

Abstract
A combination of calorie restriction (CR), dietary modification, and exercise is the recommended therapy to reverse obesity and nonalcoholic fatty liver disease. In the liver, CR shifts hepatic metabolism from lipid storage to lipid utilization pathways, such as AMP-activated protein kinase (AMPK). Perfluorooctanesulfonic acid (PFOS), a fluorosurfactant previously used in stain repellents and anti-stick materials, can increase hepatic lipids in mice following relatively low-dose exposures. To test the hypothesis that PFOS administration interferes with CR, adult male C57BL/6N mice were fed ad libitum or a 25% reduced calorie diet concomitant with either vehicle (water) or 100 μg PFOS/kg/day via oral gavage for 6 weeks. CR alone improved hepatic lipids and glucose tolerance. PFOS did not significantly alter CR-induced weight loss, white adipose tissue mass, or liver weight over 6 weeks. However, PFOS increased hepatic triglyceride accumulation, in both mice fed ad libitum and subjected to CR. This was associated with decreased phosphorylated AMPK expression in liver. Glucagon (100 nM) treatment induced glucose production in hepatocytes, which was further upregulated with PFOS (2.5 μM) co-treatment. Next, to explore whether the observed changes were related to AMPK signaling, HepG2 cells were treated with metformin or AICAR alone or in combination with PFOS (25 μM). PFOS interfered with glucose-lowering effects of metformin, and AICAR treatment partially impaired PFOS-induced increase in glucose production. In 3T3-L1 adipocytes, metformin was less effective with PFOS co-treatment. Overall, PFOS administration disrupted hepatic lipid and glucose homeostasis and interfered with beneficial glucose-lowering effects of CR and metformin.
Keywords: liver, PFOS, perfluorinated compounds, caloric restriction, AMP-activated protein kinase, NAFLD, metformin, hepatocytes, adipocytes, AICAR
Nonalcoholic fatty liver disease (NAFLD) is a complex and multifactorial disease that affects 10% of children, 30% of adults, 50% of obese, 76% of diabetics, and 100% of morbidly obese diabetics in the United States (Eguchi et al., 2013; Naik et al., 2013; Reddy and Rao, 2006). NAFLD is characterized by the accumulation of lipids in the liver greater than 5% of the total liver weight and is frequently detected in people with central obesity or diabetes (Masarone et al., 2015; Naik et al., 2013; Smith and Adams, 2011). The American Association for the Study of Liver Disease guidelines recommends dietary modification and/or weight loss as the current NAFLD therapeutic strategy (Chalasani et al., 2018; Larter et al., 2013). In overweight patients with NAFLD, dietary restriction and exercise have been shown to decreased hepatic steatosis and serum lipids in patients with NAFLD (Larson-Meyer et al., 2008; Oh et al., 2014; Schwenger and Allard, 2014). In addition, long-term calorie restriction (CR) in humans improves insulin levels, and atherosclerosis outcomes (Fontana et al., 2004). CR mimetics have also been examined as potential treatments for NAFLD. For example, metformin, a commonly used anti-diabetes drug with some CR mimetic properties has been investigated as a potential therapy to treat NAFLD with some positive results in mice (Maslak et al., 2015). Multiple factors are suggested to contribute to induction of hepatic steatosis, and environmental exposure to synthetic chemicals has been hypothesized as a potential risk factor for predisposition to NAFLD (Al-Eryani et al., 2015; Cave et al., 2007).
Per- and polyfluoroalkyl substances (PFAS) have been used for several decades, but toxicological studies have grown substantially within the past couple of years and have shown multiple detrimental effects in rodents (Fenton et al., 2021; Lau et al., 2007). According to Environmental Protection Agency (EPA), perfluorooctanesulfonic acid (PFOS) is considered to be a possible carcinogen in humans and hepatotoxicant in rodents (US EPA, 2016). There are limited data regarding PFOS effects in human related to NAFLD or liver effects. Human studies have reported positive correlations between serum PFOS and serum markers of liver damage (ie, alanine aminotransferase) (Bassler et al., 2019; Darrow et al., 2016; Gallo et al., 2012; Gleason et al., 2015; Lin et al., 2010). PFOS administration increases liver weight and expression of genes involved in fatty acid oxidation and transport in rodents (Du et al., 2009; Wan et al., 2012). Moreover, PFOS-induced steatosis has been shown to be dose- and time-dependent, as observed with increased expression of fatty acid transport genes, and decreased expression of mitochondrial β-oxidation genes (Wan et al., 2012). Liver weight increases are, in part, attributed to an increase in hepatic triglycerides (Bijland et al., 2011).
A hallmark characteristic of hepatic steatosis is the disequilibrium between de novo lipogenesis, free fatty acid uptake, oxidation, esterification, and secretion of lipids from the liver (Naik et al., 2013; Smith and Adams, 2011). AMP-activated protein kinase (AMPK) is an important signaling molecule that regulate energy metabolism. AMPK is activated by phosphorylation at the Thr 172 residue of the alpha subunit in response to increases in the cellular AMP:ATP ratio; once activated, AMPK inhibits hepatic glucose production and lipid synthesis and induces lipid oxidation to produce energy (Nerstedt et al., 2010). CR increases the cellular AMP:ATP ratio, which induces AMPK activation and is implicated for the benefits of glucose and lipid metabolism with exercise, weight loss, and use of anti-diabetes drugs (Fulco and Sartorelli, 2008; Towler and Hardie, 2007). AMPK phosphorylates sterol regulatory element binding protein-1c (SREBP-1c) at Ser-327 and inhibits SREBP1c-mediated lipogenesis, thus reducing hepatic steatosis (Li et al., 2011). CR increases phosphorylation of AMPK, which then activates Sirtuin 1 (Sirt1) deacetylase under certain redox status (Fulco and Sartorelli, 2008). In steatotic mouse livers, there is a decrease in AMPK phosphorylation (Ha et al., 2011). As AMPK promotes catabolic pathways and inhibits ATP-consuming pathways, AMPK has been shown to be an effective target type 2 diabetes (T2D) treatment (Viollet et al., 2006).
The overall goal of this project was to determine whether PFOS exposure interferes with CR to decrease hepatic lipid content. It was hypothesized that PFOS administration could interfere with the benefits of CR, potentially through targeting AMPK phosphorylation. Metformin, a T2D therapy, and activates AMPK in hepatocytes, which in turn suppresses lipogenic genes, such as SREBP1c and acetyl-CoA carboxylase (ACC) (Zhou et al., 2001; Viollet et al., 2012). 5-aminoimidazole-4-carboxamide-1-beta-d-ribofuranoside (AICAR), a direct AMPK activator, also reversed alcoholic fatty liver in rats (Tomita et al., 2005). As both metformin and AICAR act on AMPK to provoke similar effects to increase ATP-producing pathways, these compounds will be used to interrogate the mechanisms related to CR in vitro. Overall, we present novel findings illustrating that PFOS administration concurrent with a modest reduction in caloric intake thwarted CR-induced decline in hepatic lipids and improvement in glucose tolerance in vivo and interfered with metformin-induced glucose-lowering effects in vitro.
MATERIALS AND METHODS
Animals and treatments
Ten-week-old male C57BL/6N mice, weighing approximately 30 g, were purchased from Charles River Laboratories (Wilmington, Massachusetts) and randomly assigned to treatment groups distributing body weight equally. Procedures were approved by the University’s Institutional Animal Care and Use Committee, and ethical standards associated with animal care and use were followed. The mice were housed under a controlled temperature (70–73°F) with relative humidity (30%–70%), lighting (12 h, light-dark cycles) environment and acclimated for 5 weeks on the standard rodent chow to allow for additional weight gain. At 15 weeks of age, the mice were then fed a purified rodent chow (AIN-93G Growth Purified Diet, Test Diet, St. Louis, Missouri) and monitored for an additional 5 weeks for food consumption to calculate a 25% CR. At 21 weeks of age, mice were fed either ad libitum (AL) or placed on 25% CR diet. The mice were then divided into 2 subsequent groups (n = 8) that were administered tap water as vehicle (Veh) via oral gavage (5 ml/kg) or heptadeca-PFOS potassium salt (100 µg/kg, 5 ml/kg) purchased from Sigma Aldrich (>98.5% purity for linear chain, catalog no. 77282, Lot no. BCBH2834V, St. Louis, Missouri) for 6 weeks. Our rationale for this dose was that it is lower or is similar to doses that have been described for mice for no observable adverse effects (ATSDR, 2018). Our decision to euthanize the mice at 6 weeks was determined by the response to GTT and ITT, which were conducted 3 and 4 weeks after the start of the study. As a difference in GTT and ITT response was observed, it was determined that the mice would be allowed to recover from the challenge and tissues would be collected to understand underlying mechanisms contributing to the phenotype. Each treatment group had n = 8 mice/group, which was based on a power analysis at the time of study design. Body weight and food intake were monitored daily and recorded. After 6 weeks of treatment, necropsies were performed and liver tissue sections were fixed in 10% buffered formalin for a minimum of 24 h, and then processed for paraffin embedding, and sectioning (5 μm), and hematoxylin and eosin (H&E) staining. Serum, liver, white adipose tissue (WAT), and skeletal muscle from non-fasted mice were collected, snap frozen with liquid nitrogen, and stored at −70°C until analysis.
Glucose tolerance and insulin tolerance tests
Glucose tolerance test (GTT) and insulin tolerance test (ITT) were performed as described previously (More et al., 2013; Xu et al., 2012). The GTT and ITT were performed after 3 and 4 weeks of CR and PFOS treatment, respectively. For the GTT, mice were fasted for 12 h and glucose (1 g/kg, 10 ml/kg i.p.) was administered. For ITT, mice were fasted for 12 h and insulin (1 unit/kg, 10 ml/kg i.p.) was administered. Blood glucose was determined by measuring concentrations in blood collected from the tail vein at t = 0 immediately before glucose or insulin administration, and at t = 15, 30, 60, and 120 min after glucose or insulin administration using a Bayer Contour glucometer.
Measurement of cholesterol, triglyceride, free fatty acid, and glucose concentration in liver and media
Cholesterol, triglyceride, and free fatty acid quantifications were performed using colorimetric assay kits from Pointe Scientific Inc. (Canton, Michigan) according to the manufacturer’s protocol. Tissues (50 mg) were homogenized with PBS and extracted with chloroform-methanol (2:1; vol/vol). The residue was re-suspended in 1% Triton X-100 in 100% ethanol. Lipid content was normalized with tissue weight. Serum glucose concentration was measured with a glucose assay kit (Eton Bioscience Inc., San Diego, California) based on manufacturer’s protocol.
RNA isolation and quantitative real-time PCR
Total RNA was isolated from liver tissue using TRIzol reagent (Invitrogen, Camarillo, California) according to the manufacturer’s instructions. About 1 μg of total RNA was converted to cDNA, and mRNA levels were quantified by quantitative real-time PCR using a Roche LightCycler 480 System (Roche Applied Science, Mannheim, Germany). SYBR green reagent was used, and relative target gene expression was normalized to GAPDH mRNA.
Determination of relative protein expression in liver and skeletal muscle by Western blot
Tissues (approximately 50 mg) were homogenized in 1 ml of RIPA buffer containing protease and phosphatase inhibitor. Protein lysates were electrophoretically resolved using polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were resolved by SDS-PAGE (7.5%–10% resolving and 4% stacking gel) and then transblotted onto low fluorescence polyvinylidene fluoride (PVDF) membrane (Millipore, Billerica, Massachusetts) in transfer buffer at 30 V overnight. Antibody blocking conditions, dilutions, incubation conditions, and secondary antibodies are described in Supplementary Table 1. PVDF membranes were incubated in blocking solution for 1 h. Next, blots were incubated with primary antibody specific to the protein of interest diluted in blocking buffer at either 4°C overnight or room temperature for 3 h. Primary antibody was removed, and the membrane was washed with washed in Tris-buffered saline with 0.2% Tween 20 three times for 5 min at room temperature. Next, blots were incubated with secondary antibody specific diluted in blocking buffer for 1 h. The membrane was either visualized via infrared detection (LiCOR Odessey, Lincoln, Nebraska) or incubated in ECL+ chemiluminescent substrate for 5 min (GE Healthcare Life Sciences, Pittsburgh, Pennsylvania) and visualized using light-sensitive film. The relative abundance of protein in each sample was quantified by densitometric analysis and normalized to β-actin.
Primary hepatocyte and HepG2 culture and treatment
Primary mouse hepatocytes were isolated from livers of untreated adult C57BL/6 mice using a 2-step collagenase perfusion; 1 × 106 cells/well in 2 ml completed medium (William’s medium E supplied with 10% FBS) were seeded on collagen-coated 6-well plates. After cell attachment (approximately 4 h), they were cultured in serum-free William’s E Media containing 1% insulin-transferrin-selenium and dexamethasone supplement (Invitrogen, California). Media was then replaced with custom William’s Media E lacking glucose and supplemented with pyruvate. Media was collected at time zero to measure glucose output. Glucagon (100 nM), PFOS (2.5 µM) or a combination of both, were added to the hepatocytes and media was collected at 6 h to measure glucose output using a glucose assay (Eton Bioscience Inc., San Diego, California). HepG2 cells are a common cell line to study glucose output and AMPK phosphorylation (Huang et al., 2015; Sefried et al., 2018; Zhang et al., 2019; Zhao and Song, 2021). HepG2 cells are a well-accepted tool to screen antidiabetic activity of novel anti-diabetic therapies, metformin, and AMPK signaling (Pasachan et al., 2021; Qiao et al., 2019; Seo et al., 2015; Wei et al., 2020). 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR) is an analog of adenosine monophosphate (AMP) that is capable of stimulating AMP-dependent protein kinase (AMPK) activity and was also used to understand whether PFOS could act on the AMPK pathway (Wang et al., 2018). HepG2 cells were treated with AICAR (100 µM), PFOS (0.025–25 µM), or a combination of AICAR and PFOS and media glucose concentration was measured 24 h after treatment. HepG2 cells were then also treated with PFOS (25 µM), metformin (1 mM), or in combination. Media was collected after 6–10 h after treatment and media glucose concentration was assayed using the glucose assay described above. The PFOS concentrations selected for the above studies are based on previous studies performed in rat hepatocytes or HepG2 cells (Behr et al., 2020; Bjork and Wallace, 2009; Qiu et al., 2016; Yao et al., 2016).
3T3-L1 and human preadipocytes cell culture and treatment
Mouse 3T3-L1 preadipocytes were obtained from ATCC (American Type Culture Collection, Manassas, Virginia) and cultured as described previously (Xu et al., 2016). Briefly, the cells were maintained in DMEM containing 10% FBS, 100 U/ml penicillin, and 100 µg/ml streptomycin to confluency at 37°C, and the cells were then seeded in 6-well and 24-well plates. Adipocyte differentiation was induced at 2 days post-confluence (day 0), using a mixture of 0.5 mM isobutylmethylxanthine (IBMX, no. I5879, Sigma-Aldrich, St Louis, Missouri), 0.25 µM dexamethasone (no. D4902, Sigma-Aldrich, St Louis, Missouri), and 5 µg/ml insulin in DMEM containing 10% of FBS. On day 2, day 4, and day 6, this medium was replaced with DMEM containing 10% of FBS and insulin only. The medium was switched to DMEM containing 10% of FBS on day 8 for 24 h. On day 9, the differentiated 3T3-L1 cells were then treated with metformin (MET, 1 mM), PFOS (50 µM), and a combination and after 48 h, the cell culture media was collected and stored at −80°C until analysis. To assess lipid deposition, human visceral preadipocytes were also cultured as described by Xu et al. (2016). Briefly, human preadipocytes obtained from Lonza (donor no.: 24711; Lot no.: 0000313366; Cat no.: PT-5005, Lonza Walkersville, Inc., Walkersville, Maryland). Human visceral adipocytes were cultured and differentiated as per the manufacturer’s instructions. Briefly, human primary preadipocytes were grown to confluence in Preadipocyte growth medium-2 provided in 5% CO2 at 37°C. When the cells reached confluency, the medium was replaced with Preadipocyte differentiation medium supplemented with a cocktail of insulin, dexamethasone, indomethacin, and isobutyl-methylxanthine to Preadipocyte Growth Medium-2 for 10 days and the cells were monitored for lipid droplet formation. After 10 days of culture, adipocytes were treated with metformin (1 mM), PFOS (50 µM), or in combination for 24 h, before staining with Oil Red O for visualizing lipid droplets.
Adiponectin assay
The Mouse Adiponectin/Acrp30 Quantikine ELISA Kit was purchased from R&D Systems, Minneapolis, Minnesota. The protocol followed was according to the product insert. Mouse sera were diluted 1:2000 and media from cultured adipocytes was diluted 1:2000 before running in the assay.
Oil Red O staining
After treatment of the human visceral preadipocytes for 24 h, cells were fixed with 10% buffered formalin for 30 min and then stained with Oil Red O (ORO) working solution. A fresh working solution was prepared by combining 3 parts of stock solution (0.5% ORO in isopropanol) with 2 parts of deionized water and then filtered after 15 min. Cells were incubated in working solution for 15 min at room temperature and then ORO solution was removed. Wells were quickly rinsed 2 times with fresh 60% isopropanol, washed with water, and mounted in glycerin jelly. Quantification includes the addition of 1 ml isopropanol to each well then 140 μl of those wells into a 96-well plate for spectrophotometer reading.
Statistical analysis
Power analysis using the sample size calculator (http://www.jerrydallal.com/LHSP/SIZECALC.HTM) was performed with the following assumption that the difference in mean between the test and control group is 50% of the mean and the standard deviation is 25% of the mean values. Based on this analysis, the sample size used was an n = 8. Depending on the analysis, an unpaired Student’s t test or an ANOVA followed by a Duncan’s multiple range post hoc test was performed. Significance was considered to be p < .05.
RESULTS
Effect of CR and PFOS on Body Weight, Liver Weight, and Serum Chemistry
Male C57BL/6 mice were fed AL or 25% calorically restricted (CR) and administered vehicle (VEH) or 100 µg/kg PFOS daily for 5 weeks. In Figures 1A and 1B, CR reduced the percent body weight from day 0 and the AUC for body weight over time. Body weight and percent weight loss was similar between VEH and PFOS treated mice over the course of the study. CR decreased liver weight by 37% and WAT tissue by 48% (Figs. 1C and 1D). Liver weight was similar between AL-Veh and AL-PFOS groups, but liver weight was slightly higher in CR-PFOS than CR-Veh mice (Figure 1C). WAT weight was similar between VEH and PFOS treated groups (Figure 1D). Overall, PFOS administration did not significantly alter body or tissue weight, as well as response to CR-induced weight loss.
Figure 1.

Adult male mice were administered vehicle (water, VEH) or perfluorooctanesulfonate (PFOS, 100 µg/kg/day) and fed ad libitum (AL) or placed on a 25% kcal calorie restriction (CR) for approximately 6 weeks. (A) Percent change (%) in body weight from day 0 and (B) AUC of percent body weight illustrates that PFOS did not alter CR-induced percent weight loss at the time of necropsy. (C) Liver weight and (D) white adipose tissue (WAT) weights were decreased by CR. Liver weight was similar between AL-Veh and AL-PFOS groups, but liver weight was slightly increased in mice treated with PFOS (CR-PFOS) that underwent CR compared with vehicle controls (CR-VEH). Asterisks (*) represent a statistical difference in treatments compared with controls, and # represents a significant difference between CR-VEH versus CR-PFOS (p < .05).
Serum clinical markers associated with response to weight loss and CR were evaluated (Table 1). CR increased adiponectin levels by 24% compared with AL-fed mice. In AL-fed mice, PFOS also increased adiponectin by 23%. In CR-fed mice, PFOS did not significantly affect serum adiponectin levels.
Table 1.
Serum Clinical Parameters: Adiponectin, Cholesterol, Free Fatty Acids, Glucose, and Triglycerides Measurements at Time of Necropsy
| Parameters | Unit | AL-VEH | CR-VEH | AL-PFOS | CR-PFOS |
|---|---|---|---|---|---|
| Adiponectin | μg/ml | 11.01 ± 5.96 | 13.67 ± 4.60* | 13.58 ± 9.74$ | 13.57 ± 6.68# |
| Cholesterol | mg/dl | 120 ± 10.9 | 108 ± 14.9 | 114 ± 10.0 | 98.2 ± 4.64 |
| FFA | μmol/l | 0.96 ± 0.15 | 1.10 ± 0.11 | 1.15 ± 0.35 | 1.06 ± 0.10 |
| Triglycerides | mg/dl | 172 ± 42 | 131 ± 17.0 | 154 ± 17.3 | 137.5 ± 4.31 |
Eighteen-week-old C57BL/6N mice underwent treatment for approximately 6 weeks of either water (VEH) or perfluorooctanesulfonic acid (PFOS) administration (100 µg/kg/day) and fed either ad libitum (AL) or underwent 25% kcal calorie restriction (CR). After euthanization, these parameters were assayed and analyzed.
p < .05, CR-VEH compared with AL-VEH control.
p < .05, AL-PFOS compared with AL-VEH control.
p < .05, CR-PFOS compared with AL-VEH control.
PFOS Interfered With CR-Induced Improvement of Glucose Utilization
CR can improve glucose tolerance and increase insulin sensitivity (Colman et al., 2009; Fontana et al., 2010), which were assayed by GTT and ITT, respectively. Figure 2 illustrates our evaluation of PFOS on glucose and insulin tolerance. After glucose challenge, blood glucose levels increased 15 min after glucose administration and the returned to baseline 2 h later, with the rise in blood glucose being lower in CR-VEH mice (Figure 2A). Blood glucose levels in CR-PFOS mice were significantly higher than the CR-VEH mice at 1 and 2 h after glucose challenge (Figure 2A). Overall, CR decreased glucose load by 58% compared with AL fed mice (Figure 2B). In AL-fed mice, PFOS administration did not significantly affect response to glucose challenge, although the glucose load tended to increase, but did not reach statistical significance (Figure 2B). Interestingly, PFOS administration worsened the response to glucose challenge in mice that were placed on CR. The glucose load was 74% higher in CR mice administered PFOS (Figure 2B). Figure 2C depicts the effect of PFOS on insulin sensitivity by ITT. In CR- and AL-fed mice, PFOS increased the glucose load after 2.5 h after insulin challenge compared with controls (Figure 2D), suggesting that PFOS interferes with insulin suppression of hepatic glucose production. The mRNA expression for genes involved in glucose homeostasis was analyzed in liver tissues by qPCR. CR did not markedly decrease Irs1 gene expression levels in liver, however, with PFOS, CR-fed mice significantly decreased Irs1 gene expression (Figure 2E). CR-fed mice had decreased liver gene expression of Pepck and G6pase compared with AL controls (Figure 2E). In both CR- and AL-fed mice, PFOS significantly decreased Glut2 mRNA expression in liver (Figure 2E).
Figure 2.

Effect of PFOS and CR on glucose homeostasis. A, Glucose tolerance test (GTT) performed after 3 of CR and PFOS treatment. CR significantly improved glucose clearance at 0.5 and 1 h compared with AL feeding, which did not occur in mice that underwent CR with PFOS administration. B, Total glucose load was plotted as an area under the curve (AUC). PFOS significantly increased total glucose load in CR groups. C, Insulin tolerance test (ITT) performed after 4 weeks of CR and PFOS treatment. D, Glucose load with ITT was not different between groups until 2.5 h where the two PFOS groups had significantly elevated glucose levels compared with controls. E, Hepatic mRNA data from the conclusion of the study. Irs-1 and Glut2 expression were decreased in mice that underwent CR and PFOS administration. Glut2 expression was decreased in AL mice administered PFOS compared with the AL fed vehicle controls. Asterisks (*) represent a statistical difference in treatments compared with controls.
PFOS Significantly Increased Liver Lipid Content
It has been previously reported that PFOS increased liver weight (Seacat et al., 2002, 2003) and this may be attributable to an accumulation of lipids within the liver (Polyzos et al., 2009). We evaluated the liver histology with H&E staining. As anticipated, CR decreased staining associated with the presence of glycogen. Presence of vacuoles were observed in the AL-PFOS and CR-PFOS treatment groups (Figure 3A). This observation is consistent with what has been described in cells and liver following treatment with perfluorooctanoic acid (Filgo et al., 2015; Tang et al., 2018; Wu et al., 2018), which is another perfluoroalkyl substance (PFAS) that exerts many of the same biological effects as PFOS.
Figure 3.

PFOS significantly increased hepatic lipid content. A, Liver histopathology. H&E staining of liver tissue illustrates PFOS administration increased vacuolization in mice that underwent CR compared with AL feeding. Vacuoles were visualized in AL-PFOS and CR-PFOS livers, which were consistent with histopathology described for other perfluorinated chemicals (Filgo et al., 2015; Wu et al., 2019). Liver lipids were isolated and quantified via colorimetric assays. B, PFOS decreased free fatty acid (FFA) concentration in livers of AL and CR mice. C, PFOS increased liver triglyceride concentrations in AL and CR mice. D, Relative mRNA expression in liver in response to CR and PFOS treatment. Cluster of differentiation 36 (CD36), a fatty acid translocase, and Cyp4a14 were higher in CR-PFOS than CR-Veh mice. Asterisks (*) represent a statistical difference in treatments compared with controls, and # represents a significant difference between CR-Veh versus CR-PFOS (p < .05).
Hepatic free fatty acid levels were similar between AL-VEH and CR-VEH groups but were decreased by about 40% in AL-fed mice and CR-fed mice that received PFOS (Figure 3B, left panel). These data are further supported by increased liver triglyceride concentrations in PFOS-treated mice. As expected, hepatic triglycerides were 32% lower in CR mice compared with AL control (Figure 3B, right panel). Interestingly, PFOS administration increased hepatic triglycerides in both AL- and CR-fed mice by 95% compared with AL-VEH and 37.6% compared with CR-VEH, respectively (Figure 3B, right panel). Figure 3C illustrates relative gene expression changes in liver. CR increased Cluster of differentiation (CD36), Cytochrome P450 4a14 (Cyp4a14), and Enoyl-CoA Hydratase and 3-Hydroxyacyl CoA Dehydrogenase (Ehhadh), and decreased Fatty acid synthase (Fas), acetyl-CoA carboxylase (Acc1), and Glycerol-3-phosphate acyltransferases (Gpat) mRNA expression in liver. In AL mice, PFOS increased Cyp4a14 and Ehhadh mRNA expression in liver (Figure 3C). In CR-PFOS mice, CD36 and Cyp4a14 mRNA expression were at least 25% higher than the AL-Veh and CR-Veh mice.
PFOS Alters Protein Expression in Liver
To better understand whether PFOS affected response to CR at the level of protein expression, liver lysates were evaluated for changes in protein expression. Western blot was performed to quantify the relative abundance proteins involved in glucose and lipid homeostasis (Figure 4A). Densitometric analysis showed no significant changes in protein abundance of fatty acid-binding protein 1 (Fabp1), phosphorylated sterol regulatory element-binding transcription factor 1 (p-Srebp-1c), phosphorylated insulin receptor substrate 1 (p-Irs1), and Ampk protein targets with PFOS treatment, and CR compared with AL feeding. As shown in Figure 4B, PFOS treatment increased cluster of differentiation 36 (cd36), or fatty acid translocase protein levels by 6.5% in the AL group. Phosphorylated protein kinase B (p-Akt), a kinase involved in insulin signaling, was increased with PFOS treatment in the AL group by 80%, in addition PFOS treatment and CR also increased p-Akt by 72% compared with the AL-Veh group (Figure 4C). Phosphoenolpyruvate carboxykinase (Pepck; Figure 4D), the rate limiting enzyme for gluconeogenesis, was increased with CR feeding by 134% in the control group and 104% in the PFOS-treated mice. There were no significant changes observed with PFOS treatment. Stearoyl-CoA desaturase-1 (Scd-1; Figure 4E), a protein involved in fatty acid synthesis, was decreased by CR feeding by 31% in the Veh controls, and by 60% in the PFOS-treated mice. PFOS treatment also decreased Scd-1 in the CR fed mice by 47% compared with CR-Veh group. Phosphorylated Ampk (p-Ampk; Figure 4F) was increased with PFOS treatment in the AL-fed mice by 10%, and although not significant, PFOS increased p-Ampk by 3.6% in the CR-fed mice.
Figure 4.

PFOS alters liver expression of proteins involved in lipogenesis. Total protein lysates were isolated from liver, separated by SDS-PAGE, transferred to PVDF membrane, and then immunoblotted for relative protein abundance. (A) Western blots for illustrates protein abundance with mice that underwent PFOS treatment, and CR compared with AL feeding. Relative protein abundance was quantified by densitometric analysis and normalized to β-actin. Fatty acid-binding protein 1 (Fabp1), phosphorylated sterol regulatory element-binding transcription factor 1 (p-Srebp-1c), phosphorylated insulin receptor substrate 1 (p-Irs1), and AMP-activated protein kinase (AMPK) protein targets had no significant changes in protein abundance between groups. Densitometry of (B) cluster of differentiation 36 (cd36), (C) phosphorylated protein kinase B (p-Akt), (D) phosphoenolpyruvate carboxykinase (Pepck), (E) stearoyl-CoA desaturase-1 (Scd-1), and (F) phosphorylated Ampk (p-Ampk) illustrate changes to protein abundance in response to CR and PFOS treatment. Bars illustrate statistical difference between groups, where * represents p < .05 and ** represents p < .01.
PFOS Significantly Increased Glucose Content in Media of Primary Hepatocytes and HepG2 Cells
Based on our GTT observations and changes in p-Ampk in liver, the effect of PFOS on glucose production in hepatocytes was measured. Primary hepatocytes were isolated from C57BL/6 mice and were treated with glucagon, PFOS (2.5 µM), and a combination of both for 24 h. Expectedly, glucagon increased glucose within the media of the hepatocytes 13-fold compared with vehicle (Figure 5A). PFOS also increased media glucose concentration by 15-fold and when in combination, PFOS and glucagon treatment increased the glucose within the media by 19.6-fold compared with vehicle (Figure 5A). Next, we used HepG2 cells to study the effect of PFOS on glucose output and confirm these phenomena in another hepatocyte model. HepG2 cells are commonly used to study glucose output (ref). To understand if PFOS induction of glucose production could be antagonized, we treated HepG2 cells with AICAR to block hepatocyte glucose synthesis and stimulate phosphorylation of AMPK concurrent with PFOS challenge (Figure 5B). Treatment with 25 µM PFOS treatment increased media glucose concentration by 6-fold compared with the vehicle controls (Figure 5B). AICAR treatment partially blocked this PFOS-induced effect, with the media glucose concentration being only 3.5-fold higher than the vehicle controls (Figure 5B). To further confirm this finding, HepG2 cells were treated for 10 h with metformin (1 mM), PFOS (25 µM), and a combination of both. Metformin significantly decreased glucose production in the media of HepG2 cells by 67% compared with the DMSO vehicle controls (Figure 5C). PFOS co-treatment with resulted in higher glucose concentrations than metformin alone (Figure 5C), indicating that PFOS can dampen metformin-induced lowering of glucose within the media. P-Ampk protein levels in the cell lysates were analyzed by Western blot (Figure 5D). As expected, p-Ampk levels were higher in the metformin lysates than vehicle control. Compared with vehicle, PFOS (25 µM) decreased the relative amount of p-Ampk protein. The amount of p-Ampk was also lower in the metformin and PFOS co-treated cells (Figure 5D), suggesting that PFOS interferes with the actions of metformin.
Figure 5.

In vitro glucose production and AMPK phosphorylation in mouse primary hepatocytes and HepG2 cell lines treated with PFOS and CR mimetics, metformin, and AICAR. A, Media glucose measurement in primary mouse hepatocytes treated with glucagon, PFOS (2.5 µM), or in combination. Glucagon and PFOS each increased the media glucose concentration. Glucagon and PFOS co-treatment increased media glucose concentration more than either glucagon or PFOS alone. *p < .05 compared with DMSO; #p < .05 compared PFOS; &p < .05 compared with glucagon. B, Media glucose was measured in HepG2 cells with 24 h after treatment with AICAR (100 µM), PFOS (0.025–25 µM), or a combination of AICAR and PFOS. In HepG2 cells, 25 µM PFOS, but not 0.25 or 2.5 µM PFOS, increased glucose concentration in media compared with DMSO control. AICAR co-treatment with PFOS (25 µM) resulted in lower glucose production than PFOS alone (25 µM). *p < .05 compared with DMSO; #p < .05 compared PFOS. C, Media glucose concentrations were measured in HepG2 cells after metformin (1 mM), PFOS (25 µM), or a combination of metformin and PFOS treatment. After 10 h treatment, metformin decreased glucose concentration in the media, whereas PFOS treatment increased glucose production. A combination of metformin and PFOS increased media glucose concentration compared with metformin alone. *p < .05 compared with DMSO; #p < .05 compared PFOS. D, Phosphorylated Ampk (p-Ampk) was measured 10 h after HepG2 cells were treated with PFOS (25 and 50 µM) and metformin (1 mM) alone or in combination. Metformin increased p-Ampk levels, but not in cells treated with PFOS. *p < .05 compared with DMSO; #p < .05 for PFOS effect versus corresponding treatment.
PFOS Increases Glucose Within the Media of 3T3-L1 Cells and Human Adipocytes
Mature 3T3-L1 adipocytes were treated with metformin (1 mM) and PFOS (50 µM) or in combination for 12 h. PFOS significantly increased media glucose concentrations compared with the vehicle control (Figure 6A). Adiponectin levels were also measured, with only metformin inducing adiponectin and this effect was not observed in metformin and PFOS-treated adipocytes (Figure 6B). Human adipocytes were also treated with metformin (1 mM), PFOS (50 µM), or in combination for 24 h (Figs. 6C and 6D). Oil Red O staining of human adipocytes depicted more lipids with PFOS treatment compared with DMSO control (Figure 6C). Metformin decreased Oil Red O staining, which was not observed with PFOS cotreatment (Figure 6D).
Figure 6.

PFOS impairs metformin activity in mature 3T3-L1 adipocytes and human adipocytes. 3T3-L1 cells were treated with metformin (MET, 1 mM), PFOS (50 µM), and a combination. A, PFOS treatment increased glucose concentration in media of mature 3T3-L1 cells was significantly increased upon PFOS treatment. Asterisk (*) represents a statistical difference from DMSO (p < .05). B, Metformin increased adiponectin levels in mature adipocytes, which was not observed with PFOS cotreatment. Asterisk (*) represents DMSO versus PFOS (p < .05). C, D, Oil Red O staining of the human adipocytes. Mag.10×. Human adipocytes were treated with DMSO, metformin (MET, 1 mM), PFOS (50 µM), and a combination of PFOS with metformin for 10 days. Only MET decreased Oil Red O staining. Asterisks (*) represent a significant difference compared with DMSO.
DISCUSSION
NAFLD is rising worldwide and is associated with obesity and diabetes (Masarone et al., 2015). Although obesity is considered to be a predominant risk factor for NAFLD, other etiologies for NAFLD have been hypothesized, such as exposures to chemicals present in the environment (Cave et al., 2007). On the other hand, modest CR is considered to afford numerous health benefits and decreases risk for many diseases of metabolic origin. In humans, CR decreases liver steatosis and improves glucose metabolism (Johari et al., 2019; Wolf et al., 2019). Individuals who calorically restrict have a lower incidence of fatty liver disease and treatment guidelines for NAFLD include dietary modification and weight loss (Kani et al., 2017; Lam and Younossi, 2010; Schwimmer et al., 2019). The study herein addressed, not only whether PFOS administration could induce markers of steatosis and NAFLD, but also interfere with the benefits of CR, which include improved glucose metabolism and decreased liver lipid content. As we identified that PFOS could interfere with CR benefits in a mouse model, we then studied whether PFOS could interfere with metformin-induced glucose consumption and AMPK phosphorylation using mouse primary hepatocytes and HepG2 cells. Our findings are consistent with a recent study from our laboratory in mice that were subjected to weight loss by switching from a high fat diet to low fat diet (Marques et al., 2020).
CR with or without exercise decreases hepatic steatosis and lowers serum liver enzymes (Larson-Meyer et al., 2008; Straznicky et al., 2012; Tamura et al., 2005; Yoshimura et al., 2014). It has been reported that a 15%, 30%, and 40% kcal reduction in mice has beneficial implications on body weight and hepatic lipids (Fu and Klaassen, 2013). In our study, mice were placed on a 25% kcal reduction or fed AL for 6 weeks dosed with vehicle or with 100 µM/kg/day PFOS via oral gavage. Expectedly, our results are consistent with other studies showing significant decreases in body, liver, and WAT weights with a 25% kcal reduction (Fu and Klaassen, 2013; Weindruch et al., 1986), which is a less stringent kcal reduction (ranges from approximately 25% to 60%) (Koubova and Guarente, 2003). Our GTT is evidence that the CR regimen increased glucose tolerance and lowered the total glucose load. We also observed decreased serum and hepatic lipids and these results are consistent with another mouse study (Fu and Klaassen, 2013). CR activates the AMPK pathway, which inhibits hepatic glucose and lipid synthesis and induces lipid oxidation to produce energy as outline in Figure 7 (Nerstedt et al., 2010). AMPK is a heterotrimeric serine/threonine protein kinase that regulates energy homeostasis in metabolic tissues upon high AMP:ATP ratios (Nerstedt et al., 2010), and promotes hepatic lipid metabolism and energy balance by promoting catabolic pathways and inhibiting ATP-consuming pathways (Viollet et al., 2006). Expectedly, in our study, CR induced AMPK activation in livers of mice compared with AL control.
Figure 7.
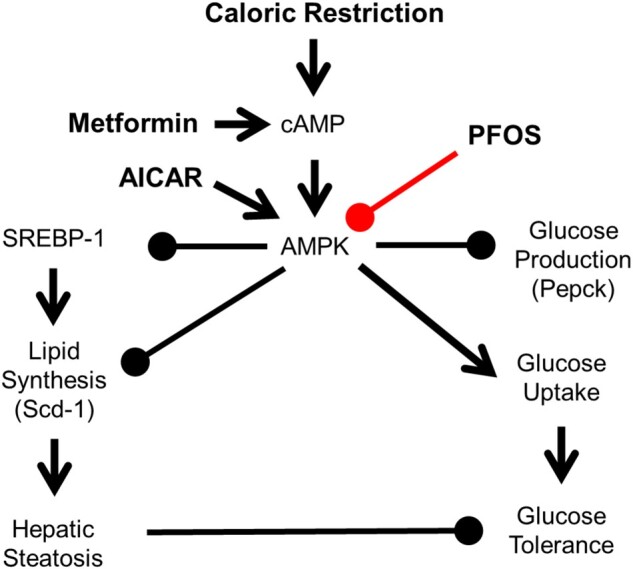
Hypothesis mechanism of PFOS effects in lipid and glucose signaling. AMPK is activated by phosphorylation in response to increases in the cellular AMP:ATP ratio, which is increased by CR or metformin. AMPK can also be directly activated by 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside (AICAR). Once activated, AMPK inhibits hepatic glucose production and lipid synthesis via SREBP-1. AMPK can induce glucose production via insulin signaling. Inhibiting AMPK activation can lead to hepatic steatosis and glucose intolerance.
In this study, we used the C57BL/6 mouse model on a purified diet that underwent CR and daily administration of PFOS to assess the effects of PFOS on a CR diet via liver physiology. PFOS administration can increase liver weight (Marques et al., 2020; Seacat et al., 2003; Wan et al., 2012), but we did not observe increased liver weight in this study for either AL or CR mice. A likely reason is that we used a relatively low daily dose for mouse (0.1 mg/kg/day), which approaches the lowest observable effect level for mouse (Luebker et al., 2005). In addition to increases in liver weight with PFOS exposure, decreases in body weight have also been observed with PFOS treatment (Marques et al., 2020; Seacat et al., 2003). Another study showed increased body weight at a lower concentration of 1 mg/kg and increased liver weight at higher concentrations of 5 and 10 mg/kg over 21 days with all concentrations increasing the liver weight attributed to the appearance of cellular vacuolations (Wan et al., 2012). We observed that PFOS administration increased hepatic triglyceride content in AL and CR mice. It is known that PFOS can induce hepatic steatosis in rats and mice, but fewer studies address effects of PFOS on hepatic lipid accumulation in humans, and only one study has found that higher plasma PFAS exposure was positively associated with more disease severity in children with NAFLD (Jin et al., 2020). Cytokeratin 18 M30, a biomarker for nonalcoholic steatohepatitis, was also found to positively associated with PFOA, perfluorohexanesulphonic acid, and perfluorononanoic acid in adults (Bassler et al., 2019). To the best of our knowledge, this is the first study that has investigated the effects of PFOS on CR and AMPK activation. Wan et al. also reported a prediabetic phenotype in mice exposed to PFOS (Wan et al., 2014). Our data are consistent with this finding. IRS-1 gene expression was decreased upon PFOS exposure in mice undergoing CR compared with our AL mouse model. Glut-2 gene expression was also decreased in both CR and AL mouse models upon PFOS exposure, which may contribute the observed glucose intolerance. PFOS has been shown to have a positive correlation with serum ALT levels (Gallo et al., 2012) and increased liver weights (Seacat et al., 2002), which may be attributable to an accumulation of lipids. In our study, we show PFOS treatment increased hepatic triglycerides and interfered with the CR-induced lipid loss. H&E staining shows PFOS with CR induces the presence of vacuoles compared with any other group, consistent with findings in other studies (Wan et al., 2012). PFOS induces PPAR-α, which is suggested to account for toxicity associated with PFOS (Takacs and Abbott, 2007), however, there are PPAR-α independent toxicities as well (Rosen et al., 2010). We demonstrate PPAR-α activation via induction of CYP4a14, a downstream target of PPAR-α, upon PFOS exposure and consistent with previous studies (Abbott et al., 2009; Takacs and Abbott, 2007).
Metformin was utilized in hepatocytes because it is used in the treatment of T2D and has been shown to suppress hepatic gluconeogenesis via AMPK activation in hepatocytes and HepG2 cells (Cao et al., 2014; Zang et al., 2004; Zhou et al., 2001). Primary hepatocytes and HepG2 cells are well described in vitro tools to study glucose production. PFOS treatment increased glucose production in both cultured mouse primary hepatocytes and HepG2 cells. This finding is consistent with a previous study that reported induction of PEPCK mRNA levels and insulin resistant in HepG2 cells treated with 100 µM PFOS (Qiu et al., 2016). We show that PFOS dampens the beneficial effects of metformin on lowering glucose production via AMPK activation. Our results indicate that PFOS can potentially interfere with AMPK phosphorylation in liver and HepG2 cells (Figure 7), which is relatively new to the field of environmental health. PFOS is transported into the liver via Oatp1a1in mouse liver (Weaver et al., 2010; Zhao et al., 2017) or OATP1B1 and 1B3 isoforms in human liver (Zhao et al., 2017). The primary transporters required for hepatic uptake of metformin are OCT1 and OCT3 (Nies et al., 2011). Because PFOS and metformin are likely not utilizing the same transporters, this suggests that PFOS may inhibit beneficial effects of metformin downstream from transporter-mediated mechanisms instead of competing for transport into the hepatocyte. We observed these alterations at a treatment concentration of 25 µM PFOS, which is similar or lower than what has been reported in the literature by others in HepG2 cells (Behr et al., 2020; Bjork and Wallace, 2009; Qiu et al., 2016). The likely reason for the insensitivity of HepG2 cells for PFOS is that HepG2 cells have relatively low expression up OATP1B1 and 1B3, which would facilitate cellular uptake (Godoy et al., 2013).
In our study, we present novel findings illustrating that PFOS administration concurrent with a modest reduction in caloric intake thwarted CR-induced decline in hepatic lipids and improvement in glucose tolerance in vivo and interfered with metformin-induced glucose-lowering effects in vitro. To the best of our knowledge, this is the first study that investigates PFOS effects on metformin and AMPK in a CR diet and few studies have identified AMPK as a target of environmental exposures (Liu et al., 2014; Wang et al., 2010). Our results indicate novel findings of lowered AMPK levels upon PFOS administration as well as PFOS interference with a major pharmacological therapeutic for diabetic intervention.
In conclusion, PFOS, a fluorosurfactant previously used as a stain repellent and anti-stick material increased hepatic lipids in mice following relatively low dose exposures. An increase in hepatic lipids leads to hepatic steatosis, which is the first hit in NAFLD. A combination of CR, dietary modification, and exercise is the recommended therapy to reverse obesity and NAFLD. The ability to mount an effective response to CR required to shift hepatic metabolism to fatty acid oxidation depends upon induction of pathways, such as AMPK. Our study showed that PFOS administration interfered with CR-induced reduction of hepatic lipids and improvement of glucose tolerance. PFOS increased hepatic lipid content and decreased the ability of mice to effectively decrease liver lipid content during CR. Our in vitro studies illustrated that PFOS interfered with agents such as metformin and AICAR that phosphorylate AMPK. In summary, PFOS decreased the beneficial effects of a CR diet and suggest that it can modulate the AMPK pathway. Our studies point to the novel idea that PFOS can impact the liver’s response to the benefits of CR and implore further exploration in humans.
DECLARATION OF CONFLICTING INTERESTS
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
Supplementary Material
ACKNOWLEDGMENTS
This work was presented in part at the Gordon Research Conference Mechanisms of Toxicity in Summer 2015 (travel award to D.S.) and Society of Toxicology 2015 annual meeting (travel award to D.S.).
FUNDING
National Institutes of Health (5R01ES016042-04, 3R01ES016042-2S2, 5K22ES013782-03); Rhode Island IDeA Network of Biomedical Research Excellence grants from the National Center for Research Resources (5P20RR016457-11); the National Institute for General Medical Science (8P20GM103430-11), components of the National Institutes of Health (NIH). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- Abbott B. D., Wolf C. J., Das K. P., Zehr R. D., Schmid J. E., Lindstrom A. B., Strynar M. J., Lau C. (2009). Developmental toxicity of perfluorooctane sulfonate (PFOS) is not dependent on expression of peroxisome proliferator activated receptor-alpha (PPAR alpha) in the mouse. Reprod. Toxicol. 27, 258–265. [DOI] [PubMed] [Google Scholar]
- Al-Eryani L., Wahlang B., Falkner K. C., Guardiola J. J., Clair H. B., Prough R. A., Cave M. (2015). Identification of environmental chemicals associated with the development of toxicant-associated fatty liver disease in rodents. Toxicol. Pathol. 43, 482–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ATSDR. (2018). Toxicological Profile for Perfluoroalkyls. Draft for Public Comment. U.S. Department of Health and Human Services, Agency for Toxic Substances and Disease Registry, Atlanta, GA. doi:10.15620/cdc:59198. Available at: https://www.atsdr.cdc.gov/toxprofiles/tp.asp?id=1117&tid=237. Accessed January 4, 2021. [PubMed] [Google Scholar]
- Bassler J., Ducatman A., Elliott M., Wen S., Wahlang B., Barnett J., Cave M. C. (2019). Environmental perfluoroalkyl acid exposures are associated with liver disease characterized by apoptosis and altered serum adipocytokines. Environ. Pollut. 247, 1055–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behr A.-C., Plinsch C., Braeuning A., Buhrke T. (2020). Activation of human nuclear receptors by perfluoroalkylated substances (PFAS). Toxicol. In Vitro 62, 104700. [DOI] [PubMed] [Google Scholar]
- *Bijland S., Rensen P. C. N., Pieterman E. J., Maas A. C. E., van der Hoorn J. W., van Erk M. J., Havekes L. M., Willems van Dijk K., Chang S.-C., Ehresman D. J., et al. (2011). Perfluoroalkyl sulfonates cause alkyl chain length-dependent hepatic steatosis and hypolipidemia mainly by impairing lipoprotein production in APOE3-Leiden CETP mice. Toxicol. Sci. 123, 290–303. [DOI] [PubMed] [Google Scholar]
- Bjork J. A., Wallace K. B. (2009). Structure-activity relationships and human relevance for perfluoroalkyl acid–induced transcriptional activation of peroxisome proliferation in liver cell cultures. Toxicol. Sci. 111, 89–99. [DOI] [PubMed] [Google Scholar]
- Cao J., Meng S., Chang E., Beckwith-Fickas K., Xiong L., Cole R. N., Radovick S., Wondisford F. E., He L. (2014). Low concentrations of metformin suppress glucose production in hepatocytes through amp-activated protein kinase (AMPK). J. Biol. Chem. 289, 20435–20446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cave M., Deaciuc I., Mendez C., Song Z., Joshi-Barve S., Barve S., McClain C. (2007). Nonalcoholic fatty liver disease: Predisposing factors and the role of nutrition. J. Nutr. Biochem. 18, 184–195. [DOI] [PubMed] [Google Scholar]
- Chalasani N., Younossi Z., Lavine J. E., Charlton M., Cusi K., Rinella M., Harrison S. A., Brunt E. M., Sanyal A. J. (2018). The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 67, 328–357. [DOI] [PubMed] [Google Scholar]
- Colman R. J., Anderson R. M., Johnson S. C., Kastman E. K., Kosmatka K. J., Beasley T. M., Allison D. B., Cruzen C., Simmons H. A., Kemnitz J. W., et al. (2009). Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 325, 201–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrow L. A., Groth A. C., Winquist A., Shin H.-M., Bartell S. M., Steenland K. (2016). Modeled perfluorooctanoic acid (PFOA) exposure and liver function in a mid-ohio valley community. Environ. Health Perspect. 124, 1227–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y., Shi X., Liu C., Yu K., Zhou B. (2009). Chronic effects of water-borne PFOS exposure on growth, survival and hepatotoxicity in zebrafish: A partial life-cycle test. Chemosphere 74, 723–729. [DOI] [PubMed] [Google Scholar]
- Eguchi A., Povero D., Alkhouri N., Feldstein A. E. (2013). Novel therapeutic targets for nonalcoholic fatty liver disease. Expert Opin. Ther. Targets 17, 773–779. [DOI] [PubMed] [Google Scholar]
- Fenton S. E., Ducatman A., Boobis A., DeWitt J. C., Lau C., Ng C., Smith J. S., Roberts S. M. (2021). Per- and polyfluoroalkyl substance toxicity and human health review: Current state of knowledge and strategies for informing future research. Environ. Toxicol. Chem. 40, 606–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filgo A. J., Quist E. M., Hoenerhoff M. J., Brix A. E., Kissling G. E., Fenton S. E. (2015). Perfluorooctanoic acid (PFOA)-induced liver lesions in two strains of mice following developmental exposures: PPARα is not required. Toxicol. Pathol. 43, 558–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana L., Klein S., Holloszy J. O. (2010). Effects of long-term calorie restriction and endurance exercise on glucose tolerance, insulin action, and adipokine production. Age 32, 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana L., Meyer T. E., Klein S., Holloszy J. O. (2004). Long-term calorie restriction is highly effective in reducing the risk for atherosclerosis in humans. Proc. Natl. Acad. Sci. U.S.A. 101, 6659–6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z. D., Klaassen C. D. (2013). Increased bile acids in enterohepatic circulation by short-term calorie restriction in male mice. Toxicol. Appl. Pharmacol. 273, 680–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco M., Sartorelli V. (2008). Comparing and contrasting the roles of AMPK and SIRT1 in metabolic tissues. Cell Cycle 7, 3669–3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo V., Leonardi G., Genser B., Lopez-Espinosa M.-J., Frisbee S. J., Karlsson L., Ducatman A. M., Fletcher T. (2012). Serum perfluorooctanoate (PFOA) and perfluorooctane sulfonate (PFOS) concentrations and liver function biomarkers in a population with elevated PFOA exposure. Environ. Health Perspect. 120, 655–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleason J. A., Post G. B., Fagliano J. A. (2015). Associations of perfluorinated chemical serum concentrations and biomarkers of liver function and uric acid in the US population (NHANES), 2007–2010. Environ. Res. 136, 8–14. [DOI] [PubMed] [Google Scholar]
- Godoy P., Hewitt N. J., Albrecht U., Andersen M. E., Ansari N., Bhattacharya S., Bode J. G., Bolleyn J., Borner C., Böttger J., et al. (2013). Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch. Toxicol. 87, 1315–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha S.-K., Kim J., Chae C. (2011). Role of AMP-activated protein kinase and adiponectin during development of hepatic steatosis in high-fat diet-induced obesity in rats. J. Comp. Pathol. 145, 88–94. [DOI] [PubMed] [Google Scholar]
- Huang Q., Chen L., Teng H., Song H., Wu X., Xu M. (2015). Phenolic compounds ameliorate the glucose uptake in HepG2 cells’ insulin resistance via activating AMPK: Anti-diabetic effect of phenolic compounds in HepG2 cells. J. Funct. Foods 19, 487–494. [Google Scholar]
- Jin R., McConnell R., Catherine C., Xu S., Walker D. I., Stratakis N., Jones D. P., Miller G. W., Peng C., Conti D. V., et al. (2020). Perfluoroalkyl substances and severity of nonalcoholic fatty liver in children: An untargeted metabolomics approach. Environ. Int. 134, 105220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johari M. I., Yusoff K., Haron J., Nadarajan C., Ibrahim K. N., Wong M. S., Hafidz M. I. A., Chua B. E., Hamid N., Arifin W. N., et al. (2019). A randomised controlled trial on the effectiveness and adherence of modified alternate-day calorie restriction in improving activity of. Non-Alcoholic Fatty Liver Disease. Sci. Rep. 9, 11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kani A. H., Alavian S. M., Esmaillzadeh A., Adibi P., Haghighatdoost F., Azadbakht L. (2017). Effects of a low-calorie, low-carbohydrate soy containing diet on systemic inflammation among patients with nonalcoholic fatty liver disease: A parallel randomized clinical trial. Horm. Metab. Res. 49, 687–692. [DOI] [PubMed] [Google Scholar]
- Koubova J., Guarente L. (2003). How does calorie restriction work? Genes Dev. 17, 313–321. [DOI] [PubMed] [Google Scholar]
- Lam B., Younossi Z. M. (2010). Treatment options for nonalcoholic fatty liver disease. Ther. Adv. Gastroenterol. 3, 121–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson-Meyer D. E., Newcomer B. R., Heilbronn L. K., Volaufova J., Smith S. R., Alfonso A. J., Lefevre M., Rood J. C., Williamson D. A., et al. (2008). Effect of 6-month calorie restriction and exercise on serum and liver lipids and markers of liver function. Obesity 16, 1355–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larter C. Z., Yeh M. M., Haigh W. G., Van Rooyen D. M., Brooling J., Heydet D., Nolan C. J., Teoh N. C., Farrell G. C. (2013). Dietary modification dampens liver inflammation and fibrosis in obesity-related fatty liver disease. Obesity 21, 1189–1199. [DOI] [PubMed] [Google Scholar]
- Lau C., Anitole K., Hodes C., Lai D., Pfahles-Hutchens A., Seed J. (2007). Perfluoroalkyl acids: A review of monitoring and toxicological findings. Toxicol. Sci. 99, 366–394. [DOI] [PubMed] [Google Scholar]
- Li Y., Xu S., Mihaylova M., Zheng B., Hou X., Jiang B., Park O., Luo Z., Lefai E., Shyy J. Y.-J., et al. (2011). AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin resistant mice. Cell Metab. 13, 376–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C.-Y., Lin L.-Y., Chiang C.-K., Wang W.-J., Su Y.-N., Hung K.-Y., Chen P.-C. (2010). Investigation of the associations between low-dose serum perfluorinated chemicals and liver enzymes in US adults. Am. J. Gastroenterol. 105, 1354–1363. [DOI] [PubMed] [Google Scholar]
- Liu C., Xu X., Bai Y., Wang T.-Y., Rao X., Wang A., Sun L., Ying Z., Gushchina L., Maiseyeu A., et al. (2014). Air pollution–mediated susceptibility to inflammation and insulin resistance: Influence of CCR2 pathways in mice. Environ. Health Perspect. 122, 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luebker D. J., York R. G., Hansen K. J., Moore J. A., Butenhoff J. L. (2005). Neonatal mortality from in utero exposure to perfluorooctanesulfonate (PFOS) in Sprague-Dawley rats: Dose-response, and biochemical and pharamacokinetic parameters. Toxicology 215, 149–169. [DOI] [PubMed] [Google Scholar]
- Marques E., Pfohl M., Auclair A., Jamwal R., Barlock B. J., Sammoura F. M., Goedken M., Akhlaghi F., Slitt A. L. (2020). Perfluorooctanesulfonic acid (PFOS) administration shifts the hepatic proteome and augments dietary outcomes related to hepatic steatosis in mice. Toxicol. Appl. Pharmacol. 408, 115250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masarone M., Federico A., Abenavoli L., Loguercio C., Persico M. (2015). Non alcoholic fatty liver: Epidemiology and natural history. Rev. Recent Clin. Trials 9, 126–133. [DOI] [PubMed] [Google Scholar]
- Maslak E., Zabielski P., Kochan K., Kus K., Jasztal A., Sitek B., Proniewski B., Wojcik T., Gula K., Kij A., et al. (2015). The liver-selective NO donor, V-PYRRO/NO, protects against liver steatosis and improves postprandial glucose tolerance in mice fed high fat diet. Biochem. Pharmacol. 93, 389–400. [DOI] [PubMed] [Google Scholar]
- More V. R., Xu J., Shimpi P. C., Belgrave C., Luyendyk J. P., Yamamoto M., Slitt A. L. (2013). Keap1 knockdown increases markers of metabolic syndrome after long-term high fat diet feeding. Free Radic. Biol. Med. 61, 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik A., Belič A., Zanger U. M., Rozman D. (2013). Molecular interactions between NAFLD and xenobiotic metabolism. Front. Genet. 4, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerstedt A., Johansson A., Andersson C. X., Cansby E., Smith U., Mahlapuu M. (2010). AMP-activated protein kinase inhibits IL-6-stimulated inflammatory response in human liver cells by suppressing phosphorylation of signal transducer and activator of transcription 3 (STAT3). Diabetologia 53, 2406–2416. [DOI] [PubMed] [Google Scholar]
- Nies A. T., Hofmann U., Resch C., Schaeffeler E., Rius M., Schwab M. (2011). Proton pump inhibitors inhibit metformin uptake by organic cation transporters (OCTs). PLoS One 6, e22163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S., Tanaka K., Tsujimoto T., So R., Shida T., Shoda J. (2014). Regular exercise coupled to diet regimen accelerates reduction of hepatic steatosis and associated pathological conditions in nonalcoholic fatty liver disease. Metab Syndr Relat. Disord. 12, 290–298. [DOI] [PubMed] [Google Scholar]
- Pasachan T., Duangjai A., Ontawong A., Amornlerdpison D., Jinakote M., Phatsara M., Soodvilai S., Srimaroeng C. (2021). Tiliacora triandra (Colebr.) diels leaf aqueous extract inhibits hepatic glucose production in HepG2 cells and type 2 diabetic rats. Molecules 26, 1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyzos S. A., Kountouras J., Zavos C. (2009). Nonalcoholic fatty liver disease: The pathogenetic roles of insulin resistance and adipocytokines. Curr. Mol. Med. 9, 299–314. [DOI] [PubMed] [Google Scholar]
- Qiao Y., Xu Q., Feng W., Tao L., Li X.-N., Liu J., Zhu H., Lu Y., Wang J., Qi C., et al. (2019). Asperpyridone A: an unusual pyridone alkaloid exerts hypoglycemic activity through the insulin signaling pathway. J. Nat. Prod. 82, 2925–2930. [DOI] [PubMed] [Google Scholar]
- Qiu T., Chen M., Sun X., Cao J., Feng C., Li D., Wu W., Jiang L., Yao X. (2016). Perfluorooctane sulfonate-induced insulin resistance is mediated by protein kinase B pathway. Biochem. Biophys. Res. Commun. 477, 781–785. [DOI] [PubMed] [Google Scholar]
- Reddy J. K., Rao M. S. (2006). Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G852–858. [DOI] [PubMed] [Google Scholar]
- Rosen M. B., Schmid J. R., Corton J. C., Zehr R. D., Das K. P., Abbott B. D., Lau C. (2010). Gene expression profiling in wild-type and PPARα-null mice exposed to perfluorooctane sulfonate reveals PPARα-independent effects. PPAR Res. 2010, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenger K. J. P., Allard J. P. (2014). Clinical approaches to non-alcoholic fatty liver disease. World J. Gastroenterol. 20, 1712–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwimmer J. B., Ugalde-Nicalo P., Welsh J. A., Angeles J. E., Cordero M., Harlow K. E., Alazraki A., Durelle J., Knight-Scott J., Newton K. P., et al. (2019). Effect of a low free sugar diet vs usual diet on nonalcoholic fatty liver disease in adolescent boys: A randomized clinical trial. JAMA 321, 256–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seacat A. M., Thomford P. J., Hansen K. J., Clemen L. A., Eldridge S. R., Elcombe C. R., Butenhoff J. L. (2003). Sub-chronic dietary toxicity of potassium perfluorooctanesulfonate in rats. Toxicology 183, 117–131. [DOI] [PubMed] [Google Scholar]
- Seacat A. M., Thomford P. J., Hansen K. J., Olsen G. W., Case M. T., Butenhoff J. L. (2002). Subchronic toxicity studies on perfluorooctanesulfonate potassium salt in cynomolgus monkeys. Toxicol. Sci. 68, 249–264. [DOI] [PubMed] [Google Scholar]
- Sefried S., Häring H.-U., Weigert C., Eckstein S. S. (2018). Suitability of hepatocyte cell lines HepG2, AML12 and THLE-2 for investigation of insulin signalling and hepatokine gene expression. Open Biol. 8, 180147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo W.-D., Lee J. H., Jia Y., Wu C., Lee S.-J. (2015). Saponarin activates AMPK in a calcium-dependent manner and suppresses gluconeogenesis and increases glucose uptake via phosphorylation of CRTC2 and HDAC5. Bioorg. Med. Chem. Lett. 25, 5237–5242. [DOI] [PubMed] [Google Scholar]
- Smith B. W., Adams L. A. (2011). Nonalcoholic fatty liver disease and diabetes mellitus: Pathogenesis and treatment. Nat. Rev. Endocrinol. 7, 456–465. [DOI] [PubMed] [Google Scholar]
- Straznicky N. E., Lambert E. A., Grima M. T., Eikelis N., Nestel P. J., Dawood T., Schlaich M. P., Masuo K., Chopra R., Sari C. I., et al. (2012). The effects of dietary weight loss with or without exercise training on liver enzymes in obese metabolic syndrome subjects. Diabetes Obes. Metab. 14, 139–148. [DOI] [PubMed] [Google Scholar]
- Takacs M. L., Abbott B. D. (2007). Activation of mouse and human peroxisome proliferator-activated receptors (alpha, beta/delta, gamma) by perfluorooctanoic acid and perfluorooctane sulfonate. Toxicol. Sci. 95, 108–117. [DOI] [PubMed] [Google Scholar]
- Tamura Y., Tanaka Y., Sato F., Choi J. B., Watada H., Niwa M., Kinoshita J., Ooka A., Kumashiro N., Igarashi Y., et al. (2005). Effects of diet and exercise on muscle and liver intracellular lipid contents and insulin sensitivity in type 2 diabetic patients. J. Clin. Endocrinol. Metab. 90, 3191–3196. [DOI] [PubMed] [Google Scholar]
- Tang J., Jia X., Gao N., Wu Y., Liu Z., Lu X., Du Q., He J., Li N., Chen B., et al. (2018). Role of the Nrf2-ARE pathway in perfluorooctanoic acid (PFOA)-induced hepatotoxicity in Rana nigromaculata. Environ. Pollut. 238, 1035–1043. [DOI] [PubMed] [Google Scholar]
- Tomita K., Tamiya G., Ando S., Kitamura N., Koizumi H., Kato S., Horie Y., Kaneko T., Azuma T., Nagata H., et al. (2005). AICAR, an AMPK activator, has protective effects on alcohol-induced fatty liver in rats. Alcohol. Clin. Exp. Res. 29, 240S–245S. [DOI] [PubMed] [Google Scholar]
- Towler M. C., Hardie D. G. (2007). AMP-activated protein kinase in metabolic control and insulin signaling. Circ. Res. 100, 328–341. [DOI] [PubMed] [Google Scholar]
- US EPA. (2016). Health Effects Support Document for Perfluorooctane Sulfonate (PFOS). U.S. Environmental Protection Agency Office of Water, Health and Ecological Criteria Division, Washington, 822-R-16–002, 245 pp. [Google Scholar]
- Viollet B., Foretz M., Guigas B., Horman S., Dentin R., Bertrand L., Hue L., Andreelli F. (2006). Activation of AMP-activated protein kinase in the liver: A new strategy for the management of metabolic hepatic disorders. J. Physiol. 574, 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viollet B., Guigas B., Sanz G. N., Leclerc J., Foretz M., Andreelli F. 2. (2012). Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. 122, 253–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan H. T., Zhao Y. G., Leung P. Y., Wong C. K. C. (2014). Perinatal exposure to perfluorooctane sulfonate affects glucose metabolism in adult offspring. PLoS One 9, e87137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan H. T., Zhao Y. G., Wei X., Hui K. Y., Giesy J. P., Wong C. K. C. (2012). PFOS-induced hepatic steatosis, the mechanistic actions on β-oxidation and lipid transport. Biochim. Biophys. Acta 1820, 1092–1101. [DOI] [PubMed] [Google Scholar]
- Wang C., Li Y., Hao M., Li W. (2018). Astragaloside IV inhibits triglyceride accumulation in insulin-resistant HepG2 cells via AMPK-induced SREBP-1c phosphorylation. Front Pharmacol. 9, 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Meng D., Chang Q., Pan J., Zhang Z., Chen G., Ke Z., Luo J., Shi X. (2010). Arsenic inhibits neurite outgrowth by inhibiting the LKB1-AMPK signaling pathway. Environ. Health Perspect. 118, 627–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver Y. M., Ehresman D. J., Butenhoff J. L., Hagenbuch B. (2010). Roles of rat renal organic anion transporters in transporting perfluorinated carboxylates with different chain lengths. Toxicol. Sci. 113, 305–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Q., Zhan Y., Chen B., Xie B., Fang T., Ravishankar S., Jiang Y. (2020). Assessment of antioxidant and antidiabetic properties of Agaricus blazei Murill extracts. Food Sci. Nutr. 8, 332–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weindruch R., Walford R. L., Fligiel S., Guthrie D. (1986). The retardation of aging in mice by dietary restriction: Longevity, cancer, immunity and lifetime energy intake. J. Nutr. 116, 641–654. [DOI] [PubMed] [Google Scholar]
- Wolf R. M., Oshima K., Canner J. K., Steele K. E. (2019). Impact of a preoperative low-calorie diet on liver histology in patients with fatty liver disease undergoing bariatric surgery. Surg. Obes. Relat. Dis. 15, 1766–1772. [DOI] [PubMed] [Google Scholar]
- Wu X., Xie G., Xu X., Wu W., Yang B. (2018). Adverse bioeffect of perfluorooctanoic acid on liver metabolic function in mice. Environ. Sci. Pollut. Res. Int. 25, 4787–4793. [DOI] [PubMed] [Google Scholar]
- Wu Y., Deng M., Jin Y., Liu X., Mai Z., You H., Mu X., He X., Alharthi R., Kostyniuk D. J., et al. (2019). Toxicokinetics and toxic effects of a Chinese PFOS alternative F-53B in adult zebrafish. Ecotoxicol. Environ. Saf. 171, 460–466. [DOI] [PubMed] [Google Scholar]
- Xu J., Kulkarni S. R., Donepudi A. C., More V. R., Slitt A. L. (2012). Enhanced Nrf2 activity worsens insulin resistance, impairs lipid accumulation in adipose tissue, and increases hepatic steatosis in leptin-deficient mice. Diabetes 61, 3208–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Shimpi P., Armstrong L., Salter D., Slitt A. L. (2016). PFOS induces adipogenesis and glucose uptake in association with activation of Nrf2 signaling pathway. Toxicol. Appl. Pharmacol. 290, 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X., Sha S., Wang Y., Sun X., Cao J., Kang J., Jiang L., Chen M., Ma Y. (2016). Perfluorooctane sulfonate induces autophagy-dependent apoptosis through spinster 1-mediated lysosomal-mitochondrial axis and impaired mitophagy. Toxicol. Sci. 153, 198–211. [DOI] [PubMed] [Google Scholar]
- Yoshimura E., Kumahara H., Tobina T., Matsuda T., Ayabe M., Kiyonaga A., Anzai K., Higaki Y., Tanaka H. (2014). Lifestyle intervention involving calorie restriction with or without aerobic exercise training improves liver fat in adults with visceral adiposity. J. Obes. 2014, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang M., Zuccollo A., Hou X., Nagata D., Walsh K., Herscovitz H., Brecher P., Ruderman N. B., Cohen R. A. (2004). AMP-activated protein kinase is required for the lipid-lowering effect of metformin in insulin-resistant human HepG2 cells. J. Biol. Chem. 279, 47898–47905. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Chen J., Zeng Y., Huang D., Xu Q. (2019). Involvement of AMPK activation in the inhibition of hepatic gluconeogenesis by Ficus carica leaf extract in diabetic mice and HepG2 cells. Biomed. Pharmacother. 109, 188–194. [DOI] [PubMed] [Google Scholar]
- Zhao H., Song L. (2021). TKP, a serine protease from trichosanthes kirilowii, inhibits cell proliferation by blocking aerobic glycolysis in hepatocellular carcinoma cells. Nutr. Cancer, 1–13. doi: 10.1080/01635581.2021.1882508. [DOI] [PubMed] [Google Scholar]
- Zhao W., Zitzow J. D., Weaver Y., Ehresman D. J., Chang S.-C., Butenhoff J. L., Hagenbuch B. (2017). Organic anion transporting polypeptides contribute to the disposition of perfluoroalkyl acids in humans and rats. Toxicol. Sci. 156, 84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G., Myers R., Li Y., Chen Y., Shen X., Fenyk-Melody J., Wu M., Ventre J., Doebber T., Fujii N., et al. (2001). Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 108, 1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.