

Abstract
The active metabolite of the chemotherapeutic irinotecan, SN-38, is detoxified through glucuronidation and then excreted into the gastrointestinal tract. Intestinal bacteria convert the glucuronidated metabolite back to the toxic SN-38 using β-glucuronidase (GUS), resulting in debilitating diarrhea. Inhibiting GUS activity may relieve this side-effect of irinotecan. In this study, we sought to determine whether any known drugs have GUS inhibitory activity. We screened a library of FDA-approved drugs with a cell-free biochemical enzyme assay using purified bacterial GUS. After triage, five drugs were confirmed to inhibit purified bacterial GUS. Three of these were the monoamine oxidase inhibitors nialamide, isocarboxazid and phenelzine with average IC50 values for inhibiting GUS of 71, 128 and 2,300 nM. The tricyclic antidepressant amoxapine (IC50 = 388 nM) and the antimalarial mefloquine (IC50 = 1.2 μM) also had activity. Nialamide, isocarboxazid and amoxapine had no significant activity against purified mammalian GUS, but showed potent activity for inhibiting endogenous GUS activity in a cell-based assay using living intact E. coli with average IC50 values of 17, 336 and 119 nM, respectively. Thus, nialamide, isocarboxazid, and amoxapine have potential to be repurposed as therapeutics to reduce diarrhea associated with irinotecan chemotherapy and warrant further investigation for this use.
Keywords: β-glucuronidase, CPT-11, irinotecan, nialamide, isocarboxazid, amoxapine, phenelzine, mefloquine
INTRODUCTION
Irinotecan (also called CPT-11) is frequently used to treat colon, lung, and brain cancer and it has also been used against refractory forms of leukemia and lymphoma.1 Irinotecan functions as a prodrug, having a carbamate-linked dipiperidino group that significantly increases its solubility and bioavailability.2 In vivo, this dipiperidino group is removed by carboxylesterases to produce the cytotoxic active metabolite, SN-38.3 Frequently, the dose-limiting side effect of irinotecan is severe diarrhea generated by its complex metabolism and its active metabolite regenerating in the intestine.4,5 The active metabolite SN-38 is glucuronidated in the liver by UDP-glucuronosyltransferase (UGT) enzymes.6 This inactivated metabolite, referred to as SN-38G7, is excreted via the biliary ducts into the gastrointestinal (GI) tract. SN-38G is a substrate for the bacterial enzyme β-glucuronidase (GUS) that is produced by bacteria, such as E. coli, normally inhabiting the GI tract. The removal of the glucuronide group of SN-38G by GUS regenerates the active and toxic SN-38.8,9 The resultant SN-38 is now present in the intestinal lumen and therefore results in toxicity to the intestinal cells causing severe diarrhea. This delayed side-effect of irinotecan increases patient suffering and prevents dose-intensification and efficacy in a significant fraction of patients undergoing irinotecan treatment.10,11
β-glucuronidase enzymes can hydrolyze glucuronic acid sugar moieties from a variety of small organic molecules.12 Commonly-used water purity tests that detect bacterial contamination do so through the measurement of GUS activity that is present in a wide range of bacteria.13 In recent work, we sought to identify potent and selective inhibitors of bacterial β-glucuronidases to inhibit the regeneration of SN-38 in the intestines.14 This approach was envisioned to reduce or eliminate the GI toxicity of irinotecan treatment without harming the normal GI bacteria required for intestinal health. From a high throughput screen for GUS inhibitors, four hits were selected for follow-up studies.14 These compounds showed potent inhibition of bacterial GUS, but no activity against the homologous mammalian enzyme. The compounds inhibited GUS activity in live bacteria with IC50 values ranging from 18 nM to 1.3 μM and no effect on bacteria viability even at 100 μM. In proof of concept studies, oral dosing of the most potent inhibitor protected mice from irinotecan-induced diarrhea.
We previously reported the development and validation of a semi-automated high throughput assay that was used to screen compounds for GUS inhibitors.14,15 This high throughput primary assay was a cell-free biochemical enzyme assay using purified bacterial GUS. As part of the validation for this GUS enzyme assay, we screened a small collection of 1,120 compounds purchased from the Prestwick Chemical company. According to the company, ~90% of these compounds are Food and Drug Administration (FDA)-approved drugs. In this study, we sought to determine whether any known drugs have genuine and potent GUS inhibitory activity. We report that five known, FDA-approved drugs have GUS inhibitory activity in a cell-free biochemical enzyme assay using commercially available purified bacterial GUS and in a whole cell GUS activity assay using living intact E. coli. Thus, these drugs may warrant further investigation as therapeutic agents to prevent irinotecan-induced diarrhea.
MATERIALS AND METHODS
Materials
All common reagents such as HEPES, Triton X-100, carbenicillin, and dimethyl sulfoxide (DMSO) were reagent-grade quality and obtained from Thermo Fisher Scientific (Waltham, MA) or Sigma-Aldrich (St. Louis, MO). 4-methylumbelliferyl glucuronide (4MUG) was obtained from Sigma-Aldrich. The solid black 96-well plates (cat# 3915) for the assay and 96-well clear plates (cat# 9017) for cytotoxicity assay were from Corning Incorporated (Corning, NY). Falcon polypropylene plates (cat# 1190) used for serial dilution of compounds were obtained from Becton Dickinson (Franklin Lake, NJ). Amoxapine, nialamide, isocarboxazid and other compounds for follow-up studies were obtained from Sigma-Aldrich. The Prestwick Chemical Collection was obtained from Prestwick Chemical Company (Washington DC). E. coli DH5α (Zymo Research, Irvine, CA) was used for the cell-based assay. Purified E. coli GUS enzyme (cat # G8420–25KU) and Bovine taurus GUS enzyme (cat G0501–100KU) was purchased from Sigma-Aldrich.
GUS Enzyme Assay
The semi-automated GUS high throughput enzyme assay was performed as previously described15 and was used to screen the Prestwick Chemical Collection. The follow-up studies were performed manually in a similar manner with the exception of plate type and volumes, as briefly outlined here. Compound stock solutions were made in 100% DMSO. Serial dilutions of compounds for IC50 determinations were initially performed in 100% DMSO in 96-well polypropylene plates, then each compound concentration stock solution was diluted into assay buffer (50 mM HEPES, pH 7.4 and 0.017% Triton X-100), producing a constant 5% DMSO in all wells. Subsequently, 20 μL of this aqueous diluted compound (or just 5% DMSO for controls) was added to the wells of a solid black 96-well plate followed by 40 μL of GUS enzyme diluted in assay buffer. After addition of enzyme, the reaction was initiated by addition of 40 μL of 4MUG substrate (312.5 μM 4MUG) diluted in 50 mM HEPES, pH 7.4. 4MUG stock solutions were prepared in the same buffer. Final concentrations in the assembled assay were 50 mM HEPES, pH 7.4, 0.01% Triton X-100, 1% DMSO, 125 μM 4MUG and 50 pM GUS. The enzyme reaction was allowed to proceed for 20 minutes at 23°C and was terminated by the addition of 40 μL of a 1M sodium carbonate solution. Fluorescence at 460 nm was determined using a 355 nm excitation wavelength with a 0.1 s/well read time in a BMG PheraStar (BMG LABTECH, Cary, NC). Fluorescence data, expressed in relative fluorescence units (RFU), were normalized to DMSO (100% activity) and “no enzyme” (0% activity) controls as maximum and minimum responses, respectively. The Bovine taurus GUS enzyme assay was performed in an identical manner except Bovine taurus GUS enzyme (1 nM) was used instead of bacterial GUS. The IC50 values and Hill slopes were calculated from concentration response data using GraphPad Prism software (GraphPad Software Inc., La Jolla, CA) employing either four-parameter or a three parameter (fixed bottom) curve fit.
GUS Cell Based Assay
Cultures of E. coli (DH5α) carrying the empty expression vector pCMV5 were grown over night in LB containing carbenicillin (50 μM) and then used to initiate fresh LB/carbenicillin cultures adjusted to an initial OD of 0.1. These cultures were allowed to reach an OD of 0.6 and then washed twice with 50 mM HEPES, pH 7.4 containing carbenicillin (50 μM) and concentrated by centrifugation to an OD of 1 for use in the assay. The GUS cell based assay was performed in an identical manner as the enzyme assay except the Triton X-100 was left out of the assay buffer to avoid cell lysis, the E. coli cells replaced the enzyme and the reaction was allowed to proceed for 2 hrs at 37°C. The resulting data was analyzed as outlined for the enzyme assay.
Toxicity Assay
Compounds were tested for cytotoxicity in E. coli cells. The cells were grown and prepared for assay as described above, and plated in clear 96-well plates. Cells were treated with 100 μM and 10 μM concentrations (1% DMSO) of test compounds and incubated for 2 hours at 37°C. Subsequently, 25uL of MTS viability reagent (CellTiter 96 Aqueous Non-Radioactive Cell Proliferation Assay Kit, Promega Corp., Madison, WS) was added to the wells and incubation continued for 5 minutes. The plates were then analyzed for absorbance at 490 nm on a SpectraMax Plus 384 (Molecular Devices, Sunnyvale, CA). Controls included DMSO only (considered 100% viability), “no cells” (representing 0% viable cells), and the cytotoxic positive control compound kanamycin at 50 μg/ml.
RESULTS
In this study, we sought to determine if any known drugs have GUS inhibitory activity. The Prestwick collection of FDA-approved drugs was screened with the GUS enzyme assay as previously reported, initially to validate the GUS enzyme assay for HTS.15 This screen of 1,120 compounds resulted in 40 actives having ≥50% inhibition for a hit rate of 3.6% and all plates had Z’-factors of ≥0.8 (average Z’-factor was 0.90). Since the collection was screened at 10 μM compound, a high concentration relative to in vivo drug levels, a cut-off of 91% inhibition was applied as criteria for selecting initial compounds for follow-up studies. This requirement allowed us to focus on the more potent actives, resulting in a short list of 7 compounds. Furthermore, antibiotics and antiseptics were eliminated since the desire is to identify drugs that do not disrupt the gut microbial flora, but instead only inhibit bacterial GUS activity. This further triaging of actives resulted in 4 compounds. We observed that two of these actives belong to the monoamine oxidase inhibitor (MAOI) class of drugs, though another MAOI while active (62% inhibition), did not quite meet the 91% inhibition criteria. So to test more examples of this class of inhibitors, we also included this compound (phenelzine) in our studies. Thus, a total of five compounds were selected for follow-up studies which included IC50 confirmation and E. coli cell-based assays.
The five compounds that remained after triage were nialamide, isocarboxazid, phenelzine, amoxapine and mefloquine (Fig. 1). Nialamide, isocarboxazid and phenelzine are all irreversible hydrazine-class MAOI drugs, though nialamide is no longer on the market. Amoxapine is a tricyclic antidepressant and mefloquine is an antimalarial drug. Concentration response data for each of the five compounds was generated using the purified E. coli GUS enzyme assay from which IC50 values and Hill slopes were calculated (Fig. 2). The average IC50 values and standard deviations (SDs) for the MAOIs nialamide, isocarboxazid and phenelzine were 71 ± 31, 128 ± 56, and 2,282 ± 1041 nM (see summary Table 1 for all compound IC50 data).
Fig. 1.
Structures of studied drugs
Fig. 2. Potency determinations using an E. coli GUS enzyme assay.
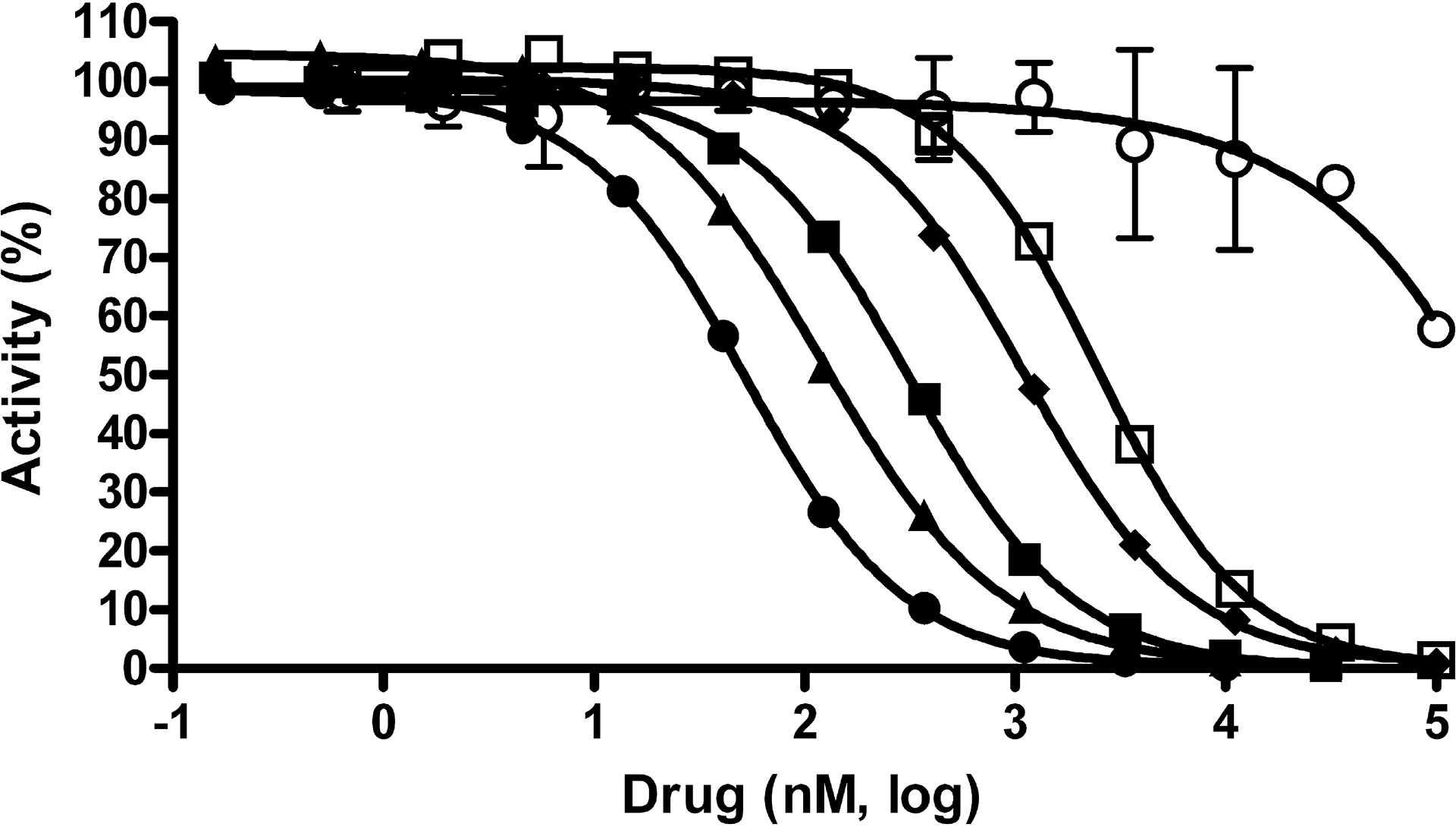
Concentration response data for compounds was normalized to controls with and without enzyme and plotted as percent activity. Potency determinations of nialamide (●), isocarboxazid (▲), phenelzine (□), amoxapine (■), loxapine (○) and mefloquine (♦) resulted in average IC50 values (nM) and SDs of 71 ± 32, 128 ± 56, 2,282 ± 1,041, 388 ± 98, >100,000 and 1,212 ± 234, respectively. The average Hill slope values derived from the IC50 curves of all active drugs ranged from 0.93 – 1.26. Data points represent the average of three determinations per concentration and error bars represent standard deviation. Data are representative of three independent experiments.
Table 1.
Summary of GUS Inhibitory Activity of Studied Drugs
| Drug | Drug Class | Currently Marketed |
E. coli GUS Enzyme Assaya |
B. taurus GUS Enzyme Assaya |
E. coli
Cell-Based Assaya |
|---|---|---|---|---|---|
| IC50 ± SD (nM) | IC50 ± SD (nM) | IC50 ± SD (nM) | |||
| Nialamide | Irreversible MAOI | No | 71 ± 32 | 74,813 ± 1,841 | 17 ± 2 |
| Isocarboxazid | Irreversible MAOI | Yes | 128 ± 56 | >100,000 | 336 ± 120 |
| Phenelzine | Irreversible MAOI | Yes | 2,282 ± 1041 | NDb | 7,123 ± 1650 |
| Amoxapine | Tricyclic Antidepressant | Yes | 388 ± 98 | >100,000 | 119 ± 61 |
| Loxapinec | Tricyclic Antidepressant | Yes | >100,000 | ND | >100,000 |
| Mefloquine | Antimalarial | Yes | 1,212 ± 234 | ND | 5,961 ± 1526 |
IC50 value determinations were performed at least three times, with average IC50 values and standard deviations (SD) shown. The range of average Hill slopes for all measurable IC50 curves (where at least 50% inhibition was obtained) was 0.84 to 1.26.
ND = not determined;
This drug was included as a study control
Amoxapine generated an average IC50 value and SD of 388 ± 98 nM in the E. coli GUS enzyme assay. Loxapine is another tricyclic antidepressant drug that has the identical structure as amoxapine, except that loxapine has a methyl group, instead of hydrogen, on the secondary amine of the piperazine ring (Fig. 1). We tested loxapine as a specificity control and this compound resulted in an average IC50 value of >100 μM in the GUS enzyme assay. Thus, the methyl group on the piperazine ring of loxapine resulted in >250-fold loss in potency. The antimalarial drug mefloquine hydrochloride generated an average IC50 value of 1,212 ± 234 nM in this GUS enzyme assay.
Compound aggregation has been reported as a common non-specific inhibitor mechanism for purified enzyme assays.16 The Hill slopes calculated from concentration response data can be used to eliminate many non-specific inhibitors in enzyme assays. For single site binding, the Hill slope of an IC50 curve should be 1.0. IC50 curves with steep slopes, i.e. significantly greater than 1.0, can be an indicator of non-specific mechanisms, including compound aggregation.16,17 The IC50 curves for all the tested compounds (with measurable IC50 values) had average Hill slope values that ranged from 0.93 to 1.26 in the E. coli GUS enzyme assay, which is close to the ideal value expected when measuring inhibition of a single enzyme. To assess whether the compounds were inhibiting signal by merely quenching fluorescence of the product formed, GUS enzyme assays were done in which compound (100 μM) was added after the enzyme reaction was stopped and then fluorescence was measured as usual. Adding the compounds at the end of the assay resulted in no inhibition of signal for any of the studied compounds (data not shown), indicating that the observed activity is not due to fluorescence quenching, color quenching or other assay artifact. Thus, these compounds produced data consistent with specific binding to a single site on GUS and not inhibition by non-specific mechanisms or assay artifact.
Tumor-derived mammalian GUS activity may be important for optimal anti-tumor efficacy of irinotecan. Recent evidence suggests that mammalian GUS may convert SN-38G back to SN-38 within the tumor and thus increase the concentration of active drug (SN-38) in the tumor.18–20 Therefore, any inhibitor of bacterial GUS used therapeutically should not inhibit the mammalian GUS since this may decrease the efficacy of irinotecan at the site of the tumor. Therefore, we tested the three most potent drugs from the screen – nialamide, isocarboxazid and amoxapine – in enzyme assays identical to the E. coli GUS enzyme assay, except for the use of mammalian GUS purified from Bovine taurus liver. Nialamide generated an average IC50 of 74.8 μM, while isocarboxazid and amoxapine had IC50 values >100 μM. Thus, nialamide was over 1,000-fold more potent against E. coli GUS than mammalian GUS, while the other two drugs where >250-fold more selective for the E. coli GUS.
An E. coli cell based assay was developed in order to assess the activity of these drugs against whole cells, instead of purified enzyme. We took advantage of the well-known specificity and sensitivity of the 4MUG substrate to detect GUS activity in E. coli cells. This assay mimicked the enzyme assay in format, with the GUS enzyme replaced by live log-phase E. coli cells and the assay incubated for a longer time (2 hr) to detect GUS activity in these un-modified cells. Four experimental results confirmed that this cell-based assay was measuring GUS activity and no other E. coli cell enzymes. First, The Km value for the substrate was determined with this cell-based assay to be 151 μM (data not shown), which is similar to the 125 μM Km value we previously reported for the purified enzyme assay.15 Secondly, the Hill slopes derived from concentration-response data for all 5 active drugs tested in this cell-based assay were in the 0.8 – 1.1 range, close to the expected value of 1.0 for inhibition of a single enzyme. Thirdly, maximal inhibition was achieved by all active compounds (Fig. 3), which would be expected if 100% of the observed activity was coming from a single enzyme rather than a mixture of enzymes. Finally, the overall rank order of potencies in the cell based assay is similar to the E. coli GUS assay and absolute IC50 values derived from the cell-based assay for the test compounds are within 5-fold of the purified bacterial GUS enzyme assay (see Table 1).
Fig. 3. Activities in an E. coli cell-based GUS activity assay.
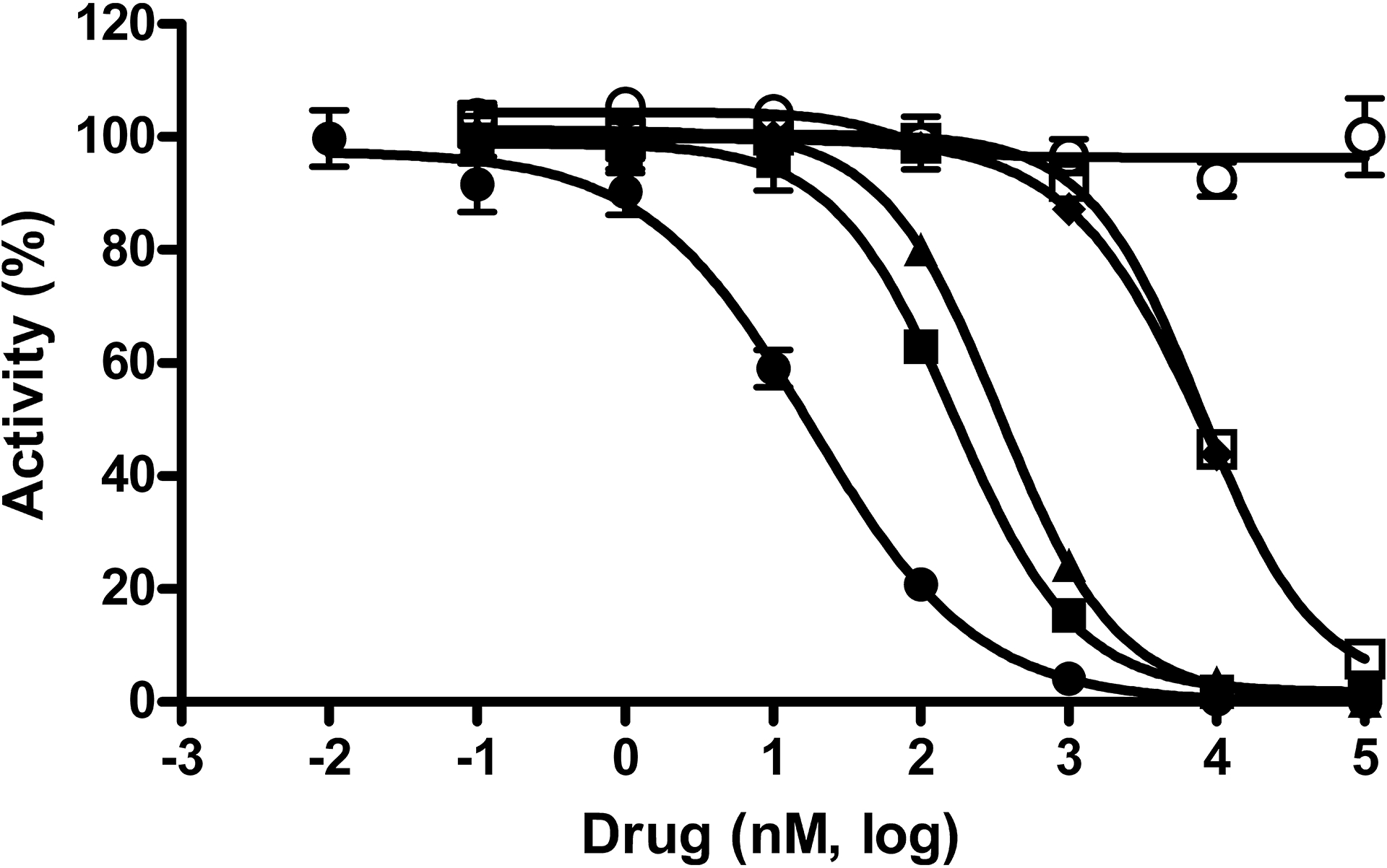
Concentration response data for compounds was normalized to controls with and without whole E. coli cells and plotted as percent activity. Potency determinations of nialamide (●), isocarboxazid (▲), phenelzine (□), amoxapine (■), loxapine (○) and mefloquine (♦) resulted in average IC50 values (nM) and SDs of 17 ± 2, 336 ± 120, 7,123 ± 1,650, 119 ± 61, >100,000 and 5,961 ± 1,526, respectively. The average Hill slope values derived from the IC50 curves of all active drugs ranged from 0.8 – 1.1. Data points represent the average of three determinations per concentration and error bars represent standard deviation. Data are representative of three independent experiments.
The potencies of the five hits from the Prestwick collection and the one control compound were determined using the E. coli cell-based assay (Fig. 3 and Table 1). In the irreversible MOAI class of drugs, nialamide potently inhibited the cell-based assay with an IC50 of 17 ± 2 nM, while isocarboxazid generated an IC50 of 336 ± 120 nM. Thus, the potency of nialamide shifted 4-fold more potent in the cell-based assay compared to the E. coli GUS enzyme assay, while isocarboxazid was about 3-fold less potent in the cell-based assay. The other irreversible MOAI drug, phenelzine, was also tested in the cell-based assay and resulted in an IC50 value of 7,123 nM, which is 2.6-fold less potent compared to the enzyme assay. Amoxapine was also tested in this assay resulting in an IC50 of 119 ± 61 nM, which is 3.3-fold more potent than the 388 nM IC50 value generated using the enzyme assay. As a specificity control, loxapine was also tested, but was completely inactive (>100 μM) in this assay, consistent with its inactivity in the enzyme assay. Mefloquine generated an average IC50 value of 5,961 ± 1,526 nM and thus shifted over 4.9-fold less potent in the cell-based assay compared to the enzyme assay.
One possible explanation for the inhibitory activity of the drugs in the cell-based assay is E. coli cell toxicity and/or bacteriostatic activity resulting in reduced GUS activity. Therefore, the viability of drug-treated E. coli cells was assessed with a metabolic viability assay (MTS kit, Promega). Each compound was tested at 100 and 10 μM for 2 hrs and data normalized to solvent (DMSO) controls with and without cells (Fig. 4). Kanamycin was used as a control to demonstrate assay sensitivity to a known bactericidal antibiotic. Kanamycin treatment (50 μg/ml) reduced activity in this assay by over 90%. Amoxapine, isocarboxazid, loxapine, and nialamide showed no signs of toxicity in this assay at up to 100 μM drug. In contrast, the 100 μM concentration of mefloquine completely inhibited viability in this assay while the 10 μM concentration was at maximum control levels (100% viability). Thus, the IC50 value of mefloquine in the cell-based GUS assay may be a blend of GUS inhibitory activity and bactericidal activity at higher concentrations. Phenelzine showed some inhibitory activity (~10 – 20%) at both the 100 and 10 μM concentrations, but not enough to explain all of its cell-based activity.
Fig. 4. Bacterial cytotoxicity assessment of studied drugs.
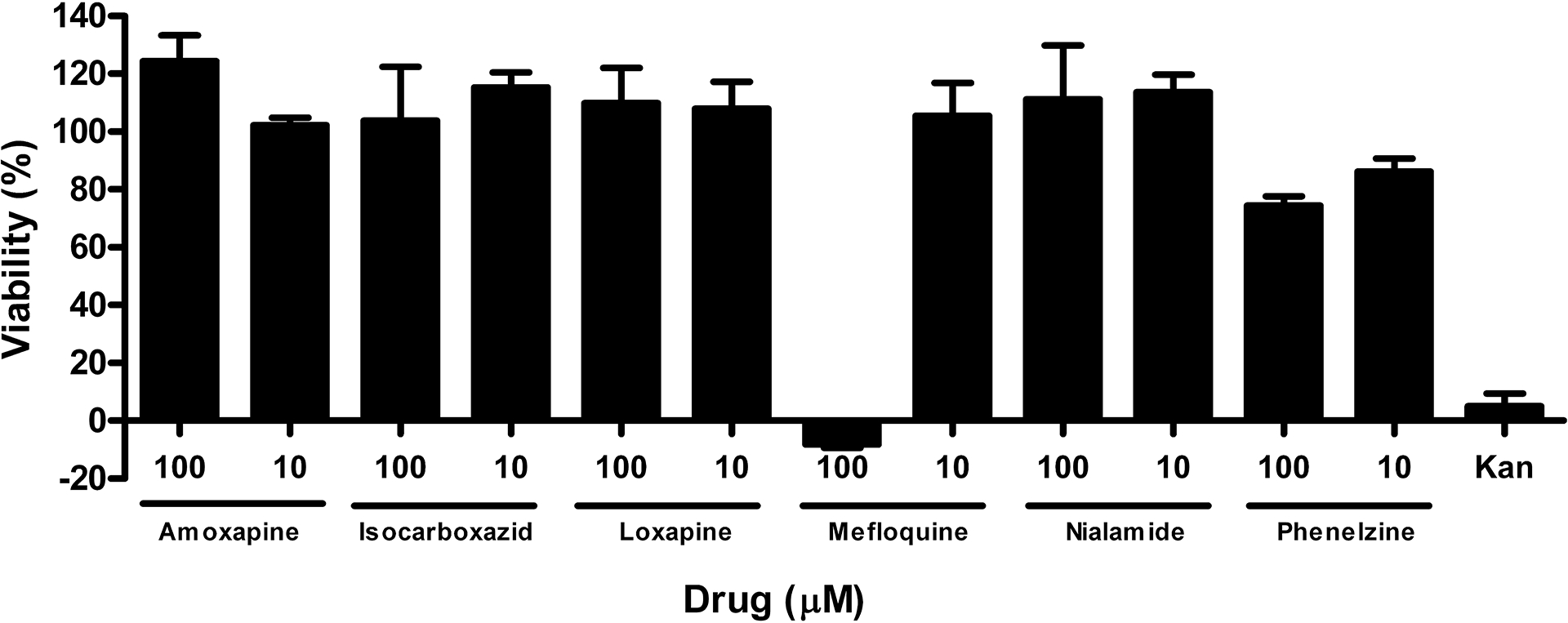
E. coli bacteria were treated with compounds at 10 and 100 μM, as indicated, for two hours. Viability was assessed with MTS viability reagent. Absorbance data was normalized to controls with and without whole E. coli cells and plotted as percent viability, with ‘no cells’ considered as 0% viability and DMSO only treated cells set at 100% viable. Kanamycin, a known cytotoxic antibiotic drug, was used as a control.
DISCUSSION
We have sought to determine if any known drugs have potent and selective GUS inhibitory activity and whether they maintained activity in cell-based assays. Here we report the follow-up studies from single-point actives obtained from screening a collection of FDA-approved drugs, the Prestwick collection, using our previously reported high throughput GUS enzyme assay.15 The hit rate was high, with 40 actives displaying ≥50% inhibition at the screening concentration of 10 μM. Raising the cut-off to 91% and elimination of antiseptics/antibiotics resulted in a short list of 4 actives for follow-up. We also included a compound (phenelzine) that did not meet the activity cut-off. It was also chosen for follow-up since it was in the same class as two on the short list and it had >50% inhibition. Thus, the actives were nialamide, isocarboxazid, phenelzine, amoxapine and mefloquine and all of these compounds confirmed by IC50 determinations in the E. coli GUS enzyme assay. The five hits can be categorized into three drug classes: irreversible MAOI, tricyclic antidepressant and antimalarial.
In the irreversible MAOI class, nialamide was a very potent inhibitor of GUS activity with an IC50 of 71 nM in the GUS enzyme assay. Surprisingly, this is more potent than its reported in vitro IC50 values of 2.6 – 13 μM for inhibiting monoamine oxidase (in rat brain homogenates), its original intended target.21,22 When tested for inhibitory potency against purified mammalian GUS, nialamide had an IC50 of approximately 75 μM. Thus, nialamide displayed a dramatic 1,000-fold selectivity for inhibiting E. coli GUS over mammalian GUS. Furthermore, nialamide had more potent activity for inhibiting endogenous GUS in the E. coli cell-based assay, generating an IC50 of 17 nM. This activity was not due to acute toxicity of nialamide. This unusual increased potency in a cell-based assay (also observed with amoxapine) may be due to a unique mechanism of action or due to compound concentration inside the bacterial cell. This same phenomenon was observed previously for some, but not all, GUS inhibitor compounds.14 The other compound in this same class, isocarboxazid, was also relatively potent with an IC50 of 128 nM, which is more potent than its reported potency of 4.8 μM IC50 for MAO in rat brain homogenates.22 Isocarboxazid was >780-fold more selective for inhibiting bacterial GUS compared to its activity against mammalian GUS, which was not measurable (>100 μM IC50). This drug also inhibited in the cell-based assay with an IC50 of 336 nM, 2.6-fold less potent compared to the purified enzyme assay. Phenelzine is also an MAOI that is structurally similar to nialamide and isocarboxazid in that it contains a hydrazine group and is irreversible against its original target. Phenelzine was a much weaker inhibitor of GUS at 2.2 μM IC50, in contrast to its IC50 for MAO that was reported to be 70 – 900 nM (depending on subclass of MAO-A or MAO-B, or total activity). Thus, the phenelzine results indicated that inhibition by the MAOIs was not solely due to the presence of a hydrazine group or the irreversible nature of these drugs. Phenelzine showed some toxicity to E. coli at 10 and 100 μM, but not enough to account for all of its GUS inhibitory activity.
The tricyclic antidepressant amoxapine potently inhibited purified GUS with an IC50 of 388 nM. In comparison, amoxapine had no measurable IC50 against mammalian GUS (>100 μM) thus resulting in a >250-fold selectivity for inhibiting bacterial GUS over mammalian GUS. Furthermore, amoxapine had more potent activity in the cell-based assay with an IC50 of 119 nM. Loxapine was used as a control compound for this class since it has an identical structure to amoxapine except that loxapine has a methyl group on the nitrogen of the piperazine group. Despite this very small structural difference, loxapine had no measurable IC50 value (>100 μM) for both the GUS enzyme assay and the cell-based assay. Thus, the amoxapine/loxapine pair served to illustrate the exquisite structural selectivity for inhibiting signal in these assays and demonstrated that a free amine in the piperazine group is critical for inhibiting GUS.
Finally, mefloquine is an antimalarial drug that was also identified in our screen. This drug had only weak activity for inhibiting purified GUS (IC50 = 1.2 μM) and its potency worsened by 5-fold when tested in the cell-based assay (IC50 = 6 μM). Since this compound resulted in complete inhibition in the toxicity assay at 100 μM, though none evident at 10 μM, it is possible that some of the cell-based activity is due to toxicity. Based on our data, we speculate that any use of mefloquine to inhibit GUS in vivo would likely require high doses and thus may function like a general antibiotic.
Nialamide, the most potent of these drugs for inhibiting bacterial GUS, has a number of issues with respect to its use as a therapeutic. First, nialamide is no longer on the market. Nialamide was withdrawn from the market in 1963 due to interactions with food products containing high levels of tyramine.23 Ingestion of certain foods high in tyramine (e.g. aged cheese) resulted in sometimes severe tyramine toxicity in patients taking nialamide, a general problem with all the non-selective irreversible MAOIs.24 Therefore, toxicity of nialamide is a major concern, even if it were available again. However, given the high potency of this drug for inhibiting GUS, it may be possible to use lower, and thus safer, doses of nialamide that would have acceptable side effects. Special diets, especially avoiding intake of tyramine-enriched foods, help reduce food toxicity side effects of MAOIs.24 It is also conceivable that nialamide could be re-formulated for low-dose time release in the intestine. The food-induced toxicity reported for nialamide is assumed to be due to inhibition of MAO in the intestine. Thus, we speculate that dosing with nialamide may result in sufficient concentrations of nialamide in the GI tract to effectively inhibit GUS. In contrast to nialamide, isocarboxazid is still on the market for treatment of major depression. Like all drugs in the MAOI class, isocarboxazid has toxicity/side effect concerns and can be problematic in combination with many other medicines due to drug-drug interactions.24 Phenelzine had relatively weak activity in our assays and so it is not clear if effective in vivo concentrations could be achieved. It should be recognized that any GUS inhibitor would only be needed short term (weeks) and perhaps even intermittently. Thus, the long term toxicity of nialamide or isocarboxazid may be avoidable with strategic, short term dosing regimens to minimize long term drug exposure with the accompanying toxicity/side effects.
Amoxapine is a marketed member of an older class of antidepressant drugs with significantly fewer toxicity concerns compared to the MAOI drug class. It also has a safer side effect profile and far fewer drug-drug interactions than the MAOI class of drugs in general. Antidepressant use in general is common in cancer patients and thus amoxapine could also treat cancer-induced depression.25 According to a recent report, amoxapine and loxapine have been discovered to be potent non-competitive inhibitors of P-glycoprotein, a transporter responsible for multidrug resistance displayed by some cancer cells.26 Thus, the use of amoxapine as a GUS inhibitor may also have the added benefit of enhancing the sensitivity of multidrug resistant cancer cells to irinotecan and/or other chemotherapeutic drugs given in combination with irinotecan. Tricyclic antidepressants typically take about three weeks to reach peak efficacy for treatment of depression.27 Unlike the long term, slow acting mechanism of amoxapine for depression, amoxapine as a GUS inhibitor will only be needed short term and should act immediately to prevent re-activation of SN-38G. Thus, some of the side effects encountered with chronic use of amoxapine may be minimized with strategic intermittent dosing. The combination of its potency for inhibiting GUS and its safer profile suggests that amoxapine may have the best potential among these studied drugs as a therapeutic treatment of irinotecan induced diarrhea. A major question concerning the use of amoxapine is whether a sufficient concentration of amoxapine (and/or active metabolites) can be safely achieved in the intestine to significantly inhibit GUS activity with acceptable side effects. Normal blood levels of amoxapine have been reported to be 0.017 to 0.21 μg/ml (54 – 669 nM), and if its 8-OH metabolite is included, then up to 1.27 μM blood concentration can be safely achieved. These blood concentrations are at or above the in vitro IC50 values we determined for amoxapine. Therefore, achieving therapeutic intestinal, rather than blood, concentration may be the primary clinical hurdle with amoxapine. It should be noted that loxapine undergoes metabolism that includes some of the drug being de-methylated at the piperazine ring – essentially generating amoxapine and amoxapine-like molecules in vivo.28,29 Thus, loxapine, though it lacked activity in our in vitro assays, may have GUS inhibitory activity in vivo due to its metabolites. This potential activity can only be assessed by in vivo animal studies with loxapine.
We have identified five known drugs that inhibit E. coli GUS activity in enzyme assays: nialamide, isocarboxazid, phenelzine, amoxapine and mefloquine. These compounds displayed IC50 values ranging from 71 nM to 2.3 μM against purified E. coli GUS. Furthermore, nialamide, isocarboxazid and amoxapine had no significant activity against purified mammalian GUS. All five compounds also had activity in an E. coli cell-based assay with IC50 values for inhibiting endogenous GUS ranging from 17 nM to 7.1 μM. In the future, mechanism of action studies will allow further characterization of these drugs as GUS inhibitors. More importantly, animal studies will be required to assess the in vivo efficacy of these compounds for preventing this irinotecan-induced side effect. Taken together, our data suggests that nialamide, isocarboxazid and amoxapine may have potential therapeutic use for preventing irinotecan induced diarrhea. As existing drugs, these molecules could be rapidly tested in clinical trials and may not only alleviate irinotecan induced diarrhea, but may also allow higher doses of irinotecan to be tolerated, thus enhancing the anti-cancer efficacy of this drug.
Acknowledgements
This work was supported in part by a grant from the Golden LEAF Foundation, funds from the State of North Carolina, National Institutes of Health (NIH) grant 1SC2GM081129 (J.E.S.).
List of abbreviations:
- GUS
β-glucuronidase
- DMSO
dimethyl sulfoxide
- RFU
relative fluorescence units
- GI
gastrointestinal
- 4MUG
4-methylumbelliferyl glucuronide
- E. coli
Escherichia coli
- B. Taurus
Bovine Taurus
REFERENCES
- 1.Pommier Y (2006) Topoisomerase I inhibitors: camptothecins and beyond, Nat Rev Cancer 6, 789–802. [DOI] [PubMed] [Google Scholar]
- 2.Pizzolato JF, and Saltz LB (2003) The camptothecins, Lancet 361, 2235–2242. [DOI] [PubMed] [Google Scholar]
- 3.Smith NF, Figg WD, and Sparreboom A (2006) Pharmacogenetics of irinotecan metabolism and transport: an update, Toxicol In Vitro 20, 163–175. [DOI] [PubMed] [Google Scholar]
- 4.Ma MK, and McLeod HL (2003) Lessons learned from the irinotecan metabolic pathway, Curr Med Chem 10, 41–49. [DOI] [PubMed] [Google Scholar]
- 5.Mathijssen RHJ, van Alphen RJ, Verweij J, Loos WJ, Nooter K, Stoter G, and Sparreboom A (2001) Clinical Pharmacokinetics and Metabolism of Irinotecan (CPT-11), Clin Cancer Res 7, 2182–2194. [PubMed] [Google Scholar]
- 6.Miley MJ, Zielinska AK, Keenan JE, Bratton SM, Radominska-Pandya A, and Redinbo MR (2007) Crystal structure of the cofactor-binding domain of the human phase II drug-metabolism enzyme UDP-glucuronosyltransferase 2B7, J Mol Biol 369, 498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagar S, and Blanchard RL (2006) Pharmacogenetics of uridine diphosphoglucuronosyltransferase (UGT) 1A family members and its role in patient response to irinotecan, Drug Metab Rev 38, 393–409. [DOI] [PubMed] [Google Scholar]
- 8.Stein A, Voigt W, and Jordan K Review: Chemotherapy-induced diarrhea: pathophysiology, frequency and guideline-based management, Therapeutic Advances in Medical Oncology, pp. 51–63 vol. 2:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tobin PJ, Dodds HM, Clarke S, Schnitzler M, and Rivory LP (2003) The relative contributions of carboxylesterase and beta-glucuronidase in the formation of SN-38 in human colorectal tumours, Oncology reports 10, 1977–1979. [PubMed] [Google Scholar]
- 10.Hu ZP, Yang XX, Chan SY, Xu AL, Duan W, Zhu YZ, Sheu FS, Boelsterli UA, Chan E, Zhang Q, Wang JC, Ee PL, Koh HL, Huang M, and Zhou SF (2006) St. John’s wort attenuates irinotecan-induced diarrhea via down-regulation of intestinal pro-inflammatory cytokines and inhibition of intestinal epithelial apoptosis, Toxicology and applied pharmacology 216, 225–237. [DOI] [PubMed] [Google Scholar]
- 11.Kurita A, Kado S, Matsumoto T, Asakawa N, Kaneda N, Kato I, Uchida K, Onoue M, and Yokokura T Streptomycin alleviates irinotecan-induced delayed-onset diarrhea in rats by a mechanism other than inhibition of beta-glucuronidase activity in intestinal lumen, Cancer chemotherapy and pharmacology. [DOI] [PubMed] [Google Scholar]
- 12.Basinska A, and Florianczyk B (2003) Beta-glucuronidase in physiology and disease, Annales Universitatis Mariae Curie-Sklodowska 58, 386–389. [PubMed] [Google Scholar]
- 13.Farnleitner AH, Hocke L, Beiwl C, Kavka GC, and Mach RL (2002) Hydrolysis of 4-methylumbelliferyl-beta-D-glucuronide in differing sample fractions of river waters and its implication for the detection of fecal pollution., Water Res. 36, 975–981. [DOI] [PubMed] [Google Scholar]
- 14.Wallace BD, Wang H, Lane KT, Scott JE, Orans J, Koo JS, Venkatesh M, Jobin C, Yeh LA, Mani S, and Redinbo MR (2010) Alleviating cancer drug toxicity by inhibiting a bacterial enzyme, Science 330, 831–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahmad S, Hughes MA, Lane KT, Redinbo MR, Yeh LA, and Scott JE (2011) A High Throughput Assay for Discovery of Bacterial beta-Glucuronidase Inhibitors, Curr Chem Genomics 5, 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McGovern SL, Caselli E, Grigorieff N, and Shoichet BK (2002) A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening, J Med Chem 45, 1712–1722. [DOI] [PubMed] [Google Scholar]
- 17.Feng BY, Simeonov A, Jadhav A, Babaoglu K, Inglese J, Shoichet BK, and Austin CP (2007) A high-throughput screen for aggregation-based inhibition in a large compound library, J Med Chem 50, 2385–2390. [DOI] [PubMed] [Google Scholar]
- 18.Tobin P, Clarke S, Seale JP, Lee S, Solomon M, Aulds S, Crawford M, Gallagher J, Eyers T, and Rivory L (2006) The in vitro metabolism of irinotecan (CPT-11) by carboxylesterase and beta-glucuronidase in human colorectal tumours, Br J Clin Pharmacol 62, 122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prijovich ZM, Chen KC, and Roffler SR (2009) Local enzymatic hydrolysis of an endogenously generated metabolite can enhance CPT-11 anticancer efficacy, Mol Cancer Ther 8, 940–946. [DOI] [PubMed] [Google Scholar]
- 20.Huang PT, Chen KC, Prijovich ZM, Cheng TL, Leu YL, and Roffler SR (2011) Enhancement of CPT-11 antitumor activity by adenovirus-mediated expression of beta-glucuronidase in tumors, Cancer Gene Ther 18, 381–389. [DOI] [PubMed] [Google Scholar]
- 21.Ulus IH, Maher TJ, and Wurtman RJ (2000) Characterization of phentermine and related compounds as monoamine oxidase (MAO) inhibitors, Biochem Pharmacol 59, 1611–1621. [DOI] [PubMed] [Google Scholar]
- 22.Maxwell DR, Gray WR, and Taylor EM (1961) Relative activity of some inhibitors of mono-amine oxidase in potentiating the action of tryptamine in vitro and in vivo, Br J Pharmacol Chemother 17, 310–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lexchin J (2005) Drug withdrawals from the Canadian market for safety reasons, 1963–2004, CMAJ 172, 765–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wimbiscus M, Kostenko O, and Malone D (2010) MAO inhibitors: risks, benefits, and lore, Cleve Clin J Med 77, 859–882. [DOI] [PubMed] [Google Scholar]
- 25.Fisch M (2004) Treatment of depression in cancer, J Natl Cancer Inst Monogr, 105–111. [DOI] [PubMed] [Google Scholar]
- 26.Palmeira A, Rodrigues F, Sousa E, Pinto M, Vasconcelos MH, and Fernandes MX (2011) New uses for old drugs: pharmacophore-based screening for the discovery of P-glycoprotein inhibitors, Chem Biol Drug Des 78, 57–72. [DOI] [PubMed] [Google Scholar]
- 27.Leonard BE (1984) Pharmacology of new antidepressants, Prog Neuropsychopharmacol Biol Psychiatry 8, 97–108. [DOI] [PubMed] [Google Scholar]
- 28.Badway MA, and Dugas JE (1984) Loxapine yields amoxapine, J Clin Psychopharmacol 4, 363–364. [DOI] [PubMed] [Google Scholar]
- 29.Cheung SW, Tang SW, and Remington G (1991) Simultaneous quantitation of loxapine, amoxapine and their 7- and 8-hydroxy metabolites in plasma by high-performance liquid chromatography, J Chromatogr 564, 213–221. [DOI] [PubMed] [Google Scholar]