

Abstract
The aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor well-known for its adaptive role as a sensor of environmental toxicants and mediator of the metabolic detoxification of xenobiotic ligands. In addition, a growing body of experimental data has provided indisputable evidence that the AHR regulates critical functions of cell physiology and embryonic development. Recent studies have shown that the naïve AHR—that is, unliganded to xenobiotics but activated endogenously—has a crucial role in maintenance of embryonic stem cell pluripotency, tissue repair, and regulation of cancer stem cell stemness. Depending on the cellular context, AHR silences the expression of pluripotency genes Oct4 and Nanog and potentiates differentiation, whereas curtailing cellular plasticity and stemness. In these processes, AHR-mediated contextual responses and outcomes are dictated by changes of interacting partners in signaling pathways, gene networks, and cell-type-specific genomic structures. In this review, we focus on AHR-mediated changes of genomic architecture as an emerging mechanism for the AHR to regulate gene expression at the transcriptional level. Collective evidence places this receptor as a physiological hub connecting multiple biological processes whose disruption impacts on embryonic development, tissue repair, and maintenance or loss of stemness.
Keywords: Ah receptor, embryonic development, stemness, tissue repair
Environmental sensing and integration of external cues to mount appropriate responses are critical for adaptation and survival of the organism. The aryl hydrocarbon receptor (AHR), a member of the large bHLH/PAS protein family of biosensors (Bersten et al., 2013), is a ligand-activated transcription factor known to sense pollutants present in the environment and coordinate adaptive responses for their detoxification. This receptor has long been studied for its role in mediating the toxicity of environmentally persistent pollutants such as polycyclic aromatic hydrocarbons, halogenated aromatic hydrocarbons, and coplanar polychlorinated biphenyls (Nebert et al., 2004). Furthermore, studies in Ahr-null mice or using endogenous ligands have suggested a number of regulatory functions in embryonic development and cellular homeostasis (Safe et al., 2018). In addition to in utero embryonic resorption and neonatal death, deletion of the Ahr causes multiorgan defects including reproductive, hepatic, cardiovascular, and immune systems (Fernandez-Salguero et al., 1997). Consistent with these observations, activation of the AHR by exposure to environmental ligands during early embryogenesis disrupts developmental trajectories and causes pathological conditions leading to defective phenotypes. Examples include cleft palate, cardiac insufficiency, hydronephrosis, and craniofacial deformities that are found in dioxin-exposed mice (Yoshioka and Tohyama, 2019). These developmental defects point at the involvement of the Ah receptor in proper ontogenetic processes, suggesting that disruption of AHR signaling derails developmental trajectories during embryogenesis resulting in organ dysfunction.
Recent studies in our laboratory have shown that timely and coordinated expression of the AHR is required to prevent pluripotency loss and cause premature differentiation of mouse embryonic stem (ES) cells. ES cells are resident cells of the embryonic inner cell mass that are capable of self-renewal and with the ability to differentiate into the three germ layers of the embryo proper (ectoderm, endoderm, and mesoderm); termed pluripotency. AHR expression, which under physiological circumstances is actively repressed by pluripotency factors OCT4, NANOG, and SOX2 (Ko et al., 2014), is quickly upregulated during ES cell differentiation, suggesting that the AHR has a regulatory function in the commitment of the differentiating cells to their cellular lineages (Ko and Puga, 2017). If untimely derepressed, however, AHR causes OCT4 and SOX2 downregulation and loss of pluripotency (Ko et al., 2016). This interaction between AHR and pluripotency factors may play a decisive role in the differentiation of preimplantation embryonic cells and be critical for the repair process of injured tissues, as shown in Ahr-null tissues, which express higher levels of SOX2, OCT4, and NANOG and regenerate faster after chemical injury than their wild-type counterparts (Morales-Hernández et al., 2017; Moreno-Marín et al., 2017). This is because AHR expression is required for the adult/somatic stem cells to decide whether to proliferate or differentiate and thus controls the rate of tissue repair (Di Giaimo et al., 2018; Metidji et al., 2018). In cancer stem cells (CSCs) and myeloblastic leukemia cells, AHR drives differentiation by repressing OCT4, NANOG, and SOX2 (Bunaciu and Yen, 2011; Cheng et al., 2015). In contrast, subsequent to AHR activation, the upregulated expression of stem-like genes promotes the expansion and tumorigenicity of breast CSCs and CS-like cells (Al-Dhfyan et al., 2017; Stanford et al., 2016). Variations in the tumor microenvironment, including the types of oncometabolites, epigenetic landscape, and signaling pathways, collectively contribute to diversify the role of the AHR in different CSCs. It is likely that the unique cell-specific interaction patterns established between ligands and the ligand-binding domain of the Ah receptor may dictate its differential functionality in regulating tissue homeostasis and cancer stemness (Giani Tagliabue et al., 2019).
In this short review, we summarize recent findings and discuss the involvement of the AHR in (1) preimplantation embryogenesis, (2) tissue repair, and (3) maintenance of stemness in cancer stem-like cells through the regulation of pluripotency factors. We highlight AHR-modulated chromatin interactions as an emerging mechanism of AHR function.
CROSSTALK BETWEEN THE AHR AND PLURIPOTENCY FACTORS REGULATES MAINTENANCE OF PLURIPOTENCY IN EARLY EMBRYOGENESIS
During differentiation, successive lineage choices orchestrated by lineage-specific transcription factors gradually restrict cellular plasticity (Kalkan and Smith, 2014). In preimplantation embryos, the high levels of Ahr expression found in fertilized eggs become heterogeneous among blastomeres by the 4-cell stage embryos. Although undetectable during cleavage, low levels of Ahr expression are found in early blastocysts and thereafter spread in organ primordia (Ko and Puga, 2017). In parallel with these in vivo observations, Ahr expression is repressed in mouse pluripotent ES cells by direct binding of core pluripotency factors OCT4, NANOG, and SOX2 on the promoter distal silencer domain of the Ahr gene (Ko et al., 2014) (Figure 1A). Release from the repressive mechanism mediated by the Polycomb Group proteins and induction of a paused RNA polymerase II complex, quickly upregulate the expression of the Ahr during differentiation, in strong support of the importance of the AHR in embryonic development (Figure 1B). On the other hand, untimely AHR expression in ES cells lengthens mitotic progression and leads to loss of the pluripotent state, suggesting that repression of the Ahr is required to prevent premature loss of pluripotency and entry in differentiation (Ko et al., 2016). Although Ahr repression in mouse ES cells may be required to maintain the pluripotent state, a recent study has shown that paracrine signaling mediated by a kynurenine/AHR complex is essential to sustain the undifferentiated state of human ES cells, to the extent that the concentration of kynurenine can serve as a marker of the undifferentiated state (Yamamoto et al., 2019). Kynurenine, a metabolite generated from the action of indoleamine 2,3-dioxygenase (IDO1) in the tryptophan catabolic pathway, is also an endogenous AHR ligand secreted by human pluripotent stem cells (PSCs). The kynurenine/AHR complex selectively binds to promoters of self-renewal genes, together with IDO1, and AHR, forming a paracrine signal loop that leads to sustained maintenance of the undifferentiated state (Yamamoto et al., 2019). In contrast, the tryptophan metabolite 2-aminoadipic acid (2-AAA), generated through the kynurenine catabolic pathway by kynurenine aminotransferase 2, has been proposed as a marker for ectodermal differentiation. Upon ectodermal commitment, human PSCs take up less tryptophan and secrete 2-AAA into the media (Yamamoto et al., 2019). Taken together, these results suggest that, AHR may fine-tune the decision between maintenance of pluripotency and differentiation of PSCs. Furthermore, upregulation of IDO1 by the AHR may be responsible for the maintenance of pluripotency at the metabolic level, as IDO1 has been shown to suppress mitochondrial activity and promote glycolysis through increase of the NAD+/NADH ratio (Liu et al., 2019).
Figure 1.

Crosstalk between the AHR and pluripotency factor in pluripotent state and during differentiation. A, In pluripotent ES cells, complexes of OCT4, NANOG, and SOX2 cooperate with Polycomb Group Repressive Complexes PRC1/2 to bind to a distal silencer domain in the Ahr upstream region and actively repress AHR expression. Unproductive RNA polymerase II is paused at the Ahr transcription start site and drives the synthesis of short abortive transcripts. B, During differentiation, AHR expression is derepressed by reversal of repressive marks in the Ahr promoter chromatin, release of pluripotency factors and PcG proteins, binding of Sp factors, establishment of histone marks of open chromatin, and engagement of active RNAPII to drive full-length RNA transcript elongation. C, In human embryonal carcinoma cells, AHR suppresses OCT4 and NANOG expression by binding to flanking Alu elements. This generates short noncoding RNA transcripts that target the degradation of OCT4 and NANOG mRNA through the RISC complex. At the NANOG locus, AHR cooperatively binds with CTCF, thus initiating chromatin looping and heterochromatinization around the NANOG gene leading to silencing of NANOG expression to allow differentiation to proceed.
The AHR controls pluripotency by regulating, even silencing altogether, the expression of the pluripotency factors. Studies in human embryonic carcinoma cells (ECCs) conducted in the Fernandez-Salguero laboratory have shown that during differentiation of human ECCs, Alu-retrotransposons located in the NANOG and OCT4 promoter domains bind AHR and are transcribed by RNA polymerase-III, repressing NANOG and OCT4 in differentiated cells by a mechanism likely to involve processing of the Alu-derived transcripts by non-coding microRNAs (Morales-Hernández et al., 2016). In addition, an interaction between AHR and CTCF was found to form a regional heterochromatin loop anchored at NANOG-flanking Alu elements that silenced NANOG expression (González-Rico et al., 2020; Morales-Hernández et al., 2016) (Figure 1C). Thus, AHR may regulate the function of pluripotency factors and differentiation through more than one mechanism.
In mouse ES cells, an interaction between AHR and the chromatin remodeling NuRD-Sall4 complex upregulates Cdx2 while downregulating Sox17 expression, promoting trophectoderm differentiation and inhibiting primitive endoderm differentiation (Gialitakis et al., 2017). The AHR-NuRD-SALL4 complex alters the expression of differentiation-specific genes that control the appearance of the first embryonic lineage occurring in 8- to 16-cell-stage mouse embryos. It appears that depending on the partner proteins with which it interacts, the AHR complex has a dual function in controlling the balance between maintenance of pluripotency and differentiation. On the one hand, the partnership with SALL4 stabilizes the pluripotent state in ES cells by repressing the expression of trophectodermal and neural genes (Miller et al., 2016; Yuri et al., 2009). On the other hand, the trophectoderm transcriptional program driven by ARID3A may maintain trophoblast self-renewal, including maintenance of Cdx2 expression, and promote further trophoblast differentiation through epigenetic HDAC1/2-dependent acetylation/deacetylation-mediated repression of Oct4 and Nanog (Rhee et al., 2014). Given the function of the NuRD complex in the control of lineage commitment and regulation of chromatin accessibility (Bornelöv et al., 2018; Reynolds et al., 2012), it is possible that the AHR could play a central role in cell fate decisions driven by cofactor-mediated epigenetic changes during early embryonic development.
INTERPLAY BETWEEN AHR AND PLURIPOTENCY FACTORS REGULATES TISSUE REPAIR AND REGENERATION
The adult stem cell population expresses the pluripotency factors OCT4, SOX2, and NANOG to maintain multi- or unipotency and self-renewal (Driessens and Blanpain, 2011; Tsai et al., 2012). A timely and well-orchestrated replenishment of differentiated cells and stroma together with the dedifferentiation of terminally differentiated cells are the main mechanisms of tissue repair and regeneration after pathological injury (Galliot et al., 2017; Merrell and Stanger, 2016). Analogous to its function in ES cells, AHR signaling appears to be involved in the maintenance of the somatic stem cell population. In several types of somatic stem cells and/or progenitors, including basal bronchiolar stem cells, intestinal stem cells, neural progenitor cells, ependymal cell, and hematopoietic stem cells (HSCs), the Ah receptor is required to restrict proliferation and maintain the cell population in a quiescent state (Boitano et al., 2010; Han et al., 2020; Metidji et al., 2018; Morales-Hernández et al., 2017; Singh et al., 2011). It is therefore not surprising that both AHR activation by TCDD and Ahr deletion disrupt proliferation, gene expression, and the repopulation capacity of HSCs (Laiosa et al., 2016; Singh et al., 2009). Indeed, chemical damage in mouse liver and lung is promptly and efficiently repaired in the absence of the AHR (Jackson et al., 2014; Mitchell et al., 2006; Morales-Hernández et al., 2017; Moreno-Marín et al., 2017), whereas AHR activation negatively regulates wound repair and fin regeneration in zebrafish (Carvajal-Gonzalez et al., 2009; Mathew et al., 2009), and liver regeneration after hepatectomy in mice (Jackson et al., 2014). In good agreement with these observations, conditional Ahr knockout in a brain ischemic model in mice has been recently shown to increase proliferation of progenitor cells while reducing inflammatory gliosis, therefore favoring restorative neurogenesis (Chen et al., 2019).
The AHR is expressed in several barrier tissues, including lung, skin, and gut epithelia (Esser and Rannug, 2015), and by regulating the expression of pluripotency factors, it may also modulate tissue repair and regeneration in these tissues. In agreement with this idea, high levels of Sox2, Oct4, and Nanog expression have been found in tissues of Ahr-null mice, including brain, liver, and lung, suggesting that lack of the AHR may expand the population of progenitor or stem-like cells and prime tissues for repair after a pathological insult (Chen et al., 2019; Morales-Hernández et al., 2017; Moreno-Marín et al., 2017) (Figure 2). Expansion of the stem cell population after diethylnitrosamine treatment has been shown to promote tumor development and progression in Ahr−/− livers (Moreno-Marín et al., 2017). This is consistent with findings that Ahr ablation favors proliferation of several types of stem cells and leads to malignant transformation of intestinal stem cells (Han et al., 2020; Metidji et al., 2018) and to loss of barrier and inefficient repair in intestine after microbial infection (Metidji et al., 2018). Therefore, although lack of the receptor allows prompt tissue repair, its presence is required to curtail uncontrolled stem cell proliferation. Consistently, initial downregulation of the Ahr followed by its upregulation has been observed during the repair process of injured zebrafish brain, allowing proliferation of ependymal cells that subsequently transdifferentiate into postmitotic neurons (Di Giaimo et al., 2018). Similarly, mice exposed to naphthalene showed facilitated lung regeneration in the absence of AHR (Morales-Hernández et al., 2017). Conversely, activation of the AHR after TCDD exposure has been shown to delay cell cycle progression by upregulating the G1-phase cyclin-dependent kinase inhibitor p27Kip1 in the regenerating mouse liver (Jackson et al., 2014) (Figure 2). Hence, the AHR may function as a restorative chronometer to control the speed of tissue repair after injury. Modulation of AHR expression may provide a unique opportunity for therapeutic tissue regeneration.
Figure 2.
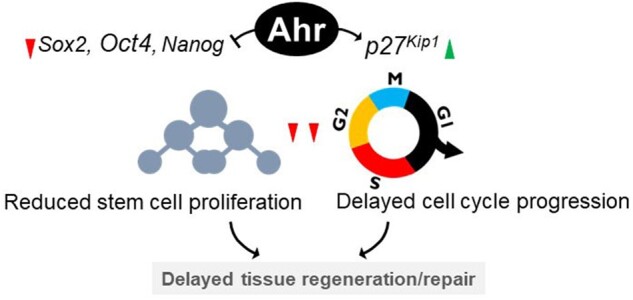
Ahr regulates tissue repair and regeneration. The activated AHR is known to induce the expression of the G1-phase cyclin-dependent kinase inhibitor p27Kip1 thus delaying the cell cycle. In addition, the pluripotency factors OCT4, SOX2, and NANOG are upregulated in the absence of AHR. These changes likely bring about stasis in cell proliferation and depletion of the stem cell pool. Thus, tissue repair proceeds swiftly in the absence of AHR, and lags upon AHR activation.
Besides its interplay with pluripotency factors, the AHR crosstalks with WNT signaling to coordinate responses after tissue damage. AHR activation by TCDD blocks caudal regeneration of zebrafish by upregulating WNT signaling and inducing the expression of R-Spondin1, a novel ligand of the WNT coreceptor (Mathew et al., 2008). Likewise, dietary derivatives that activate Ahr reduce proliferation and tumorigenesis of intestinal stem cells through downregulation of WNT target genes and upregulation of WNT inhibitor proteins (Metidji et al., 2018). Specifically, AHR upregulation suppresses FOXM1, a downstream member of WNT signaling that modulates the nuclear localization of β-catenin (Zhang et al., 2011), leading to depletion of the colon stem cell pool (Han et al., 2020). Considering the involvement of WNT/β-catenin signaling in the mobilization of the stem cell population during regeneration events (Bastakoty and Young, 2016), AHR-mediated downregulation of WNT signaling may represent yet another therapeutic target of tissue regeneration. Repression of AHR signaling may facilitate tissue repair and regeneration by regulating the size of the adult stem cell pool.
AHR REGULATES STEMNESS IN CANCER STEM CELLS IN A CONTEXT-DEPENDENT MANNER
CSCs are a tumorigenic and often chemo-resistant subpopulation of cancer cells responsible for cancer relapse. Multiple lines of evidence suggest that similar to its role in embryonic and adult stem cells, the Ah receptor regulates stemness in CSCs, but unlike in ESCs, the AHR-mediated regulation of stemness in CSCs is either suppressive or promotive depending on the cellular context. The specificity of the AHR function in CSCs depends on the cell type, on the level and type of oncometabolites present in the tumor microenvironment, and on the signaling pathways that the AHR interacts with (Safe et al., 2018).
The AHR functions as a tumor suppressor by inducing the expression of differentiation markers and reducing the expression of pluripotency factors and other CSC markers. Among these, AHR has been shown to induce the differentiation of stem-like glioblastoma cancer cells by repressing OCT4 through direct binding of the AHR to the OCT4 promoter (Cheng et al., 2015). In addition, AHR represses OCT4 and NANOG and other CSC markers, including CD133, ALDH1, BMI1, and MUSASHI-1 and either impairs clonogenic potential or induces differentiation of various CSC subtypes (Bunaciu and Yen, 2011; Prud’Homme et al., 2010; Zhao et al., 2012). Similarly, AHR ablation in melanoma cells upregulates the expression of stemness genes ALDH1A1, CD133, and CD44, and leads to melanosphere formation (Contador-Troca et al., 2013; 2015). The Ah receptor has also been shown to reduce the intranuclear levels of OCT4 in human leukemia cells, leading to loss of stemness and triggering differentiation (Bunaciu and Yen, 2011). To regulate the nuclear translocation of OCT4, the AHR may be responsible for blocking its phosphorylation on threonine residue 235 (Oct4-pT235), a post-translational modification responsible for nuclear translocation of OCT4 (Lin et al., 2012). Indeed, activation of the AHR by 2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE, a tryptophan metabolite and AHR agonist) has been shown to reduce phosphorylation at OCT4-pT235, resulting in inhibition of proliferation and limitation of tumorigenesis in xenografted human hepatocellular carcinoma cells and in human glioblastoma neurospheres (Zhao et al., 2015).
Collectively, these results indicate that the Ah receptor may function as a tumor suppressor by inhibiting the expression of pluripotency factors and markers of CSCs. Yet, it may also behave as a tumor promoter by controlling the WNT-PTEN/PI3K/AKT circuit in CSCs. A strong correlation between AHR overexpression and high levels of WNT5a and β-catenin has been observed in inflammatory CSCs from breast cancer patients (Mohamed et al., 2019). In cells from these patients, the AHR-promoted tumor expansion by activating AKT signaling and upregulating the expression of β-catenin and downstream genes of the WNT pathway while suppressing PTEN (Al-Dhfyan et al., 2017). Similarly, in stem-cell-like choriocarcinoma cells, activation of AHR by TCDD was found to increase their chemoresistance and tumorigenic potential by activating β-catenin (Wu et al., 2018). Thus, upregulation of WNT signaling appears to be required to increase the expression of pluripotency factors OCT4, NANOG, and SOX2 and maintain the CSC population (Lien and Fuchs, 2014), and ALDH1A1 (Cojoc et al., 2015; de Sousa e Melo and Vermeulen, 2016), whereas loss of PTEN activates the AKT pathway to induce expression of ABCG2 (Wang et al., 2019). In addition, stabilization of the AHR by deubiquitylation in human lung cancer cells has been shown to result in its nuclear interaction with IKKα and gene activation of KLF4, c-MYC, ABCG2, and ALDH1 (Ouyang et al., 2020) and increased the expression of ABCG2, c-MYC, KLF4, ALDH1A1, and LGR6 in radioresistant stem-like lung cancer cells by directly binding to their promoter regions (Yan et al., 2018). Apart from the interaction with signaling pathways, activation of the AHR has been shown to increase the nuclear localization of SOX2 and to drive the expansion of human breast cancer cells. By direct binding to the promoter region of SOX2, the AHR activates expression of ALDH1 and pluripotency factors and promotes the trans-differentiation of breast cancer cells (Stanford et al., 2016). Considering that expression of these genes may be activated by direct binding of the AHR to their promoter domains, the tumor-promoting function of the AHR can be the result either of a direct action on the transcription of these genes or of an indirect action through integration with the WNT and PTEN/PI3K/AKT signaling pathways.
GENOMIC ORGANIZATION AS AN EMERGING EPIGENETIC MECHANISM REGULATED BY THE AHR
A growing body of recent experimental evidence supports the contention that the activated AHR, like other ligand-activated nuclear receptors, directs a highly specific genome-wide mechanism of chromatin reorganization. By this mechanism, the AHR establishes local nucleosomal remodeling events to facilitate access to transcription factor binding sites on the promoter domain of its classical target, Cyp1a1 and other genes in the AHR battery (Fujii-Kuriyama and Mimura, 2005). Other long-range conformational changes induced by the activated AHR in the genome have been observed that result in looping of enhancer-promoter chromatin domains at the Cyp1a1 promoter, first observed by the Whitlock group 30 years ago (Elferink and Whitlock, 1990). In agreement, the AHR was shown to regulate chromatin accessibility in innate lymphoid cells at selected enhancer domains, including those in its own locus, through a positive feedback mechanism (Li et al., 2018). Evidence of the involvement of the receptor in chromatin remodeling is further strengthened by a recent study investigating the implication of smooth muscle cell-to-chondromyocyte transition in atherosclerotic tissues. The data support a model whereby the AHR plays a protective role in the maintenance of lesion cap integrity by increasing global chromatin accessibility around chondrocyte marker genes (Kim et al., 2020).
Mechanistically, the AHR partners with the ATP-dependent chromatin remodeling complexes SWI/SNF and NuRD to activate genes responsible for xenobiotic metabolism and embryonic development (Gialitakis et al., 2017; Hankinson, 2005). Furthermore, benzo-a-pyrene-activated AHR binds to the Lymphoid-Specific Helicase (LSH, also known as SMARCA6) promoter, inducing its expression in non-small-cell lung carcinoma (Mao et al., 2018). LSH is also a chromatin remodeling protein that belongs to SNF family, that not only directs DNA methylation (Zhu et al., 2006), but also orchestrates genome-wide nucleosome occupancy changes around putative enhancers and binding sites of lineage specification factors (Ren et al., 2019). It is also important to mention that recent studies conducted in differentiating multipotent bone marrow stromal cells have identified the AHR as a target regulated by super-enhancers. During differentiation of bone marrow stromal cells, 4 super-enhancer clusters on the AHR locus coordinate the downregulation of the AHR, which results in upregulation of adipocyte and osteoblast-specific differentiation genes (Gérard et al., 2019). Lastly, the AHR has also been shown to be the target of active ILC3s and Th17s cell-specific super-enhancers, which control differentiation of IL22-producing cells and their lineage specification (Koues et al., 2016).
In both ES and differentiated cells, AHR binds to genomic loci found at varying distances away from promoter regions of known target genes, suggesting that AHR may direct long-range chromatin interactions upon activation. In an AHR ChIP-seq assay using TCDD-treated MCF7 cells, peaks of AHR binding were predominantly found in intragenic and intergenic areas (Figs. 3A and 3B), indicating that to regulate expression of target genes AHR may preferentially bind to distal regulatory elements (Dere et al., 2011; Fu et al., 2019; Lo and Matthews, 2012; Yang et al., 2018). Furthermore, an exploratory analysis of an enhancer sequencing dataset using MCF7 cells showed that many of the AHR-bound enhancer regions were linked by both the CCCTC-binding factor (CTCF) and the CTCF complex cohesin component, RAD21 (Figure 3C). Bearing in mind the essential function of CTCF in chromatin organization (Merkenschlager and Nora, 2016), it is plausible that the AHR orchestrates global but highly specific enhancer-promoter interactions to facilitate appropriate gene expression changes in response to environmental and developmental stimuli. It is noteworthy that the Mediator complex, which interacts with the CTCF complex to coordinate chromatin looping, is an AHR coactivator that only binds the Cyp1a1 enhancer, but not the promoter (Kagey et al., 2010; Wang et al., 2004). Consistently, other studies have shown an insulator function for the AHR, with SLUG and CTCF cobound to a B1 short interspersed nuclear element retrotransposon (B1-X35S) to demarcate genomic boundaries (Román et al., 2011). Furthermore, during differentiation of human ECCs, AHR and CTCF have been shown to coimmunoprecipitate, cooccupying two NANOG-flanking Alu elements, initiating heterochromatinization and silencing NANOG expression (González-Rico et al., 2020). Studies examining genome-wide binding sites of the AHR showed that, in fact, CTCF was called among the top 10 motifs in AHR ChIP-seq peaks, suggesting AHR-CTCF cooccupancy or potential synergy between AHR and CTCF by AHR-directed recruitment of CTCF to is binding sites (Lo and Matthews, 2012; Yang et al., 2018). Hence, it is possible that AHR directs long-range chromatin contacts through CTCF to influence expression of target genes.
Figure 3.

Cooccupancy of AHR-binding sites by complexes of AHR and CTCF. A, Statistics summarizing locations of AHR binding sites in TCDD-treated MCF7 cells. Public sequencing data was downloaded from Geo Omnibus where it is available with the accession number GSE90550 (Yang et al. 2018). B, AHR, CTCF, and RAD21 binding profile across 47 dioxin-responsive genes in TCDD-treated MCF7 cells (Tomblin et al. 2016). Strongest AHR binding was observed in gene bodies, which were cooccupied by CTCF and RAD21. SRA database files SRR5057971, SRR10096218, and SRR10096220 were downloaded from http://ncbi.github.io/sra-tools/ and used to generate bam files. DeepTools was used to generate the 1× RPGC-normalized bigwig files from the bam files and to calculate signal values. C, AHR, CTCF, and RAD21 binding across candidate enhancer locations in MCF7 cells. AHR cooperatively binds the enhancers with CTCF and RAD21. MCF7 STARR-seq databases obtained from ENCODE (ENCFF356ZLC) and were analyzed the same way as in Figure 2B. TSS: transcription start site, TTS: transcription termination site, TES: transcription end site.
CONCLUSIONS AND FUTURE DIRECTIONS
It is crucial to explore the precise mechanisms through which the AHR regulates the expression of target genes during differentiation of early embryonic cells, during the regenerative events regulating the tissue repair processes, and during the maintenance versus loss of tumorigenic potential of CSCs. As shown in earlier studies, most of AHR-bound chromatin domains lack canonical AHR binding motifs, implying that AHR interacts with DNA though other protein complexes or by chromatin reorganization (Dere et al., 2011). Therefore, a precise AHR interactome map is needed that integrates both enhancer activity assays and genome-wide conformation data under a variety of metabolizable and nonmetabolizable ligands. Correlation between the AHR interactome map with transcriptional and chromatin changes will allow us to identify key signaling pathways directly regulated by the AHR. Furthermore, 3-dimensional chromatin conformation studies are also necessary to determine AHR-directed enhancer-promoter interactions and to delineate AHR-regulated transcriptional hubs during embryonic development, tissue repair, and tumorigenesis. This mechanistic approach will give us the needed information that will allow us to understand the role of this receptor in health and disease and open avenues of access to intervention. Another important question revolves around the role of endogenous ligands on AHR function. It remains to be known which specific ligand(s) drive AHR action during early embryonic development in tissue homeostasis and cancer biology, as well as their nature and sources. When well characterized, endogenous ligands will present an excellent therapeutic opportunity to modulate AHR function.
ACKNOWLEDGMENTS
We thank Dr Ying Xia for a critical reading of the manuscript.
FUNDING
National Institute of Environmental Health Sciences (R01 ES024744, ES010807 and P30 ES06096).
DECLARATION OF CONFLICTING INTERESTS
The authors indicate no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
REFERENCES
- Al-Dhfyan A., Alhoshani A., Korashy H. M. (2017). Aryl hydrocarbon receptor/cytochrome P450 1A1 pathway mediates breast cancer stem cells expansion through PTEN inhibition and β-Catenin and Akt activation. Mol. Cancer 16, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastakoty D., Young P. P. (2016). Wnt/β-catenin pathway in tissue injury: Roles in pathology and therapeutic opportunities for regeneration. Faseb J. 30, 3271–3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersten D. C., Sullivan A. E., Peet D. J., Whitelaw M. L. (2013). BHLH-PAS proteins in cancer. Nat. Rev. Cancer 13, 827–841. [DOI] [PubMed] [Google Scholar]
- Boitano A. E., Wang J., Romeo R., Bouchez L. C., Parker A. E., Sutton S. E., Walker J. R., Flaveny C. A., Perdew G. H., Denison M. S., et al. (2010). Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science 329, 1345–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornelöv S., Reynolds N., Xenophontos M., Gharbi S., Johnstone E., Floyd R., Ralser M., Signolet J., Loos R., Dietmann S., et al. (2018). The nucleosome remodeling and deacetylation complex modulates chromatin structure at sites of active transcription to fine-tune gene expression. Mol. Cell 71, 56–72.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunaciu R. P., Yen A. (2011). Activation of the aryl hydrocarbon receptor Ahr promotes retinoic acid-induced differentiation of myeloblastic leukemia cells by restricting expression of the stem cell transcription factor Oct4. Cancer Res. 71, 2371–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal-Gonzalez J. M., Roman A. C., Cerezo-Guisado M. I., Rico-Leo E. M., Martin-Partido G., Fernandez-Salguero P. M. (2009). Loss of dioxin-receptor expression accelerates wound healing in vivo by a mechanism involving TGFβ. J. Cell Sci. 122, 1823–1833. [DOI] [PubMed] [Google Scholar]
- Chen W. C., Chang L. H., Huang S. S., Huang Y. J., Chih C. L., Kuo H. C., Lee Y. H., Lee I. H. (2019). Aryl hydrocarbon receptor modulates stroke-induced astrogliosis and neurogenesis in the adult mouse brain. J. Neuroinflam. 16, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J., Li W., Kang B., Zhou Y., Song J., Dan S., Yang Y., Zhang X., Li J., Yin S., et al. (2015). Tryptophan derivatives regulate the transcription of Oct4 in stem-like cancer cells. Nat. Commun. 6, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cojoc M., Peitzsch C., Kurth I., Trautmann F., Kunz-Schughart L. A., Telegeev G. D., Stakhovsky E. A., Walker J. R., Simin K., Lyle S., et al. (2015). Aldehyde dehydrogenase is regulated by β-catenin/TCF and promotes radioresistance in prostate cancer progenitor cells. Cancer Res. 75, 1482–1494. [DOI] [PubMed] [Google Scholar]
- Contador-Troca M., Alvarez-Barrientos A., Barrasa E., Rico-Leo E. M., Catalina-Fernández I., Menacho-Márquez M., Bustelo X. R., García-Borrón J. C., Gómez-Durán A., Sáenz-Santamaría J., et al. (2013). The dioxin receptor has tumor suppressor activity in melanoma growth and metastasis. Carcinogenesis 34, 2683–2693. [DOI] [PubMed] [Google Scholar]
- Contador-Troca M., Alvarez-Barrientos A., Merino J. M., Morales-Hernández A., Rodríguez M. I., Rey-Barroso J., Barrasa E., Cerezo-Guisado M. I., Catalina-Fernández I., Sáenz-Santamaría J., et al. (2015). Dioxin receptor regulates aldehyde dehydrogenase to block melanoma tumorigenesis and metastasis. Mol. Cancer 14, 148–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dere E., Lo R., Celius T., Matthews J., Zacharewski T. R. (2011). Integration of genome-wide computation DRE search, AhR ChIP-chip and gene expression analyses of TCDD-elicited responses in the mouse liver. BMC Genomics 12, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Sousa e Melo F., Vermeulen L. (2016). Wnt signaling in cancer stem cell biology. Cancers 8, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giaimo R., Durovic T., Barquin P., Kociaj A., Lepko T., Aschenbroich S., Breunig C. T., Irmler M., Cernilogar F. M., Schotta G., et al. (2018). The aryl hydrocarbon receptor pathway defines the time frame for restorative neurogenesis. Cell Rep. 25, 3241–3251.e5. [DOI] [PubMed] [Google Scholar]
- Driessens G., Blanpain C. (2011). Long live Sox2: Sox2 lasts a lifetime. Cell Stem Cell 9, 283–284. [DOI] [PubMed] [Google Scholar]
- Elferink C. J., Whitlock J. P. (1990). 2,3,7,8-Tetrachlorodibenzo-p-dioxin-inducible, Ah receptor-mediated bending of enhancer DNA. J. Biol. Chem. 265, 5718–5721. [PubMed] [Google Scholar]
- Esser C., Rannug A. (2015). The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol. Rev. 67, 259–279. [DOI] [PubMed] [Google Scholar]
- Fernandez-Salguero P. M., Ward J. M., Sundberg J. P., Gonzalez F. J. (1997). Lesions of aryl-hydrocarbon receptor-deficient mice. Vet. Pathol. 34, 605–614. [DOI] [PubMed] [Google Scholar]
- Fu H., Wang L., Wang J., Bennett B. D., Li J. L., Zhao B., Hu G. (2019). Dioxin and AHR impairs mesoderm gene expression and cardiac differentiation in human embryonic stem cells. Sci. Total Environ. 651, 1038–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii-Kuriyama Y., Mimura J.. 2005. Molecular mechanisms of AhR functions in the regulation of cytochrome P450 genes. Biochem. Biophys. Res. Commun. 338, 311–317. [DOI] [PubMed]
- Galliot B., Crescenzi M., Jacinto A., Tajbakhsh S. (2017). Trends in tissue repair and regeneration. Development 144, 357–364. [DOI] [PubMed] [Google Scholar]
- Gérard D., Schmidt F., Ginolhac A., Schmitz M., Halder R., Ebert P., Schulz M. H., Sauter T., Sinkkonen L. (2019). Temporal enhancer profiling of parallel lineages identifies AHR and GLIS1 as regulators of mesenchymal multipotency. Nucleic Acids Res. 47, 1141–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gialitakis M., Tolaini M., Li Y., Pardo M., Yu L., Toribio A., Choudhary J. S., Niakan K., Papayannopoulos V., Stockinger B. (2017). Activation of the aryl hydrocarbon receptor interferes with early embryonic development. Stem Cell Rep. 9, 1377–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giani Tagliabue S., Faber S. C., Motta S., Denison M. S., Bonati L. (2019). Modeling the binding of diverse ligands within the Ah receptor ligand binding domain. Sci. Rep. 9, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Rico F. J., Vicente-García C., Fernández A., Muñoz-Santos D., Montoliu L., Morales-Hernández A., Merino J. M., Román A. C., Fernández-Salguero P. M. (2020). Alu retrotransposons modulate Nanog expression through dynamic changes in regional chromatin conformation via aryl hydrocarbon receptor. Epigenet. Chromat. 13, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H., Davidson L. A., Fan Y., Goldsby J. S., Yoon G., Jin U., Wright G. A., Landrock K. K., Weeks B. R., Wright R. C., et al. (2020). Loss of aryl hydrocarbon receptor potentiates FoxM1 signaling to enhance self‐renewal of colonic stem and progenitor cells. Embo J. 39, e104319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hankinson O. (2005). Role of coactivators in transcriptional activation by the aryl hydrocarbon receptor. Arch. Biochem. Biophys. 433, 379–386. [DOI] [PubMed] [Google Scholar]
- Jackson D. P., Li H., Mitchell K. A., Joshi A. D., Elferink C. J. (2014). Ah receptor-mediated suppression of liver regeneration through NC-XRE-driven p21Cip1 expression. Mol. Pharmacol. 85, 533–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagey M. H., Newman J. J., Bilodeau S., Zhan Y., Orlando D. A., Van Berkum N. L., Ebmeier C. C., Goossens J., Rahl P. B., Levine S. S., et al. (2010). Mediator and cohesin connect gene expression and chromatin architecture. Nature 467, 430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalkan T., Smith A. (2014). Mapping the route from naive pluripotency to lineage specification. Philos. Trans. R. Soc. B Biol. Sci. 369, e20130540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. B., Zhao Q., Nguyen T., Pjanic M., Cheng P., Wirka R., Travisano S., Nagao M., Kundu R., Quertermous T. (2020). Environment-sensing aryl hydrocarbon receptor inhibits the chondrogenic fate of modulated smooth muscle cells in atherosclerotic lesions. Circulation 142, 575–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko C. I., Fan Y., de Gannes M., Wang Q., Xia Y., Puga A. (2016). Repression of the aryl hydrocarbon receptor is required to maintain mitotic progression and prevent loss of pluripotency of embryonic stem cells. Stem Cells 34, 2825–2839. [DOI] [PubMed] [Google Scholar]
- Ko C. I., Puga A. (2017). Does the aryl hydrocarbon receptor regulate pluripotency? Curr. Opin. Toxicol. 2, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko C. I., Wang Q., Fan Y., Xia Y., Puga A. (2014). Pluripotency factors and Polycomb Group proteins repress aryl hydrocarbon receptor expression in murine embryonic stem cells. Stem Cell Res. 12, 296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koues O. I., Collins P. L., Cella M., Robinette M. L., Porter S. I., Pyfrom S. C., Payton J. E., Colonna M., Oltz E. M. (2016). Distinct gene regulatory pathways for human innate versus adaptive lymphoid cells. Cell 165, 1134–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laiosa M. D., Tate E. R., Ahrenhoerster L. S., Chen Y., Wang D. (2016). Effects of developmental activation of the aryl hydrocarbon receptor by 2,3,7,8-tetrachlorodibenzo-p-dioxin on long term self renewal of murine hematopoietic stem cells. Environ. Health Perspect. 124, 957–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Bostick J. W., Ye J., Qiu J., Zhang B., Urban J. F., Avram D., Zhou L. (2018). Aryl hydrocarbon receptor signaling cell intrinsically inhibits intestinal group 2 innate lymphoid cell function. Immunity 49, 915–928.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien W. H., Fuchs E. (2014). Wnt some lose some: Transcriptional governance of stem cells by Wnt/β-catenin signaling. Genes Dev. 28, 1517–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y., Yang Y., Li W., Chen Q., Li J., Pan X., Zhou L., Liu C., Chen C., He J., et al. (2012). Reciprocal regulation of Akt and Oct4 promotes the self-renewal and survival of embryonal carcinoma cells. Mol. Cell 48, 627–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Wang M., Jiang T., He J., Fu X., Xu Y. (2019). IDO1 maintains pluripotency of primed human embryonic stem cells by promoting glycolysis. Stem Cells 37, 1158–1165. [DOI] [PubMed] [Google Scholar]
- Lo R., Matthews J. (2012). High-resolution genome-wide Mapping of AHR and ARNT binding sites by ChIP-Seq. Toxicol. Sci. 130, 349–361. [DOI] [PubMed] [Google Scholar]
- Mao C., Wang M., Qian B., Ouyang L., Shi Y., Liu N., Chen L., Xiao D., Wang X., Cao Y., et al. (2018). Aryl hydrocarbon receptor activated by benzo (a) pyrene promotes SMARCA6 expression in NSCLC. Am. J. Cancer Res. 8, 1214–1227 [PMC free article] [PubMed] [Google Scholar]
- Mathew L. K., Sengupta S. S., LaDu J., Andreasen E. A., Tanguay R. L. (2008). Crosstalk between AHR and Wnt signaling through R‐Spondin1 impairs tissue regeneration in zebrafish. Faseb J. 22, 3087–3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew L. K., Simonich M. T., Tanguay R. L. (2009). AHR-dependent misregulation of Wnt signaling disrupts tissue regeneration. Biochem. Pharmacol. 77, 498–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkenschlager M., Nora E. P. (2016). CTCF and cohesin in genome folding and transcriptional gene regulation. Annu. Rev. Genomics Hum. Genet. 17, 17–43. [DOI] [PubMed] [Google Scholar]
- Merrell A. J., Stanger B. Z. (2016). Adult cell plasticity in vivo: De-differentiation and transdifferentiation are back in style. Nat. Rev. Mol. Cell Biol. 17, 413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metidji A., Omenetti S., Crotta S., Li Y., Nye E., Ross E., Li V., Maradana M. R., Schiering C., Stockinger B. (2018). The environmental sensor AHR protects from inflammatory damage by maintaining intestinal stem cell homeostasis and barrier integrity. Immunity 49, 353–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller A., Ralser M., Kloet S. L., Loos R., Nishinakamura R., Bertone P., Vermeulen M., Hendrich B. (2016). Sall4 controls differentiation of pluripotent cells independently of the nucleosome remodelling and deacetylation (NuRD) complex. Development 143, 3074–3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell K. A., Lockhart C. A., Huang G., Elferink C. J. (2006). Sustained aryl hydrocarbon receptor activity attenuates liver regeneration. Mol. Pharmacol. 70, 163–170. [DOI] [PubMed] [Google Scholar]
- Mohamed H. T., Gadalla R., El-Husseiny N., Hassan H., Wang Z., Ibrahim S. A., El-Shinawi M., Sherr D. H., Mohamed M. M. (2019). Inflammatory breast cancer: Activation of the aryl hydrocarbon receptor and its target CYP1B1 correlates closely with Wnt5a/b-β-catenin signalling, the stem cell phenotype and disease progression. J. Adv. Res. 16, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales-Hernández A., González-Rico F. J., Román A. C., Rico-Leo E., Alvarez-Barrientos A., Sánchez L., Macia Á., Heras S. R., García-Pérez J. L., Merino J. M., et al. (2016). Alu retrotransposons promote differentiation of human carcinoma cells through the aryl hydrocarbon receptor. Nucleic Acids Res. 44, 4665–4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales-Hernández A., Nacarino-Palma A., Moreno-Marín N., Barrasa E., Paniagua-Quiñones B., Catalina-Fernández I., Alvarez-Barrientos A., Bustelo X. R., Merino J. M., Fernández-Salguero P. M. (2017). Lung regeneration after toxic injury is improved in absence of dioxin receptor. Stem Cell Res. 25, 61–71. [DOI] [PubMed] [Google Scholar]
- Moreno-Marín N., Barrasa E., Morales-Hernández A., Paniagua B., Blanco-Fernández G., Merino J. M., Fernández-Salguero P. M. (2017). Dioxin receptor adjusts liver regeneration after acute toxic injury and protects against liver carcinogenesis. Sci. Rep. 7, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebert D. W., Dalton T. P., Okey A. B., Gonzalez F. J. (2004). Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J. Biol. Chem. 279, 23847–23850. [DOI] [PubMed] [Google Scholar]
- Ouyang L., Yan B., Liu Y., Mao C., Wang M., Liu N., Wang Z., Shouping L., Shi Y., Chen L., et al. (2020). The deubiquitylase UCHL3 maintains cancer stem-like properties by stabilizing the aryl hydrocarbon receptor. Signal Transduct. Target Ther. 5, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prud’Homme G. J., Glinka Y., Toulina A., Ace O., Subramaniam V., Jothy S. (2010). Breast cancer stem-like cells are inhibited by a non-toxic aryl hydrocarbon receptor agonist. PLoS One 5, e00113831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J., Finney R., Ni K., Cam M., Muegge K. (2019). The chromatin remodeling protein Lsh alters nucleosome occupancy at putative enhancers and modulates binding of lineage specific transcription factors. Epigenetics 14, 277–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds N., Latos P., Hynes-Allen A., Loos R., Leaford D., O'Shaughnessy A., Mosaku O., Signolet J., Brennecke P., Kalkan T., et al. (2012). NuRD suppresses pluripotency gene expression to promote transcriptional heterogeneity and lineage commitment. Cell Stem Cell 10, 583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee C., Lee B. K., Beck S., Anjum A., Cook K. R., Popowski M., Tucker H. O., Kim J. (2014). Arid3a is essential to execution of the first cell fate decision via direct embryonic and extraembryonic transcriptional regulation. Genes Dev. 28, 2219–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Román A. C., González-Rico F. J., Moltó E., Hernando H., Neto A., Vicente-Garcia C., Ballestar E., Gómez-Skarmeta J. L., Vavrova-Anderson J., White R. J., et al. (2011). Dioxin receptor and SLUG transcription factors regulate the insulator activity of B1 SINE retrotransposons via an RNA polymerase switch. Genome Res. 21, 422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safe S., Han H., Goldsby J., Mohankumar K., Chapkin R. S. (2018). Aryl hydrocarbon receptor (AhR) ligands as selective AhR modulators: Genomic studies. Curr. Opin. Toxicol. 11–12, 10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K. P., Garrett R. W., Casado F. L., Gasiewicz T. A. (2011). Aryl hydrocarbon receptor-null allele mice have hematopoietic stem/progenitor cells with abnormal characteristics and functions. Stem Cells Dev. 20, 769–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K. P., Wyman A., Casado F. L., Garrett R. W., Gasiewicz T. A. (2009). Treatment of mice with the Ah receptor agonist and human carcinogen dioxin results in altered numbers and function of hematopoietic stem cells. Carcinogenesis 30, 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanford E. A., Wang Z., Novikov O., Mulas F., Landesman-Bollag E., Monti S., Smith B. W., Seldin D. C., Murphy G. J., Sherr D. H. (2016). The role of the aryl hydrocarbon receptor in the development of cells with the molecular and functional characteristics of cancer stem-like cells. BMC Biol. 14, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomblin J. K., Arthur S., Primerano D. A., Chaudhry A. R., Fan J., Denvir J., Salisbury T. B. (2016). Aryl hydrocarbon receptor (AHR) regulation of L-type amino acid transporter 1 (LAT-1) expression in MCF-7 and MDA-MB-231 breast cancer cells. Biochem. Pharmacol. 106, 94–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai C.-C., Su P.-F., Huang Y.-F., Yew T.-L., Hung S.-C. (2012). Molecular cell Oct4 and nanog directly regulate dnmt1 to maintain self-renewal and undifferentiated state in mesenchymal stem cells. Mol. Cell 47, 169–182. [DOI] [PubMed] [Google Scholar]
- Wang L., Lin N., Li Y. (2019). The PI3K/AKT signaling pathway regulates ABCG2 expression and confers resistance to chemotherapy in human multiple myeloma. Oncol. Rep. 41, 1678–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Ge K., Roeder R. G., Hankinson O. (2004). Role of mediator in transcriptional activation by the aryl hydrocarbon receptor. J. Biol. Chem. 279, 13593–13600. [DOI] [PubMed] [Google Scholar]
- Wu C., Yu S., Tan Q., Guo P., Liu H. (2018). Role of AhR in regulating cancer stem cell–like characteristics in choriocarcinoma. Cell Cycle 17, 2309–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T., Hatabayashi K., Arita M., Yajima N., Takenaka C., Suzuki T., Takahashi M., Oshima Y., Hara K., Kagawa K., et al. (2019). Kynurenine signaling through the aryl hydrocarbon receptor maintains the undifferentiated state of human embryonic stem cells. Sci. Signal. 12, 3306. [DOI] [PubMed] [Google Scholar]
- Yan B., Liu S., Shi Y., Liu N., Chen L., Wang X., Xiao D., Liu X., Mao C., Jiang Y., et al. (2018). Activation of AhR with nuclear IKKα regulates cancer stem-like properties in the occurrence of radioresistance. Cell Death Dis. 9, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S. Y., Ahmed S., Satheesh S. V., Matthews J. (2018). Genome-wide mapping and analysis of aryl hydrocarbon receptor (AHR)- and aryl hydrocarbon receptor repressor (AHRR)-binding sites in human breast cancer cells. Arch. Toxicol. 92, 225–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka W., Tohyama C. (2019). Mechanisms of developmental toxicity of dioxins and related compounds. Int. J. Mol. Sci. 20, 617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuri S., Fujimura S., Nimura K., Takeda N., Toyooka Y., Fujimura Y.-I., Aburatani H., Ura K., Koseki H., Niwa H., et al. (2009). Sall4 Is Essential for stabilization, but not for pluripotency, of embryonic stem cells by repressing aberrant trophectoderm gene expression. Stem Cells 27, 796–805. [DOI] [PubMed] [Google Scholar]
- Zhang N., Wei P., Gong A., Chiu W. T., Lee H. T., Colman H., Huang H., Xue J., Liu M., Wang Y., et al. (2011). FoxM1 promotes β-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell 20, 427–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q. W., Zhou Y. W., Li W. X., Kang B., Zhang X. Q., Yang Y., Cheng J., Yin S. Y., Tong Y., He J. Q., et al. (2015). Akt-mediated phosphorylation of Oct4 is associated with the proliferation of stem-like cancer cells. Oncol. Rep. 33, 1621–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S., Kanno Y., Nakayama M., Makimura M., Ohara S., Inouye Y. (2012). Activation of the aryl hydrocarbon receptor represses mammosphere formation in MCF-7 cells. Cancer Lett. 317, 192–198. [DOI] [PubMed] [Google Scholar]
- Zhu H., Geiman T. M., Xi S., Jiang Q., Schmidtmann A., Chen T., Li E., Muegge K. (2006). Lsh is involved in de novo methylation of DNA. Embo J. 25, 335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]