

Abstract
We introduce a new heterogeneous CPU+GPU-enhanced DFTB approach for the routine and efficient simulation of large chemical and biological systems. Compared to homogeneous computing with conventional CPUs, heterogeneous computing approaches exhibit substantial performance with only a modest increase in power consumption, both of which are essential to upcoming exascale computing initiatives. We show that DFTB-based molecular dynamics is a natural candidate for heterogeneous computing, since the computational bottleneck in these simulations is the diagonalization of the Hamiltonian matrix, which is performed several times during a single molecular dynamics trajectory. To thoroughly test and understand the performance of our heterogeneous CPU+GPU approach, we examine a variety of algorithmic implementations, benchmarks of different hardware configurations, and applications of this methodology on several large chemical and biological systems. Finally, to demonstrate the capability of our implementation, we conclude with a large-scale DFTB MD simulation of explicitly solvated HIV protease (3974 atoms total) as a proof-of-concept example of an extremely large/complex system which, to the best of our knowledge, is the first time that an entire explicitly solvated protein has been treated at a quantum-based MD level of detail.
Graphical Abstract:
1. INTRODUCTION
Over the past decade, the scientific community has witnessed an unprecedented growth in the use of massively parallelized computing to provide critical mechanistic insight in numerous research applications. Computational chemistry, in particular, has benefited greatly from these technological advances, since predictive simulations of complex systems (particularly condensed phase systems in realistic environments) are now possible with modern computational hardware. The majority of these calculations have been carried out on homogeneous computing architectures where a single type of processor (i.e., CPUs or GPUs exclusively) is harnessed for the entire computational simulation. However, in recent years, significant attention has focused on heterogeneous computing as a promising path for meeting exascale goals and requirements, with machines capable of performing a million trillion floating-point calculations per second.1–3 Indeed, two of the most powerful US supercomputers—Oak Ridge National Laboratory’s “Summit” and Lawrence Livermore National Laboratory’s “Sierra” (both of which went into production recently in 2018)—were specifically designed with heterogeneous CPU+GPU architectures as early predecessors to approach exascale computing.4 Compared to homogeneous computing, these heterogeneous architectures exhibit substantial performance with only a modest increase in power consumption, both of which are essential to exascale computing initiatives.5–7 As such, to enable and take advantage of these upcoming computational advancements, future simulations of large, complex chemical and biological systems will need to adapt and efficiently utilize these heterogeneous architectures.
Although heterogeneous computing is less common in time-independent applications of quantum chemistry, a specific area that would significantly benefit from these computational advances is ab initio-based molecular dynamics (AIMD).8–10 This particular computational area is a natural candidate for heterogeneous (and exascale) computing, since one of the computational bottlenecks in these simulations is the diagonalization of the Hamiltonian matrix, which is performed several (typically a hundred or thousand) times during a single molecular dynamics (MD) trajectory. In addition to its suitability for heterogeneous computing, AIMD is also more widely applicable (compared to classical MD or time-independent quantum chemistry) for calculating dynamical chemical processes such as reactive processes, polarization, and hydrogen bonding in condensed phases.11–13 To this end, we have augmented the DFTB+ software package14 to utilize heterogeneous CPU+GPU hardware for accelerating Born–Oppenheimer MD calculations in extremely large chemical and biological systems. Our motivation for modifying the DFTB+ package to use heterogeneous computing is twofold: (1) The DFTB formalism scales favorably with system size, and additional computational advances will allow simulations of systems of even greater complexity (some of which are presented in this work) on heterogeneous exascale computers. (2) We already have significant familiarity and experience with this open-source software package for large chemical/material systems,15–18 and the techniques presented in this work can be used in future methodological developments such as large-scale nonadiabatic dynamics calculations.
In this work, we present a variety of algorithmic implementations, benchmarks of different hardware configurations, and applications of our heterogeneous CPU+GPU-enhanced approach for DFTB simulations of large chemical and biological systems. The specific algorithms examined in this work are comprised of three different iterative Hamiltonian diagonalization techniques (DivideAndConquer, QR, and RelativelyRobust methods) that have been parallelized with heterogeneous CPU+GPU techniques. In addition to comparing the computational performance of each of these algorithms, we evaluate their computational efficiency on several different modern hardware configurations that include conventional CPUs and heterogeneous CPU+GPU combinations with massively parallelized K80 and P100 GPUs. Finally, we present critical scaling tests for all of these configurations and conclude with a large-scale DFTB MD simulation of explicitly solvated HIV protease (comprised of 3974 atoms) as a proof-of-concept example of an extremely large/complex system used in structure-based drug design, which, to the best of our knowledge, is the first time that an entire explicitly solvated protein (as opposed to small peptides) has been treated at a quantum-based MD level of detail.
2. THEORY AND METHODOLOGY
Before proceeding to the algorithmic techniques used in our heterogeneous CPU+GPU-enhanced approach for parallelizing Hamiltonian diagonalization, it is useful to briefly review the density functional tight binding (DFTB) formalism. The DFTB method is based on the Taylor series expansion of the DFT Kohn–Sham (KS) total energy, EKS, with respect to electron density fluctuations ρ(r) = ρ0(r) + δρ(r), where ρ0(r) is a reference density of neutral atomic species. Due to the complexity of the chemical systems in this work, we have chosen to use the third-order expansion of the KS energy, referred to as DFTB3.19,20 We commence with the unmodified KS total energy
| (1) |
where ψi are the KS orbitals, Vext is the external potential, EH is the Hartree energy, EXC is the exchange-correlation (XC) energy, and EII is the ion–ion interaction energy. Rewriting eq 1 in terms of ρ(r) and expanding up to third order, we obtain the DFTB3 energy:
The first term in eq 2, EBS, corresponds to the band structure energy (i.e., the sum over the occupied orbital energies) obtained from the diagonalization of the non-self-consistent Hamiltonian, , evaluated in DFTB by
| (3) |
where {ϕi} forms a minimal Slater-type atomic basis, is the kinetic energy operator, is the reference density of neutral atom I, and veff is an effective Kohn–Sham potential. As shown in eq 3, only two-center elements are treated within the DFTB framework, which are explicitly calculated using analytical functions as per the LCAO formalism. The Hamiltonian and overlap matrix elements are pretabulated for all pairs of chemical elements as a function of the distance between atomic pairs. Thus, no integral evaluation occurs during the simulation, which significantly improves the computational efficiency of the DFTB approach. The second term in eq 2, Eγ, is the energy due to charge fluctuations, where γAB is an analytical function of interatomic distance and the Hubbard parameter U. The third term, EΓ, captures the dependence of the Hubbard parameter as a function of the atomic charge, which improves the description of systems with localized charges. The last term, Erep, is the distance-dependent diatomic repulsive potential, which includes core–electron effects, ion–ion repulsion, and a portion of exchange-correlation effects. The pairwise repulsive functions are obtained by fitting to DFT calculations using a suitable reference structure and, like the matrix elements, are pretabulated. By applying the variational principle, we obtain the Kohn–Sham equations
| (4) |
where the DFTB Hamiltonian is given by
| (5) |
with μ ∈ A and υ ∈ B. Because the atomic charges are dependent on the one-particle wave functions, ψi, eq 5 must be solved iteratively until self-consistency is reached.
Hamiltonian Diagonalization.
Since the Hamiltonian and overlap matrix elements are pretabulated within the DFTB approach, the bottleneck in DFTB-based MD simulations is the diagonalization of the Hamiltonian matrix in eq 5, which typically is performed numerous times along an MD trajectory. Diagonalization of the Hamiltonian is a generalized symmetric-definite eigenvalue problem of the type
| (6) |
where A and B are both real and symmetric, and B is positive-definite. Equation 6 can easily be reduced to a standard symmetric eigenvalue problem (A·z = λz) using a Cholesky factorization, which can further be reduced to a tridiagonal form in order to ease diagonalization. The eigenvalues and eigenvectors can then be computed with any standard diagonalization routine. Within DFTB+, there are three diagonalization routines, hereafter referred to as eigensolvers, based on standard LAPACK routines:21 QR, DivideAndConquer, and RelativelyRobust, which we briefly review below. For the QR and RelativelyRobust eigensolvers, only the Cholesky factorization and reduction-to-standard-form algorithm utilize the GPUs, while the DivideAndConquer routine is replaced entirely by a GPU-enabled routine.
QR.
The QR eigensolver is based on QR factorization, which factorizes an m × n tridiagonal matrix T as the product of an orthogonal matrix Q and an upper triangular matrix R
| (7) |
where Q is an m × m matrix, and R is an n × n matrix. The diagonalization is then performed via the standard QR algorithm:22–24

The matrices T(k) converge to a triangular matrix, the Schur form of T, with the eigenvalues on the diagonal and σk is a shift value chosen to accelerate convergence. Since the T(k) matrices are similar, these eigenvalues are also the eigenvalues of T.
DivideAndConquer.
This eigensolver is based on a divide-and-conquer approach of recursively breaking down a problem into two or more subproblems until these become simple enough to solve directly. (Note: The DivideAndConquer eigensolver described in this section should not be confused with the similarly named [but unrelated] Divide-and-Conquer technique pioneered by Yang and Lee25 for linear-scaling quantum calculations.) This algorithm is highly successful because of deflation, which occurs when an eigenpair of a submatrix of a tridiagonal matrix is an eigenpair of a larger matrix. As for all of the eigensolver routines, the first step is reduction to a block-tridiagonal form:
| (8) |
Unlike the QR eigensolver, the divide-and-conquer approach uses the fact that a tridiagonal matrix is “almost” block diagonal:26
| (9) |
The eigenvalues and eigenvectors of B are then those of T1 and T2, and solving for these two smaller problems is almost always faster than solving the original problem all at once. First, we write T as a block diagonal matrix plus a correction:
| (10) |
The remaining steps are to (1) solve for the eigenvalues of T1 and T2, which can be accomplished by recursively calling the divide-and-conquer algorithm, and (2) build the eigenvalues and eigenvectors of the original matrix T.
RelativelyRobust.
The previously described eigensolvers, QR and DivideAndConquer, take O(n3) time, where n is the size of the matrix, due to the use of explicit orthogonalization of the eigenvectors. The main advantage of the RelativelyRobust method is the ability to numerically compute orthogonal eigenvectors in O(n2) time.27,28 Within this approach, the dot product between eigenvectors is inversely proportional to the relative gap between eigenvalues. In other words, if the eigenvalues are far apart from each other, then the eigenvectors are orthogonal. In practical terms, one can compute the eigenvectors independently (since they are nearly orthogonal), allowing for straightforward and efficient parallelization.
When eigenvalues are clustered together, the RelativelyRobust method shifts the matrix toward the clustered eigenvalues so that they appear farther apart. However, it is not enough to just shift the matrix—it is also required that the new representation be robust to small elemental perturbations so that the new eigenvalues are similar to the original matrix. In other words, if the matrix element xi is perturbed to xi(1 + εi), then, for j = 1, 2, …, n,
| (11) |
| (12) |
where relgap(λj, {λk|k ≠ j}) is a measure of the spread of the eigenvalues. Such a representation is called a Relatively Robust Representation (RRR). The conditions in eqs 11 and 12 ensure that the eigenvalues and eigenvectors of the factorization are very similar to those of the original matrix. Before giving the complete algorithm, we mention two notes: (1) A positive (or negative) definite matrix is always an RRR, and (2) it is not always possible to find an RRR for all eigenvectors, but it is possible to find a partial RRR. The RelativelyRobust eigensolver is the most efficient of the three eigensolvers, both with the CPU implementation and the GPU–CPU implementation, as shown in the following sections.

3. COMPUTATIONAL DETAILS
Our initial computational benchmarks were performed on ice supercells of increasing size, as shown in Figure 1. For our large-scale DFTB Born–Oppenheimer molecular dynamics simulations, the ligand-bound (holo) HIV protease structure was obtained from the Protein Data Bank (PDB ID: 1HVR).29 Since the initial structure determined by X-ray cryptography does not have hydrogen atoms, we used the standard setup procedures with the Amber program to add the missing atoms and minimize the structure.30 We also solvated the structure with explicit water molecules within a 3 Å thick external layer of the protein using AMBER GAFF to provide initial coordinates.31 Additional Cl− ions were included to charge-neutralize the overall system.30 All calculations were performed at the DFTB3 level of theory19,20 with the DFTB+ program14 in conjunction with the 3ob-3-1 parameter set and the corresponding Hubbard derivatives.31–33 We also included DFT-D3 dispersion34,35 effects to accurately describe London dispersion interactions, which are prevalent in these large biochemical systems. Linear algebra routines from the MAGMA library36 were substituted for LAPACK routines (where available) in the DFTB+ source code. The geometry was initially relaxed with nonperiodic boundary conditions (i.e., a cluster geometry) such that all forces were less than 0.001 eV/Å, which provided the initial geometry for equilibration. NVT simulations were performed with a Nosé–Hoover thermostat37 at 50, 100, 150, 200, 250, and 300 K until the system was equilibrated. Finally, an NVE simulation was performed for 2 ps, from which all computed properties were sampled. The time step used in all of our DFTB-based MD simulations was 0.5 fs.
Figure 1.

Sample structures of the three-dimensional supercells of ice used in the timing benchmarks.
The interaction energy of HIV protease and XK263 was calculated in the presence of three selected protein regions, as shown in Figure 2. The selected peptide regions compose the so-called “eye” (residues 22–36), “loop” (residues 72–91), and “flap” (residues 43–58) sections. All of the C-terminals and N-terminals of the three protein regions are at least 10 Å away from XK263. Hydrogen atoms were added by the teLeap algorithm on both the C-terminal and N-terminal to complete protein regions with the AMBER package.38 After computing a total of 2 ps simulation time, the interaction energies were calculated with an interval of 0.1 ps and subsequently averaged.
Figure 2.

3D structure of HIV protease and XK263. Left panel: HIV protease and three selected regions: eye (gray), flap (yellow), and loop (green). Right panel: Close-up view of the binding pocket of HIVp. Residues that have close contact with XK263 are labeled with one-letter amino acid letter codes.
4. RESULTS
Timing Benchmarks.
To evaluate the speed-up gained from our heterogeneous CPU+GPU implementation, we performed single-point energy calculations on periodic ice supercells of increasing size (shown previously in Figure 1). In Figure 3, we compare the performance of the CPU, P100 GPU, and K80 GPU for a single diagonalization with the DivideAndConquer eigensolver. While both types of GPUs provide significant speed-up, the P100 calculations are the most efficient, with almost linear growth. For this reason, all subsequent GPU-enhanced calculations were performed with 4 NVIDIA P100 GPUs and 24 Intel Xeon E5-2680v3 CPUs (the homogeneous CPU-only calculations were performed with the same 24 cores). To compare the relative performance of each of the DFTB+ eigensolvers, we plot the wall time for the single-point energy calculation for each eigensolver in Figure 4. While all three GPU-enhanced implementations benefit from the GPU enhancements, we find that the heterogeneous version of the RelativelyRobust solver provides the most speed-up. It is interesting to note that only a portion of the diagonalization subroutines in this solver uses GPU-enabled routines (i.e., only the Cholesky factorization and reduction-to-standard-form algorithm in the RelativelyRobust solver utilize the GPUs), with the DivideAndConquer method utilizing a larger portion of the GPU. In other words, there is a delicate balance in offloading specific numerical operations between the CPU and GPU, which should be carefully accounted for in any type of heterogeneous computing environment. For the purposes of the DFTB-based MD simulations discussed below, the significant speed-up gained by offloading only a portion of the diagonalization in the RelativelyRobust algorithm shows extreme promise for DFTB-based simulations of larger, more complex systems.
Figure 3.
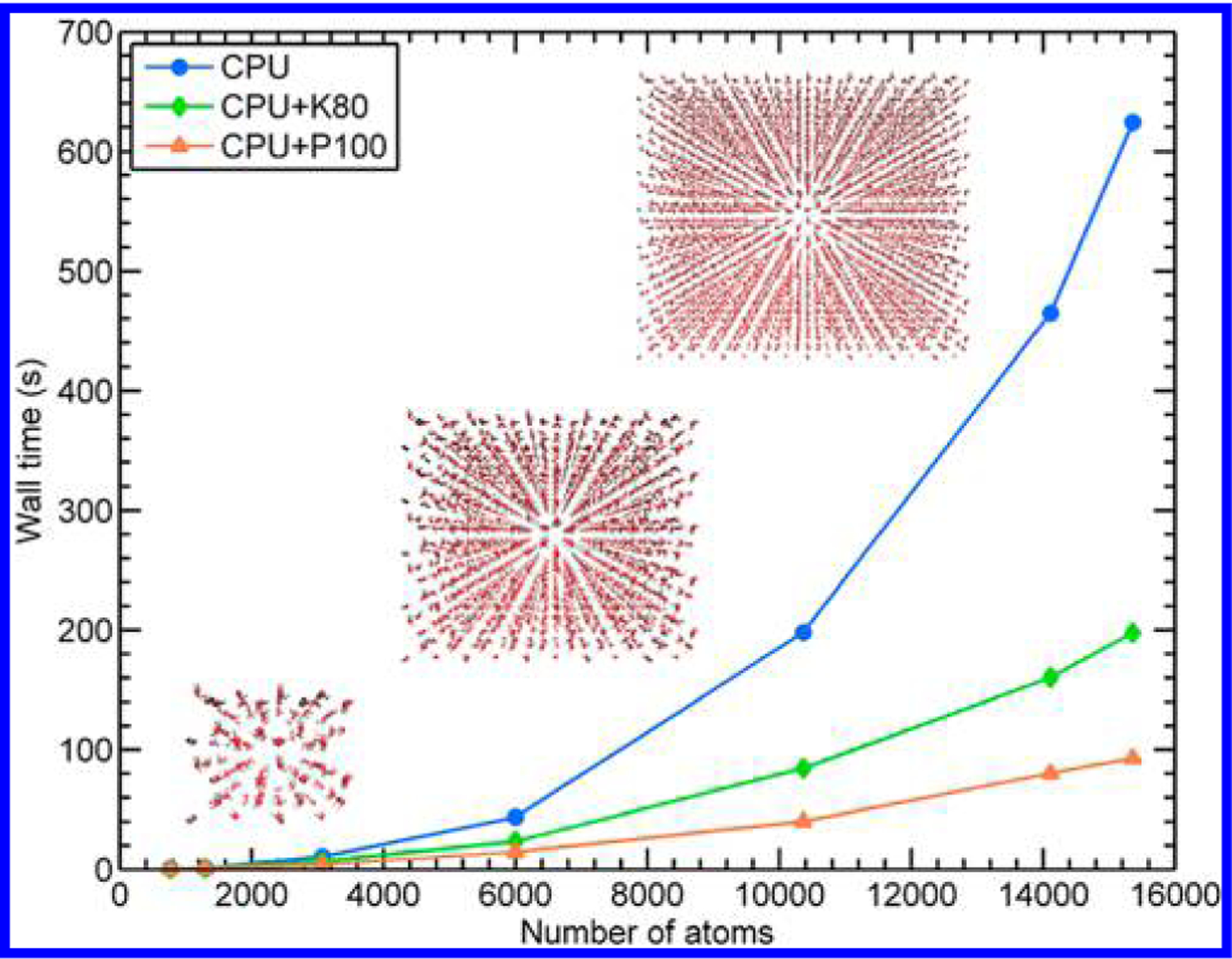
Comparison of the wall time for a single Hamiltonian diagonalization as a function of the number of atoms in each supercell of ice for the CPU, CPU/K80, and CPU/P100 implementations. While both types of GPUs offer speed-up over the CPU version, the P100 was consistently more efficient than the K80 and was used in all subsequent calculations.
Figure 4.
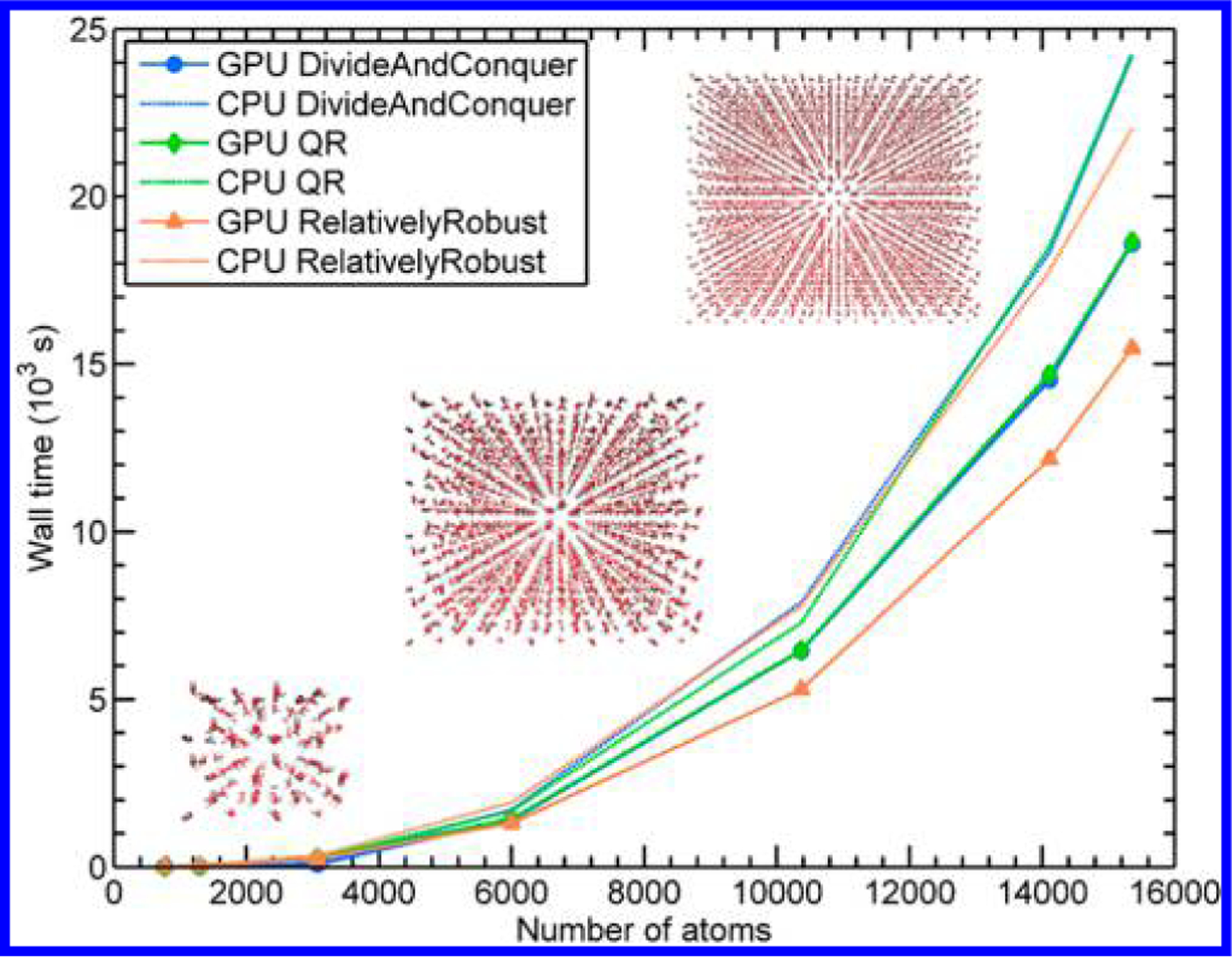
Comparison of the wall time for a single-point DFTB3 energy calculation as a function of the number of atoms in each supercell of ice for the CPU and CPU/GPU (P100) implementations of the three eigensolvers. As expected, all three eigensolvers are faster on the GPU, with RelativelyRobust being the most efficient.
Large-Scale DFTB-Based MD Simulations of Explicitly Solvated HIV Protease.
In this section, we utilize our heterogeneous CPU+GPU approach on a prototypical example of drug design with the HIV protease (HIVp) biological system, as shown in Figure 5. Broadly speaking, structure-based drug design is the creation of ligands on the basis of the structural information on their biomolecule targets to develop ideal drug candidates for clinical trials. The intricate interaction between the ligand and its surrounding protein is one of the key factors in drug design, since binding affinity predominantly affects drug potency. Although the overall ligand-binding affinity can be obtained from experiment, computational tools (such as the heterogeneous CPU+GPU approach utilized here) provide a detailed understanding into binding mechanisms that are critical to drug development, which can significantly save time and effort during the expensive drug discovery process. Here, we focus on XK263, a computer-designed chemical compound, which is composed of a ketone group that binds with HIVp via hydrogen bonding, as a demonstration of an application of our CPU+GPU-heterogeneous approach for drug design. HIVp cleaves premature polypeptides to create protein subunits of an infectious mature HIV39 and is currently one of the major drug targets for AIDS treatments. In terms of scientific interest, the HIVp protein is comprised of highly flexible “flap” regions and has served as a model system for various protein dynamics and ligand binding studies. Unlike most traditional peptidomimetic antiretroviral drugs, XK263 also has different kinetic binding mechanisms that may be of interest in future drug design.40
Figure 5.
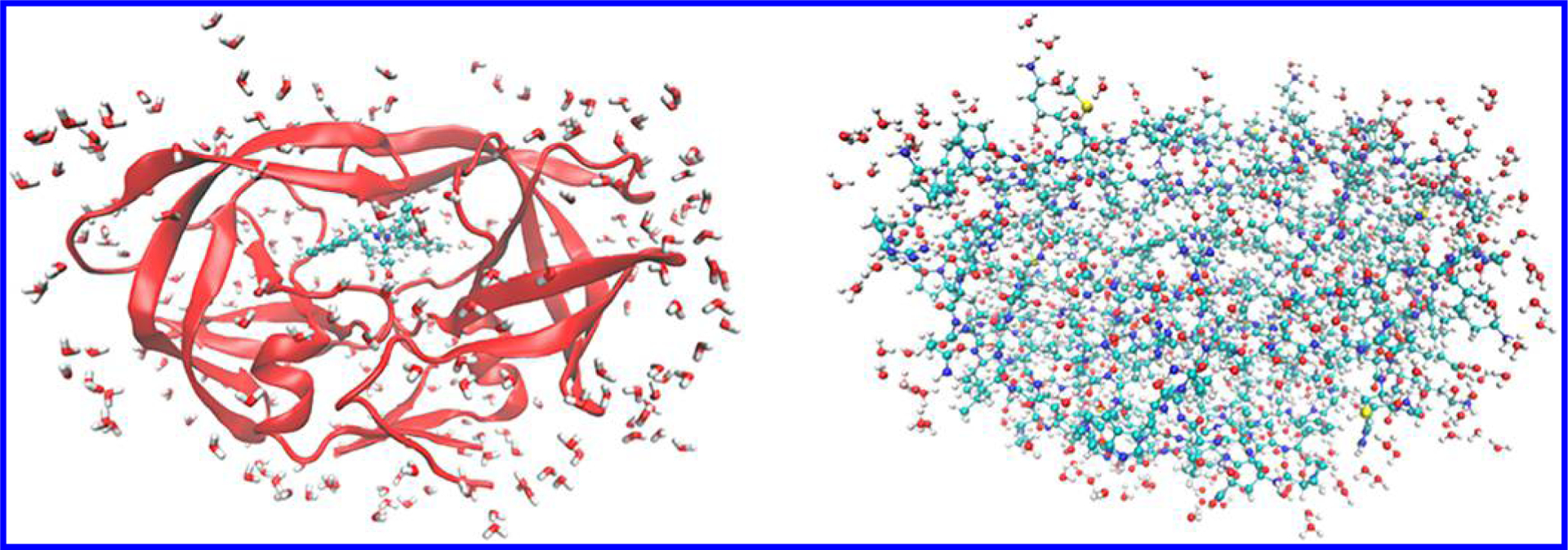
Ligand-bound (holo) structure of HIV protease complexed with XK263. (left) The protein is shown in a ribbon style (red) with the ligand (colored by element) in the binding pocket. The entire complex is surrounded by explicit molecules within 3 Å of the protein. (right) The protein, ligand, and water are all shown as atoms to emphasize the large size of this system.
Figure 6a compares the performance of the CPU against our heterogeneous CPU+GPU implementation for the initial steps of the DFTB-based molecular dynamics for the entire solvated HIVp + XK263 system (a total of 3974 atoms). It is worth noting that, even in these initial steps, the CPU+GPU heterogeneous approach already shows a sizable speed-up (~25% faster after the first 20 steps alone), and longer simulations are expected to show even larger performance gains as the DFTB energies and gradients are repeatedly computed over time. Figure 6b plots the total Born–Oppenheimer DFTB energy for the same biological system over a total period of 2 ps (384 h of wall-clock time). Both simulations utilized the DivideAndConquer eigensolver on 24 Intel Xeon E5-2680v3 CPUs and 4 NVIDIA P100 GPUs (the standard hardware configuration of a single GPU-enabled node on the Comet supercomputer41). We have chosen this particular computational resource for two reasons: (1) this specific hardware configuration is readily available to the general researcher (via the XSEDE research allocation Web site42), and (2) while we only propagate the dynamics of HIVp for only 2 ps as a proof-of-principle demonstration, we emphasize that this calculation was carried out on one single node, which demonstrates that our efficient CPU+GPU heterogeneous approach can be easily used for routine calculations of large systems with only moderate computing resources (and we anticipate that the use of heterogeneous exascale hardware described previously1–3 will enable even longer simulations of more complex systems).
Figure 6.
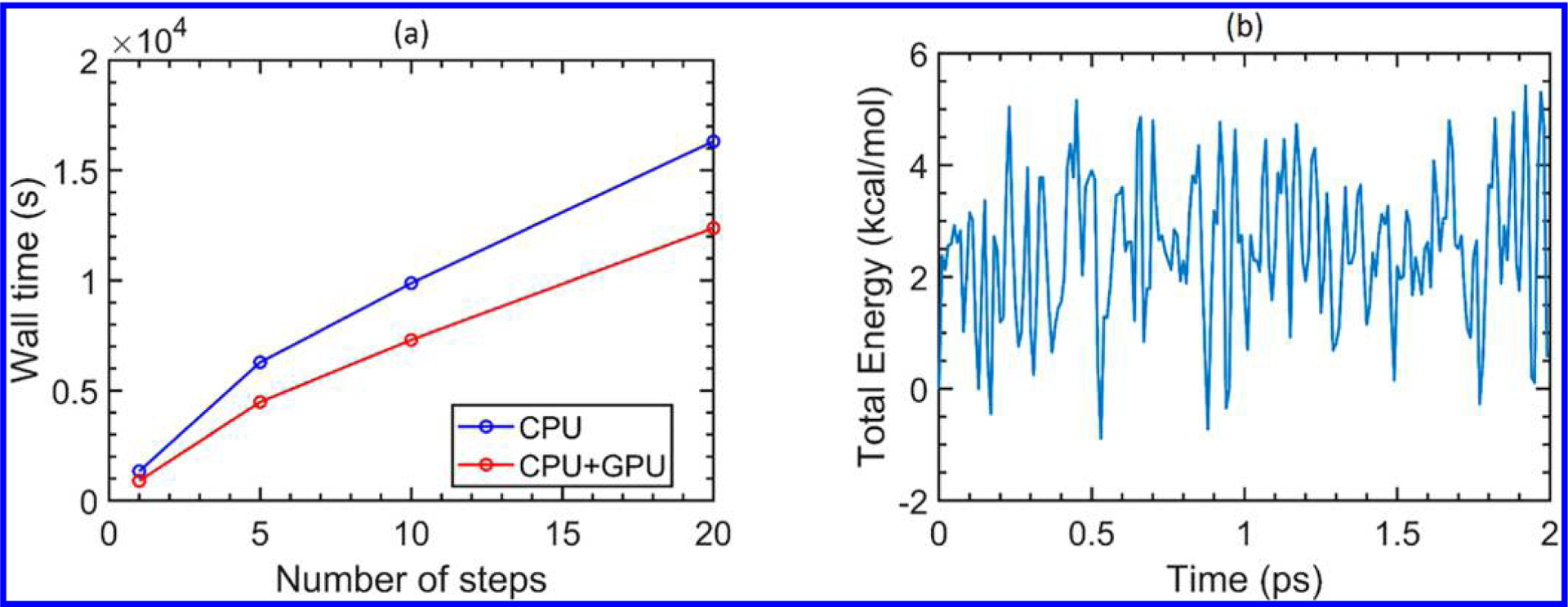
(a) Performance comparison between the CPU and our heterogeneous CPU+GPU implementation for the initial steps in the DFTB-based molecular dynamics of a solvated HIVp+XK263 system. (b) Total DFTB energy of the same biological system obtained from our CPU +GPU-enhanced DFTB code. The DFTB-based molecular dynamics were carried out on 24 Intel Xeon E5-2680v3 CPUs and 4 NVIDIA P100 GPUs on the XSEDE Comet supercomputer.
It is interesting to note that, while classical MD simulations are commonly used to probe biological systems, our CPU+GPU-enhanced DFTB approach was able to provide additional dynamical insight that could not have been obtained with classical MD alone. For example, we initially used the AMBER classical MD package to run an equilibration calculation for both HIVp and XK263 (using the Amber99SB and GAFF force fields, respectively); however, we found that the equilibrium geometry obtained with AMBER was qualitatively different than the equilibrium conformation(s) that were sampled with our CPU+GPU-enhanced DFTB approach. While the classical force field was able to find an equilibrium geometry between XK263 and the Asp25 catalytic residue (which is characterized by strong hydrogen bonds), this specific geometry is not the most stable conformation. In particular, our large-scale CPU+GPU-enhanced DFTB calculations were able to efficiently sample various conformations of other important residues in the eye regions (Leu23, Ala28, and Val32; cf. Figure 2) that are characterized by weaker nonpolar interactions between the aromatic ring of XK263 and these nonpolar residues, which were not well-captured by classical MD. More specifically, Figure 7 depicts local minima having energies of −23.9, −50.9, and −52.8 kcal/mol at 0.3, 0.6, and 1.7 ps, respectively, which correspond to the various positions of the XK263 ligand as it enters/leaves the “flap” region (which effectively acts as a gate for drug binding) in HIVp (cf. Figures 2 and 5). As mentioned previously, the first local energy minimum at 0.3 ps is characterized by hydrogen bonds between the Ile50/Ile149 groups of the flap and the ketone group in XK263 (in addition to other bonding interactions between the Ile47/Ile50 and naphthalene-2-ylmethyl groups). However, it is important to note that, while a static energy minimization for the initial structure can coax the system into the first local energy minimum at 0.3 ps, a more significant arrangement of the nonpolar residues (which can only be obtained from a dynamics calculation) was required to bring the complex to the other local minima at 0.6 or 1.7 ps (see Figures SI-1 and SI-2 in the Supporting Information). In fact, the most favorable binding interactions were obtained after 1 ps (cf. Figure 7 and Table 1), whereas the loop region only confines XK263 in the binding pocket (without forming any highly specific binding interactions), resulting in a weaker intermolecular attraction (~ −16 kcal/mol) compared to other regions in the HIVp complex. We also note that chemical modifications of the naphthalene rings in XK263 to create new and stable attractions with the loop region may also further enhance binding with HIVp (see Figure SI-3 in the Supporting Information), which we save for future computational studies. Nevertheless, our heterogeneous CPU+GPU-enhanced DFTB approach provides an efficient and accessible methodology (which is more accurate than conventional MD) for probing the energetic/dynamical effects in binding studies and drug development. Most importantly, our computational approach encompasses two significant advantages for probing large chemical and biological systems: (1) the CPU+GPU-enhanced DFTB approach is an efficient, quantum-based approach for large systems that does not rely on empirical force-field parameters, and (2) it has a significant advantage over conventional hybrid QM/MM approaches, since it is able to treat the entire system on the same theoretical footing and bypasses the requirement of manually (and arbitrarily) choosing the QM and MM regions, which can typically introduce uncontrolled artifacts/errors in large chemical/biological systems.
Figure 7.
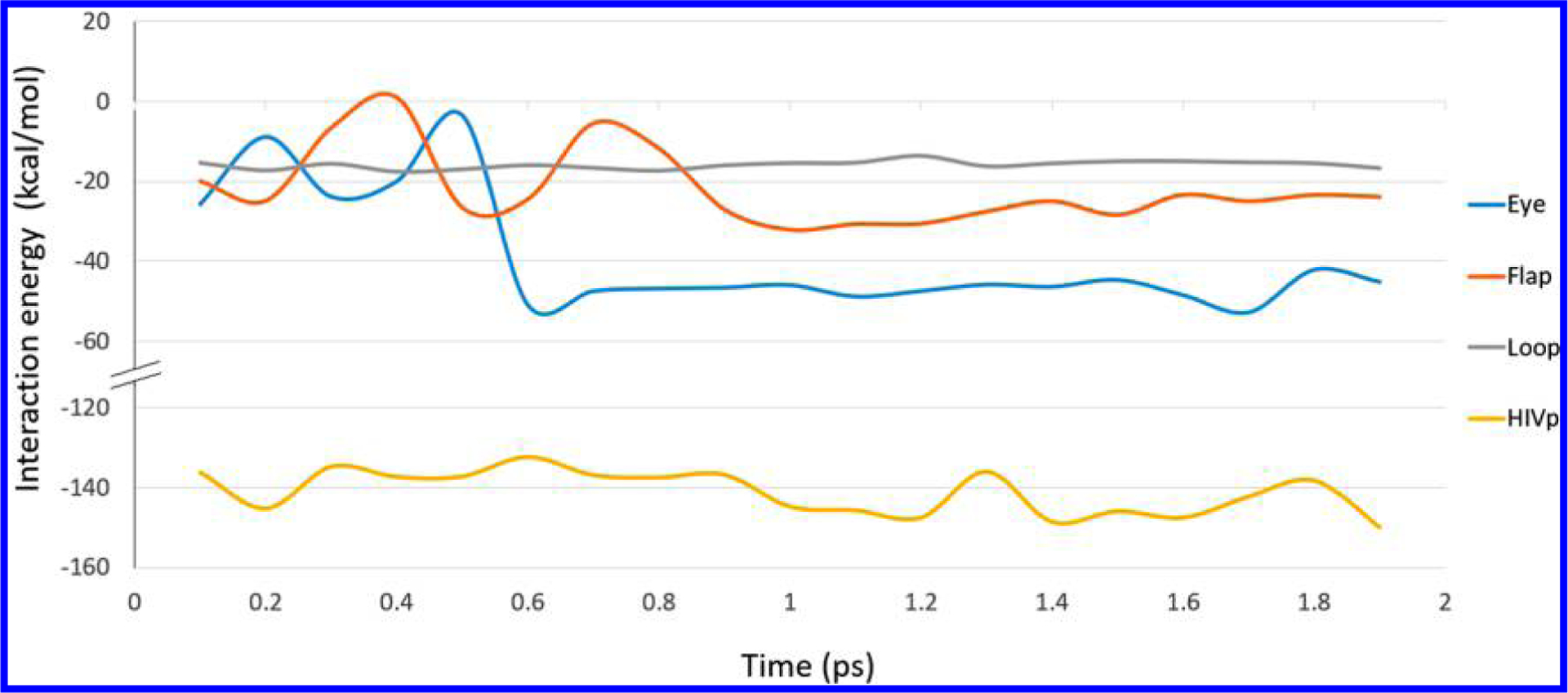
Plot of calculated interaction energies between XK263 and three HIVp regions. The total interaction energy is subdivided into blue (eye region + XK263), red (flap region + XK263), gray (loop region + XK263), and yellow (HIVp + XK263) distinct interactions. Low energy confirmations are only achieved after 1 ps of simulation time.
Table 1.
Calculated Interaction Energies between Selected Protein Regions and XK263 in kcal/mola
| protein regions | ⟨E1⟩ | ⟨E2⟩ |
|---|---|---|
| HIVp + XK263 | −141.1 ± 5.4 | −144.6 ± 4.5 |
| eye + XK263 | −39.0 ± 14.4 | −46.8 ± 2.9 |
| flap + XK263 | −21.9 ± 9.0 | −27.0 ± 3.3 |
| loop + XK263 | −15.9 ± 1.0 | −15.3 ± 0.8 |
⟨E1⟩ and ⟨E2⟩ are the average interaction energies for 0–2 and 1–2 ps of simulation time, respectively.
CONCLUSION
In closing, we have introduced a new heterogeneous CPU+GPU-enhanced DFTB approach for the routine and efficient simulation of large chemical and biological systems. We have specifically chosen to implement this heterogeneous computing approach, since CPU+GPU architectures have started to attract immense scientific attention, particularly as a promising path toward energy-efficient exascale computing initiatives.4 In terms of practical calculations that would immensely benefit from this computing approach, we show that DFTB-based molecular dynamics is a natural candidate for heterogeneous computing, since the computational bottleneck in these simulations is the diagonalization of the Hamiltonian matrix, which is performed several times during a single molecular dynamics trajectory. To thoroughly test and understand the performance of our heterogeneous CPU+GPU approach, we have examined a variety of algorithmic implementations, benchmarks of different hardware configurations, and applications of this methodology on prototypical large chemical and biological systems. Among the three GPU-enhanced Hamiltonian diagonalization routines examined in this study (QR vs DivideAndConquer vs RelativelyRobust), we find that the heterogeneous version of the RelativelyRobust solver provides the highest computational performance. Although the RelativelyRobust diagonalization routine utilizes a smaller portion (but still quite sizable) of the GPU than the DivideAndConquer method, we show that there is a delicate balance in offloading specific numerical operations between the CPU and GPU, which should be carefully accounted for in any type of heterogeneous computing environment.
Among the chemical applications examined in this study, we concluded with a large-scale DFTB MD simulation of explicitly solvated HIV protease (comprised of 3974 atoms) as a proof-of-concept example of an extremely large/complex system used in structure-based drug design—which, to the best of our knowledge, is the first time that an entire explicitly solvated protein has been treated at a quantum-based MD level of detail. This extremely large-scale calculation demonstrates the efficiency of our heterogeneous computing approach and further emphasizes the importance of DFTB-based dynamics (as opposed to static geometry optimizations) for probing large biological systems. Specifically, while it is common to carry out a quantum-mechanical optimization of biological structures initially obtained from classical MD or molecular docking methods,43–45 we show that this conventional strategy can miss important conformations, and the use of efficient and accurate dynamical approaches, such as the CPU+GPU-enhanced DFTB approach used here, can be essential. Moving forward, we anticipate that this heterogeneous CPU+GPU computational capability can also be used for other complex chemical/material systems that require long-time quantum-based dynamics, such as protein dynamics that need quantum-based methods for intricate binding interactions, nonadiabatic calculations of large, coherent, light-harvesting systems,46 or even multicomponent structural materials such as complex alloy systems.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the National Science Foundation for the use of supercomputing resources through the Extreme Science and Engineering Discovery Environment (XSEDE), Project No. TG-ENG160024.
Funding
All GPU enhancements by S.I.A., Y.S., and B.M.W. were supported by the U.S. Department of Energy, National Energy Technology Laboratory (NETL), under Award No. DE-FE0030582. All HIV protease studies by J.S. and C.-e.A.C. were supported by the National Institute of General Medical Sciences of the National Institutes of Health, under Award No. R01GM109045.
Footnotes
The authors declare no competing financial interest.
The modified DFTB routines used to carry out the heterogeneous CPU+GPU-enabled simulations in this work can be obtained upon request from the corresponding author.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jctc.8b01239.
Additional information on the detailed interactions between the eye, flap, and loop regions with XK263 (PDF)
REFERENCES
- (1).Service RF What It’ll Take to Go Exascale. Science 2012, 335, 394–396. [DOI] [PubMed] [Google Scholar]
- (2).Understanding Exascale - Exascale Computing Project. https://www.exascaleproject.org/what-is-exascale (December 6, 2018).
- (3).Lee CT; Amaro RE Exascale Computing: A New Dawn for Computational Biology. Comput. Sci. Eng 2018, 20, 18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Next-Generation U.S. Department of Energy Supercomputers Will Be GPU-Accelerated. http://www.nvidia.com/object/exascalesupercomputing.html (December 6, 2018).
- (5).Rigo A; Pinto C; Pouget K; Raho D; Dutoit D; Martinez P; Doran C; Benini L; Mavroidis I; Marazakis M; Bartsch V; Lonsdale G; Pop A; Goodacre J; Colliot A; Carpenter P; Radojković P; Pleiter D; Drouin D; Dinechin B. D. d. In Paving the Way Towards a Highly Energy-Efficient and Highly Integrated Compute Node for the Exascale Revolution: The ExaNoDe Approach, 2017 Euromicro Conference on Digital System Design (DSD), Aug 30–Sept 1, 2017; pp 486–493. [Google Scholar]
- (6).Georgopoulos K; Mavroidis I; Lavagno L; Papaefstathiou I; Bakanov K Energy-Efficient Heterogeneous Computing at exaSCALE—ECOSCALE. In Hardware Accelerators in Data Centers; Kachris C, Falsafi B, Soudris D, Eds.; Springer International Publishing: Cham, 2019; pp 199–213. [Google Scholar]
- (7).Lefèvre L; Pierson J-M Introduction to Special Issue on Sustainable Computing for Ultrascale Computing. Sustain. Comput.-Infor 2018, 17, 25–26. [Google Scholar]
- (8).Car R; Parrinello M Unified Approach for Molecular Dynamics and Density-Functional Theory. Phys. Rev. Lett 1985, 55, 2471–2474. [DOI] [PubMed] [Google Scholar]
- (9).Remler DK; Madden PA Molecular Dynamics without Effective Potentials Via the Car-Parrinello Approach. Mol. Phys 1990, 70, 921–966. [Google Scholar]
- (10).Payne MC; Teter MP; Allan DC; Arias TA; Joannopoulos JD Iterative Minimization Techniques for Ab Initio Total-Energy Calculations: Molecular Dynamics and Conjugate Gradients. Rev. Mod. Phys 1992, 64, 1045–1097. [Google Scholar]
- (11).Sponer J; Krepl M; Banas P; Kuhrova P; Zgarbova M; Jurecka P; Havrila M; Otyepka M How to Understand Atomistic Molecular Dynamics Simulations of RNA and Protein-RNA Complexes? WIREs RNA 2017, 8, e1405. [DOI] [PubMed] [Google Scholar]
- (12).Fu C; Xu L; Aquino FW; v. Cresce A; Gobet M; Greenbaum SG; Xu K; Wong BM; Guo J Correlating Li+-Solvation Structure and Its Electrochemical Reaction Kinetics with Sulfur in Subnano Confinement. J. Phys. Chem. Lett 2018, 9, 1739–1745. [DOI] [PubMed] [Google Scholar]
- (13).Tuckerman ME Ab Initio Molecular Dynamics: Basic Concepts, Current Trends and Novel Applications. J. Phys.: Condens. Matter 2002, 14, R1297. [Google Scholar]
- (14).Aradi B; Hourahine B; Frauenheim T DFTB+, a Sparse Matrix-Based Implementation of the DFTB Method. J. Phys. Chem. A 2007, 111, 5678–5684. [DOI] [PubMed] [Google Scholar]
- (15).Ilawe NV; Oviedo MB; Wong BM Real-Time Quantum Dynamics of Long-Range Electronic Excitation Transfer in Plasmonic Nanoantennas. J. Chem. Theory Comput 2017, 13, 3442–3454. [DOI] [PubMed] [Google Scholar]
- (16).Ilawe NV; Oviedo MB; Wong BM Effect of Quantum Tunneling on the Efficiency of Excitation Energy Transfer in Plasmonic Nanoparticle Chain Waveguides. J. Mater. Chem. C 2018, 6, 5857–5864. [Google Scholar]
- (17).Oviedo MB; Wong BM Real-Time Quantum Dynamics Reveals Complex, Many-Body Interactions in Solvated Nanodroplets. J. Chem. Theory Comput 2016, 12, 1862–1871. [DOI] [PubMed] [Google Scholar]
- (18).Alvarez Barragan A; Ilawe NV; Zhong L; Wong BM; Mangolini L A Non-Thermal Plasma Route to Plasmonic TiN Nanoparticles. J. Phys. Chem. C 2017, 121, 2316–2322. [Google Scholar]
- (19).Gaus M; Cui QA; Elstner M DFTB3: Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (SCC-DFTB). J. Chem. Theory Comput 2011, 7, 931–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Yang Y; Yu HB; York D; Cui Q; Elstner M Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method: Third-Order Expansion of the Density Functional Theory Total Energy and Introduction of a Modified Effective Coulomb Interaction. J. Phys. Chem. A 2007, 111, 10861–10873. [DOI] [PubMed] [Google Scholar]
- (21).Anderson EBZ; Bischof C; Blackford S; Demmel J; Dongarra J; Du Croz J; Greenbaum A; Hammarling S; Mckenney A; Sorensen D LAPACK Users’ Guide, 3rd ed.; Society for Industrial and Applied Mathematics: Philadelphia, PA, 1999. [Google Scholar]
- (22).Francis JGF QR Transformation - a Unitary Analog to LR Transformation - Part 1. Comput. J 1961, 4, 265–271. [Google Scholar]
- (23).Francis JGF The QR Transformation - Part 2. Comput. J 1962, 4, 332–345. [Google Scholar]
- (24).Kublanovskaya VN On Some Algorithms for the Solution of the Complete Eigenvalue Problem. USSR Compt. Math. Math+ 1962, 1, 637–657. [Google Scholar]
- (25).Yang W; Lee T-S A Density-Matrix Divide-and-conquer Approach for Electronic Structure Calculations of Large Molecules. J. Chem. Phys 1995, 103, 5674–5678. [Google Scholar]
- (26).Cuppen JJM A Divide and Conquer Method for the Symmetric Tridiagonal Eigenproblem. Numer. Math 1980, 36, 177–195. [Google Scholar]
- (27).Dhillon IS A New O(n2) Algorithm for the Symmetric Tridiagonal Eigenvalue/Eigenvector Problem; University of California, Berkeley: Berkeley, CA, 1989. [Google Scholar]
- (28).Parlett BN; Dhillon IS Relatively Robust Representations of Symmetric Tridiagonals. Linear Algebra Appl. 2000, 309, 121–151. [Google Scholar]
- (29).Huang YMM; Raymundo MAV; Chen W; Chang CEA Mechanism of the Association Pathways for a Pair of Fast and Slow Binding Ligands of HIV-1 Protease. Biochemistry 2017, 56, 1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Joung IS; Cheatham TE Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Gaus M; Goez A; Elstner M Parametrization and Benchmark of DFTB3 for Organic Molecules. J. Chem. Theory Comput 2013, 9, 338–354. [DOI] [PubMed] [Google Scholar]
- (32).Gaus M; Lu XY; Elstner M; Cui Q Parameterization of DFTB3/3OB for Sulfur and Phosphorus for Chemical and Biological Applications. J. Chem. Theory Comput 2014, 10, 1518–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Kubillus M; Kubar T; Gaus M; Rezac J; Elstner M Parameterization of the DFTB3 Method for Br, Ca, Cl, F, I, K, and Na in Organic and Biological Systems. J. Chem. Theory Comput 2015, 11, 332–342. [DOI] [PubMed] [Google Scholar]
- (34).Grimme S; Antony J; Ehrlich S; Krieg H A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys 2010, 132, 154104. [DOI] [PubMed] [Google Scholar]
- (35).Grimme S; Ehrlich S; Goerigk L Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem 2011, 32, 1456–1465. [DOI] [PubMed] [Google Scholar]
- (36).Tomov S; Dongarra J; Baboulin M Towards Dense Linear Algebra for Hybrid GPU Accelerated Manycore Systems. Parallel. Comput 2010, 36, 232–240. [Google Scholar]
- (37).Martyna GJ; Tuckerman ME; Tobias DJ; Klein ML Explicit Reversible Integrators for Extended Systems Dynamics. Mol. Phys 1996, 87, 1117–1157. [Google Scholar]
- (38).Case D; Ben-Shalom I; Brozell B; Cerutti D; Cheatham T; Cruzeiro V; Darden T; Duke R; Ghoreishi D; Gilson M; Gohlke H; Goetz A; Greene D; Harris R; Homeyer N; Izadi S; Kovalenko A; Kurtzman T; Lee T; LeGrand S; Li P; Lin C; Liu J; Luchko T; Luo R; Mermelstein D; Merz K; Miao Y; Monard G; Nguyen C; Nguyen H; Omelyan I; Onufriev A; Pan F; Qi R; Roe D; Roitberg A; Sagui C; Schott-Verdugo S; Shen J; Simmerling C; Smith J; Salomon-Ferrer R; Swails J; Walker R; Wang J; Wei H; Wolf R; Wu X; Xiao L; York D; Kollman P AMBER 2018; University of California: San Francisco, CA, 2018. [Google Scholar]
- (39).Kohl NE; Emini EA; Schleif WA; Davis LJ; Heimbach JC; Dixon RAF; Scolnick EM; Sigal IS Active Human Immunodeficiency Virus Protease Is Required For Viral Infectivity. Proc. Natl. Acad. Sci. U. S. A 1988, 85, 4686–4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Markgren PO; Schaal W; Hamalainen M; Karlen A; Hallberg A; Samuelsson B; Danielson UH Relationships Between Structure and Interaction Kinetics for HIV-1 Protease Inhibitors. J. Med. Chem 2002, 45, 5430–5439. [DOI] [PubMed] [Google Scholar]
- (41).Comet User Guide. http://www.sdsc.edu/support/user_guides/comet.html (December 6, 2018).
- (42).XSEDE Research Allocatrions. https://portal.xsede.org/allocations/research (December 6, 2018).
- (43).Adeniyi AA; Soliman MES Implementing QM in Docking Calculations: Is It a Waste of Computational Time? Drug Discovery Today 2017, 22, 1216–1223. [DOI] [PubMed] [Google Scholar]
- (44).Su PC; Tsai CC; Mehboob S; Hevener KE; Johnson ME Comparison of Radii Sets, Entropy, QM Methods, and Sampling on MM-PBSA, MM-GBSA, and QM/MM-GBSA Ligand Binding Energies of F-Tularensis Enoyl-ACP Reductase (FabI). J. Comput. Chem 2015, 36, 1859–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Lu HT; Huang XQ; AbdulHameed MDM; Zhan CG Binding Free Energies for Nicotine Analogs Inhibiting Cytochrome P450 2A6 by a Combined Use of Molecular Dynamics Simulations and QM/MM-PBSA Calculations. Bioorg. Med. Chem 2014, 22, 2149–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Maiuri M; Oviedo MB; Dean JC; Bishop M; Kudisch B; Toa ZSD; Wong BM; McGill SA; Scholes GD High Magnetic Field Detunes Vibronic Resonances in Photosynthetic Light Harvesting. J. Phys. Chem. Lett 2018, 9, 5548–5554. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.