

Abstract
Oxidative stress is intimately tied to neurodegenerative diseases, including Parkinson’s disease and amyotrophic lateral sclerosis, and acute injuries, such as ischemic stroke and traumatic brain injury. Acid sensing ion channel 1a (ASIC1a), a proton-gated ion channel, has been shown to be involved in the pathogenesis of these diseases. However, whether oxidative stress affects the expression of ASIC1a remains elusive. In the current study, we examined the effect of hydrogen peroxide (H2O2), a major reactive oxygen species (ROS), on ASIC1a protein expression and channel function in NS20Y cells and primary cultured mouse cortical neurons. We found that treatment of the cells with H2O2 (20 µM) for 6 h or longer increased ASIC1a protein expression and ASIC currents without causing significant cell injury. H2O2 incubation activated mitogen-activated protein kinases (MAPKs) pathways, including the extracellular signal-regulated kinase1/2 (ERK1/2), c-Jun N-terminal kinase (JNK), and p38 pathways. We found that neither inhibition of the MEK/ERK pathway by U0126 nor inhibition of the p38 pathway by SB203580 affected H2O2-induced ASIC1a expression, whereas inhibition of the JNK pathway by SP600125 potently decreased ASIC1a expression and abolished the H2O2-mediated increase in ASIC1a expression and ASIC currents. Furthermore, we found that H2O2 pretreatment increased the sensitivity of ASIC currents to the ASIC1a inhibitor PcTx1, providing additional evidence that H2O2 increases the expression of functional ASIC1a channels. Together, our data demonstrate that H2O2 increases ASIC1a expression/activation through the JNK signaling pathway, which may provide insight into the pathogenesis of neurological disorders that involve both ROS and activation of ASIC1a.
Keywords: acid-sensing ion channels (ASICs), hydrogen peroxide (H2O2), reactive oxygen species (ROS), mitogen-activated protein kinase (MAPK), c-Jun N-terminal kinase (JNK)
Introduction
Acid-sensing ion channels (ASICs) are proton-gated, voltage-insensitive cationic channels that are widely expressed in the nervous system. Four ASIC genes, namely ASIC1, ASIC2, ASIC3 and ASIC4, and six ASIC isoforms, namely ASIC1a, ASIC1b, ASIC2a, ASIC2b, ASIC3, and ASIC4, have been identified [1, 2]. ASIC1a, 2a, and 2b are expressed in both the central and peripheral nervous systems, while ASIC3 and ASIC1b are predominantly expressed in the peripheral nervous system [1, 3–5]. ASIC1a, the predominant and functional ASIC subunit in the brain, has been demonstrated to be involved in important physiological processes, including synaptic transmission, learning and memory, olfactory function, and neurological disorders, including ischemic stroke, multiple sclerosis, epileptic seizure, and Parkinson’s disease [5–12].
Oxidative stress is the result of an imbalance between pro-oxidant and antioxidant homeostasis that leads to the generation of toxic reactive oxygen or nitrogen species (ROS/NOS), including superoxide anion (.O2-), hydrogen peroxide (H2O2), hydroxyl (.OH), nitric oxide (.NO) and peroxynitrite (ONOO-) [13]. ROS may damage all components of the cell, including proteins, lipids, and DNA [14]. The human brain is susceptible to oxidative stress, which has been shown to contribute to the pathogenesis of both acute and chronic neurological disorders, including ischemic stroke, PD, ALS, and diabetic encephalopathy [15, 16]. H2O2, one of the main ROS, plays an active role in the regulation of various physiological processes, including cell proliferation, differentiation, migration and apoptosis [17, 18]. Nevertheless, its overproduction results in oxidative stress, which can lead to extensive cellular damage. Exposure of cells to H2O2 induces activation of MAPKs, including ERK1/2, JNK, and p38 kinase [19].
The current study was designed to investigate the effect of H2O2 on ASIC1a expression and ASIC channel activation and the underlying signaling pathway in both neuronal cell lines and primary cultured mouse cortical neurons.
Materials and methods
Chemicals and antibodies
H2O2 (Cat. No. 216763), SB203580 (Cat. No. S8037) and SP600125 (Cat. No. S5567) were purchased from Sigma-Aldrich (St. Louis, MO, USA). U0126 (Cat. No. 9903 S) was purchased from Cell Signaling Technology (Danvers, MA, USA). The ASIC1 antibody was a gift from Dr. Xiang-ming Zha (University of South Alabama, USA) [20]. Antibodies, including p38 MAPK (Cat. No. 8690), phospho-p38 (Cat. No. 4511), JNK (Cat. No. 9252), phospho-JNK (Cat. No. 9251), ERK1/2 (Cat. No. 4695), phospho-ERK1/2 (Cat. No. 9101), were purchased from Cell Signaling Technology. β-Actin (Cat. No. A5441) was purchased from Sigma-Aldrich.
Cell culture
NS20Y cells were purchased from Sigma-Aldrich and cultured in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA, USA) as we described previously [21]. Cells were plated on 35-mm dishes coated with poly-L-ornithine and maintained at 37 °C in a humidified incubator with 5% CO2. Mouse cortical neurons were isolated and cultured as described previously [22]. Pregnant Swiss mice were purchased from Charles River. Briefly, pregnant Swiss mice (embryonic day 16) were anesthetized with isoflurane and sacrificed by cervical dislocation. Fetal brains were quickly removed and placed in cold phosphate-buffered saline without Ca2+/Mg2+. Tissues were dissected, incubated with 0.05% trypsin-EDTA at 37 °C for 10 min, and triturated using fire-polished glass pipettes. Cortical neurons were counted and plated in poly-L-ornithine-coated 35-mm culture dishes (1 × 106 cells/dish) or 24-well plates (2 × 105 cells/well). Neurons were cultured in neurobasal medium (Invitrogen) supplemented with B-27 (Invitrogen) and glutamine and maintained in a humidified incubator with 5% CO2 at 37 °C. The culture medium was changed every 3 days, and neurons were used for experiments 12–14 days after plating.
Western blot
Protein was extracted with M-PER™ Mammalian Protein Extraction Reagent (Cat. No. 78501, Thermo Fisher Scientific) and Halt™ Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific). After centrifugation at 13,000 × g for 15 min at 4 °C, the lysates were collected and mixed with Laemmli sample buffer and then boiled for 10 min. The protein concentrations of the cell samples were measured using the Bio-Rad protein assay kit (BioRad, Hercules, CA, USA). The proteins were separated on 10% SDS-polyacrylamide gels and then transferred to PVDF membranes. After blocking, the blots were probed with primary antibodies followed by horseradish peroxidase-conjugated secondary antibodies. The signals were visualized using an ECL kit (Millipore, WBLUR0500).
Lactate dehydrogenase (LDH) assay
Cytotoxicity was measured by the LDH assay using a cytotoxicity detection kit (Cat. No. 11644793001, Roche Diagnostics) according to the manufacturer’s instructions, as described in our previous studies [22]. At the end of the experiments, 50 μL of culture medium was transferred from each well to a 96-well plate for the measurement of LDH release. To determine the maximal releasable amount of LDH, cells were incubated with Triton X-100 (final concentration of 0.5%) for 30 min at room temperature. Fifty microliters of mixed assay reagent from the cytotoxicity detection kit was added to each well and mixed in the dark for 30 min. The absorbance at 492 and 620 nm was measured with a spectrometer (SpectraMax Plus, Molecular Devices, Sunnyvale, CA, USA), and the absorbance at 492 nm was subtracted from that at 620 nm to calculate LDH release.
Electrophysiology
ASIC currents were recorded using a patch-clamp technique as described in our previous studies [11]. The pipette solution contained (in mM) 140 CsF, 1 CaCl2, 2 MgCl2, 11 EGTA, 2 tetraethylammonium chloride, 10 HEPES and 4 MgATP (pH 7.3 adjusted with CsOH, 290–300 mOsm). The extracellular fluid (ECF) contained (in mM) 140 NaCl, 5.4 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES (pH 7.4 or pH 6.0 adjusted with NaOH/HCl, 320–330 mOsm). A multibarrel perfusion system (SF-77 Warner Instruments, Hamden, CT, USA) was used to obtain rapid exchange of ECFs. Currents were recorded with an Axopatch 200B amplifier, filtered at 2 kHz, and digitized at 5 kHz using Digidata 1332 A. ASIC currents were induced by rapid perfusion of the cells with ECF, pH 6.0, for 4 s. The interval between acid applications was 90 s to allow complete recovery of the ASIC currents from desensitization. Unless otherwise stated, cells were clamped at a holding potential of −60 mV. The pipettes had a resistance of 3–5 MΩ when filled with the pipette solution.
Ethics approval and consent to participate
Mouse cortical neurons were cultured in accordance with approved animal protocols and the guidelines of the Institutional Animal Care and Use Committee of Morehouse School of Medicine.
Statistical analysis
All data are expressed as the mean ± SEM. Where applicable, groups were compared using one-way ANOVA followed by Bonferroni’s test or Student’s t test as appropriate using GraphPad Prism 8. P < 0.05 was considered statistically significant.
Results
H2O2 increases ASIC1a protein expression in NS20Y cells
We first examined the effect of H2O2, a major ROS, on the expression of ASIC1a in NS20Y cells. High concentrations of H2O2 (e.g., >100 µM) have been well demonstrated to cause acute cell injury/death [19, 23–25]; however, these effects do not mimic the pathological conditions of most chronic neurodegenerative diseases, in which the concentrations of H2O2 are normally within the range of 10–60 µM [18, 26]. To determine the optimal concentration that is pathologically relevant but nontoxic or less toxic to cells for this study, we first investigated the concentration-dependent harmful effects of H2O2 on NS20Y cells. We found that exposure of NS20Y cells to high concentrations of H2O2 (≥ 40 μM) for 24 h significantly increased cell injury, as demonstrated by increased LDH release. In contrast, exposure to H2O2 at a concentration of 20 μM or lower did not cause significant morphological changes and increased LDH release (Fig. 1a–b, n = 7–8, P< 0.001 compared with the control). We examined the effect of H2O2 at low concentrations (10 and 20 μM) on ASIC1a protein expression. As shown in Fig. 1c, exposure of NS20Y cells to 20 μM H2O2 for 24 h significantly increased ASIC1a expression. The relative protein expression was increased 1.74 ± 0.14-fold of the control level (Fig. 1d, n = 9, P < 0.001 compared with the control). The expression of ASIC1a was not significantly changed by 10 μM H2O2 (Fig. 1d, n = 12). We also examined whether high concentrations of H2O2 (≥ 40 μM) affect ASIC1a expression despite causing toxicity. More than half of the cells were killed by treatment with 40 µM H2O2, and almost all the cells were killed by treatment with 80 µM H2O2 (Supplementary Fig. 1a). These morphological data indicating cell death are consistent with the results of the LDH assay, as shown in Fig. 1b (~55% and ~96% LDH release in 40 and 80 µM H2O2-treated cells). Since more than half of the cells were killed by 40 μM H2O2, we were only able to collect less than 50% of the cells for Western blot analysis. Our data showed that compared with the control, 40 µM H2O2 increased the expression of ASIC1a; however, there was no significant difference between the effects of 20 and 40 µM H2O2 (Supplementary Fig. 1b-c). One possible explanation could be that the injury to some cells induced by 40 µM H2O2 impaired the ability of the cells to synthesize new proteins, which may have, to some extent, limited the further increase in ASIC1a expression. Because of the massive cell death caused by 80 µM H2O2, we were unable to collect samples to perform reliable protein analysis. Furthermore, we determined the time-dependent effect of H2O2 on ASIC1a expression. We found that exposure of NS20Y cells to H2O2 (20 μM) for 6 h, but not 3 h, significantly increased ASIC1a expression (Fig. 1e). Relative ASIC1a expression was increased to 1.50 ± 0.08-fold of the control level by H2O2 treatment for 6 h (Fig. 1f, n = 9, P < 0.001 compared with the control).
Fig. 1. Effect of H2O2 on cell injury and ASIC1a protein expression in NS20Y cells.

a Representative phase-contrast images of NS20Y cells treated with or without H2O2 (20 μM) for 24 h. b LDH assay showing the concentration-dependent cytotoxic effect of H2O2 on NS20Y cells (n = 8, P < 0.001 versus the control, one-way ANOVA followed by Bonferroni’s post hoc test). c Representative blots and (d) quantification of ASIC1a protein expression in the presence or absence of H2O2 (10 and 20 μM) for 24 h in NS20Y cells (n = 9–12, P < 0.001 versus the control, unpaired Student’s t test). e Representative blots and (f) quantification of ASIC1a protein expression in the presence or absence of H2O2 (20 μM) for 3 or 6 h in NS20Y cells (n = 9, P < 0.001 versus the control, unpaired Student’s t test).
In addition to total protein expression, we also examined whether the surface expression of ASIC1a protein was affected by H2O2 using a surface biotinylation assay. We found that the surface component of ASIC1a proteins was also increased by H2O2 (20 μM, 24 h), suggesting an increase in the number of ASIC1a channels on the plasma membrane (Supplementary Fig. 2).
H2O2 enhances ASIC currents
To further confirm whether increased ASIC1a protein expression by H2O2 results in an increase in the number of functional ASIC channels on the plasma membrane, we used whole-cell patch-clamp recording to determine the change in ASIC currents. As shown in Fig. 2a, pretreatment of NS20Y cells with 20 μM H2O2 for 24 h dramatically increased the amplitude of ASIC currents. Considering that variations in cell size may cause differences in whole-cell currents, we also used the current density to compare the difference in ASIC currents. H2O2 treatment increased the ASIC current density from −20.38 ± 3.16 to −34.92 ± 4.08 pA/pF (Fig. 2b, n = 21, P = 0.008 compared with the control), suggesting an increase in the number of functional channels, which is consistent with the increase in protein expression. Next, we examined whether H2O2 has a direct modulatory effect on ASIC channels. After establishment of a stable whole-cell current, we applied 20 μM H2O2 in both normal pH ECF (pH 7.4) and acidic pH ECF (pH 6.0) and acutely perfused cells with H2O2 for 5 min. Our data showed that there was no significant change in the amplitude of ASIC currents (Fig. 2c-d, n = 7, P = 0.328).
Fig. 2. Effect of H2O2 on ASIC currents in NS20Y cells.
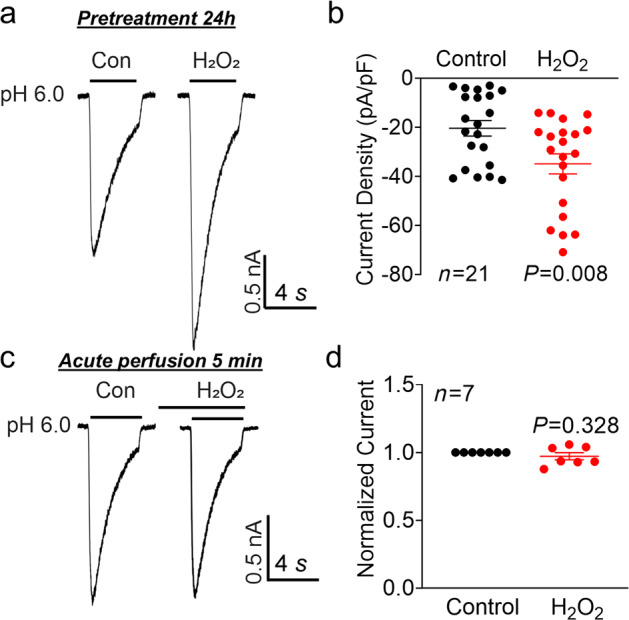
a Representative whole-cell recordings and (b) summary of the data showing increased ASIC currents in NS20Y cells after treatment with H2O2 (20 μM) for 24 h (n = 21 cells, P = 0.008 versus the control, unpaired Student’s t test). c Representative recordings of ASIC currents and (d) summary of the data showing the lack of effect of acute perfusion of NS20Y cells with H2O2 (20 μM, 5 min) on ASIC currents (n = 7 cells, paired Student’s t test).
We also examined whether H2O2 treatment (20 μM, 24 h), in addition to altering peak amplitude, affects the kinetics of ASIC currents in NS20Y cells. The activation and desensitization kinetics of ASIC currents were well fitted by a single exponential function (Supplementary Fig. 3). No significant difference was found in the time constant of activation (τact) between the control (108.51 ± 20.58 ms) and H2O2 treatment (91.18 ± 10.79 ms) groups (Supplementary Fig. 3a-b). Similarly, no significant difference was found in the time constant of desensitization (τdes) between the control (1651.96 ± 96.03 ms) and H2O2 treatment (1587.67 ± 40.48 ms) groups (Supplementary Fig. 3c-d)..
Activation of MAPK pathways by H2O2
Activation of the MAPK signaling pathways by H2O2, including the ERK, JNK, and p38 pathways, has been previously noted in several cell types, and the activation of these pathways is involved in H2O2-mediated multiple downstream processes, including changes in gene expression [27, 28]. To explore whether MAPK pathways are involved in H2O2-induced changes in ASIC1a expression, we first examined the activation of MAPK pathways by H2O2 in NS20Y cells. We found that incubation with 20 µM H2O2 dramatically activated the JNK, p38, and ERK1/2 pathways in NS20Y cells within 30–60 min (Fig. 3a-f, n = 4, P = 0.004, P < 0.001 and P = 0.016 compared with the control).
Fig. 3. Effect of H2O2 on the activation of MAPK signaling pathways in NS20Y cells.

a, c, and e Representative blots showing increased phosphorylation of JNK, p38, and ERK1/2 at 30 and 60 min following H2O2 (20 μM) treatment in NS20Y cells. b, d, and f Quantification analysis of the phosphorylation of JNK, p38, and ERK1/2 following H2O2 (20 μM) treatment in NS20Y cells (n = 4, P = 0.004, P < 0.001 and P = 0.016 versus the control, one-way ANOVA followed by Bonferroni’s post hoc test).
The JNK signaling pathway is involved in the H2O2-mediated increases in ASIC1a protein expression and ASIC currents
To explore which specific MAPK pathway is involved in H2O2-mediated changes in ASIC1a protein expression, we examined the effect of ERK, p38, and JNK pathway inhibitors on ASIC1a expression. We found that neither inhibition of the MEK/ERK pathway by U0126 (10 μM) nor inhibition of the p38 MAPK pathway by SB203580 (10 μM) affected H2O2-induced changes in ASIC1a expression (Fig. 4a–d, n = 8–9, P < 0.001 and P = 0.014 compared with the control). In contrast, inhibition of the JNK pathway by SP600125 (10 μM) dramatically decreased basal ASIC1a expression and completely abolished the H2O2-induced increase in ASIC1a expression (Fig. 4e-f, n = 9, P = 0.001, P < 0.001 compared with the control; P < 0.001 compared with H2O2 alone). Relative ASIC1a expression was decreased to 0.36 ± 0.08 and 0.30 ± 0.07 by SP600125 in the absence or presence of H2O2, respectively, and there was no significant difference between these levels. Furthermore, we examined whether the increase in surface expression of ASIC1a by H2O2 can also be affected by SP600125. Our data showed that, similar to total protein expression, the increased surface expression of ASIC1a by H2O2 was inhibited by SP600125 (Supplementary Fig. 4).
Fig. 4. Effect of MAPK signaling pathway blockade on the H2O2-mediated increase in ASIC1a expression.

a Representative blots and (b) quantification of ASIC1a protein expression in the presence or absence of H2O2 (20 μM) for 24 h in NS20Y cells and in the presence or absence of the MEK/ERK pathway inhibitor U0126 (10 μM) (n = 9, P < 0.001 versus the group not treated with H2O2 or U0126 treatment, one-way ANOVA followed by Bonferroni’s post hoc test). U0126 or vehicle (0.1% DMSO) was added 1 h prior to and during H2O2 treatment. c Representative blots and d quantification of ASIC1a protein expression in NS20Y cells in the presence or absence of H2O2 (20 μM) for 24 h and in the presence or absence of the p38 MAPK inhibitor SB203580 (10 μM) (n = 8, P = 0.014 versus the group not treated with H2O2 or SB203580 treatment). SB203580 or vehicle (0.1% DMSO) was added 1 h prior to and during H2O2 treatment. e Representative blots and (f) quantification of ASIC1a protein expression in NS20Y cells in the presence or absence of H2O2 (20 μM) for 24 h and in the presence or absence of the JNK inhibitor SP600125 (10 μM) (n = 9, P < 0.001 versus the group not treated with H2O2 or SP600125 treatment, P < 0.001 versus H2O2 alone, one-way ANOVA followed by Bonferroni’s post hoc test). SP600125 or vehicle (0.1% DMSO) was added 1 h prior to and during H2O2 treatment.
Since the surface proteins of ion channels more accurately represent functional channels, we further determined whether a decrease in surface ASIC1a protein expression by a JNK inhibitor results in decreased ASIC current density. We found that incubation with SP600125 for 24 h dramatically decreased the ASIC current density in NS20Y cells and completely abolished the H2O2-mediated increase in ASIC currents (Fig. 5a-b, n = 20–23, P = 0.04 and P = 0.004 compared with the control; P < 0.001 compared with H2O2 alone). The finding that incubation with SP600125 alone decreased the ASIC current raised the question of whether SP600125 directly regulates the gating process of ASIC1a. Before the recording of ASIC currents, we washed the cultures 3 times with ECF. It is likely that the residual SP600125 left in the dish had a negligible effect. However, we cannot absolutely exclude the possibility that the residual SP directly gated ASIC1a. To test this possibility, we performed an additional experiment to examine whether brief application of SP600125 has a direct effect on ASIC currents. We perfused NS20Y cells with 10 µM SP600125 for 5 min but did not observe significant inhibition of ASIC currents (Supplementary Fig. 5a-b). These data suggest that SP600125 does not have a direct effect on the gating process of ASIC and that the change in ASIC currents induced by long-term SP600125 treatment is likely due to the change in ASIC1a protein expression.
Fig. 5. Effect of JNK pathway blockade on H2O2-mediated changes in ASIC currents.
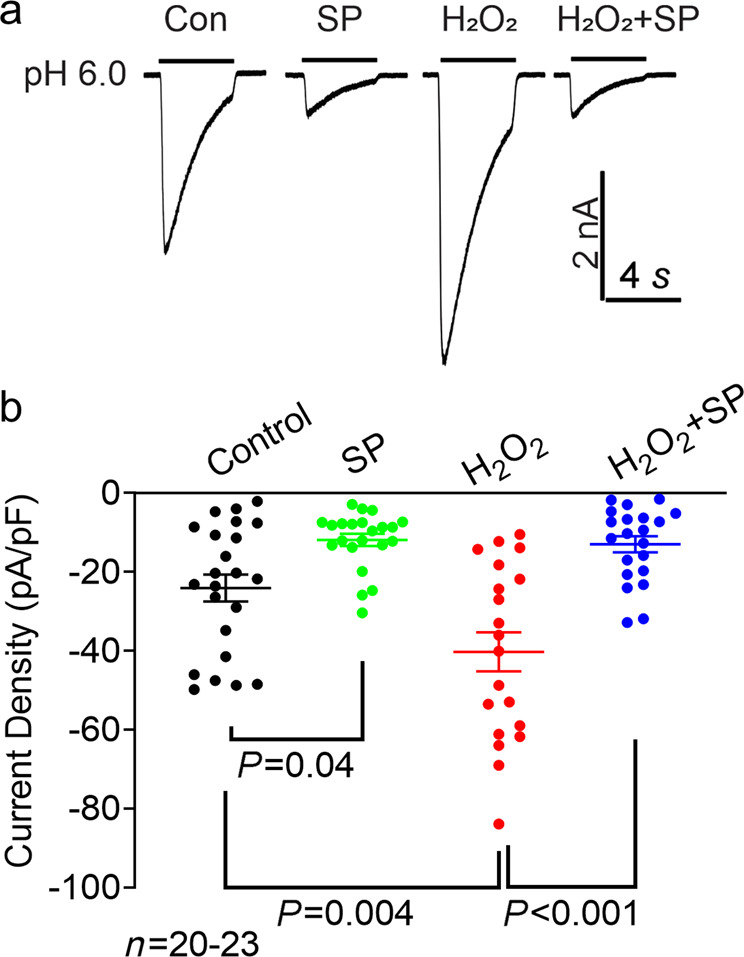
a Representative whole-cell recordings of ASIC currents in the presence or absence of H2O2 (20 μM) for 24 h in NS20Y cells and in the presence or absence of JNK inhibitor SP600125 (SP, 10 μM). b Summary of the data showing the alteration of ASIC current density by H2O2 in the presence or absence of SP600125 (n = 20–23 cells, P = 0.04 and P = 0.004 versus vehicle control, P < 0.001 versus H2O2 alone, one-way ANOVA). SP600125 or vehicle (0.1% DMSO) was added 1 h prior to and during H2O2 treatment.
H2O2 increases ASIC1a protein expression and ASIC currents in primary cultured mouse cortical neurons
We then examined whether H2O2 has a similar effect on ASIC1a expression in primary cultured mouse cortical neurons. Similar to the findings in NS20Y cells, incubation with higher concentrations of H2O2 (40 and 80 µM) caused significant neuronal injury, whereas lower concentrations of H2O2 (less than 20 μM) did not cause neuronal injury (n = 8, P < 0.001 compared with the control). We found that 20 μM H2O2 caused increases in the expression of ASIC1a and ASIC currents in cultured mouse cortical neurons (Fig. 6c and e), which is consistent with the results in NS20Y cells. Relative ASIC1a protein expression was increased 1.48-fold compared with the control level (Fig. 6d, n = 12, P < 0.001 compared with the control), and the density of ASIC currents was increased 1.41-fold compared with the control level (Fig. 6f, n = 26–29, P = 0.028 compared with the control).
Fig. 6. H2O2 increases ASIC1a protein expression and ASIC currents in primary cultured mouse cortical neurons.
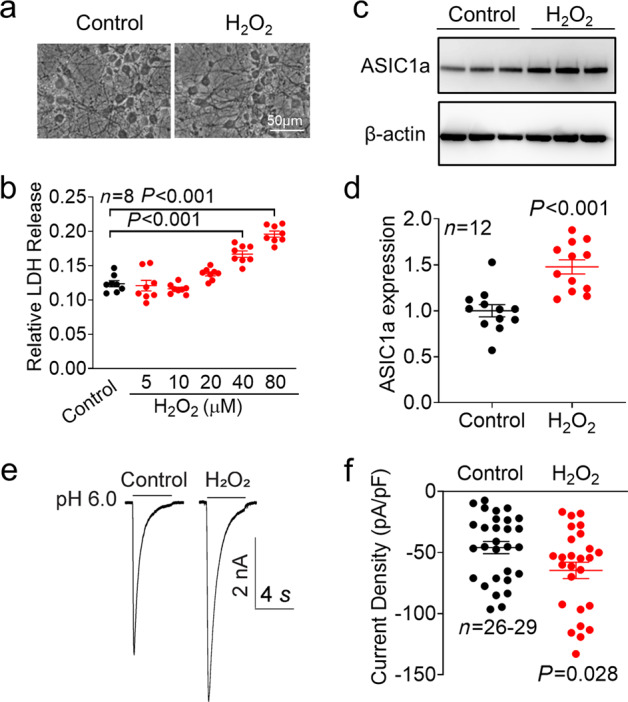
a Representative phase-contrast images showing primary cultured cortical neurons treated with or without H2O2 (20 μM) for 24 h. b LDH assay showing the concentration-dependent cytotoxic effect of H2O2 (n = 8, P < 0.001 versus the control, one-way ANOVA followed by Bonferroni’s post hoc test). c Representative blots and (d) quantification of ASIC1a protein expression in the presence or absence of H2O2 (20 μM) for 24 h in primary cultured cortical neurons (n = 12, P < 0.001 versus the control, unpaired Student’s t test). e Representative whole-cell recordings and (f) summary of the data showing an increase in ASIC currents in NS20Y cells after treatment with H2O2 (20 μM) for 24 h (n = 26–29 cells, P = 0.028 versus the control, unpaired Student’s t test).
In addition to ASIC1a, the predominant ASIC subunit, the ASIC2 gene is widely expressed in the brain. We further examined whether H2O2 affects the expression of ASIC2 in primary cultured cortical neurons. Our data showed that H2O2 treatment for 24 h did not significantly change the expression of ASIC2 (Supplementary Fig. 6a-b). We also performed a similar experiment in NS20Y cells; however, we did not observe a noticeable band between 60 and 80 kDa, suggesting that there was no expression of the ASIC2 protein in NS20Y cells (Supplementary Fig. 6c).
H2O2 increases the sensitivity of ASIC currents to PcTx1 blockade
To further determine whether the increase in ASIC currents by H2O2 is due to increased expression of ASIC1a channels, we compared the sensitivity of the ASIC currents to PcTx1 blockade with and without H2O2 treatment. PcTx1 has been shown to block ASIC1a-containing channels, including homomeric ASIC1a and heteromeric ASIC1a/2b [29, 30]. We found that there was a significant increase in the sensitivity of ASIC currents to PcTx1 blockade after H2O2 treatment for 24 h (Fig. 7a). A total of 34.20% ± 5.95% of the ASIC currents were inhibited by PcTx1 in control cells, whereas 56.27% ± 5.61% of the ASIC currents were inhibited in H2O2-treated cells (Fig. 7b). These data suggest that there are more functional ASIC1a channels distributed on the plasma membrane in the presence of H2O2 than in the absence of H2O2, providing additional evidence for our conclusion that H2O2 upregulates functional ASIC1a expression.
Fig. 7. H2O2 increases the sensitivity of ASIC currents to the inhibitory effect of PcTx1 in primary cultured mouse cortical neurons.
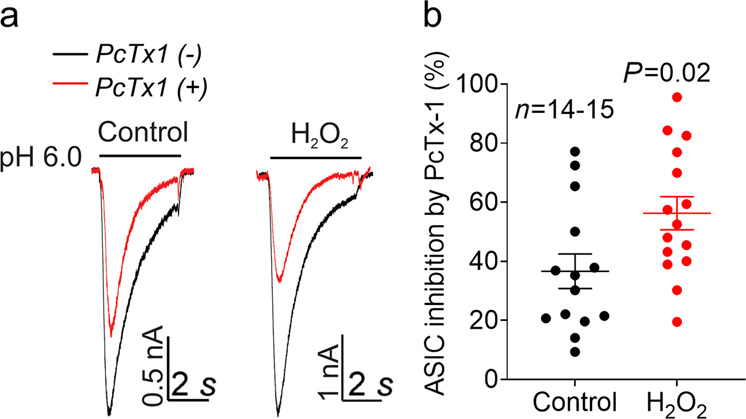
a Representative current traces and b summary of the data showing the responses of ASIC currents to the inhibitor PcTx1 (20 nM) in cortical neurons in the presence or absence of H2O2 pretreatment for 24 h (n = 14–15 cells, P = 0.012, unpaired Student’s t test). The black traces represent the currents before application of PcTx1, and the red traces represent the remaining currents after inhibition by PcTx1.
Discussion
In the current study, we explored the modulatory effect of ASIC1a by H2O2, a major ROS. We showed that H2O2 at a concentration of 20 µM significantly increases ASIC1a expression and ASIC currents in both NS20Y cells and primary cultured cortical neurons. Furthermore, we demonstrated that H2O2 activates MAPK pathways, including the ERK1/2, JNK, and p38 pathways and that inhibition of the JNK pathway, but not the ERK1/2 or p38 pathway, potently inhibits H2O2-induced changes in ASIC1a expression. Previous studies by us and others have shown that reducing and oxidizing reagents, including dithiothreitol, 5,5’-dithio-bis-(2-nitrobenzoic acid) and nitric oxide, acutely modulate ASIC currents [31, 32]. In contrast, we showed in the current study that H2O2 does not have a rapid modulatory effect on ASIC currents. Rather, it has a slow effect on ASIC1a protein expression, which indirectly affects channel function.
Oxidative stress is defined as an imbalance between pro-oxidant and antioxidant homeostasis that leads to the generation of toxic reactive oxygen or nitrogen species [13]. In the current study, we only focused on the effect of H2O2, a major ROS, on ASIC1a expression. Future studies need to be performed to explore the effects of other types of reactive oxygen or nitrogen species, including nitric oxide (.NO) and peroxynitrite (ONOO-) on ASIC. In addition, the potential effects of antioxidants such as vitamin E and glutathione on ASIC1a expression and channel function should be determined.
The concentrations of H2O2 in blood and plasma have been measured, but the exact values remain controversial due to the great variability of the results published in the literature [18, 26]. The majority of studies have suggested that the normal values of H2O2 in human blood range from 1 to 5 µM [26]. However, in chronic disease conditions, the concentration of H2O2 can reach 10–60 µM [18, 26]. For example, the level of plasma H2O2 can reach ~35 µM in rats fed a high-fat diet [33] and ~60 µM in patients with type 1 and type 2 diabetes [34]. Following acute brain ischemia, the H2O2 concentration can reach ~100 µM during reperfusion [35]. Higher concentrations of H2O2 (100 µM-2 mM) have been used widely in studies of acute cell toxicity [19, 23–25]. Interestingly, non-physiological high concentrations of H2O2 (1–10 mM) have been reported to increase the formation of intersubunit disulfide bonds, which reduces the surface expression of ASIC1a [36]. In the current study, we used 20 µM H2O2, which is a pathologically relevant concentration that occurs in chronic diseases.
ASIC1a has been implicated in the pathogenesis of PD, ischemic stroke, and ALS, and inhibition of ASIC1a channels alleviates these neurological diseases [10, 11, 37]. Intriguingly, oxidative stress has been intimately linked to the pathogenesis of these disorders. The brain, particularly, the substantia nigra and the striatum, which are critical in the development of PD, is susceptible to oxidative stress. Although the etiology of PD remains elusive, postmortem analyses of PD patients’ brains have strongly suggested the involvement of oxidative stress and mitochondrial dysfunction [38]. A previous study showed that pharmacological inhibition of ASIC1a has beneficial effects in a mouse model of PD [37], suggesting that enhanced activation of ASIC1a contributes to the pathogenesis of PD. The current finding that H2O2 upregulates ASIC1a expression may provide additional mechanistic insight into the pathogenesis of PD. A number of studies have provided evidence that increased oxidative stress occurs in diabetes or diabetic conditions in addition to PD and causes worse stroke outcomes [39–41]. ASIC1a has been shown to play a critical role in brain injury associated with cerebral ischemia [11], and whether increased expression of ASIC1a by H2O2 contributes to worse stroke outcomes in diabetic patients remains to be determined.
It is well established that exposure of cells to H2O2 induces activation of multiple MAP kinases, including ERK, JNK, and p38 MAPK [19]. In the current study, we demonstrated that inhibition of the JNK pathway, but not the ERK or p38 pathway, abolished the H2O2-induced increase in ASIC1a expression. Interestingly, we found that JNK inhibition by SP600125 dramatically reduced basal ASIC1a expression, suggesting that the JNK pathway may play a key role in regulating ASIC1a expression under physiological conditions. In this regard, it would be interesting to explore the detailed molecular mechanisms underlying the modulatory effect of the JNK pathway on ASIC expression in the future.
In conclusion, in the present study, we showed evidence that H2O2, a key player in oxidative stress, upregulates ASIC1a protein expression and increases channel function through the JNK signaling pathway. These findings will have broad implications given that oxidative stress and ASIC1a play important roles in several neurodegenerative diseases.
Supplementary information
Acknowledgements
ASIC1a antibody was a gift from Dr. Xiang-ming Zha (University of South Alabama, USA). This study was supported by National Institutes of Health (NIH) Grants SC3GM122593 (tl) and National Center for Minority Health and Health Disparities of the National Institutes of Health (U54MD007602).
Author contributions
BMW performed the electrophysiology experiments. JB and LZ performed the Western blot experiments. TY performed the neuronal culture. ZGX and TDL designed the experiment, interpreted the data and wrote the paper.
Competing interests
The authors declare no competing interests.
Contributor Information
Zhi-gang Xiong, Email: zxiong@msm.edu.
Tian-dong Leng, Email: tleng@msm.edu.
Supplementary information
The online version of this article (10.1038/s41401-020-00559-3) contains supplementary material, which is available to authorized users.
References
- 1.Leng T, Shi Y, Xiong ZG, Sun D. Proton-sensitive cation channels and ion exchangers in ischemic brain injury: new therapeutic targets for stroke? Prog Neurobiol. 2014;115:189–209. doi: 10.1016/j.pneurobio.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wemmie JA, Taugher RJ, Kreple CJ. Acid-sensing ion channels in pain and disease. Nat Rev Neurosci. 2013;14:461–71.. doi: 10.1038/nrn3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lingueglia E, de Weille JR, Bassilana F, Heurteaux C, Sakai H, Waldmann R, et al. A modulatory subunit of acid sensing ion channels in brain and dorsal root ganglion cells. J Biol Chem. 1997;272:29778–83.. doi: 10.1074/jbc.272.47.29778. [DOI] [PubMed] [Google Scholar]
- 4.Waldmann R, Champigny G, Bassilana F, Heurteaux C, Lazdunski M. A proton-gated cation channel involved in acid-sensing. Nature. 1997;386:173–7. doi: 10.1038/386173a0. [DOI] [PubMed] [Google Scholar]
- 5.Wemmie JA, Chen J, Askwith CC, Hruska-Hageman AM, Price MP, Nolan BC, et al. The acid-activated ion channel ASIC contributes to synaptic plasticity, learning, and memory. Neuron. 2002;34:463–77.. doi: 10.1016/S0896-6273(02)00661-X. [DOI] [PubMed] [Google Scholar]
- 6.Boscardin E, Alijevic O, Hummler E, Frateschi S, Kellenberger S. The function and regulation of acid-sensing ion channels (ASICs) and the epithelial Na+ channel (ENaC): IUPHAR review 19. Br J Pharmacol. 2016;173:2671–701. doi: 10.1111/bph.13533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friese MA, Craner MJ, Etzensperger R, Vergo S, Wemmie JA, Welsh MJ, et al. Acid-sensing ion channel-1 contributes to axonal degeneration in autoimmune inflammation of the central nervous system. Nat Med. 2007;13:1483–9. doi: 10.1038/nm1668. [DOI] [PubMed] [Google Scholar]
- 8.Li MH, Liu SQ, Inoue K, Lan J, Simon RP, Xiong ZG. Acid-sensing ion channels in mouse olfactory bulb M/T neurons. J Gen Physiol. 2014;143:719–31.. doi: 10.1085/jgp.201310990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vann KT, Xiong ZG. Acid-sensing ion channel 1 contributes to normal olfactory function. Behav Brain Res. 2018;337:246–51.. doi: 10.1016/j.bbr.2017.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vergo S, Craner MJ, Etzensperger R, Attfield K, Friese MA, Newcombe J, et al. Acid-sensing ion channel 1 is involved in both axonal injury and demyelination in multiple sclerosis and its animal model. Brain. 2011;134:571–84.. doi: 10.1093/brain/awq337. [DOI] [PubMed] [Google Scholar]
- 11.Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, et al. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118:687–98.. doi: 10.1016/j.cell.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 12.Ziemann AE, Schnizler MK, Albert GW, Severson MA, Howard MA, 3rd, Welsh MJ, et al. Seizure termination by acidosis depends on ASIC1a. Nat Neurosci. 2008;11:816–22.. doi: 10.1038/nn.2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emerit J, Edeas M, Bricaire F. Neurodegenerative diseases and oxidative stress. Biomed Pharmacother. 2004;58:39–46. doi: 10.1016/j.biopha.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 14.Cobley JN, Fiorello ML, Bailey DM. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018;15:490–503. doi: 10.1016/j.redox.2018.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Konyalioglu S, Armagan G, Yalcin A, Atalayin C, Dagci T. Effects of resveratrol on hydrogen peroxide-induced oxidative stress in embryonic neural stem cells. Neural Regen Res. 2013;8:485–95. doi: 10.3969/j.issn.1673-5374.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mastrocola R, Restivo F, Vercellinatto I, Danni O, Brignardello E, Aragno M, et al. Oxidative and nitrosative stress in brain mitochondria of diabetic rats. J Endocrinol. 2005;187:37–44. doi: 10.1677/joe.1.06269. [DOI] [PubMed] [Google Scholar]
- 17.Veal EA, Day AM, Morgan BA. Hydrogen peroxide sensing and signaling. Mol Cell. 2007;26:1–14. doi: 10.1016/j.molcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 18.Weinstain R, Savariar EN, Felsen CN, Tsien RY. In vivo targeting of hydrogen peroxide by activatable cell-penetrating peptides. J Am Chem Soc. 2014;136:874–7. doi: 10.1021/ja411547j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhuang S, Yan Y, Daubert RA, Han J, Schnellmann RG. ERK promotes hydrogen peroxide-induced apoptosis through caspase-3 activation and inhibition of Akt in renal epithelial cells. Am J Physiol Ren Physiol. 2007;292:F440–7. doi: 10.1152/ajprenal.00170.2006. [DOI] [PubMed] [Google Scholar]
- 20.Xu Y, Jiang YQ, Li C, He M, Rusyniak WG, Annamdevula N, et al. Human ASIC1a mediates stronger acid-induced responses as compared with mouse ASIC1a. FASEB J. 2018;32:3832–43. doi: 10.1096/fj.201701367R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Bryant Z, Leng T, Liu M, Inoue K, Vann KT, Xiong ZG. Acid sensing ion channels (ASICs) in NS20Y cells-potential role in neuronal differentiation. Mol Brain. 2016;9:68. doi: 10.1186/s13041-016-0249-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leng TD, Lin J, Sun HW, Zeng Z, O’Bryant Z, Inoue K, et al. Local anesthetic lidocaine inhibits TRPM7 current and TRPM7-mediated zinc toxicity. CNS Neurosci Ther. 2015;21:32–9. doi: 10.1111/cns.12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhat NR, Zhang P. Hydrogen peroxide activation of multiple mitogen-activated protein kinases in an oligodendrocyte cell line: role of extracellular signal-regulated kinase in hydrogen peroxide-induced cell death. J Neurochem. 1999;72:112–9. doi: 10.1046/j.1471-4159.1999.0720112.x. [DOI] [PubMed] [Google Scholar]
- 24.Chen L, Liu L, Yin J, Luo Y, Huang S. Hydrogen peroxide-induced neuronal apoptosis is associated with inhibition of protein phosphatase 2A and 5, leading to activation of MAPK pathway. Int J Biochem Cell Biol. 2009;41:1284–95.. doi: 10.1016/j.biocel.2008.10.029. [DOI] [PubMed] [Google Scholar]
- 25.Du J, Zhu Y, Chen X, Fei Z, Yang S, Yuan W, et al. Protective effect of bone morphogenetic protein-6 on neurons from H2O2 injury. Brain Res. 2007;1163:10–20. doi: 10.1016/j.brainres.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 26.Forman HJ, Bernardo A, Davies KJ. Corrigendum to “What is the concentration of hydrogen peroxide in blood and plasma?” [Arch. Biochem. Biophys. 603 (2016) 48-53] Arch Biochem Biophys. 2016;607:7. doi: 10.1016/j.abb.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 27.Guyton KZ, Liu Y, Gorospe M, Xu Q, Holbrook NJ. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J Biol Chem. 1996;271:4138–42.. doi: 10.1074/jbc.271.8.4138. [DOI] [PubMed] [Google Scholar]
- 28.Stevenson MA, Pollock SS, Coleman CN, Calderwood SK. X-irradiation, phorbol esters, and H2O2 stimulate mitogen-activated protein kinase activity in NIH-3T3 cells through the formation of reactive oxygen intermediates. Cancer Res. 1994;54:12–5. [PubMed] [Google Scholar]
- 29.Escoubas P, De Weille JR, Lecoq A, Diochot S, Waldmann R, Champigny G, et al. Isolation of a tarantula toxin specific for a class of proton-gated Na+ channels. J Biol Chem. 2000;275:25116–21.. doi: 10.1074/jbc.M003643200. [DOI] [PubMed] [Google Scholar]
- 30.Sherwood TW, Lee KG, Gormley MG, Askwith CC. Heteromeric acid-sensing ion channels (ASICs) composed of ASIC2b and ASIC1a display novel channel properties and contribute to acidosis-induced neuronal death. J Neurosci. 2011;31:9723–34.. doi: 10.1523/JNEUROSCI.1665-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cadiou H, Studer M, Jones NG, Smith ES, Ballard A, McMahon SB, et al. Modulation of acid-sensing ion channel activity by nitric oxide. J Neurosci. 2007;27:13251–60.. doi: 10.1523/JNEUROSCI.2135-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chu XP, Close N, Saugstad JA, Xiong ZG. ASIC1a-specific modulation of acid-sensing ion channels in mouse cortical neurons by redox reagents. J Neurosci. 2006;26:5329–39.. doi: 10.1523/JNEUROSCI.0938-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roberts CK, Barnard RJ, Sindhu RK, Jurczak M, Ehdaie A, Vaziri ND. A high-fat, refined-carbohydrate diet induces endothelial dysfunction and oxidant/antioxidant imbalance and depresses NOS protein expression. J Appl Physiol (1985) 2005;98:203–10. doi: 10.1152/japplphysiol.00463.2004. [DOI] [PubMed] [Google Scholar]
- 34.Wierusz-Wysocka B, Wysocki H, Byks H, Zozulińska D, Wykretowicz A, Kaźmierczak M. Metabolic control quality and free radical activity in diabetic patients. Diabetes Res Clin Pr. 1995;27:193–7. doi: 10.1016/0168-8227(95)01043-D. [DOI] [PubMed] [Google Scholar]
- 35.Hyslop PA, Zhang Z, Pearson DV, Phebus LA. Measurement of striatal H2O2 by microdialysis following global forebrain ischemia and reperfusion in the rat: correlation with the cytotoxic potential of H2O2 in vitro. Brain Res. 1995;671:181–6. doi: 10.1016/0006-8993(94)01291-O. [DOI] [PubMed] [Google Scholar]
- 36.Zha XM, Wang R, Collier DM, Snyder PM, Wemmie JA, Welsh MJ. Oxidant regulated inter-subunit disulfide bond formation between ASIC1a subunits. Proc Natl Acad Sci U S A. 2009;106:3573–8. doi: 10.1073/pnas.0813402106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arias RL, Sung ML, Vasylyev D, Zhang MY, Albinson K, Kubek K, et al. Amiloride is neuroprotective in an MPTP model of Parkinson’s disease. Neurobiol Dis. 2008;31:334–41.. doi: 10.1016/j.nbd.2008.05.008. [DOI] [PubMed] [Google Scholar]
- 38.Fahn S. Description of Parkinson’s disease as a clinical syndrome. Ann NY Acad Sci. 2003;991:1–14. doi: 10.1111/j.1749-6632.2003.tb07458.x. [DOI] [PubMed] [Google Scholar]
- 39.Kagansky N, Levy S, Knobler H. The role of hyperglycemia in acute stroke. Arch Neurol. 2001;58:1209–12. doi: 10.1001/archneur.58.8.1209. [DOI] [PubMed] [Google Scholar]
- 40.Pulsinelli WA, Levy DE, Sigsbee B, Scherer P, Plum F. Increased damage after ischemic stroke in patients with hyperglycemia with or without established diabetes mellitus. Am J Med. 1983;74:540–4. doi: 10.1016/0002-9343(83)91007-0. [DOI] [PubMed] [Google Scholar]
- 41.Pulsinelli WA, Waldman S, Rawlinson D, Plum F. Moderate hyperglycemia augments ischemic brain damage: a neuropathologic study in the rat. Neurology. 1982;32:1239–46.. doi: 10.1212/WNL.32.11.1239. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.