

Abstract
A 25-year-old man with exertional myoglobinuria had no evidence of hemolytic anemia, but he had severe parkinsonism that was responsive to levodopa. Phosphoglycerate kinase (PGK) activity was markedly decreased in muscle, and molecular analysis of the PGK1 gene identified the p.T378P mutation that was recently reported in a patient with isolated myopathy. This case reinforces the concept that PGK deficiency is a clinically heterogeneous disorder and raises the question of a relationship between PGK deficiency and idiopathic juvenile Parkinson disease.
Keywords: glycogen-storage disease type IX, myoglobinuria, parkinsonism, phosphoglycerate kinase
Human phosphoglycerate kinase (PGK1), a key enzyme in the glycolytic pathway, is a single poly-peptide (molecular weight 45 kDa) encoded by a gene located on the X chromosome and expressed in all somatic tissues. PGK deficiency is one of the relatively uncommon causes of hereditary non-spherocytic hemolytic anemia (HNSHA).1
The clinical manifestations of the disease are diverse due to the differential involvement of three tissues, erythrocytes, skeletal muscle, and the central nervous system (CNS). Thus, some patients present with HNSHA alone, others with myopathy alone, and still others with anemia and CNS symptoms, whereas the involvement of CNS and muscle or of all three tissues is distinctly less frequent.2–4 It is unclear how such different clinical manifestations can be caused by mutations in the same gene.
This conundrum is illustrated by the present patient: although he had a mutation in PGK1 that was recently reported to be associated with isolated myopathy,4 he also exhibited CNS dysfunction with early-onset parkinsonism.
CASE REPORT
This 25-year-old man had exercise intolerance, myalgias, and at least one episode of exercise-related pigmenturia (“Coca-Cola”-like urine according to the patient’s sister) since childhood. He also had developmental, language, and motor delay (he walked at 20 months). At 20 years of age, he collapsed while running and was hospitalized with documented myoglobinuria. Since then, there has been progressive decline of motor function resulting in multiple falls, and progressive dysphagia for solid and liquids. The patient also was diagnosed with bilateral cataracts.
In his late teens, he noted tremor in his right arm that was made worse by intentional movements but was also present at rest. Since then, he has become progressively stiffer and slower. He drools both during the day and at night. His hand-writing has deteriorated markedly, and he can only write a few letters with difficulty. He has trouble cutting food and needs help with dressing and hygiene. He can no longer walk without assistance. In addition to generalized muscle aches, he has numbness and possibly paresthesias in the legs. He has both urinary and fecal incontinence and episodes of constipation so severe that he is often impacted.
The patient’s mother and an older sister complain of muscle pain with exercise, and a brother and sister are asymptomatic.
At age 25, the patient presented with a 2–3-HZ tremor of the right elbow at rest, stooped posture, slow gait, and dragging of his right leg. He was unable to walk or rise from a chair unassisted. His facial expression was mask-like, and he appeared chronically ill. Neurological examination showed marked hypophonia. He was unable to copy a spiral, but what he did copy was micrographic. There was mild action tremor in both arms. Deep tendon reflexes were 2+ symmetrically, but he had no ankle jerks. He was markedly rigid in the neck and in all limbs, right more than left. He needed assistance to arise from a chair. He had moderate kyphosis, and shuffled bilaterally, more on the right, with absent arm swing on both sides. He fell back spontaneously and, while sitting, he had persistent gradual retrocollis to the point that his sister had to continually push his head forward. Strength was difficult to evaluate but appeared normal in both arms and in the left leg, while there was slight weakness of hip flexion and knee extension on the right. He was able to do deep knee bends slowly but completely.
Laboratory tests revealed increased levels of serum creatine kinase (CK; 5402 U/L; normal, <200 U/L), alanine aminotransaminase (ALT; 101 U/L; normal, 7–28 U/L) and aspartate aminotransaminase (AST; 81 U/L; normal, 12–27 U/L). His serum hemoglobin concentration was normal at 13.4 g/dl, and he had no evidence of hemolytic anemia. Computed tomography (CT) scan and magnetic resonance imaging (MRI) of the brain at 25 years of age showed no focal intracranial pathology. An electroencephalogram (EEG) was also normal. A biopsy of the right quadriceps muscle showed minimal myopathic features and no obvious glycogen accumulation.
Therapy with carbidopa–levodopa (Sinemet), 25/100 mg three times daily, was started: the parkinsonian signs improved markedly, but the patient developed psychosis. The therapy was suspended, and the dose is being readjusted.
RESULTS
Biochemical analysis of the muscle extract revealed markedly decreased PGK activity (2.1 μmol substrate utilized per minute per gram of fresh tissue; mean ± SD: 187.2 ± 30.6, in 33 controls). The activities of other glycolytic enzymes (phosphoglycerate mutase and lactate dehydrogenase) were normal. Sequencing of the PGK1 gene revealed a hemizygous c.1132A>C mutation in exon 10 (Fig. 1), which changes an evolutionarily highly conserved threonine to proline at amino acid position 378 (p.T378P) of the PGK enzyme (Fig. 1B).
FIGURE 1.
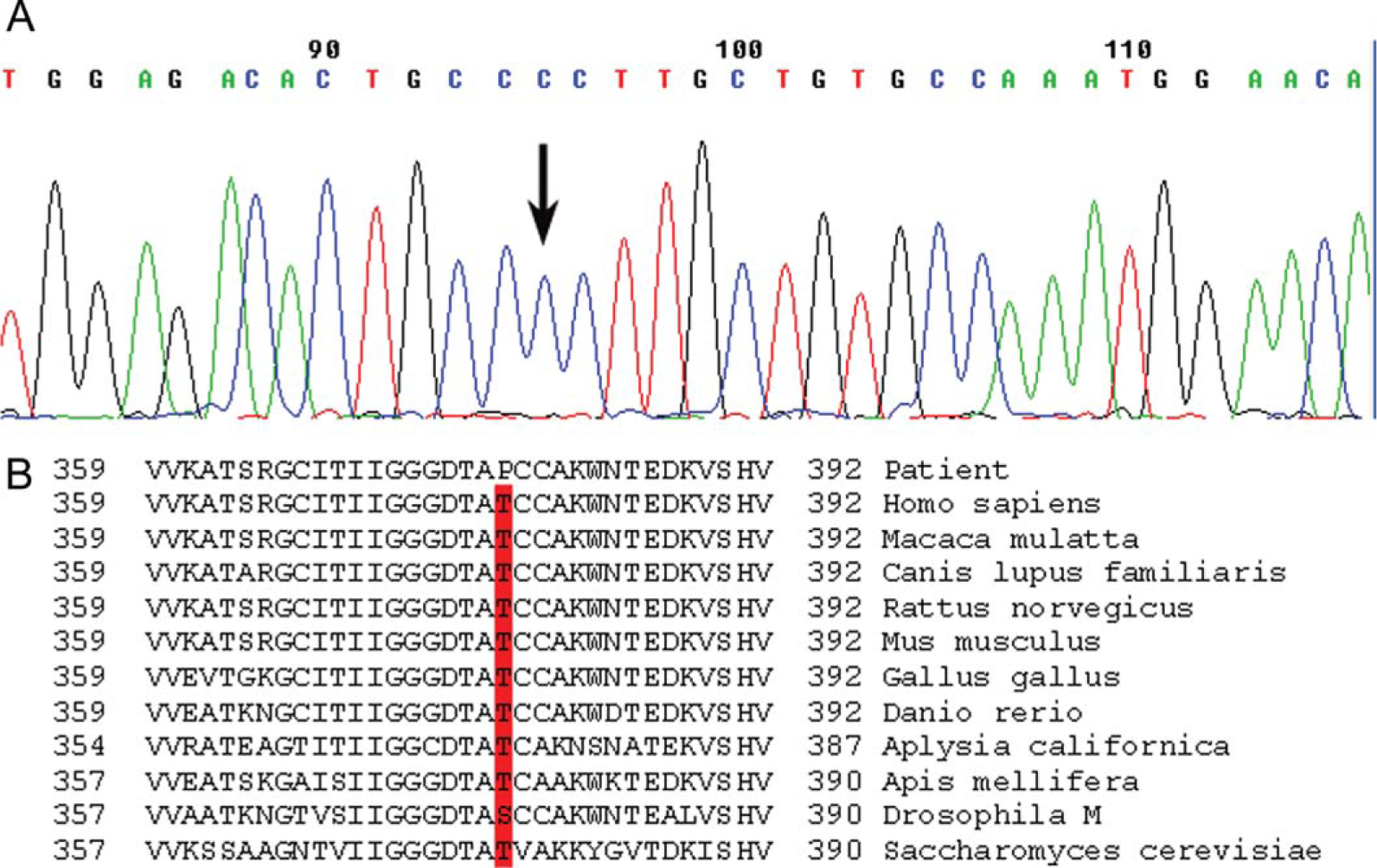
(A) Sequence of part of exon 10 of the PGK1 gene. The arrow indicates the hemizygous A>C mutation at nt position 1132 that changes an encoded threonine to a proline at amino acid position 378. (B) The region of the Pgk1 peptide containing the T378P mutation is aligned with sequences of Pgk1 proteins from other species.
DISCUSSION
PGK plays an important role in the generation of adenosine triphosphate (ATP) during glycolysis. PGK deficiency is a rare condition first described in large Chinese kindred.5 It is an X-linked genetic trait, and the disorder is usually only fully expressed in male hemizygotes. Interestingly, however, one of the very first reported cases of PGK deficiency was that of a woman in her 60s who had mild hemolytic anemia.6 Many PGK variants have been described. Some of these were associated with decreased enzyme activity, and others were detected only by abnormal electrophoretic mobility.1
Among symptomatic patients, two main clinical pictures have emerged: a syndrome characterized by hemolytic anemia and CNS involvement (seizures, mental retardation, strokes) (for review, see Spiegel et al.4); and a purely myopathic syndrome with exercise intolerance, cramps, and recurrent myoglobinuria.4 These two syndromes seem to be most common, having been described in 20 of 33 patients. Of the remaining 13 patients, 6 had isolated blood dyscrasia, 4 had myopathy and CNS dysfunction, 1 had anemia and myopathy, and 2 had involvement of all three tissues (for review, see Spiegel et al.4). The independent or combined involvement of these three tissues is difficult to explain, because PGK is a monomeric enzyme expressed in all tissues except for the testis.1
The molecular genetic basis of PGK deficiency includes 14 missense mutations, 1 single amino acid deletion, 1 10-amino-acid insertion, and 2 truncation (frameshift and termination) mutations (data obtained from the Human Gene Mutation Database at the Institute of Medical Genetics, Cardiff, UK: http://www.hgmd.cf.ac.uk/ac/index.php). Most of the single amino acid substitutions reported to date have occurred in three specific regions of the enzyme molecule, but no clear correlation could be established between the different sites of the PGK mutations and clinical phenotypes.1,4,7,8
Our patient’s clinical presentation of exercise intolerance, cramps, myoglobinuria, and elevated resting serum CK suggests a biochemical defect of glycolysis. The patient had no signs of hemolytic anemia, but he had clear signs of brain involvement dominated by parkinsonism, an uncommon clinical finding in patients with PGK deficiency. However, in 1973, Konrad et al.9 described two brothers with childhood-onset non-spherocytic hemolytic anemia, developmental delay, and generalized tremor worsened by action but not improved by medication [trihexiphenidyl (Artane) or l-dopa]. In these patients, PGK deficiency was documented in erythrocytes and leukocytes, but molecular analysis was not yet available at the time.
Notably, we recently documented the same mutation in another of our patients (p.T378P), an 18-year-old man with isolated myopathy and no signs of hemolytic anemia or brain dysfunction.4 The strikingly different clinical presentations in two individuals who harbor the same mutation illustrate the general problem underlying the varying tissue involvement in PGK deficiency. There are other examples, such as PGK Creteil,10,11 in which an amino acid substitution at position 314 caused isolated myopathy, and PGK Michigan,12 in which a substitution at position 315 caused CNS dysfunction and hemolytic anemia but no myopathy.
Our patient’s severe parkinsonism that was responsive to carbidopa–levodopa raises the possibility of a casual association between two unrelated diseases, often dubbed as “double trouble.” This appears unlikely, however, for several reasons. First, juvenile-onset Parkinson disease (PD) is very unusual. Second, there is a precedent in the literature for an association of PGK deficiency and parkinsonism,9 whereas parkinsonism is not associated with any other glycogen-storage disease. Third, brain is often involved in PGK deficiency, although parkinsonism is a most unusual presentation. Although the pathogenic mechanism remains obscure, it is reasonable to postulate a loss rather than a gain of function mechanism based on the involvement of erythrocytes and skeletal muscle. Anaerobic glycolysis is the only source of energy in mature erythrocytes due to their lack of mitochondria, and hemolytic anemia is the common presentation of glycogenoses that affect red blood cells. Similarly, a mismatch of ATP utilization and ATP regeneration has been documented in errors of glycolysis affecting muscle, including PGK deficiency, a reasonable explanation for muscle break-down.13 It is difficult to explain why parkinsonism should occur in PGK deficiency, unless in some cases the enzyme defect is especially severe in the substantia nigra. A normally low level of PGK in the substantia nigra could explain its vulnerability in PGK deficiency, but the regional distribution of PGK in normal human brain has not been studied.
Two recent studies have established associations between heterozygous pathogenic mutations in the glucocerebrosidase (GBA) gene, responsible for Gaucher disease, and sporadic PD.14,15 This suggests a paradigm shift from the common disease: common variant hypothesis to the common disease:multiple rare variant hypothesis in PD.14 We may be dealing with a similar situation, although in our case the association was with homozygous pathogenic PGK mutations, thus raising an interesting, if unorthodox, possibility. The diagnosis of PGK deficiency was made in our patient only because he suffered an episode of myoglobinuria, but this would never have been considered in any individual in whom the signs of parkinsonism over-shadow those of myopathy. Thus, is it possible that PGK deficiency may, in fact, be a cryptic cause of juvenile PD? To answer this question, as well as question of whether heterozygous PGK mutation may increase the susceptibility to PD, it may be worthwhile to sequence the PGK1 gene in a cohort of idiopathic juvenile PD cases.
Abbreviations:
- ALT
alanine aminotransaminase
- AST
aspartate aminotransaminase
- ATP
adenosine triphosphate
- CK
creatine kinase
- CNS
central nervous system
- CT
computed tomography
- EEG
electroencephalogram
- HNSHA
hereditary non-spherocytic hemolytic anemia
- MRI
magnetic resonance imaging
- PD
Parkinson disease
- PGK
phosphoglycerate kinase
REFERENCES
- 1.Beutler E PGK deficiency. Br J Haematol 2007;136:3–11. [DOI] [PubMed] [Google Scholar]
- 2.Turner G, Fletcher J, Elber J, Yanagawa Y, Davé V, Yoshida A. Molecular defect of a phosphoglycerate kinase variant associated with haemolytic anaemia and neurological disorders in a large kindred. Br J Haematol 1995;91:60–65. [DOI] [PubMed] [Google Scholar]
- 3.Noel N, Flanagan J, Kalko SG, Ramirez Bajo MJ, del Mar Manu M, Garcia Fuster JL, et al. Two new phosphoglycerate kinase mutations associated with chronic haemolytic anaemia and neurological dysfunction in two patients from Spain. Br J Haematol 2005;132: 523–529. [DOI] [PubMed] [Google Scholar]
- 4.Spiegel R, Area Gomez E, Akman HO, Krishna S, Horovitz Y, DiMauro S. Myopathic form of phosphoglycerate kinase (PGK) deficiency: a new case and pathogenic considerations. Neuromuscul Disord 2009;19:207–211. [DOI] [PubMed] [Google Scholar]
- 5.Valentine WN, Hsieh H, Paglia DE, Anderson HM, Baughan MA, Jaffe ER, et al. Hereditary hemolytic anemia associated with phosphoglycerate kinase deficiency in erythrocytes and leukocytes. N Engl J Med 1969;280:528–534. [DOI] [PubMed] [Google Scholar]
- 6.Kraus AP, Langston MF, Lynch BL. Red cell phosphoglycerate kinase deficiency. Biochem Biophys Res Commun 1968;30:173–177. [DOI] [PubMed] [Google Scholar]
- 7.Morimoto A, Ueda I, Hirashima Y, Sawai Y, Usuku T, Kano G, et al. A novel missense mutation (1060G>C) in the phosphoglycerate kinase gene in a Japanese boy with chronic hemolytic anemia, developmental delay and rhabdomyolysis. Br J Haematol 2003;122:1009–1013. [DOI] [PubMed] [Google Scholar]
- 8.Hamano T, Mutoh T, Sugie H, Koga H, Kuriyama M. Phosphoglycerate kinase deficiency: an adult myopathic form with a novel mutation. Neurology 2000;54:1188–1190. [DOI] [PubMed] [Google Scholar]
- 9.Konrad PN, McCarthy DJ, Mauer AM, Valentine WN, Paglia DL. Erythrocyte and leukocyte phosphoglycerate kinase deficiency with neurologic disease. J Pediatr 1973;82:456–460. [DOI] [PubMed] [Google Scholar]
- 10.Rosa R, George C, Fardeau M, Calvin MC, Rapin M, Rosa J. A new case of phosphoglycerate kinase deficiency: PGK Creteil associated with rhabdomyolysis and lacking hemolytic anemia. Blood 1982;60:84–91. [PubMed] [Google Scholar]
- 11.Cohen-Solal M, Valentin C, Plassa F, Guillemin G, Danze F, Jaisson F, et al. Identification of new mutations in two phosphoglycerate kinase (PGK) variants expressing different clinical syndromes: PGK Creteil and PGK Amiens. Blood 1994;84:898–903. [PubMed] [Google Scholar]
- 12.Maeda M, Bawle EV, Kulkarni R, Beutler E, Yoshida A. Molecular abnormalities of a phosphoglycerate kinase variant generated by spontaneous mutation. Blood 1992;79:2759–2762. [PubMed] [Google Scholar]
- 13.Haller RG, Vissing J. Functional evaluation of metabolic myopathies. In: Engel AG, Franzini-Armstrong C, editors. Myology. Vol. 1. New York: McGraw-Hill; 2004. p 665–679. [Google Scholar]
- 14.Mitsui J, Mizuta I, Toyoda A, Ashida R, Takahashi Y, Goto J, et al. Mutations for Gaucher disease confer high susceptibility to Parkinson disease. Arch Neurol 2009;66:571–576. [DOI] [PubMed] [Google Scholar]
- 15.Clark LN, Ross BM, Wang Y, Majia-Santana H, Harris J, Louis ED, et al. Mutations in the glucocerebroside gene are associated with early-onset Parkinson disease. Neurology 2009;69: 1270–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]