

Abstract
Increasing clinical evidence has demonstrated that the deletion or mutation of tumor suppressor genes such as the gene-encoding phosphatase and tensin homolog deleted on chromosome 10 (PTEN) in cancer cells may correlate with an immunosuppressive tumor microenvironment (TME) and poor response or resistance to immune checkpoint blockade (ICB) therapy. It is largely unknown whether the restoration of functional PTEN may modulate the TME and improve the tumor’s sensitivity to ICB therapy. Here, we demonstrate that mRNA delivery by polymeric nanoparticles can effectively induce expression of PTEN in Pten-mutated melanoma cells and Pten-null prostate cancer cells, which in turn induces autophagy and triggers cell death–associated immune activation via release of damage-associated molecular patterns. In vivo results illustrated that PTEN mRNA nanoparticles can reverse the immunosuppressive TME by promoting CD8+ T cell infiltration of the tumor tissue, enhancing the expression of proinflammatory cytokines, such as interleukin-12, tumor necrosis factor–α, and interferon-γ, and reducing regulatory T cells and myeloid-derived suppressor cells. The combination of PTEN mRNA nanoparticles with an immune checkpoint inhibitor, anti–programmed death–1 antibody, results in a highly potent antitumor effect in a subcutaneous model of Pten-mutated melanoma and an orthotopic model of Pten-null prostate cancer. Moreover, the combinatorial treatment elicits immunological memory in the Pten-null prostate cancer model.
INTRODUCTION
Since ipilimumab, an antibody that targets cytotoxic T lymphocyte–associated protein 4 (CTLA-4), became the first immune checkpoint blockade (ICB) therapy approved by the U.S. Food and Drug Administration for treatment of metastatic melanoma, a myriad of inhibitors against immune checkpoints such as programmed cell death 1 (PD-1), PD ligand 1 (PD-L1), and CTLA-4 have been developed. Some have shown promising therapeutic effects in various cancers, such as melanoma, non–small cell lung carcinoma (NSCLC), and renal cell carcinoma (RCC) (1). However, it has also been demonstrated that less than 30% of patients with NSCLC, RCC, or melanoma benefit from CTLA-4 or PD-1/PD-L1 inhibitors (2), and patients with other cancer types have shown poor response or resistance to ICB therapy (2, 3). Loss or mutation of the gene-encoding phosphatase and tensin homolog deleted on chromosome 10 (PTEN), a widely studied tumor suppressor gene, has been documented in different human cancers including prostate cancer, melanoma, glioblastoma, and others (4). Recent clinical studies demonstrated that PTEN was directly involved in the regulation of antitumor immunity. For example, loss of PTEN was markedly associated with reduced T cell infiltration at tumor sites and poor response or resistance to PD-1 blockade therapy (5–8). The loss of PTEN also contributed to the accumulation of suppressive immune cells, such as myeloid-derived suppressor cells (MDSCs) and regulatory T (Treg) cells, as well as the formation of an immunosuppressive microenvironment during tumor initiation and progression (9–11). In addition, it has also been confirmed that the expression of PTEN induced autophagy, whereas the loss of PTEN function down-regulated autophagy to effectively support cancer development (12, 13). In the context of tumor cell death, autophagy may cause the secretion of damage-associated molecular patterns (DAMPs) (14, 15), which include “find me” signals such as adenosine triphosphate (ATP), “danger” signals such as chromatin-associated high-mobility group box 1 (HMGB1) and heat-shock proteins (HSPs), and the exposure of “eat me” signals such as calreticulin (CRT) on the plasma membrane (16). DAMPs have been reported to be powerful immunological adjuvants that trigger antitumor immunity activation in many cancer therapies (17). In addition, dead cancer cells may also release autophagosomes loaded with multiple tumor antigens, which subsequently induce dendritic cell (DC) maturation and cross-presentation to T cells (18, 19). Despite these clinical and preclinical discoveries, it is not yet clear whether restoration of PTEN expression in PTEN-null or mutated tumor cells could reverse the immunosuppressive tumor microenvironment (TME) and trigger an antitumor immune response by inducing the activation of autophagy and release of DAMPs to improve ICB therapy.
Synthetic messenger RNA (mRNA) has recently shown promise in biomedical applications such as protein replacement, gene editing, and vaccination development (20–29). Unlike plasmid DNA, mRNA does not require nuclear envelope entry for effective transfection and thus has a negligible chance of integrating into the host’s genome. mRNA also provides more consistent and predictable protein expression kinetics than DNA therapeutics (30). Recently, we successfully developed a lipid-polymer hybrid nanoparticle (NP) platform for systemic delivery of PTEN mRNA to prostate cancer tumors (20). The restoration of PTEN expression by mRNA NPs markedly inhibited growth of human prostate cancer cells both in vitro and in vivo. To explore whether the tumor cell death induced by PTEN reactivation would be accompanied by the release of DAMPs that can trigger antitumor immune activation, we here developed a polymeric NP platform for PTEN mRNA delivery to several Pten-null or mutated murine tumor cells including PTEN-Cap8 and B16F10. The new PTEN mRNA NPs are self-assembled by methoxy poly(ethylene glycol)–poly(lactic-co-glycolic acid) (mPEG-PLGA) copolymers, in which the complexes of mRNA with cationic lipid–like material 1,2-epoxytetradecane–modified generation 0 polyamidoamine (G0-C14) (31) are loaded. Our results showed that these PTEN mRNA NPs not only restored the susceptibility of tumor cells to death but also led to the release of DAMPs and the activation of autophagy, which could promote the secretion of additional DAMPs and autophagosomes. In vivo results revealed that PTEN restoration elicited robust CD8+ T cell responses and reversed the immunosuppressive microenvironment by reducing the expression of Treg and monocytic MDSCs (Mo-MDSCs) and increasing the expression of proinflammatory cytokines. Furthermore, we evaluated the antitumor efficacy of the combination of PTEN mRNA NPs with anti–PD-1 immunotherapy in several Pten-null or mutated tumor models, demonstrating marked therapeutic effects and immunological memory of this combinatorial strategy. Our findings suggest that tumor suppressor restoration by mRNA nanomedicines may improve the sensitivity of ICB therapy and provide a potent combination treatment for multiple malignancies.
RESULTS
Preparation and characterization of polymeric NPs for PTEN mRNA delivery to tumor cells
We prepared a new polymeric NP platform (fig. S1) composed of mPEG-PLGA (figs. S2 and S3) and a cationic lipid–like material, G0-C14 (figs. S4 and S5), that we previously reported (31). The mPEG-PLGA copolymers are capable of self-assembly into NPs as a carrier (fig. S6), and the cationic G0-C14 was used for complexation with PTEN mRNA, which was then encapsulated into the core of the mPEG-PLGA NPs (mPTEN@NPs). An agarose gel retardation assay was performed to examine mRNA encapsulation, showing minimal mRNA leaching from the NPs, occurring at the G0-C14/mRNA mass ratio of 25 (fig. S7), and supporting its use in the mRNA NP formulation. The morphology and hydrodynamic diameter of mPTEN@NPs were assessed using transmission electron microscopy (TEM) and dynamic light scattering (DLS), respectively. The spherical structure of the mPTEN@NPs could be observed using TEM (Fig. 1A), and the hydrodynamic size of mPTEN@NPs was found to be 111.8 ± 15.3 nm (Fig. 1B). In addition, the stability of the mPTEN@NPs in a physiological environment was confirmed using long-duration monitoring of their size in phosphate-buffered saline (PBS) or cell culture medium (Fig. 1C). We also confirmed the ability of the NPs for protecting the mRNA from ribonuclease (RNase) degradation (fig. S8).
Fig. 1. Characterization of PTEN mRNA NPs (mPTEN@NPs) and expression of the tumor suppressor PTEN by treatment with mPTEN@NPs in vitro.
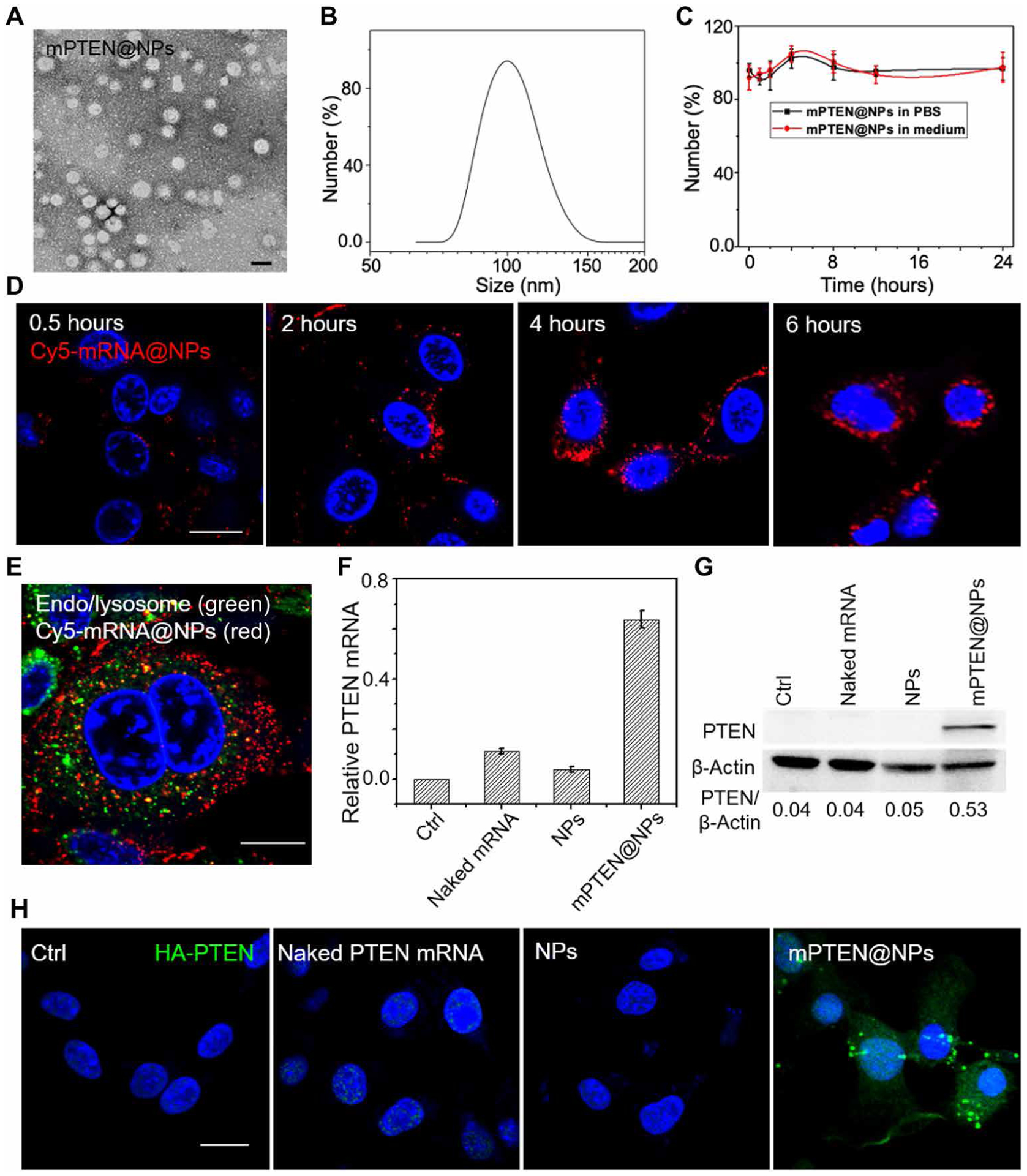
(A) TEM image of mPTEN@NPs. Scale bar, 100 nm. (B) The size distribution of mPTEN@NPs detected by DLS. (C) The size of mPTEN@NPs in PBS or medium did not change during evaluation over 24 hours. Data shown are representative of three independent experiments. (D) Cellular uptake of Cy5-mRNA@NPs (red) was characterized at different time points. (E) Cy5-mRNA@NPs (red) escape from endo/lysosomes (green). Nucleus were stained with DAPI (blue) before evaluated by confocal microscopy. (F) RT-PCR for PTEN mRNA in PTEN-Cap8 cells after indicated treatments for 48 hours. Cells without treatment (Ctrl) served as the background. Data shown are representative of three independent experiments. (G) Western blot analysis of PTEN expression in PTEN-Cap8 cells after the indicated treatments for 48 hours. Bottom: The quantitative analysis results for PTEN expression that were calculated by normalizing PTEN protein band intensity at each condition to that of β-actin by ImageJ software. (H) Immunofluorescence imaging of hemagglutinin (HA)–tagged PTEN (green) expression in B16F10 cells treated with naked PTEN mRNA, NPs, or mPTEN@NPs for 48 hours. Cells without treatment served as the control (Ctrl). Scale bars, 20 μm.
Next, we examined the intracellular uptake and trafficking of the mRNA NPs in Pten-null prostate cancer cells (PTEN-Cap8) by confocal laser scanning microscopy (CLSM). A Cy5-labeled–enhanced green fluorescent protein (EGFP) mRNA was used as a model mRNA and was loaded into the polymeric NPs (denoted as Cy5-mRNA@NPs). PTEN-Cap8 cells were incubated with Cy5-mRNA@NPs for 0.5, 2, 4, or 6 hours. The intracellular red signal from Cy5-mRNA was directly proportional to incubation time (Fig. 1D), indicating that the NPs could efficiently deliver mRNA into tumor cells. Our previous studies demonstrated that NPs containing G0-C14 had a proton-sponge effect, which may partially contribute to the cytosolic release of mRNA cargos (20, 29). Thus, we found that most of the red signal from Cy5-mRNA was not colocalized with the green signal from the endosomes or lysosomes when incubated for 6 hours (Fig. 1E and fig. S9), suggesting effective escape of the mRNA NPs into the cytoplasm. The transfection efficacy of the EGFP mRNA NPs in PTEN-Cap8 cells is shown in fig. S10.
To further study whether mutated or deleted functional genes could be restored by mRNA NPs, PTEN expression in PTEN-Cap8 and B16F10 cells was investigated via Western blot, real-time polymerase chain reaction (RT-PCR), and immunofluorescence imaging. Cells were incubated with PBS, naked PTEN mRNA, control NPs, or mPTEN@NPs for 48 hours. The mPTEN@NPs increased PTEN protein expression in PTEN-Cap8 cells (Fig. 1, F and G). For Pten-mutated B16F10 cells, PTEN expression was measured by immunofluorescence imaging for hemagglutinin (HA), which was tagged to PTEN mRNA for easy separation from endogenous PTEN. Immunofluorescence microscopy showed high expression of HA-PTEN protein after treatment with the mPTEN@NPs (Fig. 1H). Together, these findings demonstrated that our polymeric NPs effectively delivered PTEN mRNA to tumor cells for the restoration of PTEN expression.
PTEN restoration by mPTEN@NPs induces autophagy and immunogenic cell death
It has previously been reported that restoration of PTEN expression could inhibit growth of human cancer cells (20, 32, 33). Thus, we first wanted to determine whether treatment with mPTEN@NPs could also reduce viability of murine PTEN-Cap8 and B16F10 cells. These tumor cells were treated with PBS, naked PTEN mRNA, control NPs, or mPTEN@NPs at different doses for 48 hours, and the resulting cell growth inhibition was measured (Fig. 2A). Naked PTEN mRNA showed negligible toxicity against all cancer cells even at high concentration (1 μg/ml). Control NPs also showed limited cytotoxicity at the highest dose. However, after treatment with mPTEN@NPs, more dead cells were detected, and the half-maximal inhibitory concentration value was about 0.6 μg/ml for PTEN-Cap8 and about 0.7 μg/ml for B16F10. The morphology of PTEN-Cap8 cells was examined using conventional microscopic imaging after mPTEN@NP treatment, revealing increases in the number of spherical structures and cells experiencing shrinkage (fig. S11A). The results of an annexin V–fluorescein isothiocyanate (FITC)/propidium iodide (PI) assay confirmed that mPTEN@NPs treatment effectively promoted tumor cell apoptosis (fig. S11B). We also studied the expression of pro–caspase-3, cleaved caspase-3, and phospho–receptor-interacting protein kinase 1 (RIPK1)/RIPK3/mixed-lineage kinase domain–like (MLKL) axis on PTEN-Cap8 cells via Western blot. mPTEN@NPs did not induce a noticeable change in phospho-RIPK1/RIPK3/MLKL but promoted an increase of cleaved caspase-3 abundance (fig. S12).
Fig. 2. mPTEN@NP treatment induces ICD of cancer cells in vitro.
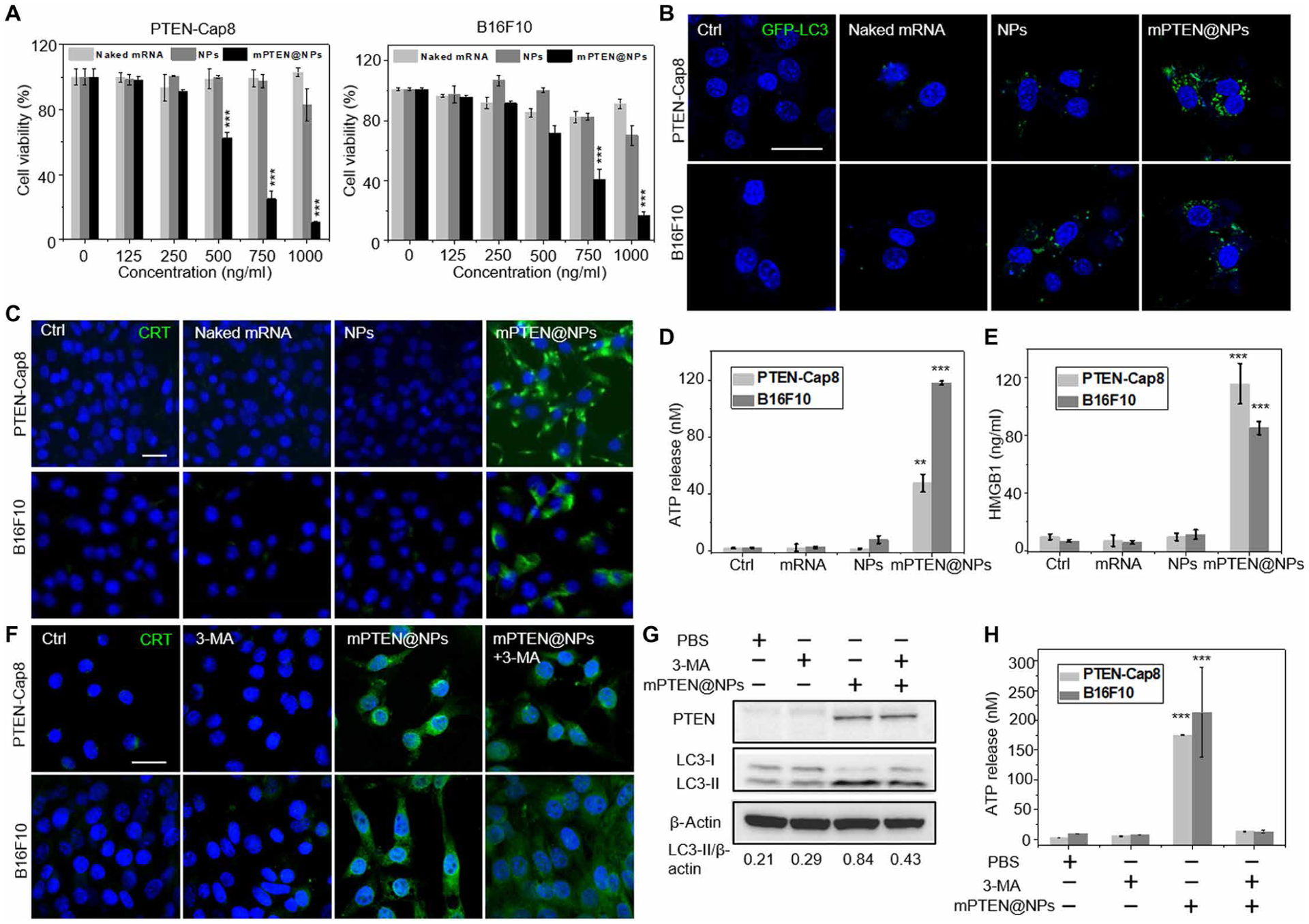
(A) Cell viability of PTEN-Cap8 and B16F10 cells after treatment with mPTEN@NPs for 48 hours. Data are presented as means ± SD (n = 3 replicates). Statistical significance was calculated using a two-tailed Student’s t test, ***P < 0.001. (B) Confocal laser scanning microscopy (CLSM) imaging for cells transfected with GFP-LC3. (C to E) Analysis of ICD markers in PTEN-Cap8 and B16F10 cells after mPTEN@NPs treatment for 48 hours. (C) CRT expression on PTEN-Cap8 and B16F10 cells was evaluated by CLSM. ATP release (D) and HMGB1 release (E) were evaluated by ELISA. (F) CRT expression was assessed by CLSM on PTEN-Cap8 and B16F10 cells that were cotreated with mPTEN@NPs and the autophagy inhibitor, 3-MA. (G) Western blot analysis for PTEN and LC3-II expressions when cotreated with mPTEN@NPs and 3-MA for 48 hours. Bottom: The quantitative analysis results for LC3-II expression that performed on LC3-II protein bands intensities at each condition were normalized with β-actin. (H) ATP release after treatment with mPTEN@NPs and 3-MA for 48 hours was evaluated by ELISA. Data in (D), (E), and (H) are presented as means ± SD (n = 3 replicates per group) and were calculated via one-way ANOVA with a Tukey post hoc test. **P < 0.01 and ***P < 0.001. Scale bars, 20 μm.
To further explore the molecular mechanisms of PTEN restoration in promoting tumor cell death, we performed phospho-array analysis for proteins from PTEN-Cap8 cells treated with PBS or mPTEN@NPs. Protein change ratios and phosphorylation ratios at 157 specific phosphorylation sites implicated in 16 signaling pathways were calculated to determine the global effects of mPTEN@NP treatment. Altered phosphorylated proteins were enriched in phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) signaling, mitogen-activated protein kinase signaling, and apoptosis pathways, indicating that PTEN restoration had a marked regulatory effect on cell growth and metabolism, ultimately contributing to cell death (fig. S13). We also found increases in several autophagy-specific proteins such as Beclin1 and HSPs such as HSP90B, which have shown potent immunostimulatory activity when exposed on the plasma membrane of dying cells (34).
It has been demonstrated that PTEN activation can lead to the induction of autophagy in many cancer cells (35, 36). To confirm the autophagy-inducing effects of mPTEN@NPs, a Western blot was used to measure the expression of autophagic marker proteins light chain 3-II (LC3-II) and p62. Naked PTEN mRNA treatment elicited no LC3-II increase, whereas the control NPs induced slight accumulation of LC3-II (fig. S14), which could be explained by the fact that various NPs could induce autophagy as reported previously (37–39). However, mPTEN@NPs treatment markedly increased LC3-II expression. mPTEN@NPs treatment also caused accumulation of p62, suggesting that mPTEN@NPs induced autophagosome formation and could result in their intracellular accumulation. Next, GFP-LC3–transfected PTEN-Cap8 and B16F10 cells were exposed to either PBS, naked PTEN mRNA, control NPs, or mPTEN@NPs for 48 hours before being imaged by CLSM. There were more GFP-LC3 dots in the tumor cells treated with mPTEN@NPs than in those treated with naked PTEN mRNA or the control NPs, further suggesting that mPTEN@NPs effectively induce autophagosome accumulation (Fig. 2B and fig. S15) (13).
The induction of autophagy could also promote the release of DAMPs and autophagosomes loaded with tumor antigens (16), triggering the antigen cross-presentation process and antitumor immune responses (40). We thus hypothesized that mPTEN@NPs might also induce DAMPs and tumor antigen release to elicit immunogenic cell death (ICD). To confirm this, three hallmark markers of ICD (41), CRT on the cell surface and release of either HMGB1 or ATP into the extracellular environment, were measured in PTEN-Cap8 and B16F10 cells after treatment with mPTEN@NPs for 48 hours. The surface exposure of CRT on cells was confirmed by immunofluorescence via CLSM or flow cytometry. Treatment with mPTEN@NPs induced CRT exposure on the plasma membrane (Fig. 2C and fig. S16). The release of HMGB1 or ATP in the cell culture supernatant was measured by an enzyme-linked immunosorbent assay (ELISA). Compared to controls, there was a remarkable increase in HMGB1 release and ATP secretion for PTEN-Cap8 and B16F10 cells treated with mPTEN@NPs (Fig. 2, D and E). In addition, ICD-related cytokines, such as interferon-α (IFN-α) and IFN-β, monocyte chemoattractant protein-1, and IFN-γ–induced protein-10 (also known as CXCL10), were shown to increase (fig. S17). We further determined ICD induction in another Pten-null mouse prostate cancer cell line (BMPC) generated from sites of metastasis of a Pten-deleted and MYC-overexpressing transgenic tumor model (42). Results showed that mPTEN@NPs (250 ng/ml) induced >50% BMPC cell death (fig. S18) and promoted DAMP release (fig. S19). To further investigate the relationship between autophagy and DAMP release, we measured the CRT expression, LC3-II expression, and ATP secretion by cotreating the tumor cells with an upstream autophagy inhibitor, 3-methyladenine (3-MA), and mPTEN@NPs. The results showed slightly lower CRT expression (Fig. 2F and fig. S20) and LC3-II expression (Fig. 2G) by tumor cells after cotreatment versus mPTEN@NPs treatment alone and a reduction of ATP release (Fig. 2H). In parallel, autophagy was inhibited by knocking down Beclin1 (BECN1), a central regulator of autophagy in mammalian cells, which resulted in a decrease in mPTEN@NPs-induced ATP and a slight decrease in CRT expression (fig. S21). Cell viability results further confirmed that 3-MA or siBECN1 prevented mPTEN@NPs-induced ICD, whereas the mechanistic target of rapamycin (mTOR) inhibitor rapamycin, an autophagy inducer, promoted cell death (figs. S22 and S23). These results demonstrated that mPTEN@NPs triggered ICD, at least partially, through the autophagy-mediated pathway.
Antitumor immune responses are induced by mPTEN@NPs in vivo
The ability of mPTEN@NPs to induce ICD of tumor cells in vitro inspired us to further explore whether PTEN restoration could activate antitumor immune responses in vivo. First, we constructed a subcutaneous B16F10 tumor model using C57BL/6 mice and measured the biodistribution of Cy5-mRNA@NPs. Cy5-mRNA@NPs or naked Cy5-mRNA, which was used as a control, were administered to mice through the lateral tail vein. After 24 hours, the tumor and major organs (heart, liver, spleen, kidneys, and lungs) were harvested and imaged. Mice treated with Cy5-mRNA@NPs showed higher fluorescence (fig. S24), suggesting that considerable Cy5-mRNA@NPs had accumulated in the tumor site. Similar biodistribution results in the BMPC genetically engineered mouse model (GEMM) of prostate cancer further confirmed NP accumulation in tumors (fig. S24). In addition, the increased tumor tissue expression of PTEN in the BMPC mice 48 hours after injection of mPTEN@NPs indicated successful delivery of PTEN mRNA to the tumor (fig. S25). The in vivo side effects of the mRNA NPs were assessed by hematological and histopathological analyses in C57BL/6 mice. Blood serum and major organs were harvested 48 hours after the last injection. We analyzed serum alanine aminotransferase and aspartate aminotransferase to assess liver function, and blood urea nitrogen to monitor kidney activity. These parameters were in the normal range after treatment with mPTEN@NPs (table S1). The results of hematoxylin and eosin (H&E) staining for major organs also showed no obvious histological differences between treatment with PBS and mPTEN@NPs (fig. S26).
Next, we evaluated antitumor immune responses using B16F10 melanoma tumor–bearing mice after mPTEN@NPs treatment. First, 4 × 105 B16F10 cells were implanted subcutaneously on the right flank of the mice to establish subcutaneous tumors. When the average tumor size had increased to ~60 mm3, the tumor-bearing mice were then randomly divided into three groups and treated with saline, control NPs, or mPTEN@NPs via tail vein injection with an mRNA dose of 700 μg/kg body weight at days 7, 10, and 13 after tumor cell implantation (Fig. 3A). At day 15 (2 days after the last injection), all the mice were euthanized and tumors and lymph nodes were harvested to assess the number and phenotype of immune cells and changes in secreted ATP, HMGB1, and cytokines. According to tumor weight, H&E staining, and terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) staining, mPTEN@NPs promoted tumor cell apoptosis and suppressed tumor growth (Fig. 3B and fig. S27). We also analyzed various stimulatory molecules expressed on lymph node–resident DCs (LNDCs) after treatment with mPTEN@NPs. The expression of various stimulatory markers including CD80, CD86, major histocompatibility complex I (MHC-I), and MHC-II on LNDCs were up-regulated after three cycles of treatment (Fig. 3C and figs. S28 and S29). Mature DCs are thought to induce antitumor immunity by engulfing and presenting tumor antigens to T cells. The percentage of CD3+CD8+ T cells within the tumor was increased (Fig. 3D and figs. S30 and S31). In addition, CD8+ effector T cells [CD8+IFN-γ+ or CD8+T-bet+ (T-box expressed in T cells)] in the tumor were also increased compared to saline-treated animals, as assessed by flow cytometry (Fig. 3E and fig. S32) and immunofluorescence staining of tumor tissues (Fig. 3F). Treg and MDSCs play important roles in tumor immune evasion (9), and their accumulation at the tumor site produces an immunosuppressive TME. Flow cytometry results revealed that mPTEN@NPs increased the frequency of type 1 T helper (CD4+IFN-γ+) cells but decreased both Treg (Foxp3+CD25+CD4+) and Mo-MDSC (CD11b+Ly6C+Ly6G−) frequency (Fig. 3, G and H, and figs. S33 to S35), indicating that mPTEN@NPs reversed the immunosuppressive TME. A comparison of cytokine release profiles (Fig. 3, I to K, and fig. S36) further suggests that mPTEN@NPs trigger antitumor immune activation and reverse the immunosuppressive TME.
Fig. 3. mPTEN@NPs induce antitumor immune responses in the Pten-mutated B16F10 tumor–bearing mouse model.
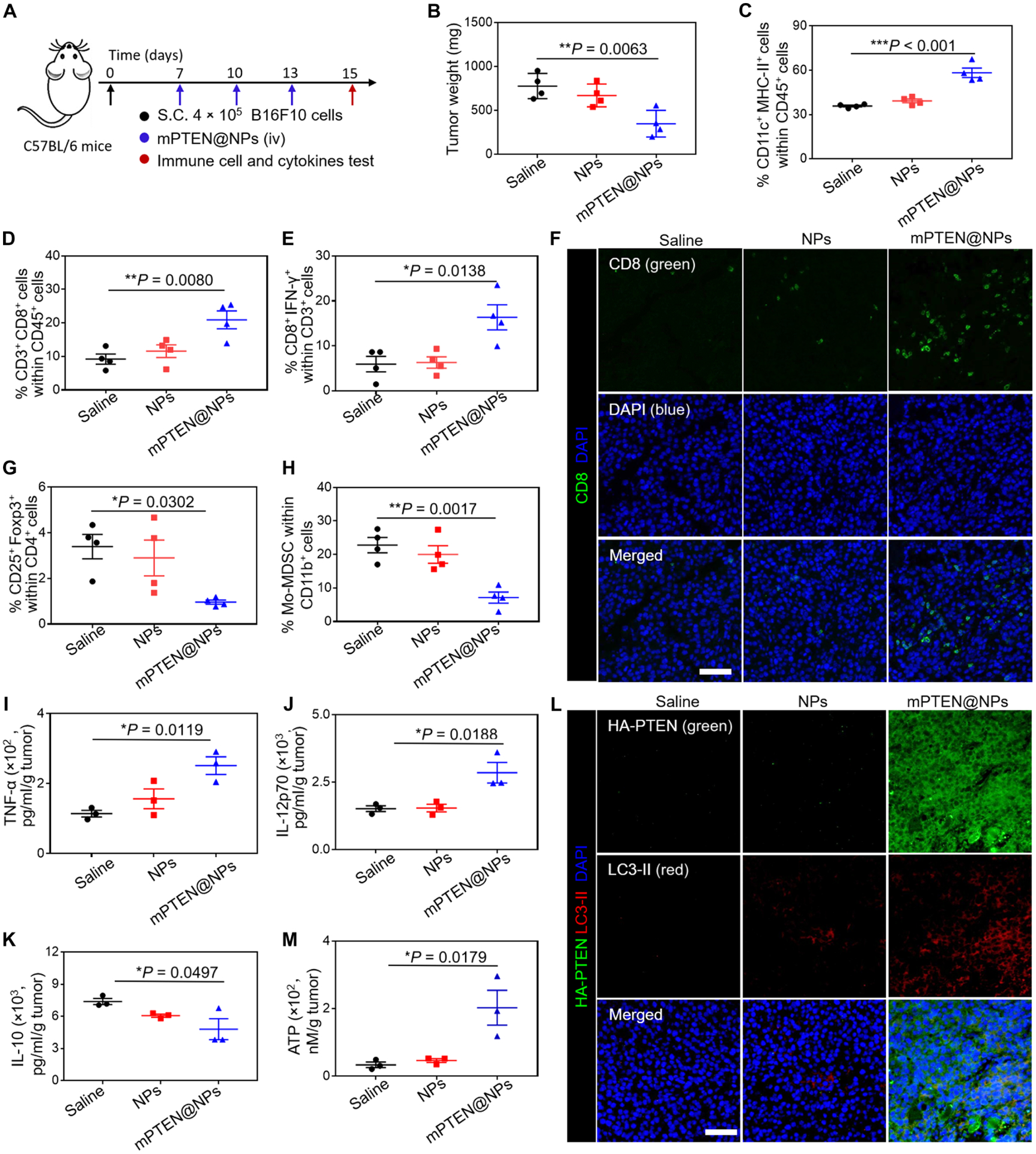
(A) Experimental timeline for treatment of B16F10 tumor–bearing mice. S.C., subcutaneous; iv, intravenous. (B) Tumor weights of B16F10 tumor–bearing mice treated with PTEN@NPs. Data are presented as means ± SD (n = 4 mice per group). (C to E) Flow cytometry analysis results of the percentage of CD11c+MHC-II+ LNDCs (C), and the percentage of CD3+CD8+ T cells (D) and IFN-γ+CD8+ T cells (E) isolated from the tumor. Data are presented as means ± SEM (n = 4 mice per group). (F) Immunofluorescence imaging from Pten-mutated B16F10 tumor tissues shows CD8+ T cell infiltration (green) after treatment with saline, NPs, or mPTEN@NPs. DAPI (blue) stains nuclei. Scale bars, 50 μm. (G and H) Flow cytometry analysis result of the percentage of Foxp3+CD25+CD4+ T cells (G) and Mo-MDSCs (H) gating on CD11b+Ly6C+Ly6G− cells. Data are presented as means ± SEM (n = 4 mice per group). (I to K) ELISA analysis results of cytokine in the supernatant of excised tumors from mice (n = 3 mice per group), including TNF-α (I), IL-12p70 (J), and IL-10 (K). (L) Immunofluorescence imaging of PTEN (green) and LC3-II (red) expression in Pten-mutated B16F10 tumor tissues after the indicated treatments. (M) Analysis of ATP release in the supernatant of excised tumors from mice treated with saline, NPs, or mPTEN@NPs (n = 3 mice per group). Statistical significance was calculated via one-way ANOVA with a Tukey post hoc test. *P < 0.05, **P < 0.01, and ***P < 0.001.
We further tested DAMPs release in vivo using an immunofluorescent method to detect the expression of HA-PTEN and CRT in tumor tissues. As compared to the control group, immunofluorescence from HA-PTEN was increased in the mPTEN@NPs treatment group (Fig. 3L). This means that mPTEN@NPs effectively delivered PTEN mRNA to the tumor site for PTEN restoration in vivo. We also observed LC3-II expression in tumors isolated from mice in the mPTEN@NP-treated group, suggesting that PTEN reactivation induced autophagy. In addition, quantification of ATP release in the tumor tissues by ELISA showed an increase after treatment of mice with mPTEN@NPs (Fig. 3M). In parallel, immunofluorescence staining revealed high CRT expression on tumors isolated from mice treated with mPTEN@NPs (fig. S37). These results collectively demonstrated that PTEN restoration via mPTEN@NPs effectively induce autophagy and the release of DAMPs in vivo.
mPTEN@NPs improve the antitumor efficacy of anti–PD-1 in a subcutaneous mouse model of Pten-mutated melanoma
Previous studies demonstrated that loss of PTEN led to a poor response to anti–PD-1 therapy in patients with melanoma or uterine leiomyosarcoma (6, 43, 44). Therefore, we explored whether the activation of antitumor immunity by PTEN restoration would improve the therapeutic efficacy of ICB in a Pten-mutated melanoma model. First, wild-type C57BL/6 mice were inoculated subcutaneously with ~4 × 105 B16F10 tumor cells on the right limb and then treated intravenously on days 4, 7, and 10 with saline, control NPs, or mPTEN@NPs (700 μg/kg body weight, 200 μl). Some mPTEN@NPs-treated mice also received intraperitoneal administration of anti–PD-1 (100 μg per mouse, 100 μl) on days 5, 8, and 11 (Fig. 4A). The combination therapy achieved greater antitumor efficacy after three cycles of treatment, in comparison to either mPTEN@NPs (P < 0.05) or anti–PD-1 alone (P < 0.01) (Fig. 4, B and C). Survival after three cycles of treatment of mPTEN@NPs + anti–PD-1 was also longer than that of the single-agent groups (fig. S38). Meanwhile, the combination of mPTEN@NPs with anti–PD-1 did not induce body weight loss (fig. S38). Although the combination of chemotherapeutic doxorubicin (Dox) with anti–PD-1 exhibited potential in inhibiting tumor growth, toxicity from Dox led to an acute body weight loss as well as splenic contraction and loss of spleen mass (fig. S39).
Fig. 4. mPTEN@NPs increase the therapeutic efficacy of anti–PD-1 in the Pten-mutated B16F10 tumor–bearing mouse model.

(A) Experimental timeline for treatment of B16F10 tumor–bearing mice. (B) Individual growth curves for mice treated as indicated. (C) The average tumor growth curves for mice treated as indicated. Data are represented as means ± SD (n = 7 mice per group). Statistical significance was calculated in (C) using a one-way ANOVA with a Tukey post hoc test. *P < 0.05, **P < 0.01, and ***P < 0.001. (D) Immunofluorescence imaging of tumor tissues showing expression of HA-PTEN (green) and LC3-II (red) after treatment with mPTEN@NPs with or without anti–PD-1. (E) Immunofluorescence staining of tumors for CD8+ T cell infiltration (green) after treatment with saline, NPs, or mPTEN@NPs. (F) Immunofluorescence staining of tumors for CRT expression (red) after indicated treatments. (G and H) ELISA analysis of ICD markers HMGB1 (G) and ATP (H) in the supernatant of tumors excised from mice treated as indicated. Data are presented as means ± SD (n = 3 mice per group). Statistical significance was calculated in (G) and (H) via one-way ANOVA with a Tukey post hoc test. *P < 0.05 and **P < 0.01. Scale bars, 50 μm.
To explore the cellular basis of the improved efficacy, immunofluorescence staining for HA-PTEN and LC3-II was performed, and the results indicated PTEN restoration and autophagy induction after combination treatment (Fig. 4D and fig. S40). The frequency of tumor-infiltrating CD8+ T cells was increased in the combination group compared to the anti–PD-1 monotherapy group (Fig. 4E and fig. S41). The flow cytometry results also demonstrated that the combination treatment led to an increase in the frequency of tumor-infiltrating CD3+ T cells, especially cytotoxic T cells (CD3+CD8+ T cells), compared to that of the saline group (fig. S42). The increase in frequency of activated LNDCs (CD80+CD86+) in the combination treatment group further confirmed the induction of an antitumor immune response (fig. S43). In addition, the percentage of CD11b+Gr-1+ MDSCs was lower in the combination treatment group compared to the anti–PD-1 group (fig. S44), suggesting that the immunosuppressive TME was improved. Quantification of cytokines and chemokines including interleukins (IL-6, IL-10, and IL-12p70), tumor necrosis factors (TNF-α and TNF-β), and IFN-γ also confirmed the improvement in the immunosuppressive TME by mPTEN@NPs with anti–PD-1 (fig. S45).
To further investigate the effect of mPTEN@NPs on antitumor immune modulation, especially the infiltration of CD8 T cells, we constructed experiments to evaluate the antitumor efficacy of mPTEN@NPs plus anti–PD-1 on CD8 T cell–depleted immunocompetent mice. Three days before receiving mPTEN@NPs and anti–PD-1, each mouse received 500 μg of anti-CD8α, which was repeated once a week thereafter. CD8+ effector T cells were effectively depleted in vivo (figs. S46 and S47), which led to reduced antitumor efficacy of mPTEN@NPs in combination with anti–PD-1. The depletion of CD8 T cells had no effect on mPTEN@NPs-mediated activation of LNDCs (fig. S48). In addition, the antitumor effect of combination treatment with anti-CD8α is comparable to that found in the group that received mPTEN@NPs alone (Fig. 4, B and C, and fig. S47). Collectively, these results suggest that, although mPTEN@NPs have direct antitumor effect, they can also induce cytotoxic T cell responses that are important for overall antitumor efficacy for the combination of mPTEN@NPs with anti–PD-1.
We also measured the ICD biomarkers of tumors. Compared to anti–PD-1 treatment, CRT expression (Fig. 4F and fig. S49), as well as the release of both HMGB1 and ATP (Fig. 4, G and H), was increased by the combination treatment. All the above results suggest that mPTEN@NPs not only can effectively trigger antitumor immune responses but also can enhance the therapeutic efficacy of anti–PD-1 therapy in Pten-mutated tumors.
mPTEN@NPs confer sensitivity to anti–PD-1 in an orthotopic mouse model of Pten-null prostate cancer
We further evaluated this combination strategy for treatment of Pten-null tumors that exhibited poor response or were intrinsically resistant to ICB therapy (5, 6, 45). Prostate cancer is one of the leading causes of cancer death for men, and both anti–PD-1 and anti–CTLA-4 monotherapy have shown limited benefits for overall survival of patients with prostate cancer (2, 45). We established an orthotopic prostate cancer model, wherein 1 × 106 PTEN-Cap8-Luc prostate cancer cells were surgically inoculated into the prostates of male C57BL/6 mice. After confirming the successful establishment of the tumor model on day 10 by in vivo bioluminescence imaging, three cycles of treatment with mPTEN@NPs in combination with anti–PD-1 were administered (Fig. 5A). Tumor imaging was performed every 5 days from initial treatment (day 10 after tumor inoculation) until day 25 (Fig. 5B). On the basis of the change in bioluminescence of PTEN-Cap8-Luc cells, it was determined that anti–PD-1 alone inhibited tumor growth very little. In comparison, the combination treatment of mPTEN@NPs with anti–PD-1 markedly slowed the change in bioluminescence signals, suggesting that mPTEN@NPs restored tumor sensitivity to anti–PD-1 (Fig. 5C). Quantitative analysis of the bioluminescence signal demonstrated an about fourfold decrease at day 25 in the combination treatment group versus anti–PD-1 alone (Fig. 5D). We also evaluated PTEN restoration and autophagy induction in the combination treatment group using immunofluorescence staining to measure HA-PTEN and LC3-II expression in tumors. Both PTEN and LC3-II signals were detected in the combination treatment group, indicating that mPTEN@NPs effectively delivered PTEN mRNA to the tumors and restored PTEN function (Fig. 5E). TUNEL staining indicated that PTEN restoration by mPTEN@NPs induced a degree of tumor cell death (fig. S50). We further evaluated tumor tissue isolated from mice bearing orthotopic prostate tumors by immunofluorescence for presence of HMGB1 and CRT. Both ICD markers were elevated after combination treatment with mPTEN@NPs and anti–PD-1, indicating that mPTEN@NPs markedly promoted tumor ICD (fig. S51). CD8 immunofluorescence imaging demonstrated an increase in CD8+ T cell infiltration into tumor tissue after combination treatment with mPTEN@NPs and anti–PD-1 (Fig. 5, F and G). Collectively, these results suggest that mPTEN@NPs improved the sensitivity of anti–PD-1 in Pten-null orthotopic prostate cancer model by improving the immunosuppressive TME and inducing ICD. Moreover, in the BMPC model of prostate cancer, the results of an increase in CD8+ T cell infiltration (fig. S52) and a decrease in CD11b+Gr-1+ MDSCs after mPTEN@NPs treatment (fig. S53) further suggest that PTEN restoration could effectively improve the immunosuppressive TME and elicit antitumor immune responses.
Fig. 5. Therapeutic efficacy and immunological memory of mPTEN@NPs with anti–PD-1 in the orthotopic mouse model of Pten-null prostate cancer.
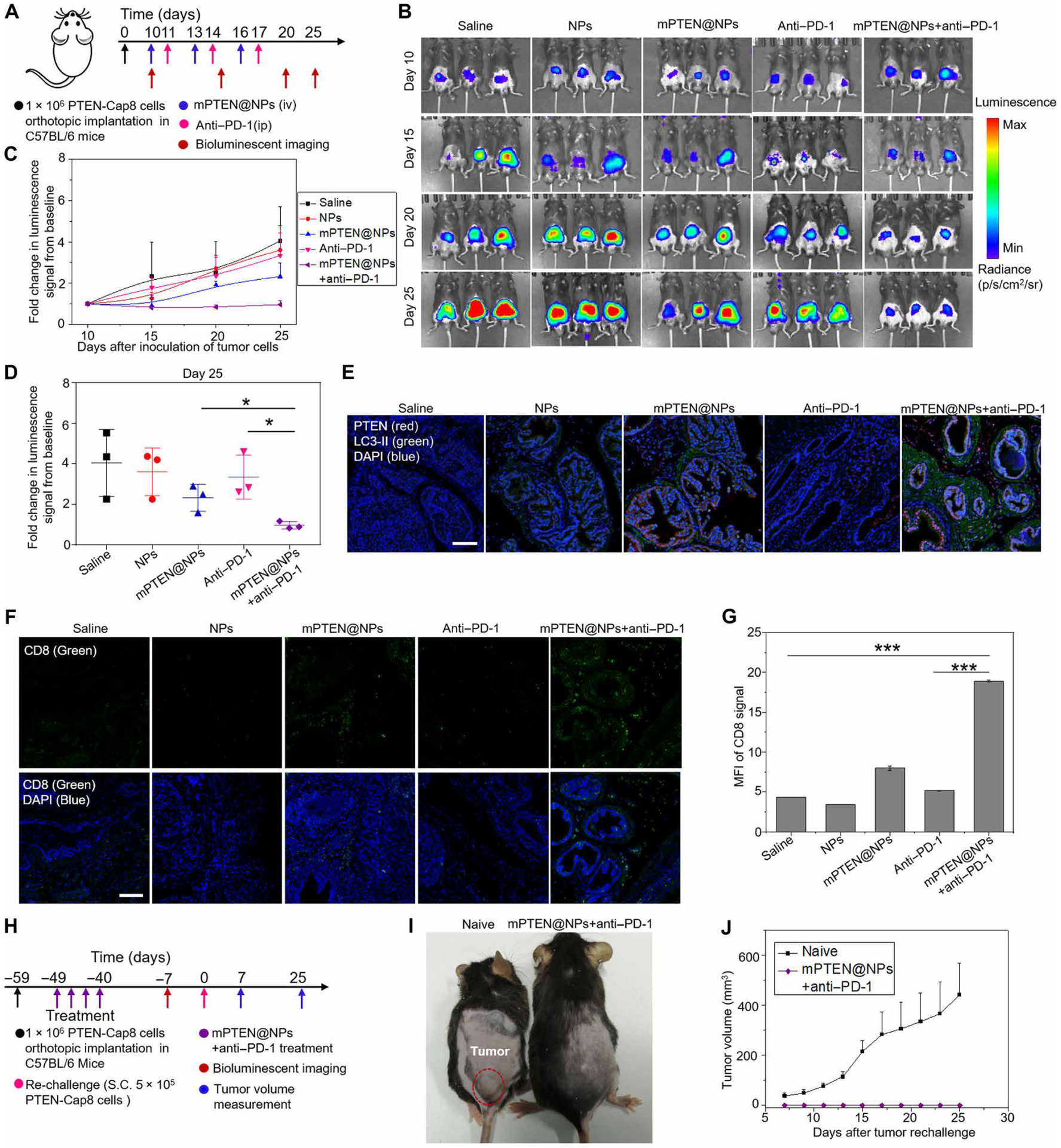
(A) Experimental timeline for treatment of Pten-null orthotopic prostate tumor–bearing mice. (B) In vivo bioluminescence imaging of Pten-null orthotopic prostate tumors from mice treated as indicated. Tumor imaging was obtained every 5 days from the initial treatment day (day 10 after tumor inoculation) until day 25. (C) The fold change in bioluminescence signals from baseline at day 10 of PTEN-Cap8-Luc tumors. (D) Quantitative analysis of the fold change in bioluminescence signals from baseline of PTEN-Cap8-Luc tumors at day 25. Data in (C) and (D) are presented as means ± SD (n = 3 mice per group). Statistical significance was calculated via one-way ANOVA with a Tukey post hoc test. *P < 0.05. (E) Immunofluorescence staining of tumors for PTEN (red) and LC3-II (green) expression at day 25 after the indicated treatments. Scale bar, 100 μm. (F) Immunofluorescence staining of tumors for CD8+ T cell infiltration (green) at day 25 after the indicated treatments. Scale bar, 100 μm. (G) Quantitative analysis of mean fluorescent intensity from (F). Data are presented as means ± SD (n = 3 mice per group). Statistical significance was calculated via one-way ANOVA with a Tukey post hoc test. ***P < 0.001. (H) Experimental timeline for treatment of Pten-null orthotopic prostate tumor–bearing mice and S.C. rechallenge. In the naive group, C57BL/6 mice were subcutaneously implanted with PTEN-Cap8 cells at day 0 without any pretreatment. (I) Representative photograph of mice from the naive group and the combination treatment group at day 25. Note that the mice in the naive group are about 7 to 8 weeks younger than those in the combination treatment group. (J) The subcutaneous tumor growth profile for the naive group and the combination treatment group (mPTEN@NPs + anti–PD-1). Data are presented as means ± SEM (n = 4 mice per group).
To address whether mPTEN@NPs + anti–PD-1 treatment elicits immunological memory to prostate cancer, mice from the orthotopic tumor model that showed complete responses to the combination treatment were rechallenged by subcutaneous injection of 5 × 105 PTEN-Cap8 cells (Fig. 5H). As a comparison, a naïve cohort of C57BL/6 mice that were 7 to 8 weeks younger than the rechallenge cohort were also subcutaneously implanted with the same number of PTEN-Cap8 cells at day 0. Tumor growth in both groups was monitored via tumor size measurements taken every other day starting on day 7. The mice pretreated with mPTEN@NPs + anti–PD-1 showed complete rejection of the rechallenged tumor cells, whereas the PTEN-Cap8 tumor grew rapidly in the naïve group (Fig. 5, I and J). These results indicate that combination therapy of mPTEN@NPs with anti–PD-1 induced immunologic memory.
DISCUSSION
The recent success of immune checkpoint inhibitors such as anti–CTLA-4 and anti–PD-1/PD-L1 as first-line therapies of choice for treatment of cancers including NSCLC and melanoma has revealed immunotherapy as a powerful treatment strategy (46). However, because of insufficient tumor immunogenicity and the immunosuppressive TME, ICB immunotherapy also suffers from a limited rate of antitumor response for many cancers, including prostate cancer (3, 47). One strategy to improve antitumor responses to ICB therapy is through combination with traditional therapies (48), such as chemotherapy (49, 50), radiotherapy (51), and photodynamic therapy (PDT) (52, 53), which could also cause ICD of tumor cells, and thereafter initiate antitumor immunity (54–56). However, chemotherapy and radiotherapy have severe side effects, and a major drawback of PDT is the limited tissue penetration of light. Thus, there is a pressing need to develop additional strategies to elicit antitumor immune responses effectively and safely that synergize with ICB therapy.
Mutation or loss of some tumor suppressor genes in cancer cells, such as PTEN and TET2, resulted in poor response or resistance to ICB therapy in mouse models (5, 57). Small-molecule inhibitors, such as PI3K inhibitors, have demonstrated a potential role in inducing partial restoration of tumor suppressor function by targeting their upstream or downstream regulators (58). However, PI3K inhibitors showed severe side effects and could not fully restore PTEN function. Given the advantages of synthetic mRNA for protein replacement (59, 60), the use of synthetic mRNA might restore the tumor suppressor’s functions without inducing substantial toxicity. In parallel, autophagy is a very important cellular “housekeeping” process for the degradation of various misfolded proteins or cytoplasmic structures, including damaged organelles and engulfed pathogens. Recent evidence has shown that autophagy not only acts as a cell-protective mechanism against internal or external stresses but also played crucial roles in stimulating immune activation (61, 62). Induction of autophagy in cancer cells could also promote the secretion of DAMPs (14, 15) and release of tumor-associated antigens (16). Autophagy-deficient mice fail to increase ATP release and cannot elicit antitumor immunity activation (63). PTEN has been shown to be a key regulator of the PI3K/Akt/mTOR signaling pathway, which in turn negatively regulates autophagy. Cumulatively, these observations suggest that reactivation of PTEN by mRNA NPs would elicit robust and safe antitumor immune responses by inducing tumor cell autophagy and release of DAMPs, thereby triggering tumor ICD and sensitizing cancers to ICB therapy.
In this work, we developed a polymeric NP platform for the in vivo delivery of PTEN mRNA to tumor cells, including Pten-null prostate cancer cells and Pten-mutated melanoma cells. Our results demonstrated that mPTEN@NPs successfully triggered antitumor immune responses by inducing autophagy activation and DAMP release. Moreover, PTEN reactivation diminished the immunosuppressive tumor environment and improved the sensitivity of Pten-null or mutated tumors to ICB therapy. We compared the antitumor efficacy of mPTEN@NPs alone, anti–PD-1 alone, and their combination in mouse models of Pten-mutated melanoma and Pten-deleted prostate cancer and validated the potent antitumor effect of the combinatorial strategy.
It should be noted that this study is limited to the PTEN tumor suppressor and it is not clear whether restoration of other tumor suppressors might have similar effects. More efforts will also be required to further explore the mutational status of tumor suppressors and their role in the immunosuppressive TME. Previous studies have also suggested that PTEN may play roles in the regulation of immune cells (64, 65). Thus, it would be interesting to explore the effects of mPTEN@NPs on the proliferation and functions of immune cells, such as T cells, in future studies. Moreover, clinical results regarding the effects of loss or mutation of different tumor suppressors on therapeutic outcomes of ICB or other immunotherapies remain very limited.
Overall, our study provides a robust and potent strategy for eliciting antitumor immune responses that may be useful against different cancer types having PTEN loss or mutation, and we expect that the combination of mRNA nanomedicine with ICB therapy could lead to development of tumor suppressor pathway–specific precision immunotherapy for effective and safe cancer treatment.
MATERIALS AND METHODS
Study design
The aim of our study was to explore whether and how the mRNA-based restoration of tumor suppressor PTEN could reverse the poor-response/resistance of PTEN-null or mutated cancers to ICB therapy. We first developed a polymeric NP to deliver PTEN mRNA and then evaluated its transfection efficacy and antitumor effects in Pten-mutated melanoma cells and Pten-null prostate cancer cells using viability assays, RT-PCR, as well as immunofluorescence and confocal imaging. For mechanistic studies, the induction of ICD and autophagy of tumor cells in vitro (n = 3 replicates per group) and changes in the number/phenotype of immune cells in vivo (n = 3 or 4 mice per group) were analyzed by flow cytometry, immunofluorescence imaging, and ELISA. To further evaluate the impact of PTEN restoration on ICB treatment, we used the subcutaneous mouse model of Pten-mutated melanoma and the orthotopic mouse model of Pten-null prostate cancer to analyze the antitumor efficacy of PTEN mRNA NPs alone, anti–PD-1 alone, and their combination (n = 3 to 7 mice per group). The animals were randomly assigned to treatment groups without blinding.
Synthesis of mPEG-PLGA copolymer
Synthesis of a block copolymer containing mPEG-PLGA with disulfide bond was performed by a straightforward three-step polymerization strategy. First, PLGA diols were prepared by the ring-opening polymerization of lactide (LA) and glycolide (GA) initiated by bis(2-hydroxyethyl) disulfide (BHD) in the presence of a stannous octoate (SnOct2) catalyst. Typically, 2.88 g of LA and 2.32 g of GA were added to a dried flask, and then 100 ml of anhydrous toluene was added into the flask to dissolve the monomer. After that, the initiator BHD (0.154 g) and catalyst SnOct2 (5%) were added to initiate the polymerization at 120°C. After reaction for 24 hours, the solution was precipitated in diethyl ether. The crude product was redissolved in dichloride methylene and precipitated in dimethyl ether again. PLGA was dried under vacuum and obtained as a white solid. Second, the prepolymer PLGA reacted with the chain extenders 1,6-Dichlorhexane and BHD at 70°C for 0.5 hours. Last, mPEG (molecular weight: 5000) was used to terminate the polymerization at 70°C for 6 hours. The final mPEG-PLGA was obtained by precipitation in diethyl ether, two dissolve-precipitation cycles in dichloride methylene-diethyl ether, and ultimately drying in a vacuum.
Preparation of polymeric NPs for PTEN mRNA delivery
We used a self-assembly method as previously described to prepare PTEN mRNA–encapsulating polymeric NPs (20). In brief, the copolymer of mPEG-PLGA and cationic molecule G0-C14 was first dissolved in 250 μl of dimethylformamide solution at a concentration of 4 mg/ml and 1 mg/ml, respectively. Then, 10 μg of mRNA (PTEN mRNA, EGFP mRNA, or Cy5-labeled EGFP mRNA) was added into the above mPEG-PLGA/G0-C14 solution. The mixture was then quickly added dropwise to the DNA/RNase-free pure water (5.0 ml) under stirring (1000 rpm). After that, the mRNA-loaded NPs were formed instantly and were kept for 30 min under 1000 rpm stirring at room temperature. The formed NPs were collected and washed three times with DNA/RNase-free pure water using an Amicon Ultra-15 centrifugal filter (molecular weight cutoff of 100 kDa, Millipore) to remove the organic solvent and free compounds. The NPs were finally dispersed in 200 μl of fresh PBS and stored at −80°C for later use in both in vitro and in vivo studies.
Physicochemical characterization of mRNA-encapsulated NPs and RNA loading efficiency study
The hydrodynamic diameter and morphological structure of mRNA-encapsulated NPs were determined by DLS (Brookhaven Instruments Corporation) and TEM [Tecnai G2 Spirit BioTWIN (FEI Company)], respectively. For TEM, the NPs were first stained with 1% uranyl acetate for 1 min and washed with pure water. To check the long-term in vitro stability of mRNA-encapsulated NPs, the NPs were incubated in PBS and cell culture medium solution at 37°C, and their hydrodynamic diameters were measured at various points (0, 2, 4, 8, 12, 24, and 48 hours). To measure the mRNA complexation ability of cationic G0-C14 and its loading efficiency, naked EGFP mRNA and EGFP mRNA complexed with different doses of G0-C14 (ratio of weight from 3.2 to 25) were run through an E-Gel 2% agarose (Invitrogen) gel for 30 min at 70 V. Last, the gel was imaged under ultraviolet (UV) light and the bands were analyzed using ImageJ software.
Cellular uptake and lysosomal escape of mRNA-encapsulated NPs
To monitor the NPs’ uptake, Cy5-mRNA@NPs were prepared. Pten-null prostate cancer cells (PTEN-Cap8 cells) were first seeded in confocal wells (Costar) at a density of 3 × 105 cells per well and incubated at 37°C in 5% CO2 for 15 hours. The cells were then incubated with medium containing Cy5-mRNA@NPs for 0.5, 2, 4, or 6 hours, respectively. The cells were then washed with PBS and analyzed using an Olympus microscope. For the detection of colocalization of Cy5-labeled mRNA with lysosomes, the cells were first treated with Cy5-labeled mRNA-encapsulated NPs for 4 or 6 hours and then stained with LysoTracker green DND-26 (Invitrogen).
In vitro cytotoxicity and apoptosis evaluation
Cancer cells were seeded in 96-well plates at a density of 3 to 5 × 104 cells per well and incubated with 100 μl of medium containing 10% fetal bovine serum (FBS, Gibco) for 15 hours. Then, the old medium was removed, and 100 μl of fresh medium containing PBS, naked PTEN mRNA, control NPS, and mPTEN@NPs at different concentrations were added and incubated for 48 hours. After washing with PBS, fresh medium containing 10% volume of alamarBlue (Thermo Fisher Scientific) was added to each well. After a 2-hour incubation, the cells were measured with a microplate reader (TECAN, Infinite M200 Pro). The measurements of UV-visible absorptions of all samples were made at 450 and 690 nm (background). Cell viability (%) was calculated using the formula: (Asample − Ablank)/(Acontrol − Ablank) × 100.
Apoptosis was also detected using annexin V–FITC/PI double-staining assay by flow cytometry. Briefly, the cells were seeded in six-well plates and then treated with PBS, naked PTEN mRNA, control NPs, or mPTEN@NPs for 48 hours. The treated cells were collected, washed with PBS, and resuspended in PBS with annexin V–FITC and PI solution at room temperature and then were analyzed by flow cytometry within 1 hour.
Western blot
Cancer cells were first seeded in a six-well plate at a density of 3 to 5 × 105 cells per well. After 15 hours, the cells were treated with 100 μl of medium containing PBS, naked PTEN mRNA, control NPs, or mPTEN@NPs for 48 hours and then collected in a 1.5-ml tube and resuspended with 100 μl of lysis buffer [50 mM tris-HCl (pH 8.0), 150 mM NaCl, and 1% (v/v) Triton X-100 and protease inhibitor]. After the protein concentration determined by a bicinchoninic acid protein assay kit (Thermo Fisher Scientific), 60 μg of the samples were run through an SDS–polyacrylamide gel electrophoresis gel and then transferred to nitrocellulose membrane blots. Next, the membrane blots were incubated with blocking buffer [5% (w/v) nonfat milk, 0.1% (v/v) Tween 20 in 0.01 M Tris-Buffered Saline and Tween (50 mM tris-HCl at pH 7.4 and 150 mM NaCl, and 0.1% Tween 20)] for 1 hour and then incubated overnight with appropriate primary antibodies at 1:1000 dilution at 4°C. The membrane blot was washed with PBS three times and incubated with horseradish peroxidase–conjugated secondary antibodies at 1:4000 dilution at room temperature for 2 hours. Last, protein signals were detected by enhanced chemiluminescence detection system (Amersham/GE Healthcare) using a Typhoon Trio Variable Mode Imager. All uncropped Western blot images are shown in figs. S54 to S58.
Cell immunofluorescent staining
Cells were seeded into confocal plates at a density of 3 × 105 cells per well and incubated with 100 μl of medium containing 10% FBS. When the cells had grown to 60 to 70% confluence, 100 μl of medium containing PBS, naked PTEN mRNA, control NPs, or mPTEN@NPs was added into each well. After 48 hours, the cells were washed with PBS and fixed with 4% paraformaldehyde for 15 min at room temperature. Next, the cells were permeabilized with 0.1% Triton X-100 PBS for 10 min and cultured with a blocking buffer containing 5% bovine serum albumin (BSA) for 1 hour at room temperature. Then, the cell samples were incubated overnight with primary antibodies, such as HA and CRT at 1:150 dilution at 4°C, washed with PBS three times, and incubated in secondary antibody (1:250) with Alexa Fluor 488 for 1 hour at room temperature. Last, the cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) (Thermo Fisher Scientific), washed with PBS, and imaged using an Olympus CLSM (FV1000). For CRT expression analysis by flow cytometry, the cells were collected after treatment and stained with Alexa Fluor 488–labeled CRT antibody (1:50) for 1 hour at 4°C, washed with PBS three times, and stained with PI. Then, the cells were analyzed by flow cytometry (FACScalibur; BD Biosciences).
Animals
C57BL/6 female and male mice about 6 weeks old were purchased from the Jackson laboratory and used for therapeutic efficacy and antitumor immune response studies. Pten-null orthotopic tumor model studies were performed in the animal facility of National Center for Nanoscience and Technology (NCNST). The BMPC mice were created in the animal facility of the University of Maryland Baltimore County. Other animal studies were performed in the animal facility of the Brigham and Women’s Hospital. All animal studies were performed under specific pathogen-free conditions and in accordance with National Institutes of Health animal care guidelines. The animal protocol was approved by the Institutional Animal Care and Use Committees at NCNST, University of Maryland Baltimore County, or Brigham and Women’s Hospital. The animals were kept in the standard laboratory conditions with 12-hour light/12-hour dark cycle, with temperature of 18° to 23°C, and with relative humidity of 40 to 60% and had free access to sterile water and food.
BMPC mice
The GEMM of prostate cancer was created as in previous study (42). Briefly, mice carrying hemizygous Hoxb13-Cre+/− were mated to mice with PtenFl/Fl (the Jackson laboratory) to generate offspring with Hoxb13-Cre+/−/PtenFl/+. These mice were interbred to generate Hoxb13-Cre+/−/PtenFl/Fl mice. Next, mice carrying Hoxb13-MYC were mated to Hoxb13-Cre+/−/PtenFl/Fl mice to generate offspring with heterozygous Hoxb13-MYC+/−/Hoxb13-Cre+/−/PtenFl/+. These mice were then interbred to generate triple transgenic mice with Hoxb13-MYC+/−/Hoxb13-Cre+/−/PtenFl/Fl genotype, referred to here as BMPC mice. When prostate tumors were detectable and had grown to about 0.5 g, the mice were used for biodistribution and antitumor immune response studies. For the biodistribution study, BMPC mice received naked Cy5-mEGFP and Cy5-mRNA@NPs (200 μl) via tail vein injection at an mRNA dose of 15 μg per animal. Twenty-four hours later, the organs (heart, liver, spleen, lung, and kidney) and tumors were harvested and imaged using the Syngene PXi Imaging System. For the antitumor immune response study, the BMPC mice were randomly divided into two groups (n = 3 mice per group) and treated with 200 μl of saline or mPTEN@NPs via tail vein injection at an mRNA dose of 700 μg/kg body weight every 3 days. After three cycles of treatment and 2 days after the last injection, the mice were sacrificed for the harvesting of their tumors to examine the PTEN expression and number/phenotype of immune cells such as CD8 T cells and MDSCs.
Antitumor immune responses induced by PTEN mRNA NPs
To prepare the B16F10-bearing tumor mouse model, about 4 × 105 cells in 100 μl of PBS were implanted subcutaneously on the right flank of 6-week-old female C57BL/6 mice. The mice were randomly divided into three groups (n = 4 mice per group), which received 200 μl of saline, control NPs, or mPTEN@NPs via tail vein at an mRNA dose of 700 μg/kg of body weight at days 7, 10, and 13 after tumor implantation. At day 15 (2 days after the last injection), the mice were euthanized and the tumors and groin lymph nodes were isolated to examine the number and phenotype of immune cells, such as T cells, DCs, and MDSCs, and concentration of secreted cytokines.
Flow cytometry for immune cells and antibodies
Antibodies for flow cytometry analysis were purchased from BioLegend and listed in table S2. Tumors and lymph nodes were washed with 1 ml of ice-cold fluorescence-activated cell sorting buffer (PBS plus 1% FBS) in a 1.5-ml microcentrifuge tube and then homogenized with a pellet pestle motor (Kontes Glass). Homogenates were then passed through a 70-μm mesh nylon cell strainer (BD Falcon) to form single-cell suspensions. For the flow cytometry analysis of surface markers, the cells were stained on ice with fluorescence-conjugated antibodies for 30 min according to the manufacturer’s instructions. For the staining of intracellular markers, such as IFN-γ, the cells were prestimulated with the cell stimulation cocktail (eBioscience) at 37°C for 6 hours and fixed and permeabilized using the intracellular staining kit (BD Biosciences) at 4°C for 30 min. Then, the cells were stained with anti–IFN-γ and other surface antibodies at 4°C for 30 min. Stained cells were finally evaluated by flow cytometry (FACScalibur; BD Biosciences) and analyzed using FlowJo software.
ATP, HMGB1, and cytokine detection
To evaluate the release of ATP and HMGB1 from tumor cells, the cells were first seeded in a six-well plate at a density of 5 × 105 cells per well and incubated with 1.5 ml of medium containing 10% FBS for 15 hours. The medium was removed, and 1.5 ml of fresh medium containing PBS, naked PTEN mRNA, control NPs, or mPTEN@NPs was added and incubated for 48 hours. The cell culture medium was collected and measured using ATP (Invitrogen) and HMGB1 (IBL International GmbH) ELISA kits according to the manufacturer’s instructions. For evaluation of intratumoral ATP, HMGB1, and cytokines such as IL-10, IL-12p70, TNF-α, IFN-γ, IL-6, and TNF-β, tumor tissues were harvested and homogenized in cold PBS buffer. The supernatant from tumor homogenates was then measured with ELISA kits (cytokine kits purchased from BioLegend) per the manufacturer’s instructions.
Immunofluorescence and H&E staining for tissues
At the end point of treatment, the mice were euthanized; the tumors and various organs (lung, heart, liver, kidney, and spleen) were harvested and fixed with 4% paraformaldehyde. All organs were embedded in paraffin and sectioned into slices at a thickness of 5 μm. The paraffin-embedded sections were deparaffinized, rehydrated in a graded ethanol series, and washed in distilled water. Sample sections were then incubated in 0.3% hydrogen peroxide for 20 min to quench the activity of endogenous peroxidase, followed by antigen retrieval in citrate buffer (10 mM, pH 6) for 30 min. After washing with PBS, the samples were incubated in blocking buffer (1% BSA and 5% normal goat serum) for 60 min. For immunofluorescence staining, samples were incubated with different primary rabbit antibodies (HA, LC3, CRT, and CD8) at a 1:50 dilution overnight at 4°C, washed with PBS, and incubated with fluorescently labeled secondary antibodies (1:1000) for 60 min at room temperature. For H&E staining, sections were stained using an assay kit (Vector Laboratories) according to the manufacturers’ protocols. Last, the slides were imaged using a confocal microscope (Olympus FluoView FV1000).
In vivo therapeutic efficacy of mPTEN@NPs + anti–PD-1 in the B16F10-bearing tumor mouse model
For in vivo therapeutic efficacy, a B16F10-bearing tumor mouse model was used as described above. Six-week-old female C57BL/6 mice were randomly divided into five groups (n = 7 per group). The groups received an injection of saline, control NPs, or mPTEN@NPs (200 μl per mouse) via tail vein at an mRNA dose of 700 μg/kg of body weight at days 4, 7, and 10 after tumor implantation, and one cohort of mice receiving mPTEN@NPs was supplemented with intraperitoneal administration of anti–PD-1 (100 μg in 100 μl saline per mouse) on days 5, 8, and 11. Tumor sizes were measured every 2 days, and the average tumor volume was calculated according to the following formula: ½(length × width × width). To investigate the effect of mPTEN@NPs on antitumor immune modulation in the CD8 T cell–depleted mice, 6-week-old C57BL/6 mice bearing B16F10 tumors were randomly divided into two groups (n = 10 per group). Three days before receiving mPTEN@NPs and anti–PD-1, each mouse was injected intraperitoneally with 500 μg of anti-CD8α in 200 μl of saline, repeated once a week thereafter. All mice received an injection of mPTEN@NPs (in 200 μl of saline) via tail vein at an mRNA dose of 700 μg/kg of body weight at days 4, 7, and 10 after tumor implantation, and anti–PD-1 (100 μg in 100 μl of saline per mouse) was given intraperitoneally on days 5, 8, and 11. The tumors were measured every 2 days, and the average tumor volume was calculated according to the following formula: ½(length × width × width).
In vivo therapeutic efficacy of mPTEN@NPs + anti–PD-1 in an orthotopic mouse model of prostate cancer
To assess the in vivo therapeutic efficacy of mPTEN@NPs + anti–PD-1 in an orthotopic mouse model of prostate tumor, the orthotopic prostate tumor mouse model was developed. In brief, 6-week-old male C57BL/6 mice were anesthetized with isoflurane (1 to 3% for maintenance via nose cone). Next, 1 × 106 PTEN-Cap8-Luc cells in 15 μl of PBS were added into equal volumes of matrigel to create a final concentration of 1 × 106 cells/30 μl and then slowly injected into the anterior prostate lobes. Carefully, the injected prostate lobe was returned to the abdomen, and the inner and outer abdominal skins were closed. Ten days after implantation, the mice were administered 100 μl luciferin at a concentration of 3 mg/ml intraperitoneally and were monitored within 10 min for tumor using an in vivo imaging system (PerkinElmer). For treatment, mice with confirmed tumors were randomly divided into five groups (n = 3 per group) and treated with 200 μl of saline, control NPs, or mPTEN@NPs via tail vein at an mRNA dose of 700 μg per kg of animal weight at days 10, 13, and 16 after tumor implantation, and some of the mPTEN@NPs-treated mice were also treated with intraperitoneal administration of anti–PD-1 (100 μg in 100 μl saline per mouse) on days 11, 14, and 17. Tumor imaging was performed every 5 days from the initial treatment day (day 10 after tumor inoculation) until day 25. Regions of interest were quantified as total flux radiance (photons per second) using an in vivo imaging system (PerkinElmer).
For the rechallenge study, C57BL/6 mice with orthotopic prostate cancer were first treated with four cycles of mPTEN@NPs with anti–PD-1 as mentioned above. Thirty-three days after the final treatment (or day −7 in Fig. 5H), orthotopic tumor imaging was performed, and four mice with complete response to the combination treatment as evaluated by undetectable bioluminescence signals were chosen to be rechallenged by subcutaneous injection of 5 × 105 PTEN-Cap8 cells. Naive C57BL/6 mice were used as controls and subcutaneously injected with the same number of PTEN-Cap8 cells at day 0 without any pretreatment. The sizes of the subcutaneous tumors were measured using a caliper every other day, from days 7 to 25, and the tumor volume was calculated according to the following formula: ½(length × width × width).
Statistical analysis
All experiments were performed in triplicate unless otherwise noted specifically, and all results are presented as the means ± SD or means ± SEM. A Student’s t test or Mann-Whitney test was used for two-group comparisons, and a one-way analysis of variance (ANOVA) with Tukey’s correction for multiple comparisons was used to compare more than two groups. All statistical analyses were carried out using GraphPad Prism 7 software, and statistical significance is indicated as *P < 0.05, **P < 0.01, and ***P < 0.001. n.s. indicates no significant difference.
Supplementary Material
Acknowledgments:
We thank the Harvard Medical School Neurobiology Imaging Facility (P30 NS072030) for confocal imaging services.
Funding:
This work was supported, in part, by the U.S. National Institutes of Health (NIH) grant CA200900.
Footnotes
Competing interests: A patent application entitled “Restoration of tumor suppression using mRNA-based delivery system” (application number 16/780, 458) related to this study has been filed. O.C.F. declares financial interests in Selecta Biosciences, Tarveda Therapeutics, Seer, Dynamics Special Purpose Corp., and PrognomIQ. All other authors declare that they have no competing interests.
Data and materials availability: All data associated with this study are present in the paper or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
stm.sciencemag.org/cgi/content/full/13/599/eaba9772/DC1
Materials and Methods
Figs. S1 to S58
Tables S1 and S2
Data file S1
View/request a protocol for this paper from Bio-protocol.
REFERENCES AND NOTES
- 1.Ribas A, Wolchok JD, Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park Y-J, Kuen D-S, Chun Y, Future prospects of immune checkpoint blockade in cancer: From response prediction to overcoming resistance. Exp. Mol. Med 50, 1–13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jenkins RW, Barbie DA, Flaherty KT, Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 118, 9–16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Milella M, Falcone I, Conciatori F, Incani UC, Del Curatolo A, Inzerilli N, Nuzzo CMA, Vaccaro V, Vari S, Cognetti F, Ciuffreda L, PTEN: Multiple functions in human malignant tumors. Front. Oncol 5, 24 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, Xu C, McKenzie JA, Zhang C, Liang X, Williams LJ, Deng W, Chen G, Mbofung R, Lazar AJ, Torres-Cabala CA, Cooper ZA, Chen P-L, Tieu TN, Spranger S, Yu X, Bernatchez C, Forget M-A, Haymaker C, Amaria R, McQuade JL, Glitza IC, Cascone T, Li HS, Kwong LN, Heffernan TP, Hu JH, Bassett RL Jr., Bosenberg MW, Woodman SE, Overwijk WW, Lizée G, Roszik J, Gajewski TF, Wargo JA, Gershenwald JE, Radvanyi L, Davies MA, Hwu P, Loss of PTEN promotes resistance to T cell–mediated immunotherapy. Cancer Discov. 6, 202–216 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.George S, Miao D, Demetri GD, Adeegbe D, Rodig SJ, Shukla S, Lipschitz M, Amin-Mansour A, Raut CP, Carter SL, Hammerman P, Freeman GJ, Wu CJ, Ott PA, Wong KK, Van Allen EM, Loss of PTEN is associated with resistance to anti-PD-1 checkpoint blockade therapy in metastatic uterine leiomyosarcoma. Immunity 46, 197–204 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barroso-Sousa R, Keenan TE, Pernas S, Exman P, Jain E, Garrido-Castro AC, Hughes M, Bychkovsky B, Umeton R, Files JL, Lindeman NI, MacConaill LE, Hodi FS, Krop IE, Dillon D, Winer EP, Wagle N, Lin NU, Mittendorf EA, Van Allen EM, Tolaney SM, Tumor mutational burden and PTEN alterations as molecular correlates of response to PD-1/L1 blockade in metastatic triple-negative breast cancer. Clin. Cancer Res 26, 2565–2572 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roh W, Chen P-L, Reuben A, Spencer CN, Prieto PA, Miller JP, Gopalakrishnan V, Wang F, Cooper ZA, Reddy SM, Gumbs C, Little L, Chang Q, Chen W-S, Wani K, De Macedo MP, Chen E, Austin-Breneman JL, Jiang H, Roszik J, Tetzlaff MT, Davies MA, Gershenwald JE, Tawbi H, Lazar AJ, Hwu P, Hwu W-J, Diab A, Glitza IC, Patel SP, Woodman SE, Amaria RN, Prieto VG, Hu J, Sharma P, Allison JP, Chin L, Zhang J, Wargo JA, Futreal PA, Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci. Transl. Med 9, eaah3560 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feng S, Cheng X, Zhang L, Lu X, Chaudhary S, Teng R, Frederickson C, Champion MM, Zhao R, Cheng L, Gong Y, Deng H, Lu X, Myeloid-derived suppressor cells inhibit T cell activation through nitrating LCK in mouse cancers. Proc. Natl. Acad. Sci 115, 10094–10099 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang R, Cai T-T, Wu X-J, Liu Y-N, He J, Zhang X-S, Ma G, Li J, Tumour YAP1 and PTEN expression correlates with tumour-associated myeloid suppressor cell expansion and reduced survival in colorectal cancer. Immunology 155, 263–272 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma MD, Shinde R, McGaha TL, Huang L, Holmgaard RB, Wolchok JD, Mautino MR, Celis E, Sharpe AH, Francisco LM, Powell JD, Yagita H, Mellor AL, Blazar BR, Munn DH, The PTEN pathway in Tregs is a critical driver of the suppressive tumor microenvironment. Sci. Adv 1, e1500845 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P, Ogier-Denis E, The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem 276, 35243–35246 (2001). [DOI] [PubMed] [Google Scholar]
- 13.Gozuacik D, Kimchi A, Autophagy as a cell death and tumor suppressor mechanism. Oncogene 23, 2891–2906 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Martins I, Ma Y, Kepp O, Galluzzi L, Kroemer G, Autophagy-dependent ATP release from dying cells via lysosomal exocytosis. Autophagy 9, 1624–1625 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, Shen S, Kepp O, Scoazec M, Mignot G, Rello-Varona S, Tailler M, Menger L, Vacchelli E, Galluzzi L, Ghiringhelli F, di Virgilio F, Zitvogel L, Kroemer G, Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 334, 1573–1577 (2011). [DOI] [PubMed] [Google Scholar]
- 16.a Y, Galluzzi L, Zitvogel L, Kroemer G, Autophagy and cellular immune responses. Immunity 39, 211–227 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P, Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 12, 860–875 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Su H, Luo Q, Xie H, Huang X, Ni Y, Mou Y, Hu Q, Therapeutic antitumor efficacy of tumor-derived autophagosome (DRibble) vaccine on head and neck cancer. Int. J. Nanomedicine 10, 1921–1930 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Viry E, Paggetti J, Baginska J, Mgrditchian T, Berchem G, Moussay E, Janji B, Autophagy: An adaptive metabolic response to stress shaping the antitumor immunity. Biochem. Pharmacol 92, 31–42 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Islam MA, Xu Y, Tao W, Ubellacker JM, Lim M, Aum D, Lee GY, Zhou K, Zope H, Yu M, Cao W, Oswald JT, Dinarvand M, Mahmoudi M, Langer R, Kantoff PW, Farokhzad OC, Zetter BR, Shi J, Restoration of tumour-growth suppression in vivo via systemic nanoparticle-mediated delivery of PTEN mRNA. Nat. Biomed. Eng 2, 850–864 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu L, Wang Y, Miao L, Liu Q, Musetti S, Li J, Huang L, Combination immunotherapy of MUC1 mRNA nano-vaccine and CTLA-4 blockade effectively inhibits growth of triple negative breast cancer. Mol. Ther 26, 45–55 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller JB, Zhang S, Kos P, Xiong H, Zhou K, Perelman SS, Zhu H, Siegwart DJ, Non-viral CRISPR/Cas gene editing in vitro and in vivo enabled by synthetic nanoparticle co-delivery of Cas9 mRNA and sgRNA. Angew. Chem. Int. Ed 56, 1059–1063 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moffett HF, Coon ME, Radtke S, Stephan SB, McKnight L, Lambert A, Stoddard BL, Kiem HP, Stephan MT, Hit-and-run programming of therapeutic cytoreagents using mRNA nanocarriers. Nat. Commun 8, 389 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schlake T, Thess A, Thran M, Jordan I, mRNA as novel technology for passive immunotherapy. Cell. Mol. Life Sci 76, 301–328 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hou X, Zhang X, Zhao W, Zeng C, Deng B, McComb DW, Du S, Zhang C, Li W, Dong Y, Vitamin lipid nanoparticles enable adoptive macrophage transfer for the treatment of multidrug-resistant bacterial sepsis. Nat. Nanotechnol 15, 41–46 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li B, Zhao W, Luo X, Zhang X, Li C, Zeng C, Dong Y, Engineering CRISPR–Cpf1 crRNAs and mRNAs to maximize genome editing efficiency. Nat. Biomed. Eng 1, 0066 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miao L, Li L, Huang Y, Delcassian D, Chahal J, Han J, Shi Y, Sadtler K, Gao W, Lin J, Doloff JC, Langer R, Anderson DG, Delivery of mRNA vaccines with heterocyclic lipids increases anti-tumor efficacy by STING-mediated immune cell activation. Nat. Biotech 37, 1174–1185 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Reinhard K, Rengstl B, Oehm P, Michel K, Billmeier A, Hayduk N, Klein O, Kuna K, Ouchan Y, Wöll S, Christ E, Weber D, Suchan M, Bukur T, Birtel M, Jahndel V, Mroz K, Hobohm K, Kranz L, Diken M, Kühlcke K, Türeci Ö, Sahin U, An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science 367, 446–453 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Kong N, Tao W, Ling X, Wang J, Xiao Y, Shi S, Ji X, Shajii A, Gan ST, Kim NY, Duda DG, Xie T, Farokhzad OC, Shi J, Synthetic mRNA nanoparticle-mediated restoration of p53 tumor suppressor sensitizes p53-deficient cancers to mTOR inhibition. Sci. Transl. Med 11, eaaw1565 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiong Q, Lee GY, Ding J, Li W, Shi J, Biomedical applications of mRNA nanomedicine. Nano Res. 11, 5281–5309 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu X, Xie K, Zhang X-Q, Pridgen EM, Park GY, Cui DS, Shi J, Wu J, Kantoff PW, Lippard SJ, Langer R, Walker GC, Farokhzad OC, Enhancing tumor cell response to chemotherapy through nanoparticle-mediated codelivery of siRNA and cisplatin prodrug. Proc. Natl. Acad. Sci 110, 18638–18643 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moses C, Nugent F, Waryah CB, Garcia-Bloj B, Harvey AR, Blancafort P, Activating PTEN tumor suppressor expression with the CRISPR/dCas9 System. Mol. Ther. Nucleic Acids 14, 287–300 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagata Y, Lan K-H, Zhou X, Tan M, Esteva FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT, Hortobagyi GN, Hung M-C, Yu D, PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 6, 117–127 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Zunino B, Rubio-Patiño C, Villa E, Meynet O, Proics E, Cornille A, Pommier S, Mondragón L, Chiche J, Bereder J-M, Carles M, Ricci J-E, Hyperthermic intraperitoneal chemotherapy leads to an anticancer immune response via exposure of cell surface heat shock protein 90. Oncogene 35, 261–268 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Maiuri MC, Tasdemir E, Criollo A, Morselli E, Vicencio JM, Carnuccio R, Kroemer G, Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ. 16, 87–93 (2009). [DOI] [PubMed] [Google Scholar]
- 36.Cai J, Li R, Xu X, Zhang L, Lian R, Fang L, Huang Y, Feng X, Liu X, Li X, Zhu X, Zhang H, Wu J, Zeng M, Song E, He Y, Yin Y, Li J, Li M, CK1α suppresses lung tumour growth by stabilizing PTEN and inducing autophagy. Nat. Cell Biol 20, 465–478 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Lin Y-X, Wang Y, Qiao S-L, An H-W, Zhang R-X, Qiao Z-Y, Rajapaksha R, Wang L, Wang H, pH-sensitive polymeric nanoparticles modulate autophagic effect via lysosome impairment. Small 12, 2921–2931 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Wang Y, Lin Y-X, Qiao Z-Y, An H-W, Qiao S-L, Wang L, Rajapaksha R, Wang H, Self-assembled autophagy-inducing polymeric nanoparticles for breast cancer interference in-vivo. Adv. Mater 27, 2627–2634 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Zabirnyk O, Yezhelyev M, Seleverstov O, Nanoparticles as a novel class of autophagy activators. Autophagy 3, 278–281 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Zhong Z, Sanchez-Lopez E, Karin M, Autophagy, inflammation, and immunity: A troika governing cancer and its treatment. Cell 166, 288–298 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, Apetoh L, Aranda F, Barnaba V, Bloy N, Bracci L, Breckpot K, Brough D, Buqué A, Castro MG, Cirone M, Colombo MI, Cremer I, Demaria S, Dini L, Eliopoulos AG, Faggioni A, Formenti SC, Fučíková J, Gabriele L, Gaipl US, Galon J, Garg A, Ghiringhelli F, Giese NA, Guo ZS, Hemminki A, Herrmann M, Hodge JW, Holdenrieder S, Honeychurch J, Hu H-M, Huang X, Illidge TM, Kono K, Korbelik M, Krysko DV, Loi S, Lowenstein PR, Lugli E, Ma YT, Madeo F, Manfredi AA, Martins I, Mavilio D, Menger L, Merendino N, Michaud M, Mignot G, Mossman KL, Multhoff G, Oehler R, Palombo F, Panaretakis T, Pol J, Proietti E, Ricci JE, Riganti C, Rovere-Querini P, Rubartelli A, Sistigu A, Smyth MJ, Sonnemann J, Spisek R, Stagg J, Sukkurwala AQ, Tartour E, Thorburn A, Thorne SH, Vandenabeele P, Velotti F, Workenhe ST, Yang HN, Zong W-X, Zitvogel L, Kroemer G, Galluzzi L, Consensus guidelines for the detection of immunogenic cell death. Onco. Targets. Ther 3, e955691 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hubbard GK, Mutton LN, Khalili M, McMullin RP, Hicks JL, Bianchi-Frias D, Horn LA, Kulac I, Moubarek MS, Nelson PS, Yegnasubramanian S, De Marzo AM, Bieberich CJ, Combined MYC activation and Pten loss are sufficient to create genomic instability and lethal metastatic prostate cancer. Cancer Res. 76, 283–292 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kakavand H, Jackett LA, Menzies AM, Gide TN, Carlino MS, Saw RP, Thompson JF, Wilmott JS, Long GV, Scolyer RA, Negative immune checkpoint regulation by VISTA: A mechanism of acquired resistance to anti-PD-1 therapy in metastatic melanoma patients. Mod. Pathol 30, 1666–1676 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, Seja E, Lomeli S, Kong X, Kelley MC, Sosman JA, Johnson DB, Ribas A, Lo RS, Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 165, 35–44 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beer TM, Kwon ED, Drake CG, Fizazi K, Logothetis C, Gravis G, Ganju V, Polikoff J, Saad F, Humanski P, Piulats JM, Gonzalez Mella P, Ng SS, Jaeger D, Parnis FX, Franke FA, Puente J, Carvajal R, Sengelov L, McHenry MB, Varma A, van den Eertwegh AJ, Gerritsen W, Randomized, double-blind, phase III trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naive castration-resistant prostate cancer. J. Clin. Oncol 35, 40–47 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Postow MA, Sidlow R, Hellmann MD, Immune-related adverse events associated with immune checkpoint blockade. N. Engl. J. Med 378, 158–168 (2018). [DOI] [PubMed] [Google Scholar]
- 47.Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, Vonderheide RH, Pittet MJ, Jain RK, Zou W, Howcroft TK, Woodhouse EC, Weinberg RA, Krummel MF, Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med 24, 541–550 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gotwals P, Cameron S, Cipolletta D, Cremasco V, Crystal A, Hewes B, Mueller B, Quaratino S, Sabatos-Peyton C, Petruzzelli L, Engelman JA, Dranoff G, Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat. Rev. Cancer 17, 286–301 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Cook AM, Lesterhuis WJ, Nowak AK, Lake RA, Chemotherapy and immunotherapy: Mapping the road ahead. Curr. Opin. Immunol 39, 23–29 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Kuai R, Yuan W, Son S, Nam J, Xu Y, Fan Y, Schwendeman A, Moon JJ, Elimination of established tumors with nanodisc-based combination chemoimmunotherapy. Sci. Adv 4, eaao1736 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sharabi AB, Lim M, DeWeese TL, Drake CG, Radiation and checkpoint blockade immunotherapy: Radiosensitisation and potential mechanisms of synergy. Lancet Oncol. 16, E498–E509 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Yang G, Xu L, Chao Y, Xu J, Sun X, Wu Y, Peng R, Liu Z, Hollow MnO2 as a tumor-microenvironment-responsive biodegradable nano-platform for combination therapy favoring antitumor immune responses. Nat. Commun 8, 902 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sang W, Zhang Z, Dai Y, Chen X, Recent advances in nanomaterial-based synergistic combination cancer immunotherapy. Chem. Soc. Rev 48, 3771–3810 (2019). [DOI] [PubMed] [Google Scholar]
- 54.Zitvogel L, Apetoh L, Ghiringhelli F, Andre F, Tesniere A, Kroemer G, The anticancer immune response: Indispensable for therapeutic success? J. Clin. Invest 118, 1991–2001 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen Q, Wang C, Zhang X, Chen G, Hu Q, Li H, Wang J, Wen D, Zhang Y, Lu Y, Yang G, Jiang C, Wang J, Dotti G, Gu Z, In situ sprayed bioresponsive immunotherapeutic gel for post-surgical cancer treatment. Nat. Nanotechnol 14, 89–97 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Nam J, Son S, Park KS, Zou W, Shea LD, Moon JJ, Cancer nanomedicine for combination cancer immunotherapy. Nat. Rev. Mater 4, 398–414 (2019). [Google Scholar]
- 57.Xu YP, Lv L, Liu Y, Smith MD, Li WC, Tan XM, Cheng M, Li Z, Bovino M, Aubé J, Xiong Y, Tumor suppressor TET2 promotes cancer immunity and immunotherapy efficacy. J. Clin. Invest 129, 4316–4331 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang J, Nie J, Ma X, Wei Y, Peng Y, Wei X, Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 18, 26 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kowalski PS, Rudra A, Miao L, Anderson DG, Delivering the messenger: Advances in technologies for therapeutic mRNA delivery. Mol. Ther 27, 710–728 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ramaswamy S, Tonnu N, Tachikawa K, Limphong P, Vega JB, Karmali PP, Chivukula P, Verma IM, Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proc. Natl. Acad. Sci. U.S.A 114, E1941–E1950 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Germic N, Frangez Z, Yousefi S, Simon H-U, Regulation of the innate immune system by autophagy: Monocytes, macrophages, dendritic cells and antigen presentation. Cell Death Differ. 26, 715–727 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hopfner KP, Hornung V, Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat. Rev. Mol. Cell Biol 21, 501–521 (2020). [DOI] [PubMed] [Google Scholar]
- 63.Castoldi F, Vacchelli E, Zitvogel L, Maiuri MC, Pietrocola F, Kroemer G, Systemic autophagy in the therapeutic response to anthracycline-based chemotherapy. Onco. Targets. Ther 8, e1498285 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hand TW, Cui W, Jung YW, Sefik E, Joshi NS, Chandele A, Liu Y, Kaech SM, Differential effects of STAT5 and PI3K/AKT signaling on effector and memory CD8 T-cell survival. Proc. Natl. Acad. Sci. U.S.A 107, 16601–16606 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen L, Guo D, The functions of tumor suppressor PTEN in innate and adaptive immunity. Cell. Mol. Immunol 14, 581–589 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.