

Abstract
Cancer cells have differential metabolic dependencies compared to their non-malignant counterparts. However, few metabolism-targeting compounds have been successful in clinical trials. Here, we investigated the metabolic vulnerabilities of triple-negative breast cancer (TNBC), in particular those metabolic perturbations that increased mitochondrial apoptotic priming and sensitivity to BH3 mimetics (drugs that antagonize anti-apoptotic proteins). We used high throughput-dynamic BH3 profiling (HT-DBP) to screen a library of metabolism-perturbing small molecules, which revealed inhibitors of the enzyme nicotinamide phosphoribosyltransferase (NAMPT) as top candidates. In some TNBC cells but not in non-malignant cells, NAMPT inhibitors increased overall apoptotic priming and induced dependencies on specific anti-apoptotic BCL-2 family members. Treatment of TNBC cells with NAMPT inhibitors sensitized them to subsequent treatment with BH3 mimetics. The combination of a NAMPT inhibitor (FK866) and an MCL-1 antagonist (S63845) reduced tumor growth in a TNBC patient-derived xenograft model in vivo. We found that NAMPT inhibition reduced NAD+ concentrations below a critical threshold that resulted in depletion of adenine, which was the metabolic trigger that primed TNBC cells for apoptosis. These findings demonstrate a close interaction between metabolic and mitochondrial apoptotic signaling pathways and reveal that exploitation of a tumor-specific metabolic vulnerability can sensitize some TNBC to BH3 mimetics.
Introduction
Altered energy metabolism has long been recognized as a hallmark of cancer1. Since cancer cells are often more dependent on specific metabolic pathways and therefore more sensitive to perturbations of these pathways than normal cells, cancer metabolism represents a potential target for therapeutic intervention2–5. However, the scope of vulnerabilities that are induced by cancer-associated metabolic reprogramming is yet to be elucidated. To identify targetable metabolic pathways, previous efforts have altered cancer cell energy metabolism by genetic or chemical inhibition and evaluated the survival of cancer cells in the absence or presence of standard of care chemotherapeutics3,5. Notwithstanding these efforts, few metabolism-perturbing small molecules (MPSM) have succeeded clinically thus far3. Reported reasons for this include insufficient target-cell activity and specificity, lack of effect of the compounds in vivo, and poor differential effect of the drug on cancer versus normal cells leading to toxicities3.
Another reason why MPSM with potential as single agents or in combination therapies may fail to be identified is that many screens are based on measuring the binary cell fate of life or death of cancer cells. An alternative is a graded measurement of how a drug increases apoptotic priming6. How “primed for death” cells are depends on the quantity and interplay between pro- and anti-apoptotic members of the BCL-2-protein family, key regulators of mitochondrial outer membrane permeabilization (MOMP), the decisive event committing a cell to apoptosis7. To measure apoptotic priming, a technique called “BH3 profiling” can be used8,9. In a BH3 profiling assay, mitochondria are exposed to synthetic peptides derived from the BH3 domains of pro-apoptotic BCL-2-protein family members and the resulting magnitude of MOMP is measured8,9. A robust MOMP response to promiscuous peptides (that is, peptides that bind to all different anti-apoptotic BCL-2 family members) indicates that the cell is close to the apoptotic threshold (“primed”), while a relatively modest response indicates that the cell is relatively far from the apoptotic threshold (“unprimed”). BH3 profiling of multiple primary patient tumor types (namely multiple myeloma, ovarian cancer, acute myeloid leukemia and acute lymphoid leukemia) has demonstrated that the pretreatment priming state of the cancer cells predicts clinical response to conventional chemotherapies10–12. Moreover, when peptides that selectively bind individual anti-apoptotic proteins such as BCL-2, BCL-XL, or MCL-1, are used for BH3 profiling, dependencies of cancer cells on anti-apoptotic BCL-2-family members could be predicted6,12–15. The BH3 profiling method was modified to a “Dynamic BH3 Profiling” (DBP) approach, which incorporates short term (16- to 48-hour) incubations of cancer cells with perturbing agents, followed by regular BH3 profiling. DBP hence measures drug-induced apoptotic signaling. DBP made it feasible to use short term ex vivo cultures to identify drugs with in vivo activity in mice and humans16,17.
In this study we used DBP to identify metabolic targets that increase apoptotic priming in Triple-Negative Breast Cancer (TNBC). In particular, we screened for metabolism perturbating agents that increased TNBC anti-apoptotic dependencies to facilitate combinations with BH3 mimetic drugs. BH3 mimetics are a class of small molecules that target the apoptotic machinery of the cancer cells by mimicking BH3-only pro-apoptotic proteins and antagonizing the pro-survival function of anti-apoptotic proteins18,19. This class of molecules has shown clinical efficacy as single agents and in combination therapies in hematologic malignancies20,21. However, in the context of solid tumors, despite reports of in vitro anti-apoptotic protein dependencies in cell lines and primary tissue22, BH3 mimetics have shown less activity23–25. In the context of TNBC, little is known about the efficacy of BH3 mimetics as single agents or in combination regiments26–28 and thus far, no clinical trials of BH3 mimetics are ongoing for TNBC (https://www.clinicaltrials.gov/). Therefore, the purpose of this study was to identify metabolic targets in TNBC to specifically increase the tumor’s anti-apoptotic dependencies and consequently increase sensitivity to BH3 mimetics.
Results
High-throughput dynamic BH3 profiling identifies metabolic compounds that increase mitochondrial apoptotic priming of TNBC cells, but not normal cells
To identify metabolic pathways that modulate apoptotic signaling and anti-apoptotic dependencies in TNBC cells, we used a high-throughput dynamic BH3 profiling (HT-DBP)17 technique to screen a library of 192 metabolism-perturbing small molecules (MPSM) with known metabolic targets29 (table S1) on the SUM149 TNBC cells. Each of the MPSM was assayed at 10-point dilutions. We looked for agents that increased mitochondrial apoptotic priming at concentrations that did not induce cell death (fig. S1A). Thirty-two MPSM matched these criteria (Fig. 1A). To enhance the potential for the hits being clinically relevant, we then performed a validation screen of the thirty-two initial hits in Human Plasma Like Medium (HPLM)30. Three compounds—terbinafine, lometrexol and pyruvinium pamoate—were excluded because they failed to induce apoptotic priming in SUM149 cells under these more physiologically relevant culture conditions (Fig. 1B).
Figure 1. HT-DBP screening identifies NAMPT inhibitors to increase mitochondrial apoptotic priming of TNBC cells, but not normal cells.
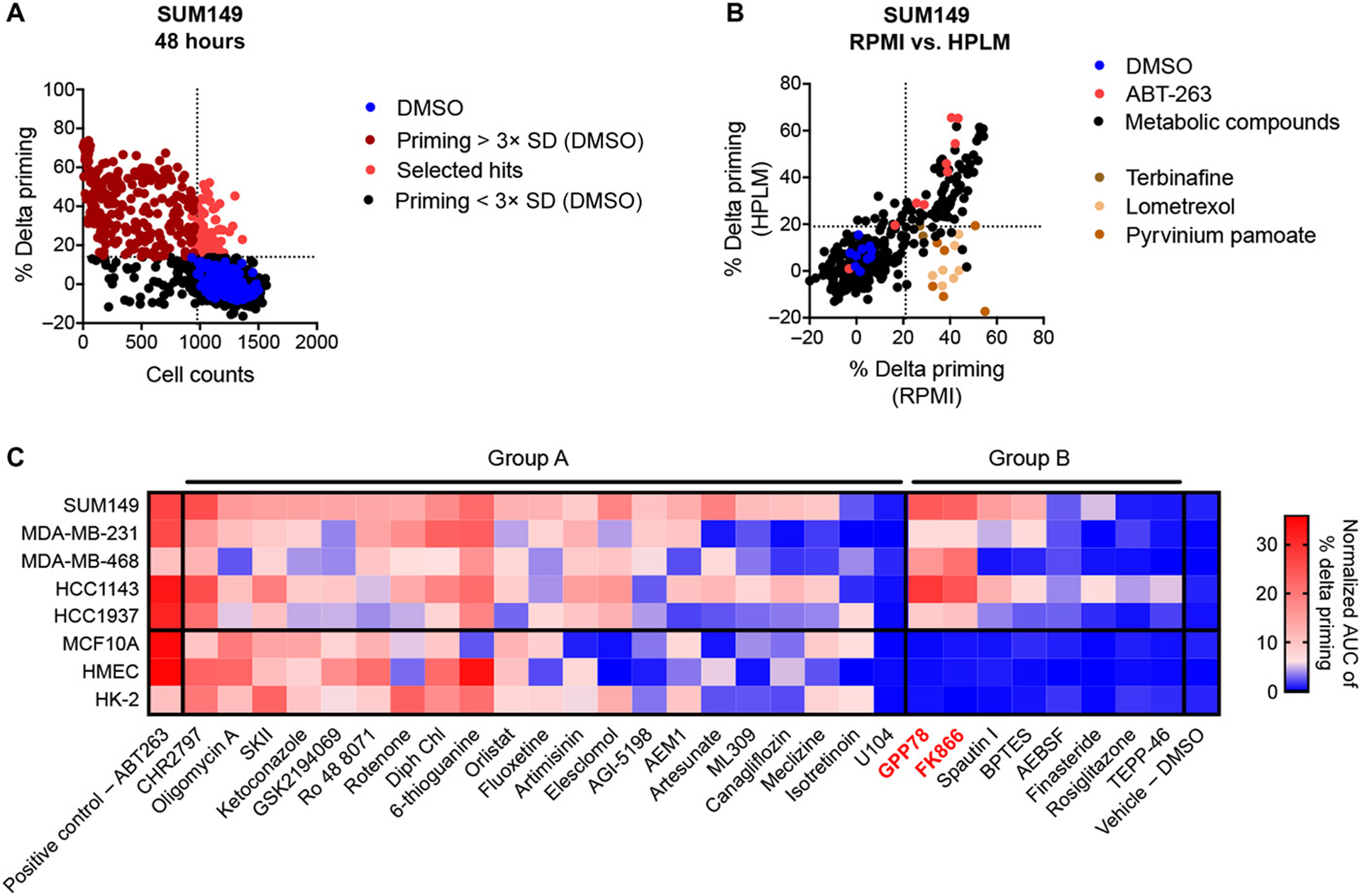
(A) HT-DBP was used to screen a library of 192 metabolism-perturbing small molecules (MPSM) on the SUM149 cells 48 hours after treatment. MPSM were assayed at 10-point dilutions (20 μM to 0.003 μM). Percent delta priming of each concentation of each MSPM was calculated by comparing cytochrome c abundance in treated cells to that of vehicle control cells. MPSM concentrations that induced priming that was more than 3x standard deviation of the DMSO treated wells (horizontal dotted line) were considered to increase the mitochondrial apoptotic priming of the cancer cells and are indicated in red. MPSM concentrations that decreased the cell count to less than 80% of the average cell count of DMSO treated cells (vertical dotted line) were considered to have an effect on cell count. MPSM concentrations that show an increased % delta priming, but no reduction in cell count were considered hits and are indicated in bright red. Control (DMSO treated) cells are indicated in blue. Data are average of technical replicates. (B) Thirty-two MPSM that increased priming of SUM149 cells after 48 hours in RPMI medium were counter screened on the SUM149 cells cultured in human plasma like medium (HPLM). Compounds that showed priming in RPMI but not in HPLM (indicated in brown) were excluded from further follow up screenings. Control (DMSO treated) cells are indicated in blue. ABT-263 treated cells (positive control) are indicated in red. Data are average of technical duplicates. (C) Metabolic compounds identified as hits in the initial screen on SUM149 were screened on MDA-MB-231, MDA-MB-468, HCC1143 and HCC1937 TNBC cells and counter screened on MCF10A, HMEC and HK-2 cells. Data are represented as average of normalized AUC measurements of the % delta priming dilution curves of the metabolic agents tested in three independent screenings. Compounds that increase apoptotic priming in both TNBC and non-malignant cells are grouped in group A, compounds that at given concentration only increase priming of TNBC cells are grouped in group B. ABT-263 is a positive control for increasing apoptotic priming, DMSO is negative control.
To test the relevance and cancer specificity of the twenty-nine remaining MPSM, we screened them on four additional TNBC cell lines (MDA-MB-231, MDA-MB-468, HCC1143 and HCC1937) and three non-malignant controls [HMEC (primary mammary epithelial cells), MCF10A (mammary epithelial cell line) and HK-2 (mouse proximal tubular cell line)] (Fig. 1C and fig. S1B). Mammary epithelial cells were selected because their metabolism closely resembles the metabolism of breast cancer cells31. Proximal tubular cells were included as a readily available non-malignant cell type often vulnerable to drug toxicities and another estimation of therapeutic index32. Validation screens confirmed the reproducibility of the screening data (Pearson correlation coefficients, fig. S1C). Based on the results of these validation and counter screenings (Fig. 1C), we classified the metabolism targeting hits into two groups according to their ability to selectively target cancer cells. Group A includes agents that similarly increased the apoptotic priming of both cancer and non-cancerous cells (Fig. 1C and fig. S1D). Group B included agents that, at least at some concentrations, increased the apoptotic priming of cancer cells but not of non-cancerous cells (Fig. 1C and fig. S1D). Since we aim to specifically increase apoptotic priming in cancer cells, we focused on compounds from group B. Of the eight compounds in group B two (GPP78 and FK866) target the Nicotinamide Phosphoribosyltransferase enzyme (NAMPT), the rate limiting enzyme in the production of nicotinamide adenine dinucleotide (NAD+) via the salvage pathway33. The other six compounds in group B all targeted different metabolic pathways (table S1). Notably, all three NAD+ metabolism targeting metabolic compounds included in our screening library were shown to increase the apoptotic priming of SUM149 cells (fig. S2). Together, these data highlight the importance of NAD+ metabolism in the regulation of mitochondrial apoptotic priming in TNBC cells.
NAMPT inhibition increases overall mitochondrial apoptotic priming, anti-apoptotic dependencies, and sensitivity of TNBC cells to BH3 mimetics
From our initial and follow up screenings we learned that NAMPT inhibitors (FK866 and GPP78) induced overall mitochondrial apoptotic priming in a subset of TNBC cell lines but not in non-malignant cells (Fig. 2, A and B). To determine if NAMPT inhibitors also induced anti-apoptotic dependencies, we performed DBP on five TNBC cell lines (SUM149, MDA-MB-468, MDA-MB-231, HCC1143, HCC1937) using the selective BH3 peptides PUMA, BAD, MS1, HRK and FS1 and several selective BH3 mimetics in addition to the promiscuous BIM peptide (Fig. 2C). In parallel, MFC10A cells were tested as a non-malignant control. The observed pattern of increase in % delta priming after NAMPT inhibition (FK866 and GPP78) induced by the BIM and PUMA peptides confirmed our initial observations that NAMPT inhibitors increase overall apoptotic priming of most TNBC cell lines tested (exception HCC1937) (Fig. 2D and fig. S3). This increase in overall apoptotic priming was not observed in the non-malignant MCF10A cells (Fig. 2D and fig. S3). Additionally, DBP revealed that NAMPT inhibition increases dependencies on anti-apoptotic proteins BCL-2, BCL-XL or MCL-1 in several of the TNBC cell lines tested (respectively, BAD and ABT-199; and HRK, A-133 and A-115; and MS1 and S63845 induced % delta priming, Fig. 2D and fig. S3). In contrast, NAMPT inhibition did not increase anti-apoptotic dependencies in MCF10A cells (Fig. 2D and fig. S3). The specific dependencies as well as the magnitude of induction of apoptotic priming and anti-apoptotic dependencies by NAMPT inhibition varied among cell lines (Fig. 2, A, B and D).
Figure 2. NAMPT inhibition increases overall mitochondrial apoptotic priming, anti-apoptotic dependencies and sensitivity of TNBC cell lines to BH3 mimetics and apoptosis inducers.
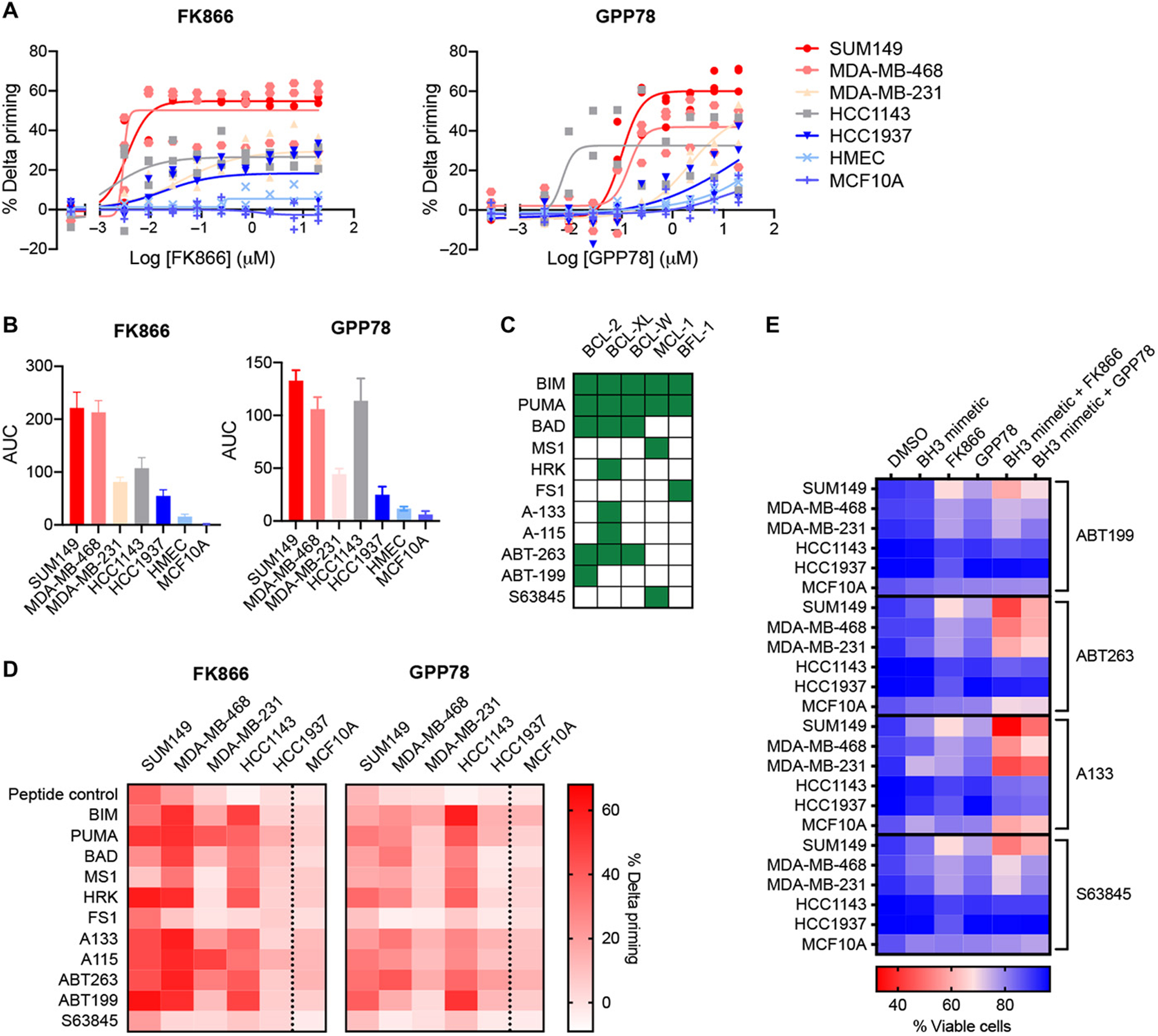
(A) Percentage delta priming of TNBC cancer cell lines and non-malignant cells (MCF10A and HMEC) after 48h exposure to NAMPT inhibitors (FK866 and GPP78) at the indicated concentrations. Priming was determined by DBP using BIM peptide. Delta priming is calculated relative to DMSO treated samples. Data are mean and SD of 2 or 3 independent experiments. (B) AUC and SD of the % delta priming dilution curves in (A). (C) Binding properties of BH3 peptides to anti-apoptotic BCL-2 family members as described previously6. Green squares indicate binding, white squares indicate no binding or an EC50 binding value > 1 μM. (D) Heatmap of the % delta priming of TNBC cell lines and MCF10A cells exposed for 48 hours to 0.01 μM FK866 or 0.1 μM GPP78. Percent delta priming is calculated by comparing cytochrome C abundance in drug treated versus vehicle treated cells. Responses to the BIM and PUMA peptide indicates overall mitochondrial priming. Responses to the BAD, HRK, MS1, FS1 peptides and BH3 mimetics indicate specific anti-apoptotic dependencies. BAD and ABT-263 indicate BCL-2, BCL-XL or Bcl-w dependency; HRK, A-133 and A-115 indicate BCL-XL dependency; ABT-199 indicates BCL-2 dependency; MS1 and S63845 indicate MCL-1 dependency. Data are average of 3 independent experiments. Data also are presented as bar graphs + SD in supplementary material (fig. S3). (E) Cell death measurements using microscopy-based AV-Hoechst cell death assay. TNBC cells and MCF10A cells were treated for 72 hours with 0.01 μM FK866 and 0.1 μM GPP78 in the presence or absence of 0.1 μM BH3 mimetics. Values are means of 3 independent experiments. Data also are presented as dot plots + SD in supplementary material (fig. S4).
To determine whether the anti-apoptotic dependencies observed by DBP translated into functional sensitization to BH3 mimetic drugs, we performed a cell death assay combining NAMPT inhibitors with different BH3 mimetics. TNBC cells were relatively insensitive to single agent treatment with BH3 mimetics or NAMPT inhibitors (FK866 and GPP78) alone (Fig. 2E and fig. S4). However, in accordance with the DBP data, treatment of TNBC cells with NAMPT inhibitors sensitized most cancer cells to selected BH3 mimetic (Fig. 2E and fig. S4). As predicted by the DBP data, the cell death induction varied between cell lines tested. In addition, as expected by the increased overall apoptotic priming of TNBC cells after NAMPT inhibition, NAMPT inhibitors also increased the sensitivity of TNBC cells to a general apoptosis inducer, staurosporine (fig. S5). Moreover, the sensitization effect to BH3 mimetics or staurosporine by NAMPT inhibitors was less observed in MCF10A cells compared to the SUM149, MDA-MB-231 and MDA-MB-468 TNBC cell lines (Fig. 2E and fig. S5).
Together these data demonstrate that the increased apoptotic priming and anti-apoptotic dependencies induced by NAMPT inhibition in a subset of TNBC cells lead to an increased sensitivity of those TNBC cells to BH3 mimetics and other apoptosis inducers in vitro.
NAMPT inhibitors increase mitochondrial apoptotic priming, anti-apoptotic dependencies, and sensitivity of a TNBC patient-derived xenograft model to BH3 mimetics
We next investigated whether NAMPT inhibition increases mitochondrial apoptotic priming and anti-apoptotic dependencies in vivo using two patient-derived xenograft (PDX) models of TNBC (designated PDX1 and PDX2)34,35. Prior to the in vivo experiment, we performed BH3 profiling on freshly isolated cancer cells from untreated PDX1 and PDX2 tumors in order to predict sensitivities to the combination of a NAMPT inhibitor and a BH3 mimetic, and to decide which BH3 mimetic to use in vivo. BH3 profiling revealed no significant differences in baseline apoptotic priming or anti-apoptotic dependencies of PDX1 and PDX2 tumor cells (fig. S6A). DBP of the PDX tumor cells after 48 hours FK866 or GPP78 exposure revealed a higher induction of mitochondrial apoptotic priming in PDX2- than in PDX1-derived tumor cells (Fig. 3, A and B). ABT-263 was used as a positive control for induction of apoptotic priming. Furthermore, DBP using a broader range of BH3 peptides—HRK, ABT-263 and A-133; and MS1 and S63845—revealed an increased dependency of PDX2 tumor cells on the anti-apoptotic proteins BCL-XL and MCL-1, respectively, after NAMPT inhibition (Fig. 3C). More than the response to HRK, ABT263 and A-133, the response to the MS1 peptide and S63845 was consistently observed after either FK866 or GPP78 exposure. These PDX data contrast with the cell line data, where we observed less of a response to the MS1 peptide and S63845 (MCL-1 dependency) than to HRK, A-133 and A-115 (BCL-XL dependency) (Fig. 2D). Unlike PDX2 tumor cells, increased anti-apoptotic dependencies were not observed in PDX1 tumor cells (Fig. 3C). Together these data predicted that PDX2 tumors would be more sensitive to the combination of a NAMPT inhibitor and a BH3 mimetic than PDX1 tumors.
Figure 3. NAMPT inhibition increases overall mitochondrial apoptotic priming, anti-apoptotic dependencies and sensitivity to S63845 of TNBC PDX2 but not PDX1.
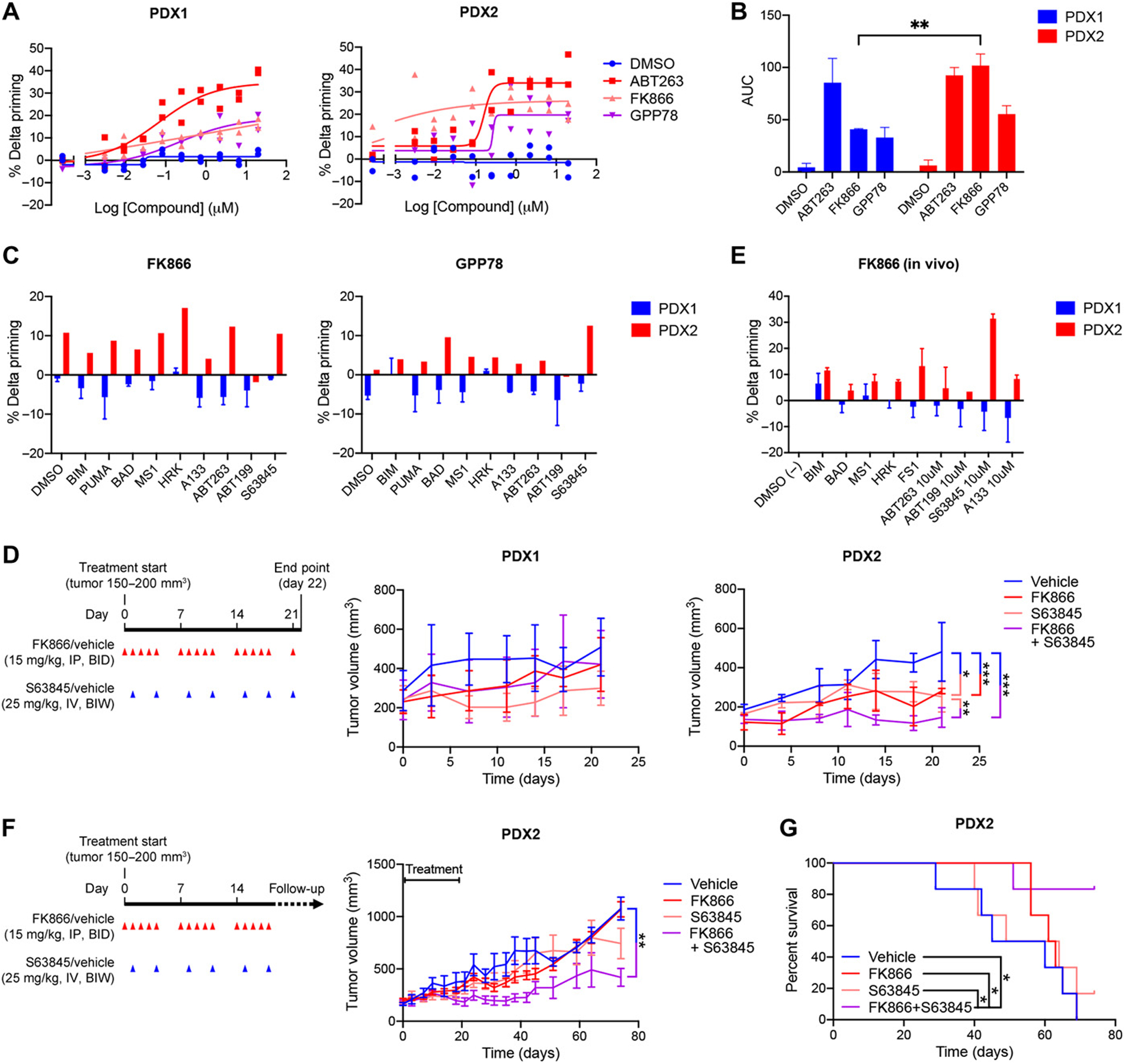
(A) Percentage delta priming of freshly isolated PDX1 and PDX2 tumor cells in vitro exposed to 0.01 μM FK866 and 0.1 μM GPP78 for 48 hours. Percent delta priming is calculated by comparing cytochrome c abundance in drug-treated versus vehicle-treated cells. ABT-263 is used as a positive control for induction of apoptotic priming, DMSO (vehicle) is used as a negative control. Data are average and SD of 2 or 3 independent experiments. (B) AUC and SD of % delta priming data shown in (A). Statistical analysis as done using two-way ANOVA with Sidak’s post-hoc test. (C) DBP of freshly isolated PDX1 and PDX2 tumor cells exposed in vitro to 0.01 μM FK866 and 0.1 μM GPP78 for 48 hours. Responses to the BIM and PUMA peptide indicates overall mitochondrial priming. Responses to the BAD, HRK, MS1, FS1 peptides and BH3 mimetics indicate specific anti-apoptotic dependencies. BAD and ABT-263 indicate BCL-2, BCL-XL or BCL-W dependency; HRK, A-133 and A-115 indicate BCL-XL dependency; ABT-199 indicates BCL-2 dependency; MS1 and S63845 indicate MCL-1 dependency. Data are average of two individual experiments. (D) Treatment schedule and volume of PDX1 and PDX2 tumors measured during the short-term efficacy study. Treatment was started when tumors reached 150–200mm3, indicated as Day 0 on x-axis. Red arrows indicate FK866 (15mg/kg) or vehicle (1% hydroxypropyl-β-cyclodextrin and 12% 1,2-propylenglycol in saline) treatment via intraperitoneal (I.P.) injection twice a day (BID). Blue arrows indicate S63845 (25 mg/kg) or vehicle (2% Vitamin E/TPGS) administration via intravenous (I.V.) injections twice a week (BIW). Mice were treated for 21 days and sacrificed on day 22. Tumor volume graphs show average and SD, n = 4 mice per treatment group. The trend in tumor volume changes by treatment across time were modeled using a linear mixed model with a random effect per subject, and a compound symmetry correlation structure using R 3.6.3. Family-wise error was adjusted via Tukey’s multiple correction method: *P <0.05; **P <0.01 and ***P <0.001; no statistical labelling indicates no statistical difference was observed. Combination treatment versus S63845, P=0.007; combination treatment versus vehicle, P<0.001; S63845 versus vehicle, P=0.021; and FK866 versus vehicle, P<0.001. (E) Percentage delta priming of PDX1 and PDX2 tumor cells at the day 22 end point of the short-term in vivo efficacy study in (D). Tumors were dissociated and BH3 profiling was performed on the freshly isolated tumor cells. Percentage delta priming refers to the difference in apoptotic priming of tumor cells derived from FK866-treated versus vehicle-treated animals. Data shown are average and SD, n = 3 or 4 tumors per treatment group. (F) Treatment schedule and PDX2 tumor volumes of mice in long-term efficacy study. Treatment was initiated when tumors reached 150–200mm3, indicated as Day 0 on x-axis. Red arrows indicate FK866 (15mg/kg) or vehicle (1% hydroxypropyl-β-cyclodextrin and 12% 1,2-propylenglycol in saline) treatment by intraperitoneal (I.P.) injection twice a day (BID). Blue arrows indicate S63845 (25mg/kg) or vehicle (2% vitamin E/TPGS) by intravenous (I.V.) injections twice a week (BIW). Mice were treated for 19 days. Animals were sacrificed when their tumors reached 1200 mm3. Study was ended when all vehicle treated animals reached tumor size of 1200 mm3 and were sacrificed (53 days post-treatment). Data are average and SD, n = 6 mice per treatment group. Statistical analysis as done using a Tukey’s multiple testing correction on a linear mixed model in R 3.6.3: **P <0.01; no statistical labelling means no statistical difference was observed. (G) Kaplan-Meier analysis of PDX2 data represented in (F). A logrank test was used to estimate a difference in survival effects across time between all treatments, multiple testing induced family-wise error was adjusted using Benjamini & Hochberg with R 3.6.3: *P <0.05.
Because we consistently observed mitochondrial response to S63845 (% delta priming of more than 10) after both FK866 and GPP78 exposure (Fig. 3C), we chose to combine FK866 with the MCL-1 antagonist S63845 for our in vivo experiments. We treated orthotopic PDX1- and PDX2-bearing animals with FK866, S63845 or the combination according to the treatment schedule described above (Fig. 3D), in four mice per treatment arm. In agreement with the DBP data, we observed that for the PDX2 bearing animals the tumor growth, measured by calipers, was significantly lower in the combination treatment group (FK866 and S63845) compared to S63845- and vehicle-treated group (Fig. 3D and fig. S6B). Less significant differences were observed for the tumor growth of the single agents compared to the vehicle treatment arms S63845 and FK866 (Fig. 3D and fig. S6B). Comparison of tumor volumes at endpoint (day 22) also showed the most significant decrease in tumor volume for the combination therapy (fig. S6, C and D). No significant differences in tumor growth or volumes between the different treatment groups were observed in the PDX1 bearing animals (fig. S6, C and D).
We hypothesized that the greater sensitivity of the PDX2 tumors to the combination would be associated with a greater increase in mitochondrial apoptotic priming of PDX2 compared to PDX1 following in vivo exposure to FK866. To test this, we performed BH3 profiling on cells derived from tumors collected at the endpoint of the treatment (treatment schedule in Fig. 3D). BH3 profiling comparing cancer cells isolated from tumors from vehicle-versus single agent FK866-treated animals revealed similar profiles as in vitro DBP of treatment-naive tumors: in vivo treatment with FK866 increased the apoptotic priming and anti-apoptotic dependencies of PDX2 tumor cells but failed to prime PDX1 tumor cells (Fig. 3E).
Subsequently, to test if short-term targeting of NAMPT can have a prolonged effect on the tumor volume after treatment stop, we repeated the efficacy study on the responsive PDX2 model. This time we tested single agents or combination treatment in tumor bearing animals and continued to monitor tumor growth after the 19-day treatment period. Over the 55 days following cessation of the treatment a significant reduction in tumor growth was observed between the combination-treated (FK866 and S63845), but not the single agents-, and the vehicle-treated animals (Fig. 3F and fig. S6E). Twenty-one days after treatment cessation there was still a significant difference in tumor volume between the combination (FK866 and S63845) and vehicle treatment groups (fig. S6F). By 55 days following treatment, tumor volume was significantly different in the combination arm compared to the single agent S63845, the single agent FK866 and vehicle arms (fig. S6F). Combination-treated animals also lived longer than single agent- and vehicle-treated animals before study endpoints (tumor volume of 1200 mm3 or all vehicle-treated animals taken off the study) were reached (Fig. 3G).
Throughout the in vivo studies, we monitored animal weight as an estimate of safety and tolerability associated with the treatments. Except for one FK866-treated animal euthanized due to a 20% body weight reduction observed immediately after the first two injections, no significant changes in weight were observed (fig. S7, A to F).
NAD+ levels need to drop below a critical level for apoptotic priming and anti-apoptotic dependencies to be increased in TNBC cells
To understand why NAMPT inhibition induces apoptotic priming and sensitivity for BH3 mimetics in some TNBC cells but not in others, we first looked at what happened to the NAD+ levels after NAMPT inhibition in the different cell lines tested. Based on the results of the combination treatment of TNBC cells with BH3 mimetics (Fig. 2E) we classified the TNBC cells as sensitive, intermediately sensitive, or insensitive to BH3 mimetics after NAMPT inhibition (Fig. 4A). When looking at the NAD+ levels after treatment with a NAMPT inhibitor relative to vehicle-treated conditions within each cell line we observed a drop in all the TNBC cell lines (fig. S8A). However, the NAD+ levels dropped much lower in the sensitive and intermediately sensitive compared to in the insensitive cell lines (Fig. 4B). Similarly, a drop in NAD+ was observed in both the sensitive (PDX2) and the insensitive (PDX1) PDX models after in vivo treatment with FK866 (fig. S8B), but the NAD+ levels dropped lower in the sensitive compared to the insensitive model (Fig. 4C). These results imply that a critical level of NAD+ needs to be maintained in the TNBC cells and the in vivo models to prevent the induction of apoptotic priming and anti-apoptotic dependencies. There were no observable differences in the NAD/NADH ratio that could explain the difference between sensitive and insensitive models (fig. S8, C and D).
Figure 4. NAD+ levels need to drop below a critical level for apoptotic priming and anti-apoptotic dependencies to be increased in TNBC cells after NAMPT inhibition.
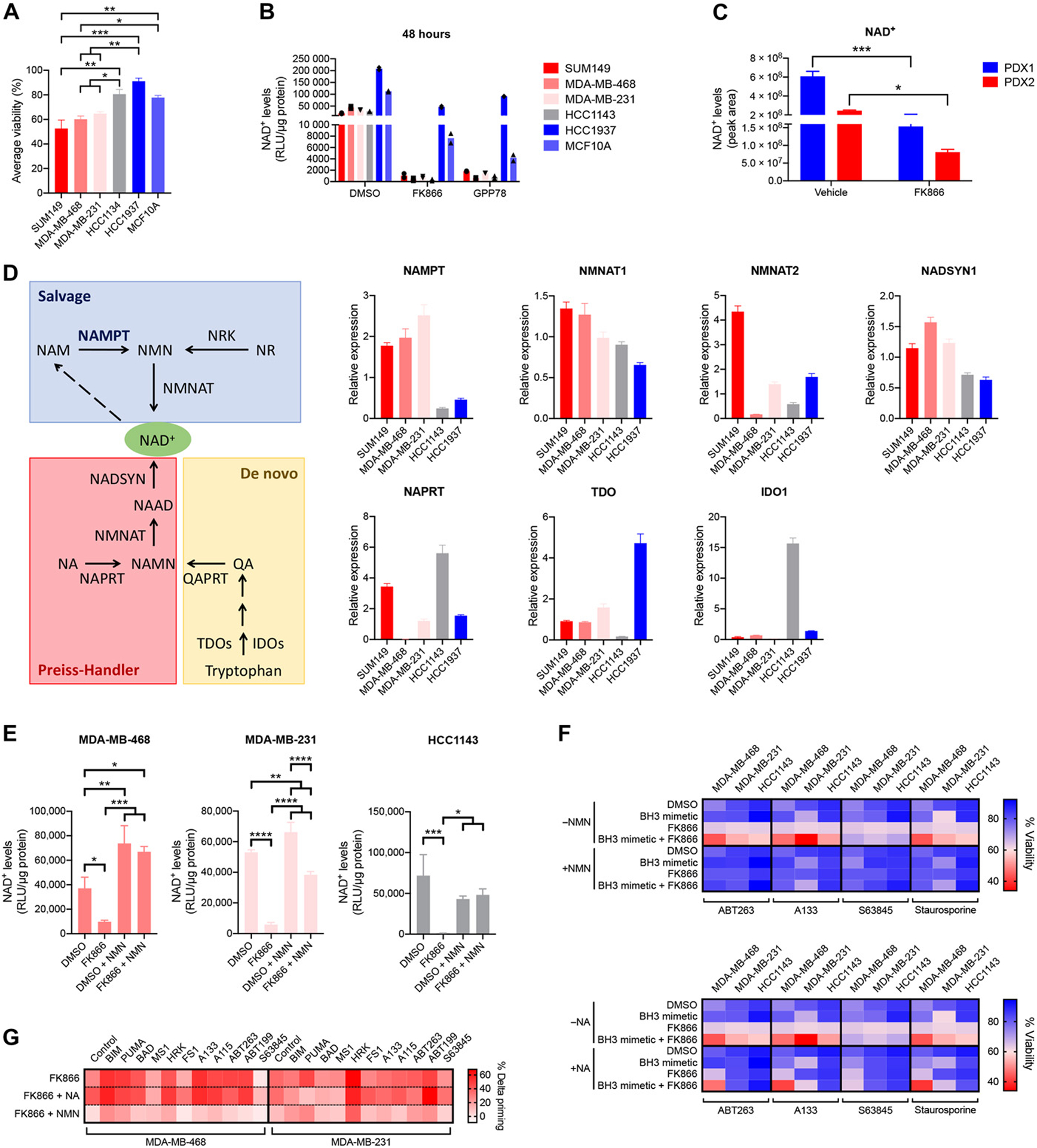
(A) Classification of TNBC cell lines according to their sensitivity for BH3 mimetics after NAMPT inhibition, based on the “average viability” calculated from the data presented in Fig. 2E and fig. S5:
Cell lines are indicated in shades of red for sensitive, shades of gray for intermediately sensitive, and shades of blue for insensitive lines. Statistical analysis as done using one-way ANOVA with Tukey’s post-hoc test. (B) NAD+ levels in indicated cell lines after 48 hours treatment with 0.01 μM FK866 or 0.1 μM GPP78, measured using the NAD/NADH kit (Promega). Data are average of 2 independent experiments. (C) NAD+ levels in PDX tumors after in vivo treatment with single-agent FK866 as described in Fig. 3D, measured using untargeted metabolomics and expressed as peak areas. Data are average and SD of n = 3 or 4 animals per treatment arm. Statistical analysis as done using one-way ANOVA with Tukey’s post-hoc test. (D) Schematics of different NAD+ synthesis pathways and expression levels of rate-limiting enzymes: de novo synthesis in yellow, Preiss-Handler pathway in red, and salvage pathway in blue. Abbreviations not yet defined:; NMNAT: NMN adenylyl transferase; NRKs: nicotinamide riboside kinases; NR: nicotinamide riboside; NADSYN: NAD synthetase; NAAD: nicotinic acid adenine dinucleotide; NAMN: nicotinic acid mononucleotide; QART: quinolinic acid phosphoribosyltransferase; QA: quinolinic acid. Expression levels of NAD+ synthesis enzymes are determined at baseline. Data are mean and SE of 3 independent experiments. (E) NAD+ levels in indicated cell lines after 24 hours treatment with 0.01 μM FK866 in the presence or absence of 25μM NMN, measured using the NAD/NADH kit (Promega). Data are mean and SD of 3 independent experiments. Statistical analysis as done using one-way ANOVA with Tukey’s post-hoc test. (F) Viability data of indicated cell lines treated for 72 hours with 0.01 μM FK866, 0.1 μM BH3 mimetics, 0.03 μM staurosporine or combination in the presence or absence of 25 μM nicotinamide mononucleotide (NMN) or 25 μM nicotinic acid (NA). Data are means of 3 independent experiments. Data also are presented as dot plots of average and SD in supplementary material (fig. S10). (G) Percentage delta priming of sensitive cell lines MDA-MB-468 and MDA-MB-231 after 48 hours exposure to 0.01 μM FK866 in the presence or absence of NA or NMN (25 μM). Data are means of 3 independent experiments. Data are also presented as bar graphs of average + SD in supplementary material (fig. S11).
Cells can produce NAD+ via different pathways (Fig. 4D): through de novo synthesis from tryptophan with indoleamine 2,3-dioxygenase 1 (IDO1) or tryptophan 2,3-dioxygenase (TDO) as rate-limiting enzymes; through the Preiss-Handler pathway from nicotinic acid (NA) with NA phosphoribosyltransferase (NAPRT) being the rate-limiting enzyme in this pathway; or through the salvage pathway with nicotinamide phosphoribosyltransferase (NAMPT) being the rate-limiting enzyme. We tested whether the differential drop in NAD+ levels and sensitization to BH3 mimetics after NAMPT inhibition could be explained by differential dependencies of the TNBC cell lines on the different pathways of NAD+ synthesis. We measured the expression levels of rate-limiting and downstream enzymes of the different NAD+ synthesis pathways (Fig. 4D). Supporting the differential sensitivity to NAMPT inhibitors, we detected high expression of NAMPT in sensitive cell lines SUM149, MDA-MB-231 and MDA-MB-468, while the intermediate and insensitive cells lines HCC1143 and HCC1937 exhibited low expression of NAMPT but high expression of enzymes involved in de novo NAD+ synthesis (TDO, IDO1). In contrast to what was published before36, we found no correlation between the expression levels of the enzymes of the Preiss-Handler pathway and the sensitivity classes (Fig. 4D). Moreover, inhibition of NAPRT, using 2-hydroxinicotinic acid (2-HNA), did not make the intermediate sensitive HCC1143 or the insensitive HCC1937 cells sensitive to the combination of FK866 + BH3 mimetics or staurosporine (fig. S9), straightening our hypothesis that HCC1143 an HCC1937 are resistant to FK866-induced sensitization to BH3 mimetics, because they have an overexpression and activation of the de novo NAD+ synthesis pathway rather than the Preiss-Handler pathway.
Addition of nicotinamide mononucleotide (NMN), the downstream metabolite of the NAMPT enzyme, to the culture medium restored cellular NAD+ levels (Fig. 4E) and obviated the FK866-induced sensitization to BH3 mimetics and staurosporine (Fig. 4F and fig. S10). In contrast, supplementation with nicotinic acid (NA), the substrate for the Preiss-Handler pathway, had no effect (Fig. 4F and fig. S10). Likewise, supplementation of the medium with NMN, but not NA, rescued the increased mitochondrial apoptotic priming observed after FK866 treatment (Fig. 4G). NMN addition could not rescue cells from apoptotic stress that does not selectively interferes with the NAD+ synthesis (staurosporine treatment; fig. S11). Additionally, we did not observe an association of sensitivity with BRCA or homologous recombination repair status of the TNBC models (table S2). These results indicate first that the sensitive TNBC lines mainly use the salvage pathway to synthesize NAD+ and, second, that it is the on-target inhibition of the NAMPT enzyme that causes the increased sensitization to BH3 mimetics after FK866 treatment.
NAD+ depletion induces apoptotic priming and anti-apoptotic dependencies in TNBC by dysregulation of adenine synthesis rather than via overall metabolic collapse
Next, we investigated the mechanism by which a drop in NAD+ levels can induce apoptotic priming and anti-apoptotic dependencies in TNBC cells and PDX models. Because NAD+ is a cofactor and substrate for over 200 metabolic enzymes, many metabolic pathways are known to be affected by the cellular NAD+ levels37. To test whether an overall metabolic collapse38 or the dysregulation of one particular metabolic pathway is responsible for the sensitization of TNBC cells to BH3 mimetics after NAMPT inhibition, we started by first visualizing exactly which metabolic pathways were affected in our TNBC models after NAMPT inhibition. To do this we performed metabolomic analysis on sensitive, intermediately sensitive and insensitive TNBC cell lines after NAMPT inhibition and on the TNBC PDX tumors after 21 days in vivo treatment (treatment schedule in Fig. 3D). Results indicated a reduction of NAD+ levels in all the TNBC models after treatment with a NAMPT inhibitor compared to vehicle treated samples (Fig. 5, A and B). These data confirmed our earlier NAD+ measurements (Fig. 4B). Moreover, they underscore that the NAMPT inhibitors hit their target in both sensitive and insensitive models and that failure of apoptotic priming induction in PDX1 tumors in vivo (Fig. 3E) was not caused by a failure of FK866 compound reaching the tumors. Additionally, the metabolomics data revealed that NAMPT inhibitors, both in the cell lines and in the in vivo treated PDX models, caused an increase of metabolites of the upper glycolytic pathway and a decrease of the intermediates of the lower glycolytic pathway and tricarboxylic acid (TCA) cycle; an increase in the pentose phosphate pathway metabolites; a drop of some intermediates of the nucleotide synthesis; and an increased oxidative stress signature (Fig. 5, A and B). Changes in amino acid levels, urea cycle intermediates, lipids, vitamins and cofactors, and glycosylation intermediates after NAMPT inhibition were less prominent (fig. S13, A and B). Overall, these observed metabolic changes after NAMPT inhibition were less pronounced in the insensitive compared to the sensitive models signature (Fig. 5, A and B) and were not as strong at 24 hours (fig. S13C) compared to 48 hours after in vitro treatment (Fig. 5A).
Figure 5. NAD+ depletion induces apoptotic priming and anti-apoptotic dependencies in TNBC by dysregulation of specific metabolic pathways rather than an overall metabolic collapse.
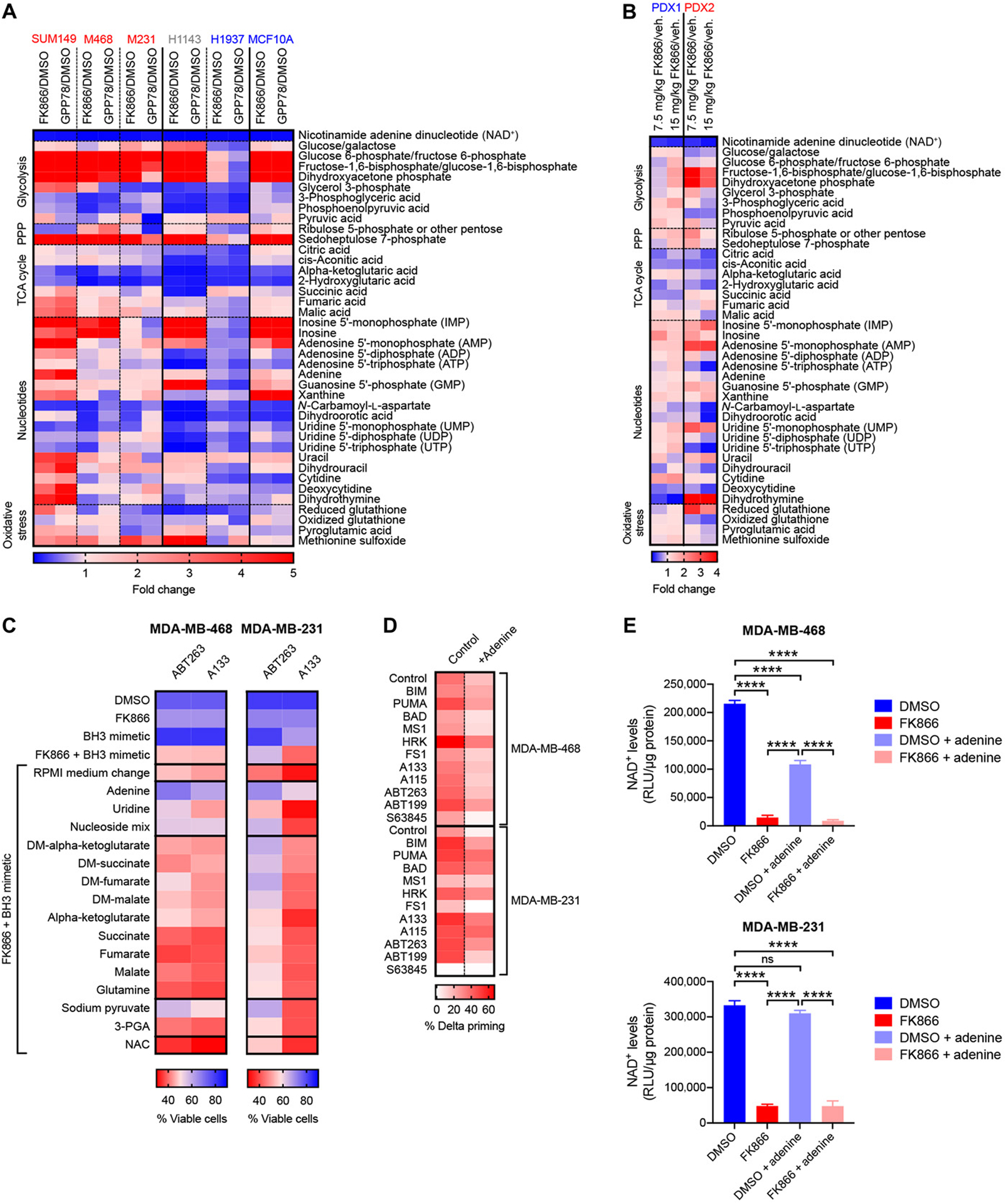
(A) Metabolomics data of intermediates of the glycolysis, pentose phosphate pathway (PPP), tricarboxylic acid (TCA) cycle, nucleoside synthesis and the oxidative stress pathways extracted from cell lines treated for 48h with 0.01 μM FK866 or 0.1 μM GPP78. Data are expressed as fold changes compared to vehicle treated samples. Data are average of technical duplicates. Sensitive models are indicated in red, intermediately sensitive models are indicated in gray, insensitive models are indicated in blue. (B) Metabolomics data of intermediates of the glycolysis, pentose phosphate pathway (PPP), tricarboxylic acid (TCA) cycle, nucleoside synthesis and the oxidative stress pathways extracted from PDX tumors treated in vivo with 7.5 mg/kg or 15 mg/kg FK866 according to treatment schedule in Fig. 3D. Data are expressed as fold changes compared to vehicle-treated samples. Data are average of n = 2 to 4 mice per treatment arm. (C) Microscopy-based annexin V/Hoechst cell viability data of MDA-MB-468 and MDA-MB-231 cells co-treated for 72 hours with 0.01 μM FK866 and 0.1 μM BH3 mimetics in the presence or absence of different metabolites. Data are means of 3 independent experiments. (D) Percentage delta priming of TNBC cells after 48 hours exposure to 0.01 μM FK866 in the absence or presence of 25 μM adenine. Data are average of 3 independent experiments. (E) NAD+ levels after 48 hours NAMPT inhibition (0.01 μM FK866) in presence or absence of 25 μM adenine. Data are average and standard deviation of 3 independent experiments. Statistical analysis as done using one-way ANOVA with Tukey’s post-hoc test: ****P <0.0001; no statistical labelling indicates no statistical difference was observed.
To test whether a decreased activity of one of the affected metabolic pathways alone could be responsible for the increased apoptotic priming and anti-apoptotic dependencies of TNBC cells after NAMPT inhibition we performed rescue experiments on two sensitive TNBC cell lines (MDA-MB-468 and MDA-MB-231). In these experiments, we supplemented the medium with intermediates of the different metabolic pathways that were affected by NAMPT inhibition and looked which intermediates could rescues the cell death induction after combination treatment of FK866 plus BH3 mimetics (Fig. 5C). We did not observe a rescue from cell death by adding uridine (precursor pyrimidine synthesis), TCA cycle intermediates, glycolysis intermediates or the ROS scavenger N-acetyl-cysteine (NAC). Addition of a nucleoside mix or sodium pyruvate could prevent cell death induction by combination treatment of FK866 plus ABT-263 but did not prevent cell death after FK866 plus A133 treatment. In contrast, exogenous adenine addition rescued the cell death induction after FK866 plus ABT-263 and FK866 plus A133. Moreover, exogenous adenine addition also rescued the increased apoptotic priming after NAMPT inhibition (Fig. 5D). However, adenine supplementation did not lead to an increase in NAD+ levels (Fig. 5E), suggesting adenine does not cause a rescue by re-constituting the depleted NAD+ stores of the cancer cells. Together these data suggest that it is not the overall metabolic collapse after NAMPT inhibition, but rather the specific dysregulation of adenine synthesis that sensitizes TNBC cells to BH3 mimetics (Fig. 6).
Figure 6. Mechanism of action NAMPT inhibitors plus BH3 mimetics.
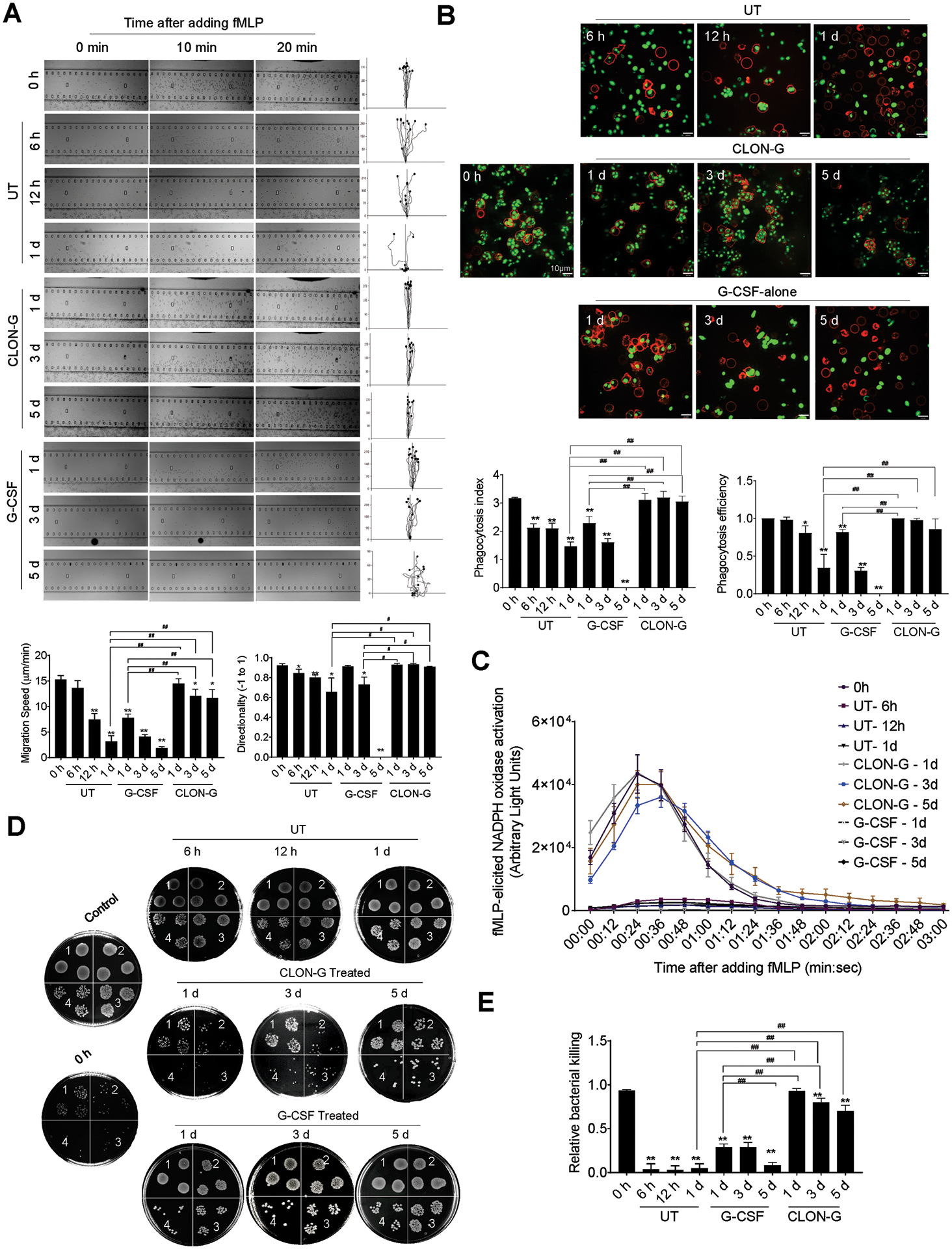
TNBC cells that are sensitive to the combination treatment of a NAMPT inhibitor and a BH3 mimetic are reliant on NAMPT to maintain critical levels of NAD+. In these cells, other NAD+ synthesis pathways, with NAPRT and TDO and IDO as rate-limiting enzymes, cannot compensate for NAMPT inhibition and NAD+ levels drop below a critical threshold. While several metabolic pathways are dysregulated, it is adenine depletion in particular that increases the mitochondrial apoptotic priming. TNBC cells that are insensitive to the combination treatment activate compensatory NAD+ synthesis pathways upon NAMPT inhibition. While NAD+ levels drop, they remain above the critical threshold and no increased mitochondrial apoptotic priming is observed.
Discussion
In this study, we identified a metabolic vulnerability to target to selectively increase sensitivity of a subset of TNBC to BH3 mimetics. We found that inhibition of the rate-limiting enzyme of the NAD+ salvage pathway, NAMPT, leads to an increased dependency of some TNBC cells on specific anti-apoptotic BCL-2 family members and sensitizes them to the corresponding BH3 mimetics.
NAMPT has been explored as a target for cancer therapy in the past33,39. However, despite promising in vitro and pre-clinical data in both solid and hematologic cancers40–42, the results of phase I and phase II clinical trials with NAMPT inhibitors were discouraging. There was a lack of single-agent drug efficacy, and some trials had to be stopped due to toxicities40,41. One of the main toxicities observed in patients treated with NAMPT inhibitors is lymphopenia. In agreement, it is shown from in vitro work that activated T-cells are particularly vulnerable to NAMPT inhibition43,44. The toxic effect of FK866 on activated T-cells could, however, be rescued by addition of nicotinamide (NAM) and nicotinic acid (NA)43, whereas in our experiments NA could not rescue the sensitized TNBC cells from cell death induction after treatment with FK866 plus an apoptosis inducer. Thus in theory, the lymphopenia could be ameliorated by increasing dietary intake or by pharmacological administration of NA, without affecting the specific toxicity of a NAMPT inhibitor + apoptosis inducers observed in a subset of TNBC cells. Moreover, in this study, BH3 profiling revealed that even at doses that do not induce cell death, NAMPT inhibitors (FK866 and GPP78) increase mitochondrial apoptotic priming and anti-apoptotic dependencies specifically in TNBC cancer cells, but not in non-malignant counterparts. In addition, because BH3 profiling allowed us to rationally identify BH3 mimetics to use in combination with NAMPT inhibition, we here propose that NAMPT inhibitors might be re-purposed and used at more tolerable doses in combination with BH3 mimetics to improve the therapeutic index.
BH3 profiling correctly identified which PDX model would be sensitized to BH3 mimetics by NAMPT inhibition, suggesting that BH3 profiling might be a good functional predictive biomarker for this combination treatment. Previous reports have described other predictive biomarkers for the use of NAMPT inhibitors as single agent or in combination therapy, varying from NAPRT and NAMPT expression in the context of single-agent use36, to the BRCA and homologous repair status in the context of the combination of NAMPT and PARP inhibitors45. In our study we did not observe an association of NAPRT expression status nor BRCA or homologous repair status of the TNBC models with their sensitivity to a NAMPT inhibitor plus a BH3 mimetic. On the other hand, high NAMPT expression was associated with a response of the TNBC models to a combination of NAMPT inhibitors and BH3 mimetics. In addition to predicting which models would respond to combination treatment of a NAMPT inhibitor and a BH3 mimetic in vivo, BH3 profiling also helped in prioritizing which BH3 mimetic to use in the combination in vivo. Based on the DBP data of PDX tumor cells we choose in this study to combine FK866 with the MCL-1 antagonist S63845 for in vivo PDX model treatments. The cell line data, on the other hand, suggest that the combination of a NAMPT inhibitor with a BCL-XL antagonist might also be interesting in the context of TNBC. We anticipate that in some cases of TNBC, combination of a NAMPT inhibitor with a BCL-XL antagonist might be efficacious. BH3 profiling could potentially be used to choose a BH3 mimetic and individualize the treatment strategy. Additionally, since NAMPT is overexpressed in a wide range of solid and hematologic cancers46, this study represents a platform that can be repeated in many other cancer types to identify potentially efficacious combination therapies using BH3 mimetics.
It would be interesting to test which pool of NAD+ is influencing the apoptotic priming of the cells the most: the cytosolic/nucleus, the mitochondrial, the endoplasmic reticulum or the peroxisomal pool or the combination of different NAD pools47. In the metabolomics experiment we performed after NAMPT inhibition, we had observed that both cytosolic and mitochondrial enzymatic reactions were affected, suggesting that NAMPT inhibition probably affects more than just one cellular NAD pool. However, these experiments do not allow us to draw conclusion on which NAD pool is affecting mitochondrial apoptotic priming.
In addition, the exact mechanism through which decreased NAD+ levels lead to an increased apoptotic priming and increased anti-apoptotic dependencies remains to be determined. Since NAD+ is an important co-factor and substrate for many metabolic enzymes, we tested which metabolic pathways were affect by the decrease in NAD+ levels after NAMPT inhibitor treatment. Performing rescue experiments, adding different intermediates of the affected metabolic pathways, we found that adenine addition sufficed to rescue the NAMPT-inhibitor induced mitochondrial apoptotic priming and sensitization to BH3 mimetics. How adenine addition rescued mitochondrial apoptotic priming needs further investigation. One possibility is that the purinosome and mTOR28 are involved in the regulation of apoptotic priming after NAMPT inhibition. We note that mTOR activity is selectively regulated by cellular adenine levels48. Moreover, mTOR was shown to control the purinosome formation and its localization to the mitochondrial outer membrane49, the same location where the BCL-2 family members interact to decide on the cell fate50. A recent publication confirmed the localization of the purinosome at the mitochondria51. Another explanation why adenine supplementation might rescue the increased sensitization to BH3 mimetics after NAMPT inhibition, might be because of a direct effect of adenine on the enzymes from the de novo NAD+ synthesis pathway and pyridine salvage pathway as reported in yeast52. In future studies it will thus be of interest to investigate if mTOR is involved in the induction of mitochondrial apoptotic priming and anti-apoptotic dependencies upon NAMPT inhibition. Moreover, it will be interesting to test whether there is a direct interaction between BCL-2 family members and the purinosome or whether adenine rescues because of its effect on the NAD+ production. Additionally, next to its role in energy metabolism, NAD+ is also an important co-enzyme and substrate for enzymes like sirtuins, poly(ADP-ribose) polymerases (PARPs) and cyclic ADP-ribose synthases (cADPRSs)53.Therefore, it would be interesting to test in follow-up studies what the contribution of dysregulation of these NAD+ dependent enzymes is to the induced anti-apoptotic dependencies and apoptotic priming observed after NAMPT-inhibition.
Lastly, it needs to be taken into account that several resistance mechanisms to single agent NAMPT inhibitors have been published, among which—in line with our observations—upregulation of the expression of enzymes of the de novo NAD+ synthesis pathway54. In addition, the upregulation of the NARPT enzyme, NAMPT mutations and the upregulation of the multi-drug resistance protein 1 (MDRP1), epigenetic and cellular signaling changes affecting the function of sirtuins, PARPs and cADPRS, have been described as resistance mechanisms in literature. These mechanisms might contribute to the resistance to the combination of NAMPT inhibitors plus pro-apoptotic drugs as well and need to be investigated in follow-up studies. However, as we tested here, we would propose that greatest efficacy will be found when used in combination with other active agents.
Here we showed that a functional assay could detect a heretofore unappreciated metabolic vulnerability in TNBC. We identified a novel pathway by which NAMPT inhibition can lead to critical NAD+ and adenine depletion (Fig. 6). Adenine supplementation not only rescued the viability of TNBC cells, it also rescued the increased mitochondrial priming. Our results therefore demonstrate the novel communication of a metabolic pathway to the mitochondrial apoptotic pathway in a cancer cell. The use of a broad panel of drugs of known target in our primary screen allowed us not only to discover new biology, but also to rapidly construct active combinations. We anticipate that similar methods can be applied to identify analogous metabolic vulnerabilities in other cancer contexts which can be exploited similarly by combination with the correct BH3 mimetic.
Materials and Methods
Materials
BH3 mimetic drugs used in this study were: ABT-236 (MedChem Express), ABT-199 (MedChem Express), A-1331852 (Chemietek, A-133), A-1155463 (Chemietek, A-115), and S63845 (MedChem Express). FK866, GPP78 and staurosporine were purchased from MedChem Express.
Cell Culture
Cancer cell lines HCC1143, HCC1937, MDA-MB-231, and MDA-MB-468 were obtained from ATCC. Cancer cell line SUM149 was obtained from Asterand. All cancer cells were grown in RPMI-1640 medium supplemented with 10% heat inactivated fetal bovine serum (HI-FBS, Gibco), no antibiotics or other additives were used. Normal cell lines MCF10A (mammary epithelial cell line) and HK-2 (mouse kidney cell line) were obtained from ATCC. HMEC human mammary epithelial cells (HMECs) were obtained from Lonza. Hk-2 were cultured in DMEM medium, in the presence of 1x penicillin/streptomycin and 1% L-glutamine (Gibco) and 10% HI-FBS. MCF10A and HMEC cells were cultured in Mammary Epithelial Growth Medium (MEGM = MEBM™ Mammary Epithelial Basal Medium with Mammary Epithelial Growth Medium SingleQuots™ Supplements added, Lonza). Freshly isolated triple-negative breast cancer PDX tumor cells were cultured in Advanced DMEM/F12 medium (Gibco) + N2 supplement (Gibco) + B27 supplement (Gibco) + 1% L-glutamine (Gibco) + 10% HI-FBS (Gibco). All cells were kept at 37°C under 5% CO2. Human plasma like medium was prepared as previously described30 and supplemented with dialyzed FBS (Gibco). Cell line authentication was performed on all cell lines used in the manuscript by means of Short Tandem Repeat (STR) analysis by the Molecular Diagnostics Core at the Dana-Farber Cancer Institute. All cell lines tested negative for mycoplasma.
Library screening
HT-DBP was used to screen a library of 192 metabolism-perturbing small molecules as previously described17. In brief, TNBC cell lines were seeded at a density of 1000–2000 cells/well in 30 μL medium/well. After attachment, the cells were drugged with a library of metabolism-perturbing small molecules developed and described previously by Harris et al29. Drugs were added using pin transfer and incubated for 48 hours before performing microscopy based BH3 profiling. For BH3 profiling, BIM peptide dilution curves were generated on untreated tumor cells. A concentration of BIM peptide that corresponds to 10% of cytochrome c negative cells was selected based on this dilution curves and was used to preform BH3 profiling on the drug treated cells. Treated cells were washed 3 times with PBS using the BioTek 406EL plate washer (BioTek). Consequently, the BIM peptide was added using the HP D300e Digital Dispenser (Hewlett-Packard Development Company) at the concentration decided on based on the dilution curves (see above). The cells were incubated in BH3 profiling buffer containing 0.002% digitonin for 1h. Cells were fixed in paraformaldehyde for 15 min and the fixative was neutralized using a tris/glycine buffer. Cells were stained overnight with Hoechst33342 (Nuclei, Invitrogen) and anti-cytochrome c-Alexa Fluor 647 antibody (BioLegend). Prior to imaging, stain solution was washed out using the BioTek 406EL plate washer. Fluorescent microscope images from HT-DBP plates were acquired using the IXM XLS high content widefield microscope (Molecular Devices) at the ICCB Longwood Screening Facility. The cytochrome c positive cells were quantified using the Multi Wavelength Cell Scoring module in Metamorph software. Metabolic agents that increase overall mitochondrial apoptotic priming will increase the BIM peptide-induced cytochrome c release compared to DMSO treated wells. The difference in cytochrome c release between drug- and DMSO-treated wells is quantified and referred to as % delta priming16. Cell counts/well information is obtained based on Hoechst staining. Dose-response curves of percent delta priming data for all individual MPSM were used to calculate the Area Under the Curve (AUC) in the Graphpad Prism software (Total peak area). For the representation of the AUC of the hits from the library screen the AUC was normalized to the number of data points (concentrations) that were used to calculate the AUC. Validation and counter screenings were performed in the same way, but instead of using a pin transfer, the selected metabolism targeting agents were added using the HP D300e Digital Dispenser (Hewlett-Packard Development Company).
BH3 profiling
For this study both “baseline BH3 profiling” and “dynamic BH3 profiling” techniques were used as described previously8,9,16. In brief, baseline BH3 profiling was performed on treatment naïve PDX1 and PDX2 derived tumor cells following the iBH3 technique9. Single cell tumor cells were obtained as described under PDX Tumor Dissociation. Cells were exposed to different BH3 peptides for 50 min in MEB-2 buffer containing 0.001% digitonin. After BH3 peptide exposure cells were fixed for 10 min in paraformaldehyde. Fixation was stopped by addition of tris/glycine buffer. Cells were stained overnight with DAPI (Nuclei, Biolegend) and anti-cytochrome c-Alexa Fluor 647 antibody (BioLegend) to be able to quantify cytochrome c release. Additionally, PDX tumor derived cells were stained with human specific anti-cytokeratin (Biolegend) and human specific anti-vimentin (Biolegend) to discriminate tumor from stroma cells. Cytochrome c–positive tumor cells were quantified by flow cytometry.
Dynamic BH3 profiling (DBP) was done in two different ways: on TNBC cancer cell lines and treatment naïve PDX tumor-derived cells after 48 hours in vitro exposure; or on PDX tumor-derived cells after in vivo treatment. Either way, cells were exposed to treatment for the indicated times, after which BH3 profiling was performed. BH3 profiling of cancer cell lines and treatment naïve PDX tumor derived cells was done via microscopy based BH3 profiling as described under the Library Screening section, with the modification that instead of one BIM concentrations, different BH3 peptides and BH3 mimetics at different concentrations were tested. BH3 profiling after in vivo treatment was done via iBH3 as described above. Priming difference (% delta priming) was calculated by comparing cytochrome C abundance in treated versus vehicle treated control cells16.
Rescue experiments
Cells were seeded in 384-well plates at concentration of 1000–2000 cells/well and allowed to adhere overnight. The next morning growth medium was replaced by fresh growth medium (RPMI-1640+ 10% HI-FBS) containing the following rescue intermediates: nicotinamide mononucleotide (NMN, 25 μM), nicotinic acid (NA, 25 μM), adenine (100 μM, Sigma-Aldrich), uridine (100 μM, Sigma-Aldrich), nucleoside mix (1x, EmbryoMax Nucleosides), dimethyl-alpha-ketoglutarate (50 μM, Sigma-Aldrich), alpha-ketoglutarate (50 μM, Sigma-Aldrich), dimethyl-succinate (50 μM, Sigma-Aldrich), succinate (50 μM, Sigma-Aldrich), dimethyl-fumarate (50 μM, Sigma-Aldrich), fumarate (50 μM, Sigma-Aldrich), dimethyl-malate (50 μM, Sigma-Aldrich), malate (50 μM, Sigma-Aldrich), glutamine (2mM, Gibco) and sodium pyruvate (1mM, Gibco), 3-phosphoglyceric acid (50 μM, 3-PGA, Sigma-Aldrich), N-acetylcysteine (10mM, NAC, Sigma-Aldrich). After replacing the medium with fresh growth medium in presence or absence of rescue metabolites, FK866 and BH3 mimetic drugs were added using the HP D300e Digital Dispenser (Hewlett-Packard Development Company). Cells were treated for 72 h before annexin-V-Hoechst staining was performed as described under measurements of cell death.
Measurements of Cell Death
Cells were seeded and treated in 384-well plates for the indicated times. After treatment, annexin-V-Hoechst staining of the cells was performed for 15 min in annexin-V binding buffer. Cells were fixed with paraformaldehyde and glutaraldehyde for 10 min after which the fixatives were neutralized with a tris/glycine buffer. Cells were washed and imaged using an IXM XLS high content widefield microscope, and the fraction of annexin-V positive cells were quantified using the Multi Wavelength Cell Scoring module in Metamorph software.
Patient-derived xenografts
Patient-derived xenograft (PDX) models of triple-negative breast cancer were originally established by Dr. Alana Welm34 (Huntsman Cancer Institute) and provided through a collaborative agreement with J.S.B. PDX models were renamed for this study: PDX1 refers to HCI-010 in the original paper34; PDX2 refers to HCI-02535. Briefly, tumor fragments were orthotopically transplanted into female NOD.SCID mice (Charles River Labs). Transplantations were completed with J.J.Z. at Harvard Medical School in accordance with Institutional Animal Care and Use Committee (IACUC) protocol #IS00000990 (J.S.B.) and Harvard University ARCM policies. Once tumors were palpable, tumor growth was monitored by biweekly in vivo caliper measurements in two dimensions. Caliper-based measurements of length (l) and width (w) were used to calculate tumor volume via ½(l×w2). Once the tumors reached 150–200 mm3, mice were randomized according to pre-treatment tumor volumes and treatment was initiated. Treatment regimens are described in the Figure 3 (D and F). FK866 (MedChemExpress) was administered via intraperitoneal (I.P.) injection twice a day (BID) at 7.5 mg/kg or 15 mg/kg as. FK866 was prepared and dosed as previously described42 in saline containing 1% hydroxypropyl-β-cyclodextrin (Sigma Aldrich) and 12% 1,2-propylenglycol (Sigma Aldrich). S63845 (MedChemExpress) was administered 25 mg/kg via intravenous (I.V.) injections twice a week. S63845 was prepared extemporaneously in 2% Vitamin E/TPGS (Sigma-Aldrich) in 0.9% NaCl (w/v) and protected from light and dosed as described55. Matched animals received the vehicle(s) corresponding to the drug-treated mice. Number of animals used per group is stated in the figure legends. Drug tolerability was assessed by daily measurements of body weight and monitoring of animal behavior. Experimental endpoints are indicated in the legend of Figure 3. Humane endpoints included body weight reductions >20% and tumor volumes >1200 mm3.
PDX tumor dissociation
PDX tumors were dissociated using a combination of mechanical and enzymatic disruption. PDX tumors were diced into small pieces using a scalpel and the tumor pieces were resuspended in digestion medium composed of DMEM/F12 (Gibco) + DNase I recombinant grade I (130U/ml, Roche) + Hyaluronidase from bovine testes (0.1KU/ml, Sigma) + Collagenase Type I from rat tail (2mg/ml, Gibco). Tumor samples were incubated in dissociation medium at 37C under continuous shaking for 45–90 min until a single cell suspension was obtained.
NAD+ and NADH measurement
NAD+ and NADH levels were determined by untargeted metabolomics analysis (see below) or using the NAD/NADH Glo Assay kit from Promega according to the manufacturer’s guidelines and as described previously56. In brief, cells were treated for the indicated time with NAMPT inhibitors or vehicle in the presence or absence of 25μM NMN. Next, samples were put on dry ice and washed 3 times with ice-cold saline and scraped in ice-cold lysis buffer (1% dodecyltrimethyl ammonium bromide (DTAB) in 0.2 N NaCl diluted 1:1 with phosphate buffered saline (PBS))51. Samples were transferred to Eppendorf tubes and stored immediately at −80°C until further analysis using the NAD/NADH kit. Measurements were normalized to the protein content of the samples. Protein content was determine using the BioRad Bradford Protein assay according to the manufacturer’s guidelines.
Untargeted metabolomics analysis
For the collection of metabolomics samples from cell lines, cells were first washed in ice cold saline, lysed in 70% methanol and stored at −80. For PDX tumor sample preparation, tumors were collected, snap frozen and crushed in liquid nitrogen. Polar metabolites were extracted using methanol-chloroform phase separation (methanol:water:chloroform in 2:1:2 ratio). Samples were consecutively dried under nitrogen flow, resuspended in 70% acetonitrile in water containing 2.5 μM of an internal standard (13C-, 15N-labeled amino acid mix; Cambridge Isotope Laboratories) and run on a ThermoFisher Q-exactive equipped with Zic-pHILIC column (150×2.1 mm, 5 μm; Merck). A volume of 5 μl was injected and the full mass spectrum was obtained in both positive and negative mode (0 to 45 min, resolution 70000, AGC target 3e6, m/z range 66 to 990). Mobile phase A for chromatography consisted of 20 mM ammonium carbonate, 0.1% ammonium hydroxide, in water and mobile phase B of 97% acetonitrile in water. A pooled sample was created from all samples and used for MS/MS runs for metabolite identification. Data were processed with Compound Discoverer 3.0 (ThermoFisher Scientific) using the mz Cloud mass spectral library for compound annotation. For compounds for which an MS/MS spectrum was not available, the Kyoto Encyclopedia of Genes and Genomes (KEGG) Compound database (www.genome.jp/kegg; Kanehisa and Goto, 2000) and human metabolome database (HMDB; www.hmdb.ca;57,58) were used for putative compound annotation based on the monoisotopic molecular weight.
Quantitative real-time reverse transcription PCR analysis
Total RNA from untreated TNBC cell lines was isolated using RNeasy Mini Kit from Qiagen according to the manufacturer’s instructions. RNA was quantified with Nanodrop 2000 (Thermo Fisher) and the RNA was reversed transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Quantitative reverse transcription PCR (RT-qPCR) was performed using pre-designed primers and corresponding Taqman probes (Thermo Fisher): Human NAMPT: Hs00237184_m1; human NAPRT Hs00403474_g1; human IDO: Hs00984148_m1; human TDO: Hs00194611_m1; human NADSYN1: Hs00216808_m1; Human NMNAT1: Hs00978910_g1; Human NMNAT2: Hs00322752_m1; Human SDHA: Hs00188166_m1; human HMBS: Hs00609296_g1; human HPRT1: Hs02800695_m1; human YWHAZ: Hs01122445_g1; human TBP: Hs00427620_m1. Real-time PCRs were carried out in a QuantStudio™ 6 Flex System. For normalization, the geometric mean of the two most stable reference genes out of at least three was used, calculated using geNorm.
Data Analysis
Analysis of the HT-DBP images was performed using the Metamorph software. The Multi Wavelength Cell Scoring Module was used to quantify the fraction of cytochrome c positive tumor cells per well. Cell Scoring was used to analyze the fraction of cytochrome c positive cell line cells. Analysis of the flow cytometry data were performed using FlowJo. All subsequent data analysis was performed in Excel Microsoft Office 365 or Version 8 of Graphpad Prism software. Area under the curve (AUC) was calculated using the Graphpad Prism software (Total peak area). Appropriate statistical analysis for the in vivo experiments was performed depending on the comparisons made and as referred to in figure legends. Statistical analysis was performed in Graphpad Prism or R 3.6.3. software and P-values are designated as * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001. All graphs show averages and standard deviations (SD) or standard error (SE).
Supplementary Material
Acknowledgements:
We thank Charles Vidoudez and Sunia Trauger of the Harvard FAS Small Molecule Mass Spectrometry Facility for the metabolomics analyses, Longwood ICCB Screening facility for the help with the Metabolic compound library screening, the Laboratory of Systems Pharmacology for the use of the high-throughput equipment, and the Molecular Diagnostics Core at the Dana-Farber Cancer Institute for cell line identify verification.
Funding:
V.W.D. and A.L were supported by NIH/NCI under award number R35 CA242427, the Ludwig center at Harvard Award (03/01/15-09/30/19) and Terri Brodeur Breast Cancer Foundation (01/01/2019-12/31/2020). N.v.G. was supported by a fellowship from Alex’s Lemonade Stand Foundation and Tap Cancer Out. V.G.F. was supported by CNPq, National Council for Scientific and Technological Development - Brazil, process 202424/2017-0. I. S. H. is supported by The Breast Cancer Research Foundation-AACR (20-20-26-HARR) and Breast Cancer Coalition of Rochester. J.S. and K.A.S were supported by Alex’s Lemonade Stand Foundation and NIH/NCI R00-CA188679. D.T.S. was supported by the Ludwig Center at Harvard.
Footnotes
Competing interests: D.T.S. is a director and equity holder of Agios Pharmaceuticals, Magenta Therapeutics, Editas Medicines, ClearCreekBio, and Life-VaultBio; he is a founder of Fate Therapeutics and Magenta Therapeutics and a consultant to FOG Pharma, VCanBio, and Bone Therapeutics. J.S.B. discloses SAB for Agios Pharmaceuticals,for Effector Pharmaceuticals and for Frontier Medicines. A.L. is paid advisor and/or equity holder status for AbbVie, Novartis, AstraZeneca, Gilead, Flash Therapeutics, Zentalis and Dielectic. All other authors declare that they have no competing interests. I.S.H. is a consultant for Ono Pharma USA.
Data and materials availability: PDX models of triple-negative breast cancer were originally established by Dr. Alana Welm34 and used in this manuscript under MTA between Huntsman Cancer Institute and Dana-Farber Cancer Institute. The mass spectrometry metabolomics data have been deposited to the EMBL-EBI MetaboLights database59 with the dataset identifier MTBLS2471 (DOI: 10.1093/nar/gkz1019, PMID:31691833). The complete dataset can be accessed here https://www.ebi.ac.uk/metabolights/MTBLS2471. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
Supplementary Materials
Figures S1 to S13.
Tables S1 and S2.
References and Notes
- 1.Hanahan D & Weinberg RA Hallmarks of cancer: the next generation. Cell 144, 646–674, doi: 10.1016/j.cell.2011.02.013 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Keenan MM & Chi JT Alternative fuels for cancer cells. Cancer J 21, 49–55, doi: 10.1097/PPO.0000000000000104 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martinez-Outschoorn UE, Peiris-Pagés M, Pestell RG, Sotgia F & Lisanti MP Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol, doi: 10.1038/nrclinonc.2016.60 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Long JP, Li XN & Zhang F Targeting metabolism in breast cancer: How far we can go? World J Clin Oncol 7, 122–130, doi: 10.5306/wjco.v7.i1.122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao Y, Butler EB & Tan M Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis 4, e532, doi: 10.1038/cddis.2013.60 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Certo M et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 9, 351–365, doi: 10.1016/j.ccr.2006.03.027 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Brunelle JK & Letai A Control of mitochondrial apoptosis by the Bcl-2 family. J Cell Sci 122, 437–441, doi: 10.1242/jcs.031682 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ryan J & Letai A BH3 profiling in whole cells by fluorimeter or FACS. Methods 61, 156–164, doi: 10.1016/j.ymeth.2013.04.006 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ryan J, Montero J, Rocco J & Letai A iBH3: simple, fixable BH3 profiling to determine apoptotic priming in primary tissue by flow cytometry. Biol Chem 397, 671–678, doi: 10.1515/hsz-2016-0107 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Ni Chonghaile T et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science 334, 1129–1133, doi: 10.1126/science.1206727 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vo TT et al. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell 151, 344–355, doi: 10.1016/j.cell.2012.08.038 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anderson MA et al. The BCL2 selective inhibitor venetoclax induces rapid onset apoptosis of CLL cells in patients via a TP53-independent mechanism. Blood 127, 3215–3224, doi: 10.1182/blood-2016-01-688796 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deng J et al. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell 12, 171–185, doi: 10.1016/j.ccr.2007.07.001 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Konopleva M et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov 6, 1106–1117, doi: 10.1158/2159-8290.CD-16-0313 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Del Gaizo Moore V, Schlis KD, Sallan SE, Armstrong SA & Letai A BCL-2 dependence and ABT-737 sensitivity in acute lymphoblastic leukemia. Blood 111, 2300–2309, doi: 10.1182/blood-2007-06-098012 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Montero J et al. Drug-induced death signaling strategy rapidly predicts cancer response to chemotherapy. Cell 160, 977–989, doi: 10.1016/j.cell.2015.01.042 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhola P, A. E, Guerriero JL, Sicinska E, Su E, Lavrova E, Ni J, Chipashvili O, Hagan T, Pioso MS, McQueeney1 K, Ng K, Aguirre, Cleary J, Cocozziello D, Sotayo A, Ryan J, Zhao JJ, Letai A. High-throughput dynamic BH3 profiling (HT-DBP) can quickly and accurately predict effective therapies in solid tumors. Science Signaling in press, doi: 10.1126/scisignal.aay1451. (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Montero J & Letai A Why do BCL-2 inhibitors work and where should we use them in the clinic? Cell Death Differ 25, 56–64, doi: 10.1038/cdd.2017.183 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Villalobos-Ortiz M, Ryan J, Mashaka TN, Opferman JT & Letai A BH3 profiling discriminates on-target small molecule BH3 mimetics from putative mimetics. Cell Death Differ 27, 999–1007, doi: 10.1038/s41418-019-0391-9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tolcher AW et al. Safety, efficacy, and pharmacokinetics of navitoclax (ABT-263) in combination with irinotecan: results of an open-label, phase 1 study. Cancer Chemother Pharmacol 76, 1041–1049, doi: 10.1007/s00280-015-2882-9 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Stilgenbauer S et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol 17, 768–778, doi: 10.1016/S1470-2045(16)30019-5 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Soderquist RS et al. Systematic mapping of BCL-2 gene dependencies in cancer reveals molecular determinants of BH3 mimetic sensitivity. Nat Commun 9, 3513, doi: 10.1038/s41467-018-05815-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Merino D et al. BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines. Cancer Cell 34, 879–891, doi: 10.1016/j.ccell.2018.11.004 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Cragg MS, Harris C, Strasser A & Scott CL Unleashing the power of inhibitors of oncogenic kinases through BH3 mimetics. Nat Rev Cancer 9, 321–326, doi: 10.1038/nrc2615 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Gandhi L et al. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol 29, 909–916, doi: 10.1200/JCO.2010.31.6208 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Panayotopoulou EG et al. Targeting of apoptotic pathways by SMAC or BH3 mimetics distinctly sensitizes paclitaxel-resistant triple negative breast cancer cells. Oncotarget 8, 45088–45104, doi: 10.18632/oncotarget.15125 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Merino D et al. Synergistic action of the MCL-1 inhibitor S63845 with current therapies in preclinical models of triple-negative and HER2-amplified breast cancer. Sci Transl Med 9, doi: 10.1126/scitranslmed.aam7049 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Li H, Liu L, Chang H, Zou Z & Xing D Downregulation of MCL-1 and upregulation of PUMA using mTOR inhibitors enhance antitumor efficacy of BH3 mimetics in triple-negative breast cancer. Cell Death Dis 9, 137, doi: 10.1038/s41419-017-0169-2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harris IS et al. Deubiquitinases Maintain Protein Homeostasis and Survival of Cancer Cells upon Glutathione Depletion. Cell Metab 29, 1166–1181 e1166, doi: 10.1016/j.cmet.2019.01.020 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cantor JR et al. Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell 169, 258–272 e217, doi: 10.1016/j.cell.2017.03.023 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu J et al. Heterogeneity of tumor-induced gene expression changes in the human metabolic network. Nat Biotechnol 31, 522–529, doi: 10.1038/nbt.2530 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu Y et al. Multiplexed assay panel of cytotoxicity in HK-2 cells for detection of renal proximal tubule injury potential of compounds. Toxicol In Vitro 23, 1170–1178, doi: 10.1016/j.tiv.2009.06.003 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Garten A et al. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat Rev Endocrinol 11, 535–546, doi: 10.1038/nrendo.2015.117 (2015). [DOI] [PubMed] [Google Scholar]
- 34.DeRose YS et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med 17, 1514–1520, doi: 10.1038/nm.2454 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zoeller JJ, V. A, Daniels VW, Taneja K, Tan BY, DeRose YS, Fujita M, Welm AL, Letai A, Leverson JD, Blot V, Bronson RT, & Dillon DA, B. J Navitoclax enhances the effectiveness of EGFR-targeted antibody-drug conjugates in PDX models of EGFRexpressing triple-negative breast cancer. Manuscript under AbbVie scientific and legal review in preparation for submission. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chowdhry S et al. NAD metabolic dependency in cancer is shaped by gene amplification and enhancer remodelling. Nature 569, 570–575, doi: 10.1038/s41586-019-1150-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Canto C, Menzies KJ & Auwerx J NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab 22, 31–53, doi: 10.1016/j.cmet.2015.05.023 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin GL et al. Therapeutic strategies for diffuse midline glioma from high-throughput combination drug screening. Sci Transl Med 11, doi: 10.1126/scitranslmed.aaw0064 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chiarugi A, Dolle C, Felici R & Ziegler M The NAD metabolome--a key determinant of cancer cell biology. Nat Rev Cancer 12, 741–752, doi: 10.1038/nrc3340 (2012). [DOI] [PubMed] [Google Scholar]
- 40.von Heideman A, Berglund A, Larsson R & Nygren P Safety and efficacy of NAD depleting cancer drugs: results of a phase I clinical trial of CHS 828 and overview of published data. Cancer Chemother Pharmacol 65, 1165–1172, doi: 10.1007/s00280-009-1125-3 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Goldinger SM et al. Efficacy and Safety of APO866 in Patients With Refractory or Relapsed Cutaneous T-Cell Lymphoma: A Phase 2 Clinical Trial. JAMA Dermatol 152, 837–839, doi: 10.1001/jamadermatol.2016.0401 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Chini CC et al. Targeting of NAD metabolism in pancreatic cancer cells: potential novel therapy for pancreatic tumors. Clin Cancer Res 20, 120–130, doi: 10.1158/1078-0432.CCR-13-0150 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bruzzone S et al. Catastrophic NAD+ depletion in activated T lymphocytes through nampt inhibition reduces demyelination and disability in EAE. PLoS One 2009 November 19;4(11) :e7897. doi: 10.1371/journal.pone.0007897 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Drijvers JM et al. Pharmacologic screening identifies metabolic vulnerabilities of CD8+ T Cells. Cancer Immunol Res 9(2):184–199. doi: 10.1158/2326-6066.CIR-20-0384 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bajrami I et al. Synthetic lethality of PARP and NAMPT inhibition in triple-negative breast cancer cells. EMBO Mol Med 4, 1087–1096, doi: 10.1002/emmm.201201250 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yaku K, Okabe K, Hikosaka K & Nakagawa T NAD Metabolism in Cancer Therapeutics. Front Oncol 8, 622, doi: 10.3389/fonc.2018.00622 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nikiforov A, Kulikova V, Zeigler M, The human NAD metabolome: functions, metabolism and compartmentalization. Crit Rev Biochem Mol Biol. 50(4):1–14 doi: 10.3109/10409238.2015.1028612 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hoxhaj G et al. The mTORC1 Signaling Network Senses Changes in Cellular Purine Nucleotide Levels. Cell Rep 21, 1331–1346, doi: 10.1016/j.celrep.2017.10.029 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pedley AM & Benkovic SJ A New View into the Regulation of Purine Metabolism: The Purinosome. Trends Biochem Sci 42, 141–154, doi: 10.1016/j.tibs.2016.09.009 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bhola PD & Letai A Mitochondria-Judges and Executioners of Cell Death Sentences. Mol Cell 61, 695–704, doi: 10.1016/j.molcel.2016.02.019 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pareek V, Tian H, Winograd N & Benkovic SJ Metabolomics and mass spectrometry imaging reveal channeled de novo purine synthesis in cells. Science 368, 283–290, doi: 10.1126/science.aaz6465 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pinson B, Ceschin J, Saint-Marc C & Daignan-Fornier B Dual control of NAD(+) synthesis by purine metabolites in yeast. Elife 8, doi: 10.7554/eLife.43808 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Navas LE, Carnero A NAD+ metabolism, stemness, the immune response, and cancer. Signal Transduct Target Ther 6:2 doi: 10.1038/s41392-020-00354-w (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guo J, et al. Identification of novel resistance mechanisnms to NAMPT inhition via the de novo NAD+ biosynthesis pathway and NAMPT mutation. Biochem Biophys Res Commun 23;491(3):681–686 dio: 10.1016/j.bbrc.2017.07.143 (2017) [DOI] [PubMed] [Google Scholar]
- 55.Brennan MS et al. Humanized Mcl-1 mice enable accurate preclinical evaluation of MCL-1 inhibitors destined for clinical use. Blood 132, 1573–1583, doi: 10.1182/blood-2018-06-859405 (2018). [DOI] [PubMed] [Google Scholar]
- 56.Gui DY et al. Environment Dictates Dependence on Mitochondrial Complex I for NAD+ and Aspartate Production and Determines Cancer Cell Sensitivity to Metformin. Cell Metab 24, 716–727, doi: 10.1016/j.cmet.2016.09.006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wishart DS et al. HMDB: the Human Metabolome Database. Nucleic Acids Res 35, D521–526, doi: 10.1093/nar/gkl923 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wishart DS et al. HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res 46, D608–D617, doi: 10.1093/nar/gkx1089 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Haug K et al. MetaboLights: a resource evolving in response to the needs of its scientific community. Nucleic Acids Res 8;48(D1):D440–D444. doi: 10.1093/nar/gkz1019 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.