BE, and Woychik NA (2016). tRNA is a new target for cleavage by a MazF toxin. Nucleic Acids Res 44, 1256–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short FL, Pei XY, Blower TR, Ong S-L, Fineran PC, Luisi BF, and Salmond GPC (2013). Selectivity and self-assembly in the control of a bacterial toxin by an antitoxic noncoding RNA pseudoknot. Proc National Acad Sci 110, E241–E249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short FL, Akusobi C, Broadhurst WR, and Salmond GPC (2018). The bacterial Type III toxin-antitoxin system, ToxIN, is a dynamic protein-RNA complex with stability-dependent antiviral abortive infection activity. Sci Rep-Uk 8, 1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder LR (1972). An RNA polymerase mutant of Escherichia coli defective in the T4 viral transcription program. Virology 50, 396–403. [DOI] [PubMed] [Google Scholar]
- Song S, and Wood TK (2020). A Primary Physiological Role of Toxin/Antitoxin Systems Is Phage Inhibition. Front Microbiol 11, 1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data is available at GEO – accession number GSE161756. Software used for RNA-seq analysis is available at https://github.com/peterculviner/genomearray. All images presented in the figures are available in Mendeley Data at doi:10.17632/d9dphh55cj.1.