

Abstract
DNA polymerase β (Pol β) plays a vital role in DNA repair and has been closely linked to cancer. Selective inhibitors of this enzyme are lacking. Inspired by DNA lesions produced by antitumor agents that inactivate Pol β, we have undertaken the development of covalent small-molecule inhibitors of this enzyme. Using a two-stage process involving chemically synthesized libraries, we identified a potent irreversible inhibitor (14) of Pol β (KI = 1.8 ± 0.45 μM, kinact = (7.0 ± 1.0) × 10−3 s−1). Inhibitor 14 selectively inactivates Pol β over other DNA polymerases. LC-MS/MS analysis of trypsin digests of Pol β treated with 14 identified two lysines within the polymerase binding site that are covalently modified, one of which was previously determined to play a role in DNA binding. Fluorescence anisotropy experiments show that pretreatment of Pol β with 14 prevents DNA binding. Experiments using a pro-inhibitor (pro-14) in wild type mouse embryonic fibroblasts (MEFs) indicate that the inhibitor (5 μM) is itself not cytotoxic but works synergistically with the DNA alkylating agent, methylmethanesulfonate (MMS), to kill cells. Moreover, experiments in Pol β null MEFs indicate that pro-14 is selective for the target enzyme. Finally, pro-14 also works synergistically with MMS and bleomycin to kill HeLa cells. The results suggest that pro-14 is a potentially useful tool in studies of the role of Pol β in disease.
Graphical Abstract

INTRODUCTION
DNA damage and repair have significant biological consequences on aging and diseases.1–4 Consequently, the DNA damage response system is an increasingly popular inhibition target. Additional incentive for developing repair enzyme inhibitors is provided by a recent report that ascribes exceptional responses to chemotherapy by some cancer patients to defects in DNA repair.5 Poly(ADP-ribose) polymerase (PARP) inhibitors, of which four are FDA approved for treating BRCA1-deficient cancers, are leading the way.6–8 A variety of other enzymes involved in DNA repair, including glycosylases,9–13 phosphodiesterases,14,15 and polymerases,16–24 are also attractive targets. DNA repair inhibitors can work in conjunction with damaging agents to kill cells. Alternatively, as in the case of the PARP inhibitors, some enzymes can be targeted to exploit a synthetic lethal relationship involving a second enzyme or pathway that is defective in a cell to selectively kill them.
DNA polymerase β (Pol β) plays a vital role in base excision repair (BER, Scheme 1) in the nucleus and mitochondria.25–29 Pol β also contributes to double strand break repair via the alternative nonhomologous end joining pathway.30 The enzyme is up-regulated and/or mutated in many human cancers, such as colon cancer, where the mutation rate reaches ~40%.31 Pol β has been postulated to exhibit a synthetic lethal relationship with homologous recombination, but this has not been verified experimentally.32 Although Pol β inhibitors have been reported, there is a need for molecules that are selective and efficacious in cells.16–24 Here, we report a selective, covalent Pol β inhibitor that acts on the enzyme in mammalian cells.
Scheme 1.
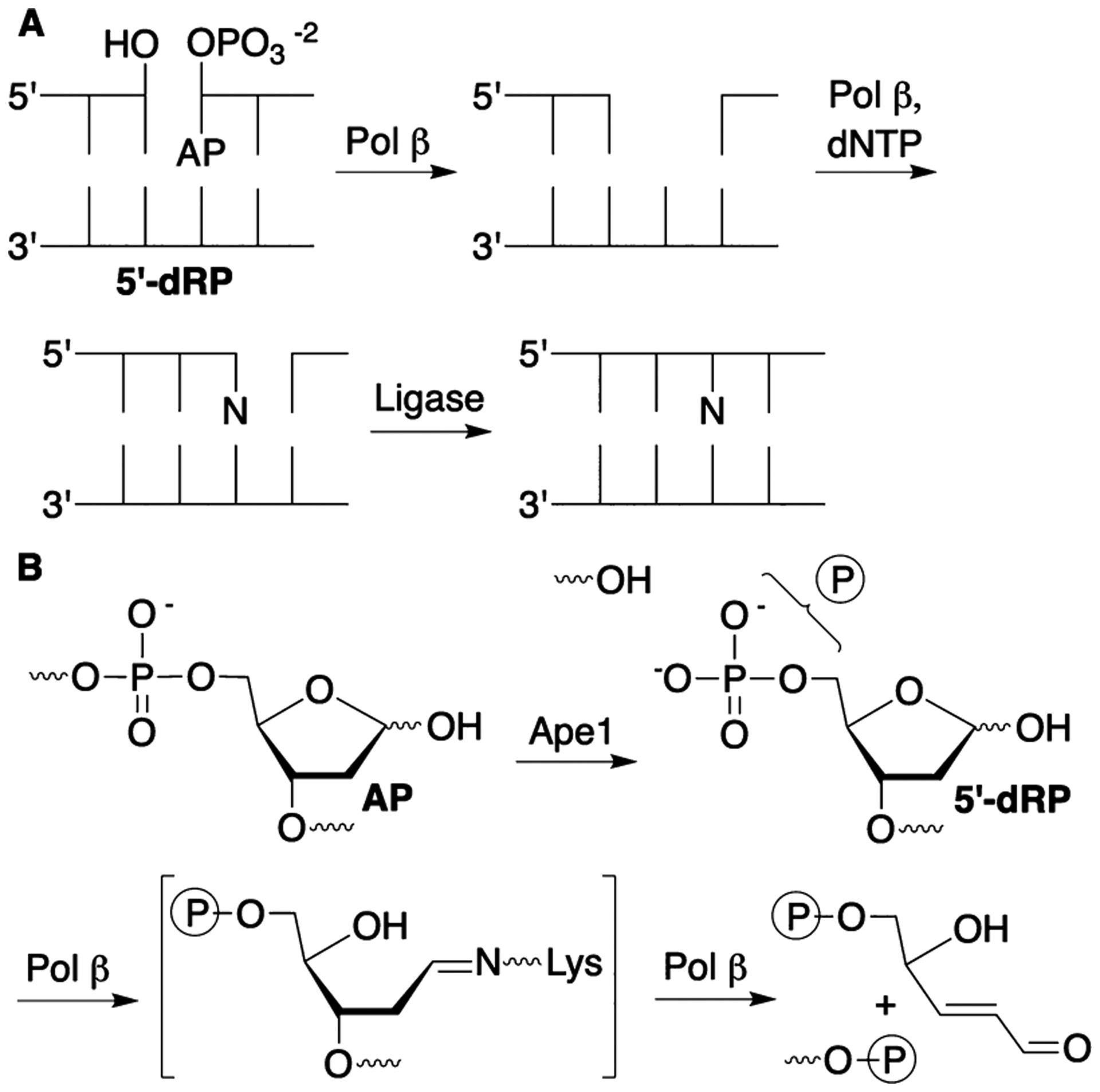
Base Excision Repair
Pol β is also one of a handful of bifunctional DNA polymerases (Scheme 1A) possessing polymerase and lyase activity (Scheme 1B). The activities are associated with separate binding sites, and 39 kDa Pol β can be divided into a 31 kDa polymerase domain and a shorter 8 kDa amino terminus domain that harbors the lyase activity. In short patch BER, Pol β reacts with the 5′-deoxyribose phosphate (5′-dRP) generated by incision of the 5′-phosphate of an abasic site (AP) by apurinic endonuclease 1 (Ape1, Scheme 1). The enzyme carries out a lyase reaction via Schiff base formation. When Pol β binds oxidized abasic sites, such as DOB, attempted excision via initial nucleophilic attack results in covalently modified, inactivated enzyme (Scheme 2).33–35
Scheme 2.

Pol β Inactivation by a DNA Lesion
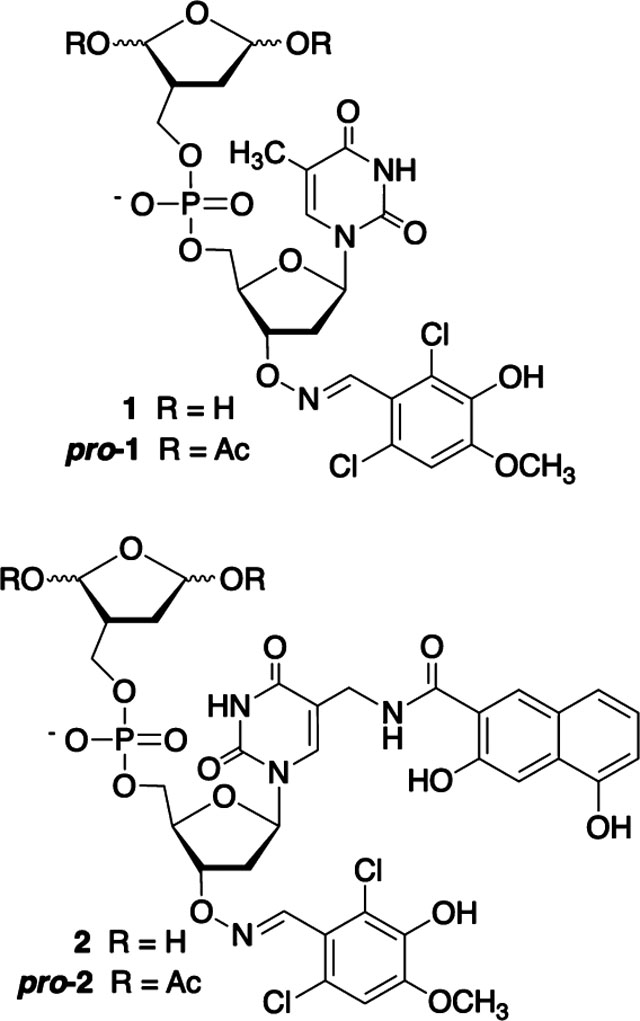
Inactivation of Pol β by oxidized abasic sites served as an inspiration for the design of mechanism-based irreversible inhibitors (1, 2).21–23 These molecules contain a 1,4-dioxobutane group linked to a 5′-phosphorylated thymidine (1) or C5-derivative (2) via a methylene linker that reduces β-elimination from the dicarbonyl component. The substituents at the 3′- and C5-positions of the thymidine were obtained by screening libraries of molecules (<350). The corresponding bis-acetates (pro-1, pro-2) undergo hydrolysis in cell lysates to the inhibitors.21 Pro-1 and pro-2 function as pro-inhibitors in cells and work synergistically with DNA damaging agents to kill mammalian cells. Although 2 is ~50-times more active against Pol β than is 1, it is even more effective at inactivating Pol λ. Pol λ has biological function that is independent of Pol β, but the enzymes have overlapping activity and the former is frequently thought of as a back-up to Pol β in BER.36–39 It is desirable to identify inhibitors that are selective for one enzyme over another, and this was one goal of the current study.
RESULTS AND DISCUSSION
Inhibitor Identification Strategy.
We previously identified a high nanomolar irreversible inhibitor of Pol β via a two-step process.21–23 A library consisting of oximes at the 3′-terminus of a nucleotide was screened in step one. The inhibitor (1) identified from this procedure was then used to synthesize a library of molecules in which structural diversity was introduced at the C5-methyl position of a thymine in the form of amides. To streamline the synthesis process and maximize the use of a stable carboxylic acid library, we proposed to start from readily available AZT (Scheme 3). In contrast to previous investigations that yielded 1 and 2, the 3′-recognition element in this study is appended to the nucleotide core via an amide bond.
Scheme 3.
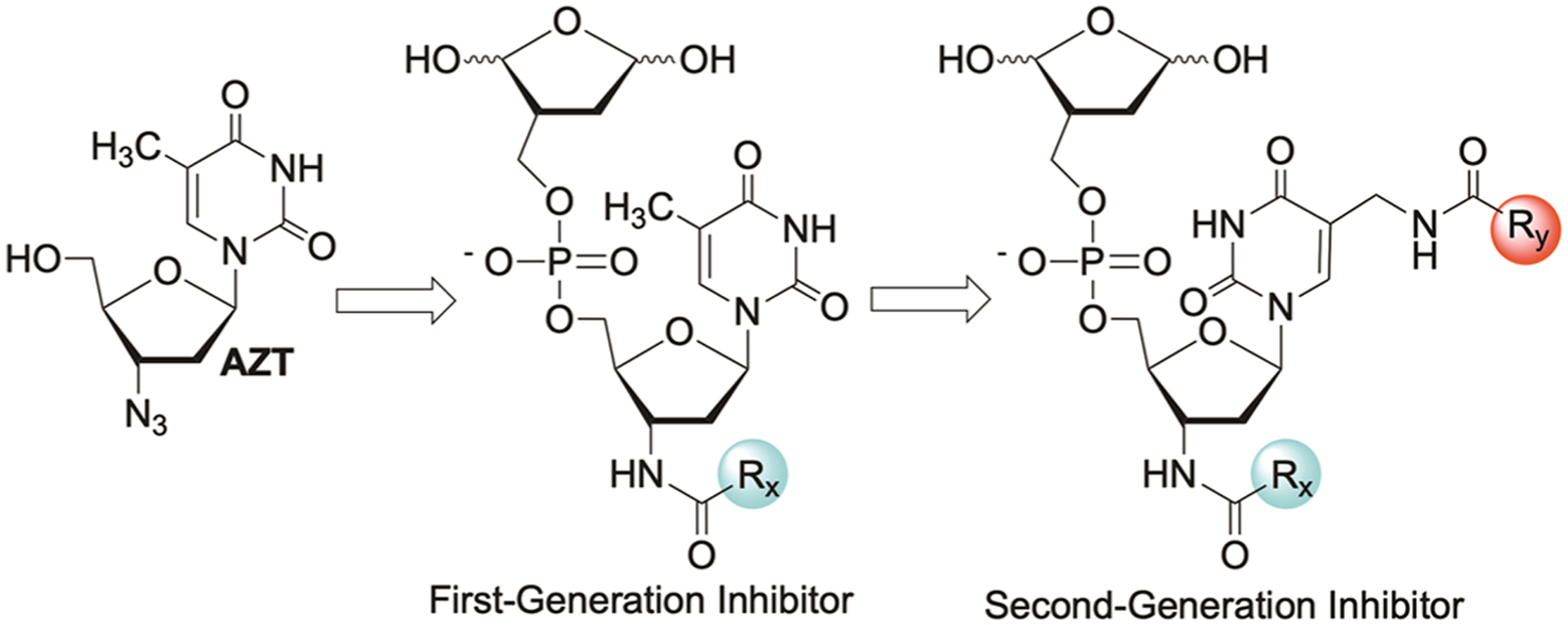
Development of First- and Second-Generation Inhibitors from AZT
Identification of a First-Generation Irreversible Inhibitor.
The precursor (6) for preparing the library was rapidly synthesized from AZT (Scheme 4). The azide was reduced and the resulting amine was protected as the trifluoroacetamide (3) prior to coupling with phosphoramidite 4, which bears the protected 1,4-dioxobutane warhead. The bis-pentene acetal was used to mask the warhead, because it could be cleaved rapidly under mild conditions that are compatible with other functional groups in the molecules that make up the library.21 Although it would have been more direct to reduce the azide after forming the phosphate bond, the slightly longer method avoided competing azide reduction by the phosphoramidite during coupling. Phosphoramidite 4 was prepared via phosphitylation of the corresponding alcohol, which was previously reported.40 A mixture of diastereomers of 5 was deprotected using concentrated aqueous ammonium hydroxide at room temperature. Crude 6 was used to produce the library of first-generation inhibitor candidates. Utilizing a mixture of diastereomers of the DOB component was not a concern because it was expected to epimerize in water, following bis-pentene cleavage.
Scheme 4.
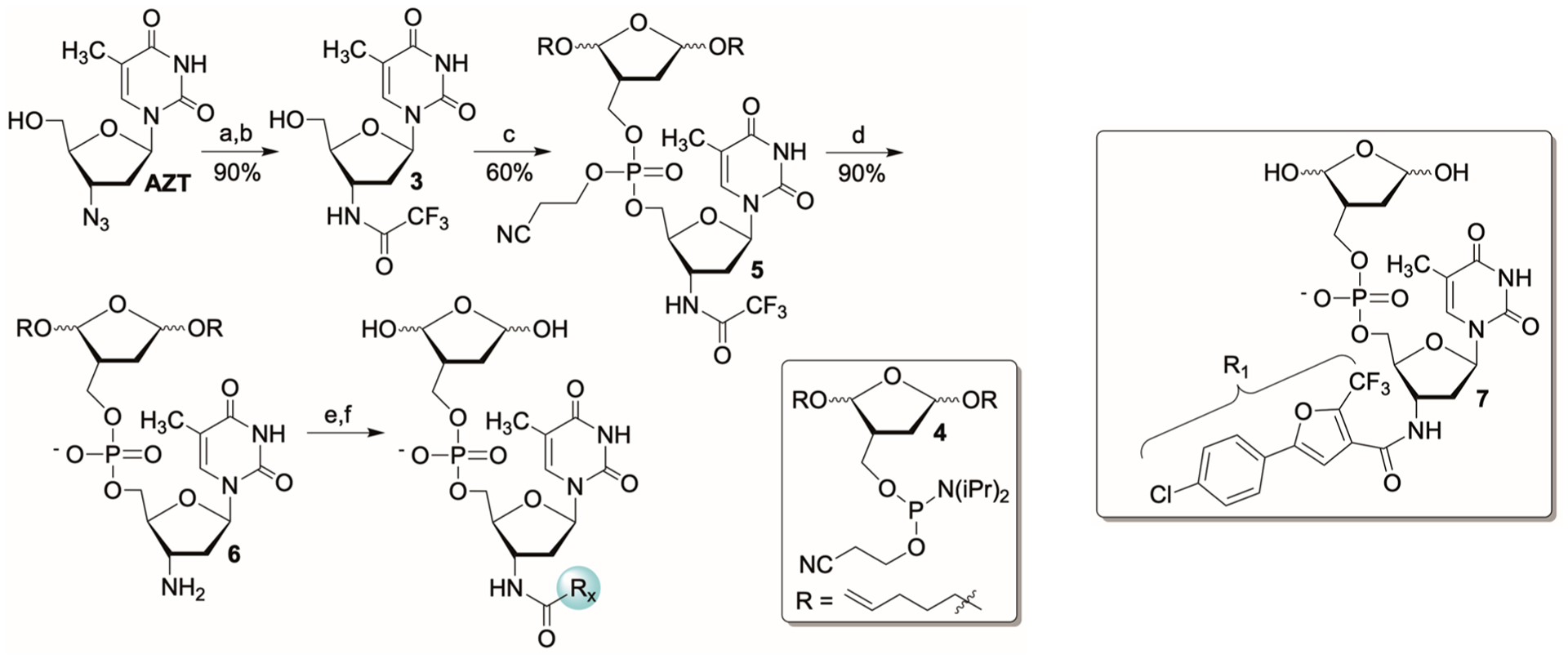
Synthesis of a First-Generation Inhibitor Librarya
aKey: (a) H2, Pd/C; (b) TFAA; (c) (i) S-Ethyl tetrazole, 4; (ii) t-BuOOH; (d) NH4OH; (e) RxCO2H (X = 1−325), HBTU, HOBt; (f) NBS.
The bis-acetal protected, first-generation library (325 members, Chart S1) was prepared in 384-well plates by coupling 6 with 1.4 equiv of HBTU, HOBt, and the corresponding carboxylic acids. The crude amides were then rapidly deprotected (5 min) in a different 384-well plate using 2.5 equiv of N-bromosuccinimide at 4 °C and quenched with sodium thiosulfate. The inhibitor candidates were immediately screened for Pol β inhibition using a strand displacement assay in which the displaced oligonucleotide was fluorescently labeled at its 3′-teminus with TAMRA, and the template was labeled at its 5′-terminus with black hole quencher (Scheme 5, Figure S1a).21,40,41 Screening reactions containing Pol β (100 nM) and inhibitor (25 μM) were preincubated (30 min) prior to diluting 10-fold and reacting with the ternary DNA substrate and dTTP (Figure S49). Initial hits were identified based upon their ability to prevent an increase in fluorescence. These molecules (3) were resynthesized from 6 and screened side-by-side with a control experiment containing all reagents except 6 to eliminate false positives (Figure S1b).
Scheme 5.

Fluorescence Screen for Identifying Inhibitor Leads
Product 7 was identified as the most promising inhibitor, and its bis-pentene acetal was resynthesized and purified to confirm its activity (Figure S2a).40 Complete loss of Pol β strand displacement activity was observed following a 20 min preincubation with 15 μM 7. The diminution of strand displacement activity was dependent upon preincubation time, indicating that 7 irreversibly inhibits Pol β (Figure S2b), and was carried forward in the search for a second-generation inhibitor (Scheme 3).
Identification of a Second-Generation Irreversible Inhibitor.
The primary amine designated for introducing structural diversity at the C5-pyrimidine position was incorporated by reacting the crude bromide obtained from 8 with concentrated aqueous ammonium hydroxide in ethanol cosolvent (Scheme 6). Following protection of the primary amine as the trifluoroacetamide, the azide in 9 was reduced and the amine coupled with the carboxylic acid (11) that provided first-generation inhibitor 7. Upon deprotection of the C5′-alcohol in 10, the nucleoside was coupled with phosphoramidite 4. The nucleoside was used in slight excess (1.2 equiv) to minimize phosphoramidite byproducts, which were difficult to separate from 12. Cleavage of the trifluoroacetamide and β-cyanoethyl protecting groups, as described for preparing 6 (Scheme 4), provided the substrate (13) for preparing the second-generation inhibitor library, and was purified by column chromatography (Scheme 6).
Scheme 6.
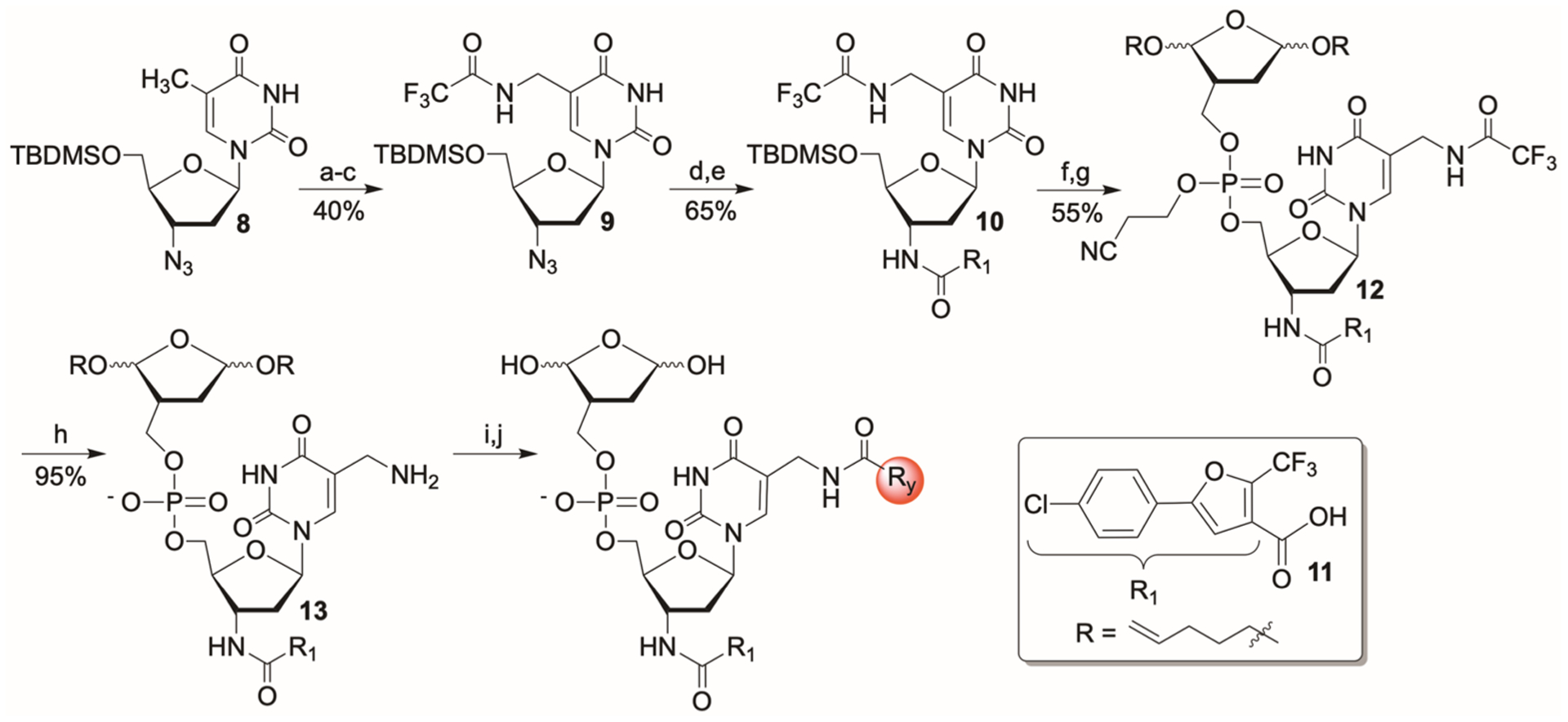
Synthesis of a Second-Generation Inhibitor Librarya
aKey: (a) NBS; (b) NH3; (c) Ethyl trifluoroacetate; (d) H2, Pd/C; (e) 11, HBTU, HOBt; (f) Et3N·3HF; (g) (i) S-Ethyl tetrazole, 4 (Scheme 4);(ii) t-BuOOH; (h) NH4OH; (i) RyCO2H (y = 1–375), HBTU, HOBt; (j) NBS.
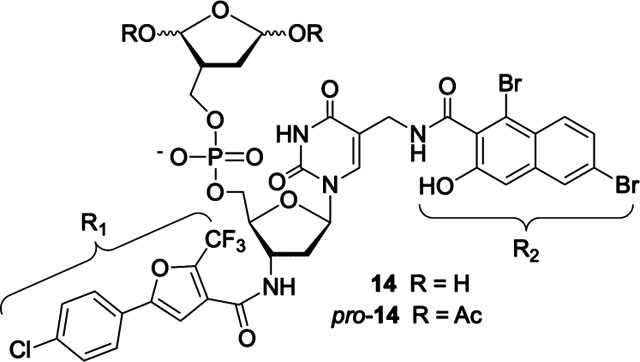
Precursor 13 was coupled with a library of carboxylic acids (375, Chart S1), followed by removal of the pentene acetal protecting groups, as described above for screening the first-generation library. Inhibitor screening was carried out at 700 nM (crude) inhibitor candidates, as opposed to 25 μM for the first-generation inhibitor. From this library, 14 was selected as the most promising candidate, after carrying out the control experiments described for the evaluation of the first-generation library (Figure S50). Inhibitor 14 was independently synthesized from 13.40 Coupling was carried out using the corresponding NHS-ester (45%) because reaction of the corresponding free acid provided the desired product in low yield. Following purification of the bis-pentenyl acetal precursor, 14 was generated on an as-needed basis.
Irreversible Inhibition of Pol β by 14.
The quantitative effect of 14 on Pol β activity at pH 7.4 was determined using the strand displacement assay employed to identify it. The time-dependent curves generated from the fluorescence-based assays were fit to an exponential growth region followed by a plateau.42,43 Rate constants extracted from these data were used to determine relative rates of enzyme activity with and without inhibitor. The effect of 14 on polymerase activity was examined between 100 and 750 nM following preincubation with Pol β for between 2 and 20 min.
The KI (1.8 ± 0.45 μM) and kinact ((7.0 ± 1.0) × 10−3 s−1) for 14 were extracted from a Kitz–Wilson plot (Figure 1A). To our knowledge, 14 and the less active 1 and 2 are the only irreversible inhibitors of Pol β. Hence, we cannot compare kinact to other molecules. However, the KI compares favorably to molecules such as honokiol, which is not selective for Pol β over Pol λ.17 Other molecules are more potent, but their selectivity is unsatisfactory or unknown.24 The IC50 (Figure 1B) for Pol β inactivation by 14 was determined using 10 (458 nM) and 12 (409 nM) min preincubation times. In comparison, 2 exhibited an IC50 value of 400 nM following 30 min preincubation.22,23 The IC50 values of 2 and 14 cannot be compared directly because they were determined under different conditions (i.e., preincubation times and extent dilution following preincubation). To make a direct comparison, 14 was analyzed under the conditions employed for measuring the IC50 of 2. Under the identical conditions, the IC50 for 14 (204 nM) was approximately one-half that of 2 (Figure S3). Additional evidence for irreversible inhibition of Pol β by 14 was obtained by comparing the polymerase activity before and after dialysis (24 h, 4 °C), which revealed that the enzyme did not regain function following dialysis (Figure 2A).
Figure 1.
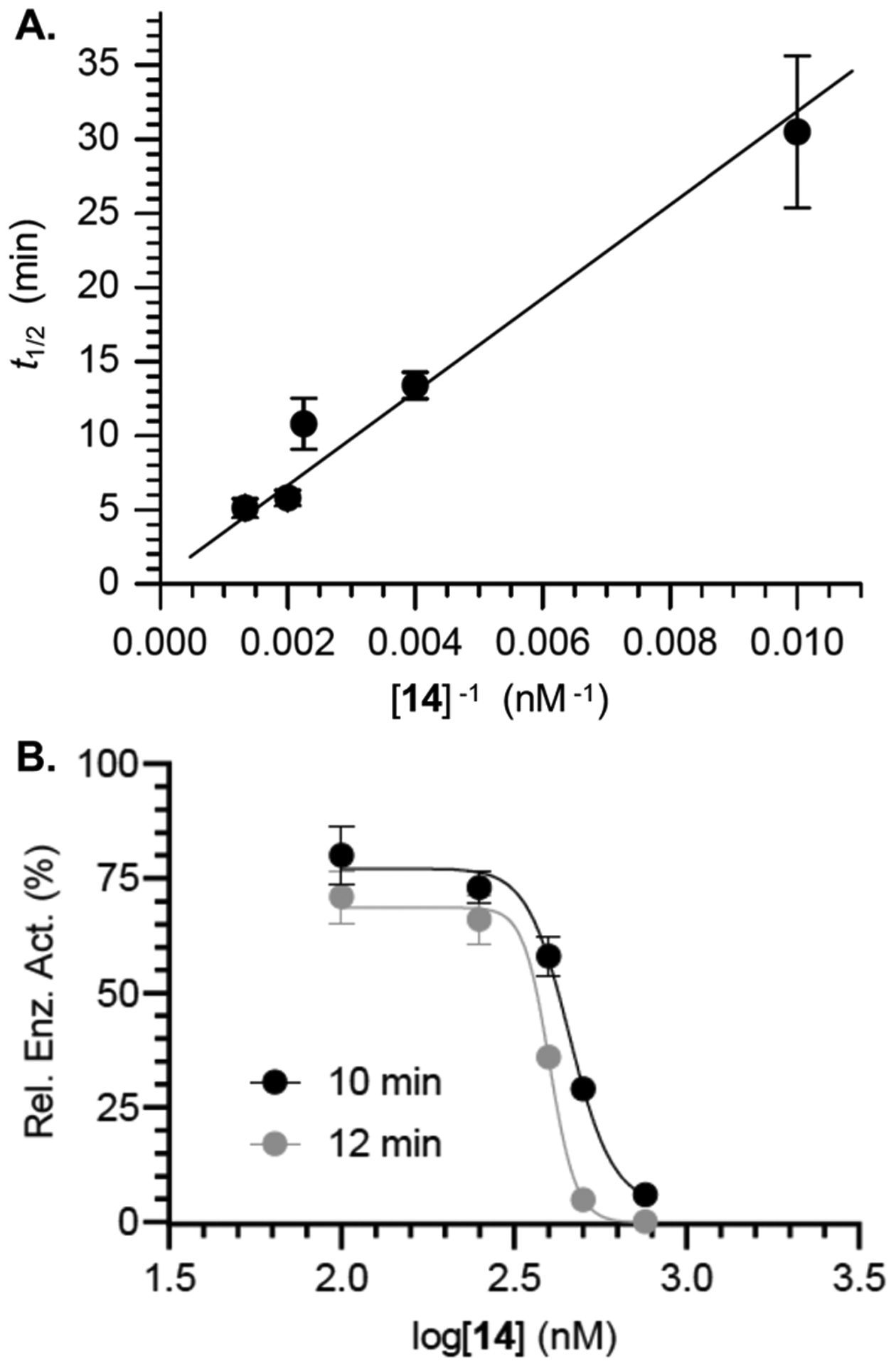
Inhibition of DNA polymerase β by 14. (A) Kitz–Wilson plot of irreversible inhibition by 14. (B) IC50 of 14 as a function of preincubation time. Data are the ave. ± std. dev. of 3 replicates.
Figure 2.
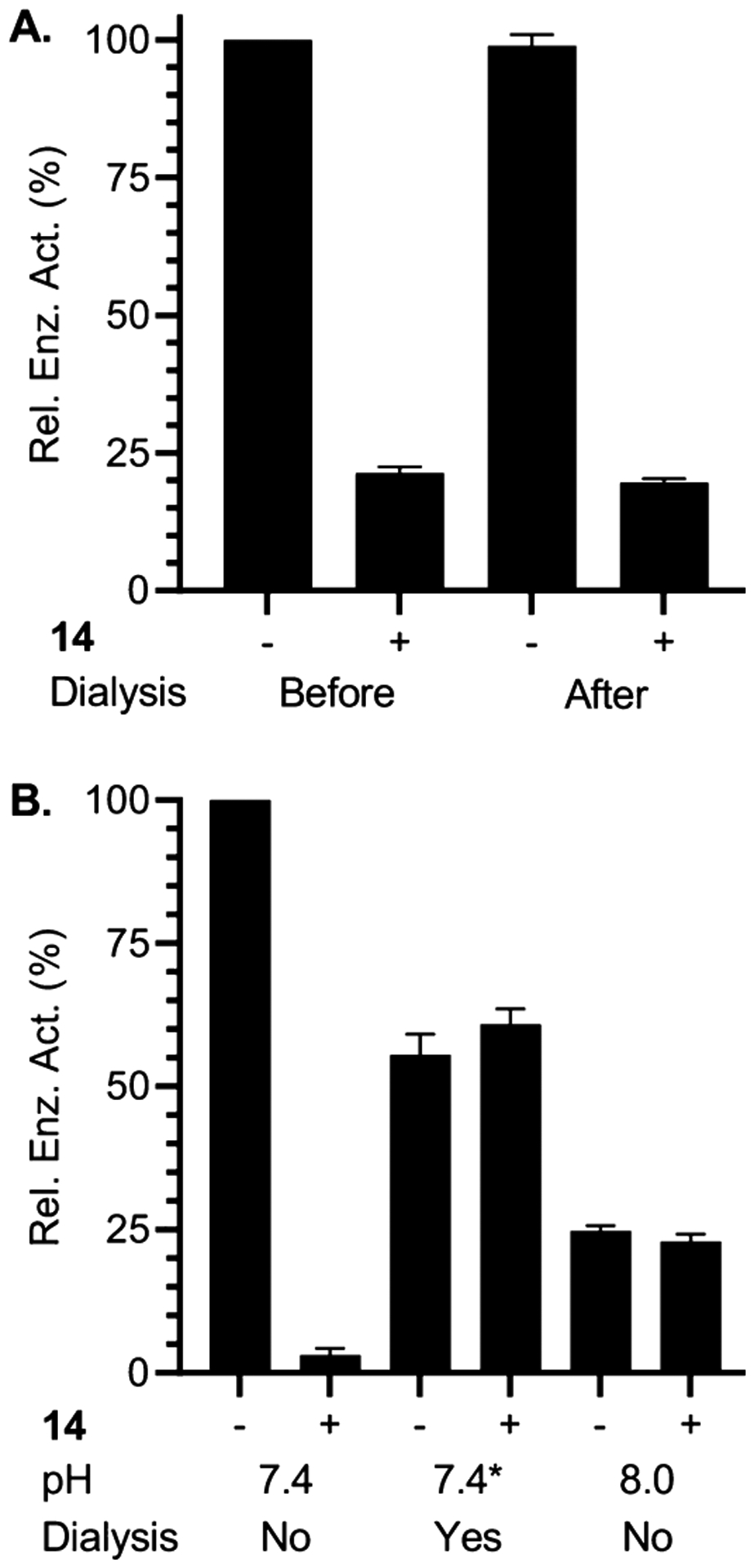
Effect of dialysis and pH on inactivation by 14. (A) Pol β strand displacement activity before and after dialysis with and without 14 (750 nM, 20 min preincubation). (B) pH Effect on inactivation by 14 (500 nM, 20 min) before and after dialysis. *Preincubation was carried out at pH 7.4, followed by dialysis at pH8.0. Data are the ave. ± std. dev. of 3 replicates.
Inhibitor 14 modifies lysine residues in the polymerase domain and prevents DNA binding by Pol β.
The residue(s) modified on Pol β by 14 were identified via LC-MS/MS analysis following trypsin digest of enzyme that was preincubated with 14 (300 nM, 30 min). Trypsin digestion was carried out under suboptimal conditions (pH 6.5) due to adduct instability at higher pH (see below). Fragment ion analysis of the two observed modified tryptic peptides revealed that Lys113 and Lys234 formed adducts with 14 (Figure 3A).40 Peptide 1 contains a single internal lysine residue and an expected fragment containing a modification on K113 (y8*, Peptide 1, Figure S51) was detected. Peptide 2 (Figure 3A) contains four internal lysine residues (K230, K234, K244, and K248) and required MS/MS fragmentation to identify the modified lysine (Figure S52). Unmodified fragments containing K230 (b13), K244 (y18), and K248 (y9, y18) indicated these residues did not react with 14. A fragment containing a single modification was observed that contained K230 and K234 (b21*). In addition, another fragment that contained one modification and included K234, K244, and K248 (y21*). When considered in total, these data indicated K234 was the modified lysine in Peptide 2.
Figure 3.
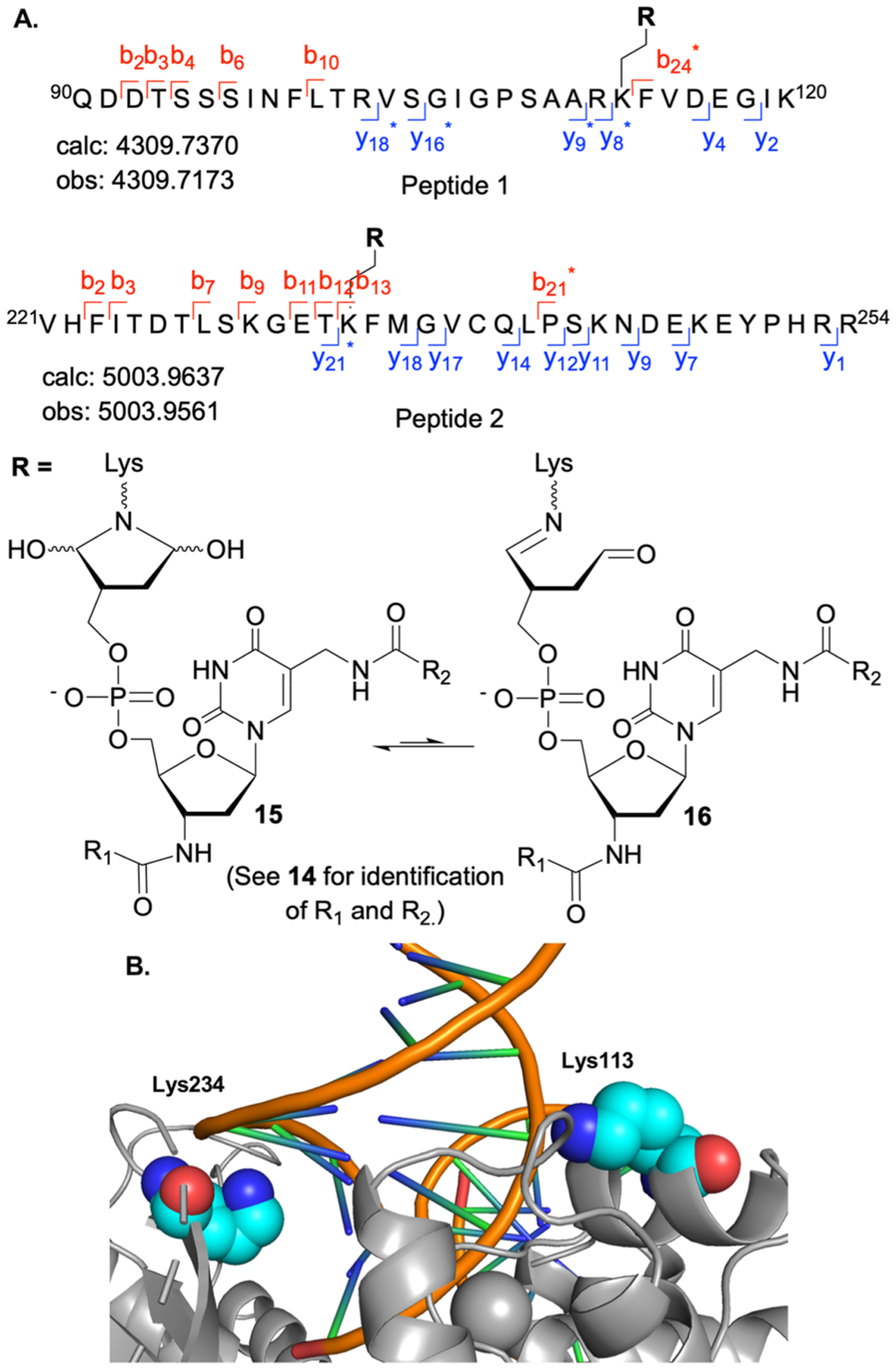
Covalent modification of Pol β by 14. (A) Modified peptides observed in trypsin digest of Pol β preincubated with 14 (300 nM, 30 min). (B) Pol β crystal structure with modified residues (cyan spheres) (PDB: 1PBX).
The fragments observed in the gas phase correspond to the ring opened dehydrated forms (16), which would be expected to exist in equilibrium with the ring closed bis-hemiaminal isomers (15) in solution (Figure 3A). (Imine formation is shown involving the C1-aldehyde, but the data do not distinguish between this adduct or one involving the C4-aldehyde.) Based upon previous experiments that established the necessity of the 1,4-dicarbonyl moiety for inactivation, ring closed isomer 15 is believed to dominate in solution.21
Support for the proposed structure of the lysine adduct(s) is gleaned by examining the effect of pH on inactivation by 14 (Figure 2B). Preincubation of Pol β with 14 at pH 7.4 completely inactivated the enzyme (Figure 2B, columns 1, 2). In contrast, preincubation at pH 8 resulted in no inactivation (Figure 2B, columns 5, 6). Furthermore, when Pol β was preincubated with 14 at pH 7.4, complete activity loss resulted, but when then dialyzed at pH 8, the inhibitor again had no effect on enzyme activity (Figure 2B, columns 3, 4). However, activity is not recovered if dialysis is carried out at pH 7.4 (Figure 2A). Based upon these observations, we suggest that the adduct(s) is unstable at pH 8, consistent with the expected behavior of 15.
Identification of the modified lysines provided a mechanistic rationale for how 14 inactivates Pol β. Lys234 is important for DNA binding and is invariant across multiple species of Pol β.44–46 In addition, analysis of the crystal structure of Pol β when complexed with DNA (PDB: 1BPX) indicates that Lys113 is within 10 Å of the DNA backbone (Figure 3B).44–46 We used a fluorescently labeled ternary substrate containing a stable abasic site analogue (Table S1) to determine if Pol β incubation with 14 affected DNA binding (Figure 4).47 Fluorescence anisotropy measurements indicated DNA binding was significantly reduced following preincubation with 14 (2 μM). DNA binding interactions were most effectively disrupted at lower Pol β concentrations. Complete DNA binding was not observed until ~800 equiv of protein were added. Together, fluorescence anisotropy and trypsin digestion experiments indicate that 14 irreversibly inhibits Pol β by covalently modifying lysine residues in the polymerase domain that result in compromised DNA binding.
Figure 4.
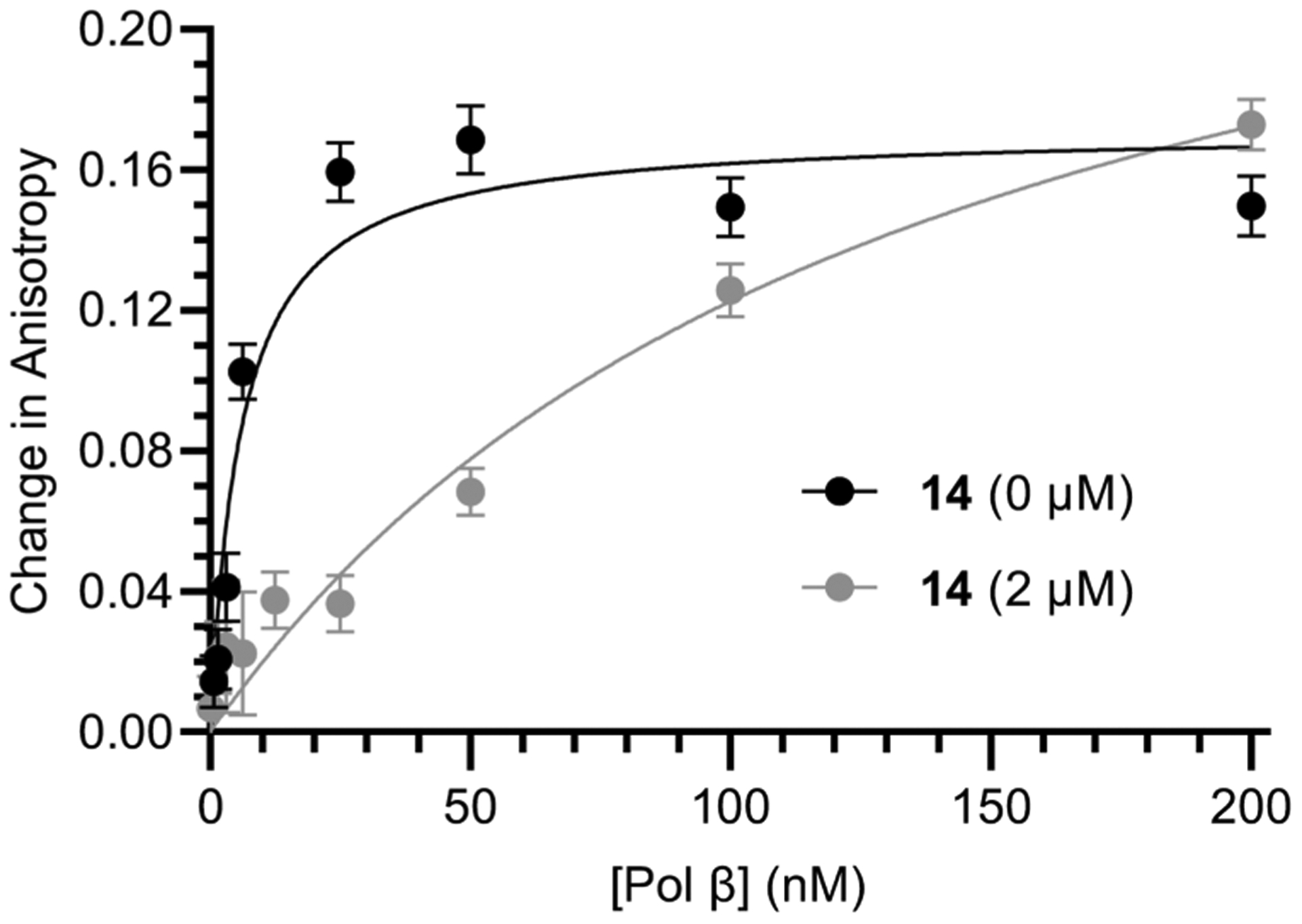
Effect of inhibitor 14 on DNA (0.25 nM) binding by Pol β. Data are the ave. ± std. dev. of 3 replicates.
Selective Inactivation of Pol β.
Selective DNA polymerase inhibition is challenging. For instance, 2 is a more potent inhibitor of Pol λ than Pol β.22 The lack of selectivity between the two X-family polymerases has been observed in other reported Pol β inhibitors.17 The two enzymes share 32% sequence homology, which contributes to the difficulty in inhibiting one over the other.48 The potency of 14 for inhibiting Pol β was compared to its effect on a model replicative polymerase (Klenow exo−) and three mammalian polymerases involved in DNA repair, Pol θ, Pol η, and Pol λ (Figure 5). The effect of 14 on the respective enzymes was ascertained by determining the relative enzyme activity following preincubation with inhibitor versus in the absence of inhibitor.
Figure 5.
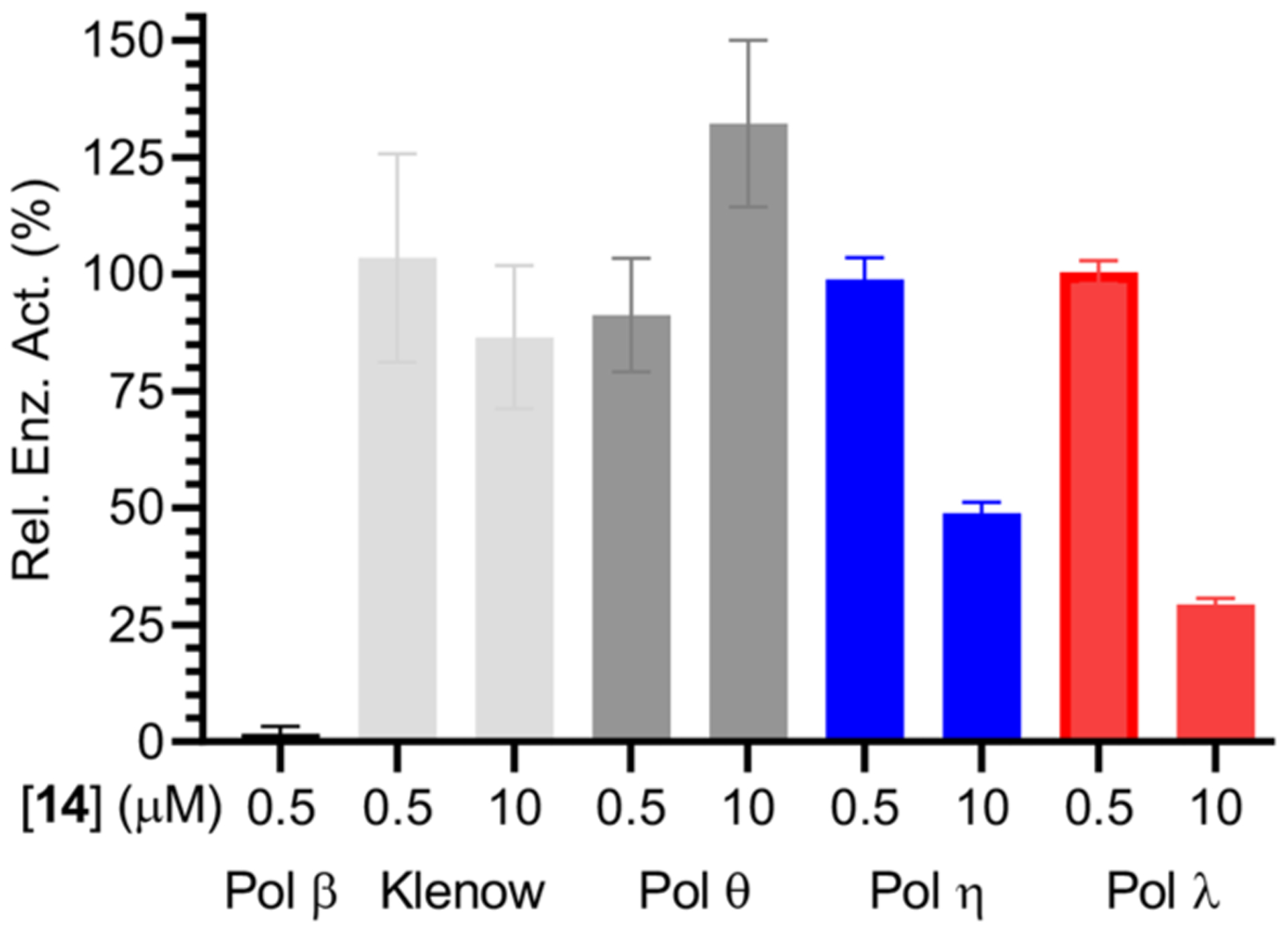
Inhibition of DNA polymerases by 14. Data are the ave. ± std. dev. of 3 replicates.
Under conditions in which 14 (500 nM, 20 min preincubation) essentially completely inactivated Pol β, it had no effect (within experimental error) on Klenow exo−, Pol θ, Pol η, or Pol λ. Increasing the concentration of 14 20-fold (10 μM) still had no effect on Klenow exo− or Pol θ activity. Pol η and Pol λ polymerase activities are reduced at 10 μM 14. However, Pol η retains approximately 50% of its activity at this concentration and the activity of Pol λ is slightly more than 25% relative to untreated enzyme. Despite these effects, the existence of significant activity at 20 times the concentration of inhibitor 14 at which Pol β is completely inactivated indicates significant selectivity for this enzyme. It is not possible to rigorously compare the selectivity of 14 to many other Pol β inhibitors because it is a covalent inhibitor, while others such as honokiol are reversible inhibitors.17 However, these data (Figure 5) clearly indicate that 14 selectively inactivates Pol β over these 4 other polymerases.
Pro-14 selectively targets Pol β in cells and works synergistically with DNA damaging agents.
Pro-14 (5 μM) killed fewer than 5% of wild type mouse embryonic fibroblasts (Pol β WT, MEFs) (Figure 6a). However, the pro-inhibitor enhanced the cytotoxicity of methylmethanesulfonate (MMS) when the concentration of alkylating agent was varied up to 1.5 mM. MMS activates BER by alkylating purines.49 The data indicate that pro-14 and MMS have a synergistic effect on MEF cytotoxicity. This is consistent with the ability of similarly designed pro-inhibitors to act synergistically with MMS.21–23
Figure 6.

Treatment mouse embryonic fibroblasts with MMS and pro-14. (A) MMS cytotoxicity with or without pro-14 in the presence or absence of Pol β. (B) Effect of greater pro-14 concentration. Data are the ave. ± std. dev. of 3 replicates.
MMS was considerably more toxic to MEFs lacking Pol β (Pol β −/−). However, the presence of pro-14 did not result in additional cell death (Figure 6A). Similarly, pro-14 did not increase the cytotoxicity of MMS in MEFs lacking pol β and/or pol λ (Figures S4a,b). Importantly, these data indicate that pro-14 does not enhance MMS cytotoxicity by targeting enzymes other than Pol β.
The selectivity of pro-14 on Pol β was examined at higher pro-14 concentrations in MEFs treated with 0.2 mM MMS. While the cytotoxicity was enhanced upon increasing pro-14 from 5 to 15 μM in WT MEFs, no additional cell death was observed in Pol β null (Pol β −/−) cells (Figure 6b). Furthermore, even in the presence of 25 μM of pro-14, no additional cytotoxicity was detected in WT MEFs or Pol β null cells (Figure S5). These data indicate that pro-14 does not target proteins that enhance MMS cytotoxicity when present at significantly higher concentrations than needed to synergistically kill cells containing Pol β.
The synergistic effect of pro-14 on the cytotoxicity of MMS and the antitumor agent bleomycin (BLM) in HeLa cells was also examined (Figure 7). pro-14 (5 μM) exhibited <7% cytotoxicity to the HeLa cells. However, at this concentration pro-14 enhanced the cytotoxicity of MMS (0.2 mM) 2.5-fold. Similarly, the cytotoxicity of bleomycin (2 μM) increased from 22% to 62%, almost 3-fold, under these conditions.
Figure 7.
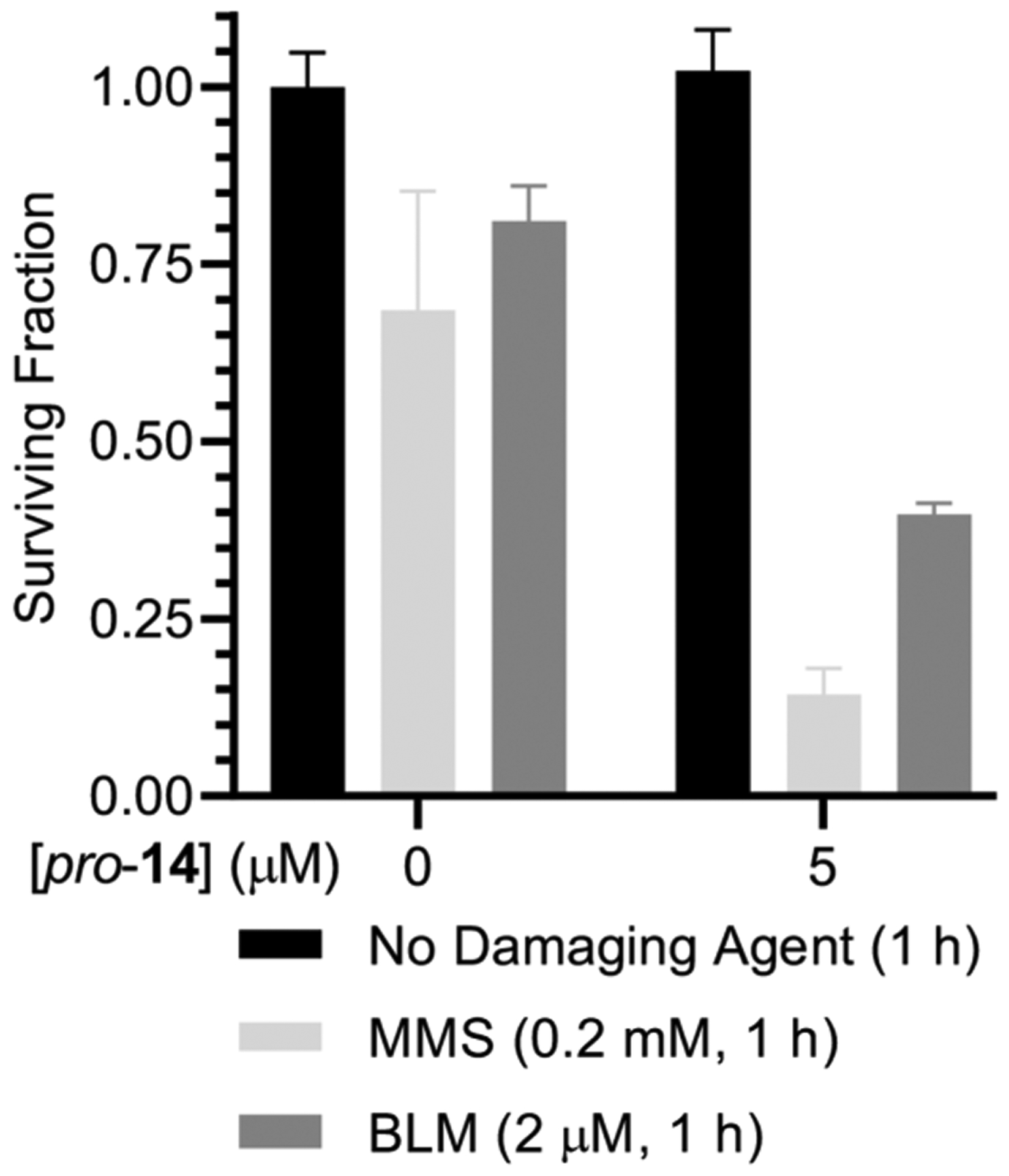
Synergistic effect of DNA damaging agents with pro-14 in HeLa cells. Data are the ave. ± std. dev. of 3 replicates.
CONCLUSIONS
Inhibitors that are selective for one DNA polymerase over another are uncommon. Covalent inhibitors, including those that react with lysine, are employed with increasing frequency to target proteins.50–52 Roughly 30% of marketed drugs are covalent in nature.53 Covalent inhibitors often lead to greater potency and longevity of effects, when they are also irreversible. Depending on the electrophilicity of the warhead, covalent inhibitors can enhance selectivity toward certain nucleophiles or residues they modify. To our knowledge, the dioxobutane family of molecules, such as those presented here, are the only examples of molecules that irreversibly inhibit DNA polymerase β. The molecule described here (14) selectively inactivates Pol β. The corresponding pro-inhibitor (pro-14) selectively targets this enzyme in mouse embryonic fibroblasts. Although pro-14 works synergistically with DNA damaging agents to kill mouse embryonic fibroblasts and HeLa cells, it is itself not highly toxic under the same conditions. This suggests that pro-14 will be a useful tool for studying the effects of Pol β inhibition in cells. Furthermore, the approach described here and elsewhere21−23 for identifying Pol β inhibitors may be useful for targeting other polymerases.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful for support from the National Institute of General Medical Sciences (GM-131736). We thank the JHU Biomolecular NMR Center, for support of the 600 and 800 MHz NMR instruments, and Katie Tripp, for assistance with the fluorescence anisotropy experiments. We thank Donna Stefanik and Sam Wilson for providing the MEF cells, as well as plasmids for expressing the 31 kDa and 8 kDa domains of Pol β. We thank Zucai Suo for providing the plasmid for expressing Pol λ.
Footnotes
The authors declare the following competing financial interest(s): S.Y. and M.M.G. are inventors on a patent application related to this work.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c02453.
All experimental details synthesis, screening, enzyme inhibition, fluorescence anisotropy, peptide digestion and LC-MS/MS analysis, and intracellular experiments (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.1c02453
Contributor Information
Ananya Majumdar, Johns Hopkins University, Biomolecular NMR Center, Baltimore, Maryland 21218, United States.
Marc M. Greenberg, Johns Hopkins University, Department of Chemistry, Baltimore, Maryland 21218, United States.
REFERENCES
- (1).Zhao L; Sumberaz P Mitochondrial DNA Damage: Prevalence, Biological Consequence, and Emerging Pathways. Chem. Res. Toxicol 2020, 33, 2491–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Nelson BC; Dizdaroglu M Implications of DNA Damage and DNA Repair on Human Diseases. Mutagenesis 2020, 35, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Thompson PS; Cortez D New Insights into Abasic Site Repair and Tolerance. DNA Repair 2020, 90, 102866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Lindahl T The Intrinsic Fragility of DNA. Angew. Chem., Int. Ed 2016, 55, 8528–8534. [DOI] [PubMed] [Google Scholar]
- (5).Wheeler DA; Takebe N; Hinoue T; Hoadley KA; Cardenas MF; Hamilton AM; Laird PW; Wang L; Johnson A; Dewal N; Miller V; Pineyro D; Castro de Moura M; Esteller M; Shen H; Zenklusen JC; Tarnuzzer R; McShane LM; Tricoli JV; Williams PM; Lubensky I; O’Sullivan-Coyne G; Kohn EC; Little RF; White J; Malik S; Harris L; Weil C; Chen AP; Karlovich C; Rodgers B; Shankar L; Jacobs P; Nolan T; Hu J; Muzny DM; Doddapaneni H; Korchina V; Gastier-Foster J; Bowen J; Leraas K; Edmondson EF; Doroshow JH; Conley BA; Ivy SP; Staudt LM Molecular Features of Cancers Exhibiting Exceptional Responses to Treatment. Cancer Cell 2021, 39, 38–53e.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Farmer H; McCabe N; Lord CJ; Tutt ANJ; Johnson DA; Richardson TB; Santarosa M; Dillon KJ; Hickson I; Knights C; Martin NMB; Jackson SP; Smith GCM; Ashworth A Targeting the DNA Repair Defect in Brca Mutant Cells as a Therapeutic Strategy. Nature 2005, 434, 917–921. [DOI] [PubMed] [Google Scholar]
- (7).Rottenberg S; Jaspers JE; Kersbergen A; van der Burg E; Nygren AOH; Zander SAL; Derksen PWB; de Bruin M; Zevenhoven J; Lau A; Boulter R; Cranston A; O’Connor MJ; Martin NMB; Borst P; Jonkers J High Sensitivity of Brca1-Deficient Mammary Tumors to the Parp Inhibitor Azd2281 Alone and in Combination with Platinum Drugs. Proc. Natl. Acad. Sci. U. S.A 2008, 105, 17079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lord C; Ashworth S Parp Inhibitors: Synthetic Lethality in the Clinic. Science 2017, 355, 1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Tahara Y.-k.; Kietrys AM; Hebenbrock M; Lee Y; Wilson DL; Kool ET Dual Inhibitors of 8-Oxoguanine Surveillance by Ogg1 and Nudt1. ACS Chem. Biol 2019, 14, 2606–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Tahara Y.-k.; Auld D; Ji D; Beharry AA; Kietrys AM; Wilson DL; Jimenez M; King D; Nguyen Z; Kool ET Potent and Selective Inhibitors of 8-Oxoguanine DNA Glycosylase. J. Am. Chem. Soc 2018, 140, 2105–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Visnes T; Cázares-Körner A; Hao W; Wallner O; Masuyer G; Loseva O; Mortusewicz O; Wiita E; Sarno A; Manoilov A; Astorga-Wells J; Jemth A-S; Pan L; Sanjiv K; Karsten S; Gokturk C; Grube M; Homan EJ; Hanna BMF; Paulin CBJ; Pham T; Rasti A; Berglund UW; von Nicolai C; Benitez-Buelga C; Koolmeister T; Ivanic D; Iliev P; Scobie M; Krokan HE; Baranczewski P; Artursson P; Altun M; Jensen AJ; Kalderén C; Ba X; Zubarev RA; Stenmark P; Boldogh I; Helleday T Small-Molecule Inhibitor of Ogg1 Suppresses Proinflammatory Gene Expression and Inflammation. Science 2018, 362, 834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Donley N; Jaruga P; Coskun E; Dizdaroglu M; McCullough AK; Lloyd RS Small Molecule Inhibitors of 8-Oxoguanine DNA Glycosylase-1 (Ogg1). ACS Chem. Biol 2015, 10, 2334–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Huang H; Stivers JT; Greenberg MM Competitive Inhibition of Uracil DNA Glycosylase by a Modified Nucleotide Whose Triphosphate Is a Substrate for DNA Polymerase. J. Am. Chem. Soc 2009, 131, 1344–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Rai G; Vyjayanti VN; Dorjsuren D; Simeonov A; Jadhav A; Wilson DM; Maloney DJ Synthesis, Biological Evaluation, and Structure-Activity Relationships of a Novel Class of Apurinic/Apyrimidinic Endonuclease 1 Inhibitors. J. Med. Chem 2012, 55, 3101–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Dorjsuren D; Kim D; Vyjayanti VN; Maloney DJ; Jadhav A; Wilson DM III Simeonov A. Diverse Small Molecule Inhibitors of Human Apurinic/Apyrimidinic Endonuclease Ape1 Identified from a Screen of a Large Public Collection. PLoS One 2012, 7, e47974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wojtaszek JL; Chatterjee N; Najeeb J; Ramos A; Lee M; Bian K; Xue JY; Fenton BA; Park H; Li D; Hemann MT; Hong J; Walker GC; Zhou P A Small Molecule Targeting Mutagenic Translesion Synthesis Improves Chemotherapy. Cell 2019, 178, 152–159.e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Gowda ASP; Suo Z; Spratt TE Honokiol Inhibits DNA Polymerases B and Λ and Increases Bleomycin Sensitivity of Human Cancer Cells. Chem. Res. Toxicol 2017, 30, 715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Strittmatter T; Bareth B; Immel TA; Huhn T; Mayer TU; Marx A Small Molecule Inhibitors of Human DNA Polymerase λ. ACS Chem. Biol 2011, 6, 314–319. [DOI] [PubMed] [Google Scholar]
- (19).Strittmatter T; Brockmann A; Pott M; Hantusch A; Brunner T; Marx A Expanding the Scope of Human DNA Polymerase λ and β Inhibitors. ACS Chem. Biol 2014, 9, 282–290. [DOI] [PubMed] [Google Scholar]
- (20).Jaiswal AS; Panda H; Law BK; Sharma J; Jani J; Hromas R; Narayan S Nsc666715 and Its Analogs Inhibit Strand-Displacement Activity of DNA Polymerase β and Potentiate Temozolomide-Induced DNA Damage, Senescence and Apoptosis in Colorectal Cancer Cells. PLoS One 2015, 10, e0123808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Arian D; Hedayati M; Zhou H; Bilis Z; Chen K; DeWeese TL; Greenberg MM Irreversible Inhibition of DNA Polymerase β by Small-Molecule Mimics of a DNA Lesion. J. Am. Chem. Soc 2014, 136, 3176–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Paul R; Banerjee S; Greenberg MM Synergistic Effects of an Irreversible DNA Polymerase Inhibitor and DNA Damaging Agents on Hela Cells. ACS Chem. Biol 2017, 12, 1576–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Paul R; Banerjee S; Greenberg MM Synergistic Effects of an Irreversible DNA Polymerase Inhibitor and DNA Damaging Agents on Hela Cells. ACS Chem. Biol 2018, 13, 832. [DOI] [PubMed] [Google Scholar]
- (24).Goellner EM; Svilar D; Almeida KH; Sobol RW Targeting DNA Polymerase β for Therapeutic Intervention. Curr. Mol. Pharmacol 2012, 5, 68–87. [PMC free article] [PubMed] [Google Scholar]
- (25).Friedberg EC; Walker GC; Siede W; Wood RD; Schultz RA; Ellenberger T DNA Repair and Mutagenesis, 2nd ed.; ASM Press: Washington, D.C., 2006. [Google Scholar]
- (26).Wu S; Beard WA; Pedersen LG; Wilson SH Structural Comparison of DNA Polymerase Architecture Suggests a Nucleotide Gateway to the Polymerase Active Site. Chem. Rev 2014, 114, 2759–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Beard WA; Wilson SH Structure and Mechanism of DNA Polymerase B. Biochemistry 2014, 53, 2768–2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Prasad R;Çaǧlayan M; Dai D-P; Nadalutti CA; Zhao M-L; Gassman NR; Janoshazi AK; Stefanick DF; Horton JK; Krasich R; Longley MJ; Copeland WC; Griffith JD; Wilson SH DNA Polymerase B: A Missing Link of the Base Excision Repair Machinery in Mammalian Mitochondria. DNA Repair 2017, 60, 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Sykora P; Kanno S; Akbari M; Kulikowicz T; Baptiste BA; Leandro GS; Lu H; Tian J; May A; Becker KA; Croteau DL; Wilson DM; Sobol RW; Yasui A; Bohr VA DNA Polymerase Beta Participates in Mitochondrial DNA Repair. Mol. Cell. Biol 2017, 37, e00237–00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ray S; Breuer G; DeVeaux M; Zelterman D; Bindra R; Sweasy JB DNA Polymerase Beta Participates in DNA End-Joining. Nucleic Acids Res. 2018, 46, 242–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Donigan KA; Sun K.-w.; Nemec AA; Murphy DL; Cong X; Northrup V; Zelterman D; Sweasy JB Human Polb Gene Is Mutated in High Percentage of Colorectal Tumors. J. Biol. Chem 2012, 287, 23830–23839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Nickoloff JA; Jones D; Lee S-H; Williamson EA; Hromas R Drugging the Cancers Addicted to DNA Repair J. National Cancer Institute 2017, 109, DOI: 10.1093/jnci/djx059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Guan L; Greenberg MM Irreversible Inhibition of DNA Polymerase β by an Oxidized Abasic Lesion. J. Am. Chem. Soc 2010, 132, 5004–5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Guan L; Bebenek K; Kunkel TA; Greenberg MM Inhibition of Short Patch and Long Patch Base Excision Repair by an Oxidized Abasic Site. Biochemistry 2010, 49, 9904–9910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Jacobs AC; Kreller CR; Greenberg MM Long Patch Base Excision Repair Compensates for DNA Polymerase β Inactivation by the C4’-Oxidized Abasic Site. Biochemistry 2011, 50, 136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Garcia-Diaz M; Bebenek K; Kunkel TA; Blanco L Identification of an Intrinsic 5 ‘-Deoxyribose-5-Phosphate Lyase Activity in Human DNA Polymerase λ. J. Biol. Chem 2001, 276, 34659–34663. [DOI] [PubMed] [Google Scholar]
- (37).Braithwaite EK; Kedar PS; Stumpo DJ; Bertocci B; Freedman JH; Samson LD; Wilson SH DNA Polymerases β and λ Mediate Overlapping and Independent Roles in Base Excision Repair in Mouse Embryonic Fibroblasts. PLoS One 2010, 5, e12229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Braithwaite EK; Prasad R; Shock DD; Hou EW; Beard WA; Wilson SH DNA Polymerase Lambda Mediates a Back-up Base Excision Repair Activity in Extracts of Mouse Embryonic Fibroblasts. J. Biol. Chem 2005, 280, 18469–18475. [DOI] [PubMed] [Google Scholar]
- (39).Stevens AJ; Guan L; Bebenek K; Kunkel TA; Greenberg MM DNA Polymerase λ Inactivation by Oxidized Abasic Sites. Biochemistry 2013, 52, 975–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40). See Supporting Information.
- (41).Dorjsuren D; Wilson DM; Beard WA; McDonald JP; Austin CP; Woodgate R; Wilson SH; Simeonov A A Real-Time Fluorescence Method for Enzymatic Characterization of Specialized Human DNA Polymerases. Nucleic Acids Res. 2009, 37, e128–e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Zhang D; Seelig G Dynamic DNA Nanotechnology Using Strand-Displacement Reactions. Nat. Chem 2011, 3, 103–113. [DOI] [PubMed] [Google Scholar]
- (43).Olson X; Kotani S; Yurke B; Graugnard E; Hughes W Kinetics of DNA Strand Displacement Systems with Locked Nucleic Acids. J. Phys. Chem. B 2017, 121, 2594–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Sawaya MR; Pelletier H; Kumar A; Wilson SH; Kraut J Crystal Structure of Rat DNA Polymerase Beta: Evidence for a Common Polymerase Mechanism. Science 1994, 264, 1930–1935. [DOI] [PubMed] [Google Scholar]
- (45).Yang L; Beard WA; Wilson SH; Broyde S; Schlick T Highly Organized but Pliant Active Site of DNA Polymerase β: Compensatory Mechanisms in Mutant Enzymes Revealed by Dynamics Simulations and Energy Analyses. Biophys. J 2004, 86, 3392–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Sawaya M; Prasad R; Wilson S; Kraut J; Pelletier H Crystal Structures of Human DNA Polymerase Beta Complexed with Gapped and Nicked DNA: Evidence for an Induced Fit Mechanism. Biochemistry 1997, 36, 11205–11215. [DOI] [PubMed] [Google Scholar]
- (47).Laverty DJ; Mortimer IP; Greenberg MM Mechanistic Insight through Irreversible Inhibition: DNA Polymerase Θ Uses a Common Active Site for Polymerase and Lyase Activities. J. Am. Chem. Soc 2018, 140, 9034–9037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Garcia-Diaz M; DomInguez O; Lopez-Fernandez LA; de Lera LT; SanIger ML; Ruiz JF; Parraga M; GarcIa-Ortiz MJ; Kirchhoff T; del Mazo J. s.; Bernad A; Blanco L DNA Polymerase Lambda, a Novel Eukaryotic DNA Polymerase with a Potential Role in Meiosis. J. Mol. Biol 2000, 301, 851–867. [DOI] [PubMed] [Google Scholar]
- (49).Fu D; Calvo JA; Samson LD Balancing Repair and Tolerance of DNA Damage Caused by Alkylating Agents. Nat. Rev. Cancer 2012, 12, 104–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Cuesta A; Taunton J Lysine-Targeted Inhibitors and Chemoproteomic Probes. Annu. Rev. Biochem 2019, 88, 365–381. [DOI] [PubMed] [Google Scholar]
- (51).Wan X; Yang T; Cuesta A; Pang X; Balius TE; Irwin JJ; Shoichet BK; Taunton J Discovery of Lysine-Targeted Eif4e Inhibitors through Covalent Docking. J. Am. Chem. Soc 2020, 142, 4960–4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Pettinger J; Jones K; Cheeseman MD Lysine-Targeting Covalent Inhibitors. Angew. Chem., Int. Ed 2017, 56, 15200–15209. [DOI] [PubMed] [Google Scholar]
- (53).Sutanto F; Konstantinidou M; Domling A Covalent Inhibitors: A Rational Approach of Drug Discovery. RSC Med. Chem 2020, 11, 876–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.