

Keywords: CIPO, cytoskeleton, MMIHS, visceral myopathy
Abstract
Visceral smooth muscle is a crucial component of the walls of hollow organs like the gut, bladder, and uterus. This specialized smooth muscle has unique properties that distinguish it from other muscle types and facilitate robust dilation and contraction. Visceral myopathies are diseases where severe visceral smooth muscle dysfunction prevents efficient movement of air and nutrients through the bowel, impairs bladder emptying, and affects normal uterine contraction and relaxation, particularly during pregnancy. Disease severity exists along a spectrum. The most debilitating defects cause highly dysfunctional bowel, reduced intrauterine colon growth (microcolon), and bladder-emptying defects requiring catheterization, a condition called megacystis-microcolon-intestinal hypoperistalsis syndrome (MMIHS). People with MMIHS often die early in childhood. When the bowel is the main organ affected and microcolon is absent, the condition is known as myopathic chronic intestinal pseudo-obstruction (CIPO). Visceral myopathies like MMIHS and myopathic CIPO are most commonly caused by mutations in contractile apparatus cytoskeletal proteins. Here, we review visceral myopathy-causing mutations and normal functions of these disease-associated proteins. We propose molecular, cellular, and tissue-level models that may explain clinical and histopathological features of visceral myopathy and hope these observations prompt new mechanistic studies.
INTRODUCTION
Visceral myopathies are debilitating, life-threatening disorders caused by smooth muscle weakness in the bowel, bladder, and uterus. This muscle weakness manifests as massive bowel distension, feeding intolerance, vomiting, intractable constipation, dramatic bladder dilation with obstructive uropathy, and uterine atony. Patients with severe gastrointestinal dysmotility due to smooth muscle dysfunction are diagnosed with myopathic chronic intestinal pseudo-obstruction (CIPO). In the most severe cases, gastrointestinal (GI) and genitourinary (GU) tracts are dysfunctional at birth and intrauterine colon growth is impaired, a condition called megacystis-microcolon-intestinal hypoperistalsis syndrome (MMIHS). This early disease presentation is associated with more severe pathology and worse outcomes. In MMIHS, intestinal dysfunction can be deadly. Concurrent bladder dysfunction is common, even though it may be less obvious than bowel symptoms (1). Curiously, in familial forms of visceral myopathy, one individual may have severe muscle dysfunction and others with the same disease-causing mutation exhibit only mild constipation or difficulty progressing through childbirth due to weak uterine contractions (2). Uterine presentations are less commonly reported than bowel and bladder symptoms but are important to consider. For example, if an infant has intestinal dysmotility and megacystis, a complicated birth history with failure to progress through labor or prolonged bleeding after childbirth may indicate an inherited visceral myopathy (2, 3). This variability in disease severity indicates that visceral myopathies are multifactorial disorders with genetic, epigenetic, and environmental modulators that are not yet well understood.
Visceral myopathies are defined by the presence of smooth muscle dysfunction. Smooth muscle, like other muscle types, relies on contractile apparatus cytoskeletal proteins to generate force. Unsurprisingly, mutations in genes encoding these contractile apparatus proteins are common genetic causes of visceral myopathy. Here, we summarize data about disease pathophysiology and cell biology, linking clinical manifestations in people with myopathic CIPO with basic science data about known mutations in smooth muscle cytoskeletal genes. Based on these data, we propose hypotheses for future investigation that could improve treatment outcomes (see Tables 2 and 3 for key findings).
Table 2.
New hypotheses to understand visceral myopathy
| ACTG2 variants that impair actin polymerization may increase cytoplasmic sequestration of MRTFs by G-actin. Reduced nuclear MRTFs can decrease SMC contractile gene expression and increase expression of ECM and promitogenic genes. |
| Stiffening of bowel and bladder due to fibrosis and pathological stretch may promote nuclear localization of YAP, which inhibits myocardin and downregulates expression of SMC contractile genes. |
| NF-κB activation by pro-inflammatory cytokines may inhibit SMC contractile gene expression because NF-κB inhibits myocardin. |
| Cytoskeletal gene mutations that reduce F-actin interactions with myosin or interactions between actin and focal adhesions may reduce 1) force generation, 2) force transmission to the ECM, and 3) the ability of muscle to resist passive stretch. Cytoskeletal gene mutations may impair force transmission to the nucleus via the Linker of Nucleoskeleton and Cytoskeleton (LINC) complex, further reducing SMC contractile gene expression in response to extracellular forces. |
| Drastically increased protein production in synthetic phenotype SMCs may trigger an unfolded protein response (UPR), an ER stress response, reinforcing a vicious cycle that further reduces SMC contractile gene expression. |
| Rapamycin and Lovastatin may increase expression of SMC contractile genes, enhancing SMC force generation. Antifibrotics may make the ECM more pliable and reduce the stiffness of the SMC microenvironment. Gene targeting approaches can be considered to silence mutations or correct them. |
ACTG2, actin gamma 2; ECM, extracellular matrix; ER, endoplasmic reticulum; G-actin, globular actin; MRTFs, myocardin-related transcription factors; SMC, smooth muscle cell; YAP, Yes-associated protein.
Table 3.
The physics of visceral myopathy
| Megacystis (massive bladder dilation) and pathological bowel dilation (in CIPO) can be modeled using Laplace’s law (Fig. 3). |
| Laplace’s law: wall tension is proportional to intraluminal radial pressure and radius and is inversely proportional to wall thickness. |
| With increased bowel and bladder distension (radius), greater wall tension is required to maintain intraluminal pressure. |
| Distension causes a decrease in wall thickness, necessitating even more wall tension to maintain intraluminal pressure. |
| Cytoskeletal mutations that impair force generation and transmission will also reduce the maximum wall tension that can be developed by the bowel and bladder. Bowel and bladder distension are likely progressive and irreversible as weakened SMCs in bowel and bladder are unable to generate the wall tension necessary to resist distension. |
CIPO, chronic intestinal pseudo-obstruction; SMC, smooth muscle cell.
CLINICAL OVERVIEW OF CIPO
CIPO is a rare disease that may first manifest in adults or in children. In the United States, there are ∼100 newborns newly diagnosed with CIPO each year (4) (incidence 0.21/0.24 per 100,000 infants) (5). A Japanese study estimated a CIPO prevalence of 0.37 per 100,000 in those younger than 15 yr of age, with over half developing CIPO as neonates (6). People with CIPO experience nausea, vomiting, constipation, massive abdominal distension, and malnutrition because they are unable to tolerate oral feeding. Symptom severity varies among affected individuals and varies over time in each individual. Most affected people have multiple abdominal surgeries and inpatient hospital admissions to optimize therapeutic regimens or to treat massive abdominal distension that may be exacerbated by many types of infection. As a consequence of bowel dysfunction, many people with CIPO require total parenteral nutrition (TPN) for growth and survival at least intermittently (1). TPN complications include life-threatening line infections and long-term liver damage. Current treatments are largely supportive, with no mechanism-based therapies for myopathic CIPO. Multi-visceral transplantation, involving bowel, liver, and other intestinal organs, is one treatment strategy with a 5-yr survival of 58% (7). Long inpatient hospital stays and the need for TPN make CIPO care expensive. A 2020 study of children with CIPO in the United States showed that the median duration for each hospitalization was 6 days and median cost per hospitalization was $52,079 (8). Costs were lower when gastrostomies and ileostomies were present before hospitalization. TPN and central line infections increased healthcare costs.
CAUSES OF CIPO
CIPO is broadly classified into neuropathic, myopathic, and mesenchymopathic forms that are due to defects in the enteric nervous system, visceral smooth muscle, or interstitial cells of Cajal (ICC) respectively. These can be further classified as primary or secondary when the cause is known, and idiopathic when no etiology is discernible. Pediatric CIPO is usually primary with genetic defects causing disease manifestations. Adult CIPO is often secondary to autoimmune disease, opiates, or infectious etiologies. Because of the differences in etiology between pediatric and adult-onset CIPO, a recent report suggested using the term pediatric intestinal pseudo-obstruction (PIPO) for childhood-onset CIPO (1). Several excellent reviews discuss the etiologies, disease manifestations, and treatments for CIPO and PIPO (1, 6, 9, 10). Here, we focus on contractile apparatus cytoskeletal proteins that can cause primary myopathic CIPO when mutated.
CAUSES OF MYOPATHIC CIPO
Cytoskeletal protein genes implicated in myopathic CIPO include ACTG2 (11–27), ACTA2 (21, 28), MYH11 (12, 21, 29–34), MYLK (35), LMOD1 (35), MYL9 (12, 36), and FLNA (37). Myotonic dystrophy type 1, Duchenne muscular dystrophy, and oculogastrointestinal muscular dystrophy also cause CIPO as do scleroderma, systemic lupus erythematosus, dermatomyositis, polymyositis, amyloidosis, ceroidosis, and mitochondrial diseases such as mitochondrial neuro-gastrointestinal-encephalomyopathy (MNGIE) (1). Most CIPO cases remain idiopathic, highlighting our limited understanding of visceral smooth muscle dysfunction. Only by defining underlying disease mechanisms, we can hope to develop robust targeted therapies.
Mutations affecting contractile apparatus cytoskeletal proteins are the most common cause of myopathic CIPO, with ACTG2 mutations accounting for ∼44% of reported cases (18). The prevailing hypothesis is that these mutations cause smooth muscle weakness by disrupting the contractile apparatus, which is composed of cytoskeletal proteins. However, basic science studies of how mutations cause myopathy remain limited. In addition, variations in symptom severity even in people who share the same gene defect strongly suggest factors beyond putative causative mutations impact disease severity. If identified, these disease-modifying genetic or non-genetic second-site modifiers could lead to new therapeutic strategies.
CYTOSKELETAL AND CONTRACTILE PROTEINS IN SMOOTH MUSCLE CELLS IMPLICATED IN VISCERAL MYOPATHY
Smooth muscle cells (SMCs) contain contractile filaments of actin and myosin, the main force-producing proteins in SMCs. These contractile filaments are obliquely arranged in smooth muscle cells, with filamentous actin (F-actin) anchored by dense bodies and connected to force-transmitting structures in the extracellular matrix (ECM) (38). Actin filaments are formed through polymerization of globular actin (G-actin) monomers. The F-actin network is regulated by proteins that nucleate, elongate, or cleave F-actin, bundle filaments into stress fibers, or organize F-actin in branching networks to generate force in conjunction with myosin heavy chains (MHCs).
In humans, there are six highly homologous isoforms of actin with >93% sequence identity (39). These conserved actin proteins are critical for cell contraction, migration, polarity, endocytosis, and signaling. Cytoplasmic actins (ACTB, ACTG1) are present in all cells. In addition, muscle cells express specialized actin isoforms: ACTA1 (skeletal muscle), ACTC1 (cardiac muscle), and ACTA2 and ACTG2 (vascular and visceral smooth muscle) (40). The myosin complex in visceral smooth muscle consists of two myosin heavy chains (myosin heavy chain 11, MYH11) and four light chains [two essential myosin light chain 6 (MYL6), plus two regulatory myosin light chain 9 (MYL9)]. Interaction of actin with myosin in smooth muscle is regulated by myosin light chain kinase (MYLK), which phosphorylates MYL9. MYL9 phosphorylation enables myosin head domain interaction with actin filaments, leading to smooth muscle contraction and force generation.
Gamma Smooth Muscle Actin
Gamma smooth muscle actin (actin gamma 2, smooth muscle; ACTG2) is the predominant actin isoform in visceral (bowel, bladder, and uterine) SMCs (41–43) and is thought to be responsible for actin-myosin-mediated visceral SMC contractility. In contrast, ACTA2 is thought to play a similar role in vascular SMC. However, only a few papers directly examine ACTG2 function in cells, even though ACTG2 mutations are the leading cause of visceral myopathies (11, 18, 26).
Most disease-causing ACTG2 mutations are heterozygous missense mutations, suggesting these mutations are dominant-negative. Large homozygous ACTG2 deletions have also been reported to cause severe visceral myopathy (17, 20). There are published cases of inherited ACTG2 mutations, but most disease-causing ACTG2 mutations arise de novo. These de novo ACTG2 mutations have been noted to cause more severe symptoms than inherited cases, which might occur because severe phenotypes reduce the likelihood of having children. Currently, 35 unique ACTG2 missense mutations have been reported in people with CIPO or MMIHS, causing changes in 26 amino acids (Table 1). These mutations are found throughout the protein, suggesting many ways to disrupt normal ACTG2 function. Arginine is the most commonly mutated amino acid with R178 and R257 mutations in over half of published CIPO or MMIHS probands (11). A meta-analysis showed that R178 mutations typically cause the most severe disease (MMIHS) where defects may be detected before birth (11). R257 mutations usually cause severe pediatric-onset CIPO (PIPO) with megacystis. R148 mutations cause less severe (but still highly debilitating) adult-onset CIPO that often affects multiple family members. In addition, single individuals with R148C and R148H mutations are in the Genome Aggregation Database (gnomAD) that is supposed to contain data for healthy individuals. These limited observations suggest that disease severity might be predicted based on ACTG2 genetics.
Table 1.
Summary of known cytoskeletal mutations associated with visceral myopathy
| Gene | Function in SMCs | Myopathy-Associated Mutations | Inheritance Pattern | Clinical Phenotype | Disease Mechanisms Determined by basic science studies | Ref. |
|---|---|---|---|---|---|---|
| ACTG2 | Predominant actin isoform in visceral SMCs. Exact role in cells is unknown. May contract SMCs or control compliance. | R38H, P39L, P39R, R40H, R40C, H41Q, V44A, M45T, R63G, R63Q, P113S, Y134N, L143F, Y144F, G147C, R148S, R148L, T149R, R178H, R178L, R178C, R178S, D185Y, T195I, E196D, G198D, A205T, R211Q, D245G, R257C, R257H, G269E, *c. 502C>T; p. R168Ter, **c.187C>T; p.R63Ter, ***c. 1006C>T; p.R336W | Mostly autosomal dominant | MMIHS, PIPO, or familial CIPO; highly variable presentation | -Impaired actin polymerization -Decreased cell contraction in collagen gel contraction assay with some mutations |
(2, 11–13, 15–22, 24, 25, 27, 44–47) |
| ACTA2 | Predominant actin isoform in vascular SMCs. Also present in visceral SMCs. Required for contractility. | R179C, R179H All other ACTA2 mutations cause vascular disease, but not visceral myopathy. |
Autosomal dominant | Multisystemic smooth muscle dysfunction: MMIHS, vascular disease, heart defects, congenital mydriasis | -Impaired actin polymerization with increased severing of R179H F-actin by cofilin, increased binding of actin to profilin and formin -Decreased movement of R179H F-actin along myosin may explain decrease in contractility - ACTA2 knockout mouse has hypocontractile bladder in vitro |
(28, 48,49) |
| MYH11 | Smooth muscle-specific myosin heavy chain (MHC). Four isoforms (SM1A, SM1B, SM2A, SM2B) by alternative splicing. Required for SMC contraction. Different isoforms may play specific roles in SMC contraction in various tissues and across developmental time points. | Compound heterozygous mutations: -2 bp deletion in exon 22 (c.2809_2810del, p.R937Gfs*7, paternal) and 49 bp deletion in exon 26 (c.3422_3470del, p.K1141Tfs*20, maternal) -c.2051 G > A, p.R684H and c.3540_3541delinsTT, p.E1180D, Q1181Ter - Heterozygous missense variant c.379C>T, P127S and a heterozygous 1.3 Mb deletion in 16q13.11 Homozygous mutations:-c.3598 A > T, p.K1200Ter -c.1591C>T, p.R531TerHeterozygous:-c.5819delC, p.P1940HfsTer91 heterozygous frameshift variant -c.5819_5820insCA, p.Q1941Nfs*91 2 bp insertion with addition of 90 unique amino acids before the stop codon |
Compound heterozygous and homozygous mutations are recessive. Heterozygous mutations are autosomal dominant. |
Homozygous and compound heterozygous mutations: MMIHS, multisystemic smooth muscle dysfunctionHeterozygous autosomal dominant: CIPO and severe esophageal dysmotility | -No basic science studies on the specific mutations identified in humans. -Several animal studies on different Myh11 isoform-specific knockout mice have been used to understand the roles of individual isoforms as well as Myh11 knockouts that affect all isoforms (50–56). SM2 knockout mice have bowel and bladder distension, urinary retention, hydronephrosis, and early death (51). -Bladder muscle strips in SM2 knockouts are hypercontractile, suggesting that SM2 is a negative regulator of SMC contraction. |
(29–34) |
| MYL9 | Regulatory myosin light chain (rMLC) of class II myosins in smooth muscle. Critical for actin-myosin interaction by increasing ATPase activity of MHC. | Homozygous deletion including the last exons Two mutations: 9 bp deletion removes canonical splice donor site at exon 2 (c.184 + 2_184 + 10del), and loss of exon 4 |
Autosomal recessive Compound heterozygous |
MMIHS with mild mydriasis MMHIS, mydriasis, broncho-pulmonary dysplasia, diffuse microvascular disease, progressive white matter loss |
-Homozygous deletion of MYL9 is hypothesized to reduce actin-myosin binding. | (12, 36, 57) |
| MYLK | Myosin light chain kinase (MLCK) phosphorylates regulatory MLC needed for actin-myosin interaction. | -7 bp duplication (c.3838_3844dupGAAAGCG, p.E1282Gfs*51) -Putative splice-site variant (c.3985 + 5C>A) causes a frameshift and creates a premature termination codon (PTC) |
Autosomal recessive | MMIHS | -Unproven hypothesis that lack of phosphorylation of the rMLC prevents myosin activation and interaction with actin, impairing contractility. -Mylk smooth-muscle specific knockout mice have severe gut dysmotility, bladder dysfunction, and hypotension due to inhibition of RLC phosphorylation (58). -Less severe vascular phenotypes could be due to compensation by other kinases. |
(35) |
| LMOD1 | Actin-binding protein that nucleates new actin filaments. | Premature termination codon (PTC) in Exon 2 of LMOD1 c.1108C>T, p.R370Ter). | Autosomal recessive | MMIHS |
-Lmod1 knockout mouse with PTC in exon 1. Stomach and bladder dilation, but no microcolon. Jejunal rings show lower passive tensile strength and weaker contractility in response to agonists. Reduction in F-actin by phalloidin staining and fractionation of monomeric and filamentous actin. No filament changes at ultrastructural level, but elongation of dense bodies in knockout mice. -siRNA knockdown of LMOD1 in human intestinal SMCs led to decrease in collagen gel contraction. |
(59) |
| FLNA | Actin-binding protein that crosslinks F-actin networks. May be important for cell shape and motility. | -Exon skipping due to 4-bp deletion in exon 40 with translation of mutant FLNA missing 41 amino acids. -Partial duplication including first 28 exons -Hemizygous nonsense mutation c.7021C>T; Q2341Ter -2-bp deletion in exon 2 with frameshift leading to PTC - no-stop FLNA mutation (c.7941_7942delCT, p.('2648Sext' 100)) |
X-linked recessive | CIPO, cardiovascular defects, brain malformation, periventricular nodular heterotopia (PVNH) | -Previously believed to be only neuropathic in origin, but detailed immunohisto-chemical analysis demonstrated diffuse abnormal layering (DAL) of intestinal smooth muscle with no enteric neuron involvement (37). -FLNA has two isoforms (60). The longer isoform is the predominant isoform in intestinal smooth muscle, and mutations in this isoform cause CIPO with PVNH in the brain. |
(37, 61–63) |
ACTA2, actin alpha 2; ACTG2, actin gamma 2; CIPO, chronic intestinal pseudo-obstruction; ECM, extracellular matrix; ER, endoplasmic reticulum; F-actin, filamentous actin; FLNA, Filamin A; G-actin, globular actin; LMOD1, Leiomodin 1; MHC, myosin heavy chain; MLC, myosin light chain; MMIHS, megacystis-microcolon-intestinal hypoperistalsis syndrome; MRTFs, myocardin-related transcription factors; MYH11, myosin heavy chain 11; MYL9, myosin light chain 9; MYLK, myosin light chain kinase; PIPO, pediatric intestinal pseudo-obstruction; SMC, smooth muscle cell; YAP, Yes-associated protein.
*One reported autosomal recessive null mutation.
**One reported heterozygous null mutation.
***One reported autosomal recessive missense mutation.
One intriguing observation is that disease severity and presentation can vary dramatically even in family members with the same ACTG2 mutation. For example, two siblings with MMIHS had an ACTG2 R178C mutation inherited from their mother. The siblings’ microcolon indicates intrauterine disease. In contrast, their mother did not have significant bowel or bladder dysfunction, but did have uterine atony postpartum (11). This raises two questions: First, what factors might modify disease severity in people with potentially pathogenic ACTG2 mutations? Second, how often do we miss ACTG2 variants that cause mild disease, since genetic testing is only likely to be performed in people with severe disease or affected relatives? To answer these questions, we need to know more about disease mechanisms.
To date, only four studies (15, 24, 44, 64) examined the phenotype of disease-causing ACTG2 variants using immortalized cancer cell lines. There are no published animal models. These studies showed that tagged ACTG2 variants tested formed fewer filaments than tagged wild-type (WT) ACTG2 in cells. Furthermore, disease-causing ACTG2 variants appeared to reduce phalloidin-stained F-actin although quantitative data were not provided. Some ACTG2 variants made cells less capable of contracting collagen gels (R148S, R178C, R178L), whereas R40C and R63Q did not significantly affect collagen gel contraction compared with WT (15, 24, 44). Although these studies provide preliminary evidence that ACTG2 mutations may affect F-actin formation, they also indicate that disease-causing mutations have heterogeneous effects. One additional caveat is that these studies were performed in cells that do not normally express ACTG2 and may lack other smooth muscle proteins likely to be relevant to disease pathogenesis.
To increase biological relevance, our recent study of the ACTG2 R257C mutation employed human intestinal smooth muscle cells (HISMCs) and included extensive quantitative analysis (64). We demonstrated that ACTG2 R257C formed fewer, shorter, thinner, and less branched ACTG2-containing filament bundles compared with WT ACTG2. However, similar quantitative analysis of all actin filament bundles using phalloidin-labeling showed no significant differences between HISMCs expressing ACTG2 R257C versus WT ACTG2. Ultrastructural analysis using platinum replica electron microscopy also revealed no appreciable difference in filament structure between ACTG2 WT- or R257C-expressing HISMCs. In addition, there were no differences in collagen gel contraction by ACTG2 WT- or R257C-expressing HISMCs. However, ACTG2 R257C-expressing HISMCs spread more quickly and were more migratory than WT ACTG2-expressing HISMCs. This suggests that, at least in vitro, HISMCs may be capable of upregulating other actin isoforms to compensate for some of the deficits caused by the ACTG2 R257C mutation. We are now pursuing studies with other ACTG2 variants.
Our understanding of how ACTG2 mutations cause disease is hampered by limited data about the normal role of ACTG2 compared with other actin isoforms. This is important because ACTG2 and ACTA2 are both expressed in visceral and vascular SMCs, albeit in different proportions depending on cell type. A single study attempted to delineate distinct roles of ACTG2 and ACTA2 in SMCs (41) using a newly generated antibody that distinguishes ACTG2 (not commercially available) from other actin isoforms. Western blots showed that chick aorta expresses ACTA2 but very little ACTG2, whereas stomach, intestine, uterus, and bladder had robust expression of both ACTG2 and ACTA2 (41). Using paraffin-embedded sections, they confirmed that human artery SMC had abundant ACTA2 expression and little ACTG2. In contrast, both ACTA2 and ACTG2 were robustly expressed in human veins and in esophagus. Using porcine vascular SMCs in vitro, they showed via immunohistochemistry that ACTA2-containing fibers were found throughout the cell, extending into vinculin-labeled focal adhesions. In contrast, ACTG2-containing fibers were restricted to the central part of the cells. Most interestingly, siRNA knockdown of ACTA2 decreased collagen gel contraction by the porcine vascular SMCs, whereas knockdown of ACTG2 did not reduce collagen gel contraction. These observations suggest that unlike ACTA2, the ACTG2 isoform may not be needed for vascular SMC contraction but might be responsible for “compliance” (Note: compliance here means resistance to passive stretch. In contrast, in the adult motility literature, compliance refers to the volume response to an imposed intraluminal pressure). They highlighted the absence of ACTG2 from myofibroblasts in vitro and from fibrotic tissue in vivo, and noted that in addition to bowel smooth muscle, ACTG2 is expressed at high levels in mammary glands, uterus (especially during pregnancy), and in veins. All of these are tissues that dramatically dilate as part of their physiological function. If ACTA2 and ACTG2 had similar functions, we would expect mutations in either of them to cause systemic smooth muscle dysfunction affecting all the tissues where both are robustly expressed. In fact, disease presentations are quite distinct, with ACTG2 mutations causing visceral myopathy involving gut, bladder, and uterus, whereas most ACTA2 mutations cause vascular diseases, as discussed in Alpha Smooth Muscle Actin. Collectively, these observations suggest that ACTG2 and ACTA2 may have unique roles in smooth muscle cells, but more work is required to understand the roles of these proteins in normal biology and disease.
Alpha Smooth Muscle Actin
Alpha smooth muscle actin (actin alpha 2, smooth muscle; ACTA2) is the predominant smooth muscle actin isoform in vascular smooth muscle, but ACTA2 is also expressed in visceral smooth muscle (41–43, 65, 66). ACTA2 thin filaments assemble with MYH11 thick filaments to form contractile units in SMCs (67). Like ACTG2 mutations, heterozygous missense mutations in ACTA2 cause smooth muscle disease (68). The majority of ACTA2 mutations predispose to vascular disease, including thoracic aortic aneurysms and dissections (TAADs), early-onset coronary artery disease (CAD), cerebrovascular events (strokes), and patent ductus arteriosus (PDA). ACTA2 mutations have also been associated with atrial septal defects (ASD), which might occur because of high ACTA2 expression in the developing heart. ACTA2 differs from ACTG2 by two amino acids at the NH2-terminus, the most divergent portion of human actin proteins, and ACTA2 has an extra glutamic acid at the NH2-terminus. The extra NH2-terminal glutamate in ACTA2 means that corresponding amino acids are one number lower in ACTG2 versus ACTA2 (39).
Although vascular disease-causing mutations in ACTA2 are found in all protein domains, mutations in ACTA2 R179 cause both visceral and vascular smooth muscle disease. This broad smooth muscle dysfunction is called multisystemic smooth muscle dysfunction syndrome (MSMDS) (49). MSMDS presents at birth or in utero and has a particularly poor prognosis. Widespread severe smooth muscle dysfunction causes developmental delay and seizures due to cerebrovascular events, pulmonary hypertension, dyspnea and recurrent respiratory infections, bladder hypotonia, intestinal malrotation, and hypoperistalsis. Since gastrointestinal and genitourinary symptoms can present before vascular disease complications, some patients with ACTA2 R179H are initially diagnosed with MMIHS or CIPO (69). All patients with the ACTA2 R179H mutation have congenital mydriasis (fixed dilation of the pupils), a symptom that may perhaps distinguish ACTA2 from ACTG2 mutation phenotypes.
It is not fully established why ACTA2 R179 mutations can cause both visceral and vascular smooth muscle disease, whereas other ACTA2 mutations cause only vascular disease. Biochemical analyses show ACTA2 R179H has a higher rate of depolymerization than WT ACTA2 and a 40-fold higher critical concentration for polymerization (28). ACTA2 R179H actin filaments are more easily severed by cofilin, and ACTA2 R179H is more tightly bound to profilin and formin than WT ACTA2. For these reasons, ACTA2 R179H is more likely to exist as monomeric G-actin as opposed to filamentous F-actin. F-actin made from ACTA2 R179H also moves more slowly along myosin. Collectively, these biochemical defects may explain the profound smooth muscle dysfunction in individuals with ACTA2 R179H mutations and demonstrate that ACTA2 defects can impair visceral as well as vascular smooth muscle function. Consistent with a role for ACTA2 in visceral smooth muscle, Acta2 knockout (KO) in mouse bladder reduced contraction strength in response to electric field stimulation or after KCl treatment ex vivo (70) compared with WT mice, but bladder function in vivo appeared normal in the Acta2 KO mouse. Although differences in actin isoform expression may explain the divergent phenotypes caused by ACTA2 and ACTG2 mutations, more work is needed to determine if ACTA2 and ACTG2 have different roles in smooth muscle cells, as Arnoldi et al. (41) suggest, since divergent function might explain differential gene expression.
Smooth Muscle Myosin Heavy Chain
To date, 10 distinct smooth muscle myosin heavy chain (MYH11) mutations have been reported in association with visceral myopathies (29–34). In contrast to the autosomal dominant ACTG2 and ACTA2 mutations, most visceral myopathy-causing MYH11 mutations are autosomal recessive (29, 31). Myosin heavy chain 11 (MYH11) is a component of a hexameric smooth muscle myosin complex in the myosin II family that generates force by pulling on F-actin. Myosins consist of six subunits: two myosin heavy chains (MHC) that dimerize via their long coiled-coil tails and two pairs of myosin light chains (MLC) that bind to the regulatory neck region of MHC (71). SMCs have a single smooth muscle MHC gene (MYH11) that encodes four unique MHC molecules via alternative splicing. Alternative splicing at the 5′ end determines the presence or absence of a 7-amino acid insertion in the S1 head region for SMB and SMA isoforms, respectively. Alternative splicing at the 3′ end determines the presence or absence of 34 amino acids in the COOH-terminus to produce SM1 or SM2. The four possible MHC proteins, therefore, are SM1A, SM1B, SM2A, and SM2B (71). SM1/2 tail isoforms have similar expression levels across most smooth muscle-containing tissues, with some changes during development. There are also reported isoform expression differences between different layers of hollow visceral organs. For example, SM1 is present in both circular and longitudinal layers of rabbit myometrium, but SM2 is only present in longitudinal muscle (72). SMA/B head isoform expression is not believed to correlate with expression of SM1/2 tail isoforms (71). SMB is the primary head isoform in bladder and stomach antrum, whereas SMA is the main head isoform in the stomach fundus and uterus (71). Furthermore, there is evidence that MHC isoform localization within individual cells may differ and affect function (71).
Mapped mutations are usually not in alternatively spliced regions, so most of them affect all MHC isoforms. MYH11 mutation-induced visceral myopathy can be severe, causing MMIHS when both alleles are mutated (32, 34). However, recently reported autosomal dominant mutations, that only affect SM2 isoforms, cause less severe visceral muscle disease (29, 31) (Table 1). Curiously some specific MYH11 mutations appear to cause visceral myopathy without vascular disease, and a different set of MYH11 mutations cause vascular disease. It is possible that these cell-type specific MYH11 mutation effects reflect distinct biochemical interactions in different types of muscle cells, but more work is needed to explain these findings.
Biochemical and cellular data for human MYH11 mutations are not yet available for visceral myopathy phenotypes. However, several mouse models of mutations in different MHC isoforms have been published that provide interesting insights into the specific functions of these isoforms. Complete Myh11 knockout caused death within three days of birth (56). Knockout animals had distended bladders, did not gain weight after birth, and had dilated cardiomyopathy. Knockout pups were able to feed but had milk retention in the stomach and upper duodenum. This is consistent with the observation that the pharynx and esophagus have skeletal muscle, which expresses MYH2, but stomach and other bowel regions have visceral smooth muscle that expresses MYH11. Further supporting a role for MYH11 in force generation, bladder strips from Myh11−/− mice lacked the initial transient state (phase 1) of high force generation with maximal shortening velocity when treated with KCl. Interestingly, however, Myh11−/− bladder smooth muscle could produce sustained contractile force (phase 2) by recruiting nonmuscle myosin heavy chain to compensate for the missing smooth muscle myosin heavy chain (MYH11). Electron micrographs of bladder smooth muscle from the Myh11−/− mice confirmed myosin filament formation by nonmuscle MHC. Consistent with force generation by nonmuscle MHC, maximum shortening velocity in Myh11−/− bladder was more sensitive to increased ADP, and active force was less inhibited by inorganic phosphate than in WT bladder muscle. Early death of Myh11−/− mice, however, suggests that phase 1 force generation mediated by smooth muscle MHC (MYH11) is critical for survival and cannot be compensated for by nonmuscle MHC.
Although the Myh11−/− mouse phenotypes are consistent with multisystem smooth muscle dysfunction, it remains unclear why some MYH11 mutations cause visceral myopathy and other MYH11 mutations cause vascular smooth muscle disease in humans. SM1 and SM2 tail isoforms are highly conserved across species, with SM1 appearing early in development and SM2 expressed close to birth (73). SM2 homozygous knockout mice have classic MMIHS symptoms, including bowel and bladder distension, urinary retention, and end-stage hydronephrosis, with death occurring within 30 days of birth (51). SM2 heterozygous animals appeared healthy. Interestingly, SM2 knockout mice also had reduced SM1 in bladder smooth muscle with a reduction in the density and diameter of thick filaments. Although this might be predicted to reduce muscle contractile strength, bladder muscle strips from SM2 knockout mice were hypercontractile with increased calcium sensitivity, suggesting SM2 negatively regulates force development. This bladder muscle hypercontractility was also surprising because SM2 KO mice have a large bladder. One possible explanation for these apparently contradictory in vitro and in vivo findings is that MYH11 isoforms are differentially expressed even within single-organ systems. For example, SMA/B head isoforms are differentially expressed in stomach antrum and fundus (71), and SM2 is more highly expressed in the urethra than in the bladder. Based on this observation, Chi et al. (51) postulated that SM2 knockout increases urethral smooth muscle contraction more than bladder smooth muscle contraction, resulting in urinary retention. We need in vitro and in vivo models for each of the different MYH11 mutations to unravel the cellular and biochemical differences underlying the pathophysiology of MYH11-mediated vascular and visceral myopathies.
Myosin Light Chain 9
Myosin light chain 9 (MYL9) encodes the regulatory myosin light chain (rMLC) of myosin class II in smooth muscle. Smooth muscle myosin function is regulated in part by phosphorylation of rMLCs by myosin light chain kinase (MYLK). Phosphorylation of the rMLC by MYLK activates actin-myosin binding and force generation. Dephosphorylation of rMLC leads to muscle relaxation. Therefore, a mutation that eliminates or disrupts MYL9 would likely reduce SMC contractility. There are two reported MYL9 mutations associated with MMIHS (12, 36, 57). One mutation is a large homozygous deletion that removes the last exon (exon 4) from the two MYL9 transcripts (36). The second is a biallelic compound heterozygous mutation that removes exon 4 on one allele and causes a 9 bp deletion that removes canonical splice donor site at exon 2 from the second allele (57). It is not yet known if these MYL9 mutations lead to loss of rMLC function or reduced rMLC abundance.
Myosin Light Chain Kinase
There are three major myosin light chain kinase (MYLK) isoforms: a long-nonmuscle isoform, a short-smooth muscle isoform, and a very short isoform (telokin) that lacks kinase activity (74). In smooth muscle, MYLK phosphorylates rMLC (MYL9) to activate actin-myosin interaction and induce contraction. Consistent with a critical role for MYLK in smooth muscle contraction, three individuals with MMIHS from two families have MYLK mutations including a seven base pair duplication and a putative splice variant in exon 23. These mutations create frameshifts and premature stop codons (35). The individual with a putative splice variant in MYLK exon 23 presented with malrotation at two days of age (35). Siblings of the individual with a seven-base duplication in MYLK exon 23 also had adverse outcomes. One pregnancy was terminated in utero at 15-wk gestation with MMIHS confirmed on autopsy. Another sibling with bladder distension and anhydramnios was born prematurely at 31 wk and died shortly after birth from respiratory distress. A third sibling died in utero at 30 wk and was found to have a distended bladder (35).
Similar to human disease phenotypes, Mylk conditional knockout mice with deletion of exons 23–25 using a smooth muscle-22α (TAGLN) Cre-ERT2 have MMIHS-like bowel and bladder dysfunction (58). Ileal strips from these mice exhibited only 13% of the tension in the control ileum and lacked the initial contraction peak expected with KCl stimulation. Furthermore, conditional Mylk mutant ileum had only 20%–28% of the force of controls in response to acetylcholine. Consistent with these findings, Mylk deletion prevented rMLC phosphorylation in smooth muscle. Additional phenotypes included lower blood pressure in Mylk mutant mice, but this was not reported in the three humans with MYLK mutations. These data would suggest that Mylk mutations cause visceral myopathy through diminished activation of smooth muscle myosin.
Leiomodin 1
Leiomodin (LMOD) is an actin binding protein that nucleates new actin filaments in cells (75). There are three LMOD isoforms: Leiomodin 1 (LMOD1) (smooth muscle), LMOD2 (cardiac muscle), and LMOD3 (skeletal muscle). The only disease-causing LMOD1 mutation reported is an autosomal recessive homozygous mutation leading to a premature termination codon in exon 2 (59). This individual displayed classic MMIHS symptoms. Immunohistochemical analysis of LMOD1 in normal human embryonic tissue showed robust staining in bladder and intestine visceral and vascular SMCs at different stages of embryonic development. In contrast, LMOD1 mRNA and protein were dramatically reduced in patient dermal fibroblasts, indicating a loss-of-function mechanism for LMOD1-mediated MMIHS.
Lmod1 knockout mice with a premature termination codon in exon 1 have visceral myopathy with bowel and bladder dysfunction, but lack microcolon (59). Immunohistochemistry confirmed LMOD1 was absent from smooth muscle layers in the bladder, stomach, and intestine. Bladder and stomach muscle was thin in Lmod1−/− mice, but the small intestine appeared morphologically normal. Nonetheless, intestinal rings had decreased passive tensile strength and reduced agonist-induced contractility. Consistent with the role of LMOD1 in nucleating actin filaments, Lmod1 knockout mice had less F-actin in the intestine and bladder by Western blot and phalloidin staining. Surprisingly, individual actin filaments appeared normal at the ultrastructural level, but there were half as many dense bodies in the bladder smooth muscle. Curiously, aorta and esophagus appeared normal in Lmod1−/− mice, which is unexpected because LMOD1 is expressed in vascular and visceral smooth muscle. Consistent with an important role in SMC function, primary human intestinal smooth muscle cells treated with siRNA to knockdown LMOD1 had a 40% reduction in contractile activity (59). These observations provide a reasonable explanation for how LMOD1 mutations might cause visceral myopathy.
Filamin A
FLNA encodes Filamin A (FLNA), an F-actin binding protein that crosslinks actin networks into three-dimensional webs, connecting to cell membranes and governing cell shape and motility (76). FLNA is expressed in the central nervous system, smooth muscle, and many other tissues. The gene is located on the X chromosome and FLNA mutations cause X-linked CIPO. Intestinal dysfunction associated with FLNA mutations has been attributed to central nervous system defects (61). However, a study published by Kapur et al. (37) reported diffuse abnormal layering (DAL) of small intestinal smooth muscle in five patients with CIPO who had FLNA mutations. In DAL, smooth muscle layers are haphazardly organized in multiple layers instead of the two perpendicular layers of circular and longitudinal muscle normally found in the intestine. One individual with an FLNA mutation also had multinucleated SMCs near the submucosal side of the muscularis propria. Consistent with the hypothesis that FLNA mutations cause CIPO, people with FLNA mutations had little FLNA immunoreactivity in small intestinal smooth muscle, whereas control bowels had robust FLNA protein staining. In contrast to smooth muscle, the enteric nervous system was not FLNA immunoreactive in either controls or affected patients suggesting FLNA X-linked CIPO has a myopathic basis.
In addition to bowel dysmotility, females with protein-truncating FLNA mutations typically present with periventricular nodular heterotopia (PVNH), whereas most males die in infancy. However, some males with FLNA mutations retain residual FLNA expression and survive with manifestations including cardiovascular malformations, CIPO, and central nervous system (CNS) defects. Recently, FLNA was found to have two different isoforms that differ by 28 residues because of an alternative translational start site (60). The longer isoform predominates in intestinal smooth muscle and mutations that affect solely this isoform are believed to cause CIPO without CNS defects. Mutations that affect both isoforms are believed to cause problems in heart, intestine, and brain, providing the first mechanistic explanation for different FLNA mutation phenotypes.
OTHER ANIMAL MODELS OF VISCERAL MYOPATHY
Srf Knockout Mouse
Serum response factor (SRF) is a critical regulator of SMC contractile gene expression that binds to a CArG box (CC[A/T]6GG), a 10-bp DNA sequence located upstream of all SMC contractile genes. Conditional Srf knockout (KO) mice crossed to a smooth muscle-specific conditional CRE displayed symptoms of severe dysmotility consistent with CIPO by 13 days after tamoxifen-induction (77). These mice had dramatic reductions in expression of ACTA2, MYH11, and smoothelin (SMTN), as well as a reduction in ratio of filamentous to monomeric actin. Conditional SMC-restricted Srf KO in adult mice resulted in SMC myopathy and cell death due to loss of critical microRNAs that suppress apoptosis (78). Conditional Srf KO mice also had less SRF-dependent myotonic dystrophy protein kinase (DMPK) and subsequent reduction in L-type calcium channel (CACNA1C) expression (79). CACNA1C downregulation reduced intracellular calcium spikes in SMCs, disrupted cell-to-cell coupling, and reduced contractility. Germline Srf KO mice died in utero with severe GI and cardiac development abnormalities (80). The cardiac defects may be secondary to impaired vascularization of the developing heart. Collectively, these observations suggest that hypomorphic SRF mutations might cause visceral myopathy.
Myocardin Knockout Mouse
Myocardin is a transcriptional coactivator that binds to SRF and is necessary for expression of SMC contractile genes. In mice, myocardin-deficient SMCs also produce more extracellular matrix proteins and activate endoplasmic reticulum (ER) stress and autophagy pathways, consistent with transition to a synthetic SMC phenotype. Mutant SMCs undergo apoptosis leading to vascular and visceral disease (81). Human loss-of-function myocardin variants have recently been identified (82). Monoallelic variants were associated with male-restricted congenital megabladder, whereas biallelic variants caused megabladder and cardiovascular defects in both sexes. The mechanism for monoallelic variants causing megabladder only in males is unclear. One affected male was diagnosed with megabladder and ventricular septal defect prenatally. Hematoxylin and eosin stained sections showed his colon had circular smooth muscle but no longitudinal smooth muscle, while the small intestine appeared normal with two smooth muscle layers. The human megabladder phenotype was recapitulated in mice that have one null Myocardin (Myocd) allele and one Myocd allele with mutations in the LZ homodimerization domain (82). It is unclear why the aorta and heart of these mice appeared relatively normal. In contrast, when Myh11-Cre-ERT2 was used to knock out Myocd in the smooth muscle of adult mice, the mice died within six months with dramatic disruption of SMC architecture of the great arteries, bowel, and bladder (81). Problems included arterial aneurysms and dissections prone to rupture, consistent with the phenotypes seen with TAADs. These mutant mice also had bladder and bowel dilation, similar to MMIHS. Constitutive loss of Myocd is embryonic lethal, causing an underdeveloped dorsal aorta and complete absence of ACTA2-positive cells in the vasculature (83). Surprisingly, the heart develops normally in Myocd−/− mice, and embryonic lethality is due to a failure of SMC differentiation in the vasculature.
NEW HYPOTHESES TO UNDERSTAND VISCERAL MYOPATHY WITH IMPLICATIONS FOR THERAPEUTICS
ACTG2 Mutations and the Myocardin Family of Transcription Factors
The Myocardin family of transcription factors, which include Myocardin (MYOCD) and Myocardin-related transcription factors (MRTFs), are critical for SMC differentiation and maintenance of the contractile phenotype (81). MRTFs also bind G-actin and can be sequestered in the cytoplasm by actin monomers. When actin polymerizes, the cytoplasmic G-actin pool is reduced, releasing MRTFs that then translocate to the nucleus and bind SRF to induce SMC differentiation and contractile gene expression. Actin variants that do not polymerize could therefore reduce SMC contractile gene expression by sequestering MRTFs in the cytoplasm (84).
Mechanical properties of the cell’s environment also dramatically influence SMC differentiation. For example, SMCs become less contractile when cultured on hard surfaces or when tissues stiffen because the mechanosensing Yes-associated protein (YAP) enters the nucleus and binds MYOCD, the master regulator of SMC differentiation. The MYOCD-YAP interaction keeps MYOCD from binding SRF, which is needed for SMC contractile gene transcription (85). Inhibition of SRF interaction with MYOCD and MRTFs enhances SRF interaction with ternary complex cofactors (TCFs), including ELK-1, to activate mitogen-induced gene expression. These SRF-TCF interactions increase production of ECM proteins that may further stiffen tissues (86).
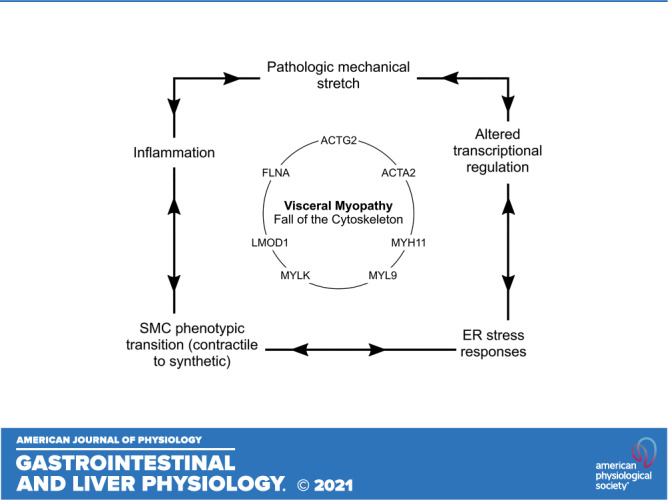
Thus, ACTG2 mutations that cause visceral myopathy might impair SMC function in several ways. First, mutations could directly affect F-actin levels or interactions in the contractile apparatus, reducing the muscle’s ability to generate force or resist passive stretch. Second, if a mutation increases cytoplasmic G-actin, the G-actin could sequester MRTFs in the cytoplasm, keeping MRTFs from activating SMC contractile gene expression. Either mechanism would make SMCs less able to resist forces that distend hollow organs. As organs stretch, increased tension would induce YAP translocation to the nucleus where YAP binds MYOCD and prevents MYOCD from transactivating SMC contractile genes in concert with SRF. The unbound SRF interacts with TCFs to increase ECM production. Collectively, these mechanisms lead to dilated bowel and bladder, muscle cell de-differentiation, and increased ECM production. The ensuing fibrosis and bowel wall stiffening further impair contraction of hollow organs leading to more permanent distension. Mutations in other genes that reduce SMC strength or impair actin polymerization could similarly induce SMCs to transition from a contractile to a synthetic phenotype, through dysregulation of the Myocardin family-SRF signaling axis (Fig. 1). Table 2 summarizes new hypotheses about how mutations cause visceral myopathy.
Figure 1.

Effects of actin gamma 2 (ACTG2) mutations and inflammation on transcriptional regulation in visceral myopathy. We propose that ACTG2 variants polymerize less efficiently than wild-type (WT) ACTG2, resulting in increased monomeric globular actin (G-actin). The increased G-actin may sequester myocardin-related transcription factors (MRTFs) in the cytoplasm, downregulating smooth muscle cell (SMC) contractile gene expression. With less MRTF in the nucleus, serum response factor (SRF) is free to bind with ELK-1, leading to increased expression of extracellular matrix (ECM) and promitogenic genes. Increased ECM deposition leads to a stiffer SMC microenvironment. This increase in stiffness along with pathologic levels of stretch due to abnormal bowel or bladder distension may increase tension on focal adhesions and release Yes-associated protein (YAP), allowing YAP to translocate to the nucleus. Nuclear YAP inhibits the interaction of myocardin with SRF, further downregulating the SMC contractile phenotype. Finally, NF-κB activation downstream of proinflammatory cytokines may further inhibit SMC contractile gene expression by NF-κB-myocardin binding. F-actin, filamentous actin.
Inflammation and Visceral Myopathy
The role of inflammation in visceral myopathy is not understood, but could contribute to “waxing and waning” symptoms experienced in people with visceral myopathy and the tremendous variability in symptom severity in people with the same underlying ACTG2 mutation (1, 11, 26). We have observed that minor illnesses like upper respiratory infections can lead to pronounced deterioration in bowel function and that improvement to prior baseline can take weeks to months (Clinical observations—Dr. Robert O. Heuckeroth, Children’s Hospital of Philadelphia). Moreno et al. (21) also reported acute exacerbation of CIPO symptoms during an episode of varicella zoster in an individual with an ACTG2 T195I mutation. Consistent with these observations, people with CIPO often have prolonged postoperative ileus (1).
One mechanistic hypothesis is that infections and postoperative ileus increase proinflammatory cytokines like IL-1β that activate NF-κB (87). Activated NF-κB can bind Myocardin and prevent SRF-dependent expression of SMC contractile genes (Fig. 1). Free SRF binds ELK-1 to activate the synthesis of ECM proteins. Reduced SMC contractile force and resistance to passive stretch lead to bowel and bladder distension. Increased organ wall tension releases YAP which binds MYOCD, further reducing SMC contractile phenotype gene expression and increasing ECM synthesis. Recovery may be slow if SMCs need to transition back from a synthetic to a contractile phenotype. If this mechanism is correct, bowel decompression could enhance contractile state SMC differentiation by reducing nuclear YAP sequestration of MYOCD.
Signaling Effects of Mechanical Stretch and Stress Responses in Smooth Muscle
Contractility in smooth muscle is less well-understood than contractility in striated muscle. In the absence of rigidly aligned sarcomeres, SMCs can generate active tension over a great range of lengths. To do this, myosin head groups interact with networks of actin filaments to generate force. However, in visceral myopathies due to cytoskeletal protein mutations (ACTG2, ACTA2, MYH11, MYL9, MYLK, LMOD1, FLNA) actin filaments may be shortened, myosin filaments may be disrupted, or actin-myosin interactions may be disturbed. Cytoskeletal mutations may also disrupt the interactions of SMCs with the ECM that are necessary for development of passive tension, force transmission, and force sensing. As distension increases, there may be less overlap between actin and myosin filaments, further reducing the ability of SMCs to contract (Fig. 2, A and B).
Figure 2.

Effects of mechanical stretch and endoplasmic reticulum (ER) stress responses. A: pathological stretch induces a phenotypic transition from contractile to synthetic (proliferative) phenotype in smooth muscle cells (SMCs). B: mutations that impair the actin cytoskeleton reduce force generation and inhibit development of passive tension and force transmission through the extracellular matrix (ECM). Mechanical signals are transmitted from the cell surface, through the cytoskeleton and contractile apparatus via the Linker of Nucleoskeleton and Cytoskeleton (LINC) complex to the genome, thereby affecting gene expression. If this force transmission is impaired by mutations affecting cytoskeletal filaments, physiological SMC gene expression in response to extracellular forces may be disrupted. C: endoplasmic reticulum (ER) is greatly expanded in the SMC synthetic phenotype to increase production of extracellular matrix (ECM) proteins and matrix metalloproteases (MMPs). If the cell cannot meet the increased protein production demand, an unfolded protein response (UPR) may be triggered as part of the ER stress response. This figure uses icons derived from the Reactome Icon Library by CSHL, OICR and EBI. F-actin, filamentous actin.
Smooth muscle cells (SMCs) are acutely mechanosensitive, detecting changes in pressure, stretch, and ECM stiffness to modulate activity of hollow organs. There are several unanswered questions about the effects of visceral myopathy-causing mutations on SMC mechanosensitivity: 1) Is the linkage between ECM and visceral SMCs disrupted by certain mutations, as is the case in thoracic aortic aneurysm and dissection (TAAD)-causing mutations that affect vascular SMCs (88)? 2) Does the increase in intraluminal pressure associated with pseudo-obstruction result in activation of stretch pathways with upregulation of ECM proteins and matrix metalloproteinases (MMPs), similar to mechanisms proposed for vascular SMC dysfunction in TAADs (68)? If so, this may explain the bowel fibrosis seen in some individuals with CIPO/MMIHS (89, 90).
Considering the clinical and basic science findings for gene mutations associated with visceral myopathies and the data available for vascular myopathies, we propose the following model for the signaling effects of mechanical stretch and stress responses in visceral myopathies (Figs. 1 and 2): 1) Impaired tonic and/or phasic contractions of visceral SMCs prevents the bowel from maintaining its dimensions and from moving food effectively, resulting in distension with pathological pressure on the bowel wall. 2) This pressure is detected by SMCs that respond by secretion of ECM proteins and MMPs to remodel the bowel wall and make it more resistant to the pathological stretch. 3) SMC contractile apparatus and cytoskeleton interact with the Linker of Nucleoskeleton and Cytoskeleton (LINC) complex to directly regulate gene expression by transmission of mechanical signals to the nucleus (91). Impaired actin filament formation disrupts interactions between the cytoskeletal/contractile apparatus and LINC complex, preventing visceral SMCs from upregulating contractile protein expression that would normally increase force generation to counteract the raised intraluminal pressure. 4) Inability to efficiently generate force and increasing stiffness of the bowel wall due to ECM deposition is a self-perpetuating cycle that makes visceral SMCs increasingly ineffective as bowel distension worsens.
Lastly, as noted in Inflammation and visceral myopathy, pathological mechanical stress, like that induced by bowel obstruction or marked distension and inflammation, causes SMCs to change from a contractile to a synthetic (proliferative) phenotype (92). In the synthetic state, SMCs increase ECM protein synthesis and significantly expand endoplasmic reticulum (ER). When ER capacity to fold ECM proteins is exceeded, the unfolded protein response (UPR) may be induced, and UPR activation further reduces contractile gene expression via inositol-requiring enzyme 1α (IRE1α)/X-box binding protein 1 (XBP1) (93). Interestingly, there is a negative feedback loop that reduces UPR in the SMC contractile state. MYOCD and MRTF-A downregulate the UPR (93) and Myocd deletion induces ER stress and increases the UPR (81). Consistent with the hypothesis that the UPR is activated in MMIHS, vacuolar degeneration, a possible manifestation of ER stress, has been reported in visceral SMCs of people with MMIHS (89, 90). SMC stressors also upregulate repair pathways, increasing production of trophic factors, including IGF-1 (68). These trophic factors further enhance SMC proliferation and increase ECM production, resulting in pathological thickening of the bowel wall. If these hypotheses are true, agents that inhibit the UPR, such as the IRE1α inhibitor 4μ8C (93), may enhance the SMC contractile state and counteract some of the effects of cytoskeletal mutations that cause visceral myopathies.
The Physics of Visceral Myopathy
Massive bowel or bladder distension is common in people with visceral myopathy. Unfortunately, as hollow organs distend, more circumferential force is required at any given tension to reduce organ cross-sectional area. This is due to a physical principle known as LaPlace’s law. The same principle is thought to be important in aneurysms, uterine contractions during labor, and for peripheral edema (94). LaPlace’s law, which applies to thin walled vessels, describes the relationship between transmural pressure (P), mean radius (R), wall thickness (t), and wall tension (T) as follows:
Thus, when bowel or bladder are distended, the increase in radius (R) means that visceral SMCs must generate more tension (T) to maintain the same transmural pressure (P) (Fig. 3, Table 3). Furthermore, as hollow organs stretch, the wall thickness (t) decreases, further increasing the tension (T) needed to resist any particular pressure. This is a problem in visceral myopathy since SMC strength is already reduced, making the hollow organ more susceptible to distension. From a therapeutic standpoint, avoiding gas-producing food [fermentable oligosaccharides, disaccharides, monosaccharides, and polyols (FODMAPs)] or medicines (e.g., lactulose) might therefore be beneficial in people with visceral myopathy. Ostomies to release gas or fluid without the need to overcome sphincter skeletal muscle can also significantly reduce bowel or bladder distension, allowing weak muscles to more efficiently resist intraluminal pressure.
Figure 3.

The physics of visceral myopathy. Megacystis (massive bladder dilation) and pathological bowel dilation (in chronic intestinal pseudo-obstruction, CIPO) can be modeled using Laplace’s law.
TARGETING THERAPEUTICS FOR VISCERAL MYOPATHIES
Incomplete understanding of underlying disease mechanisms makes it difficult to design targeted therapy for visceral myopathies. In the absence of mechanistic therapy, current treatments attempt to relieve symptoms with prokinetic medicines, continuous feeding, ostomy or bladder catheterization, intravenous nutrition, and in some cases, multi-visceral organ transplantation. Limited mechanistic data suggest a few strategies worth investigating (Fig. 4). One observation is that bowel from people with CIPO/MMIHS can have reduced expression of SMC contractile proteins and increased fibrosis (89), as might be expected based on the mechanisms described in New Hypotheses To Understand Visceral Myopathy With Implications For Therapeutics. One way to address this issue might be medicines like Rapamycin or Lovastatin that increase contractile gene expression in dedifferentiated vascular SMCs (95, 96). Anti-fibrotics should also be tested in model systems to determine whether bowel and bladder fibrosis associated with visceral myopathies can be alleviated.
Figure 4.

Hypothetical therapeutic approaches for visceral myopathies. Rapamycin and Lovastatin may increase the expression of smooth muscle cell (SMC) contractile genes, enhancing SMC force generation. Antifibrotics may make the extracellular matrix (ECM) more pliable and reduce the stiffness of the SMC microenvironment. Gene targeting approaches can be considered to silence mutations or correct them, where feasible. This figure uses icons derived from the IGI Glossary Icon Collection by Christine Liu of Two Photon Art for the Innovative Genomics Institute, and the Reactome Icon Library by CSHL, OICR and EBI.
Finally, we should consider genetic strategies to enhance SMC function in people with visceral myopathy. In Duchenne’s muscular dystrophy, a life-threatening skeletal muscle disease caused by dystrophin mutations, several gene-based therapies have been tested. The drug Golodirsen is FDA approved for people with dystrophin mutations amenable to exon 53 skipping. This medicine is a phosphorodiamidate-modified antisense oligonucleotide of the morpholino oligomer (PMO) subclass that binds exon 53 pre-mRNA, excluding this exon from the final mRNA, which leads to production of a truncated dystrophin protein (97). Adeno-associated virus (AAV)-based vectors have also been used to deliver the dystrophin-related protein utrophin to skeletal muscle. Utrophin retains most structural and binding properties of dystrophin, and AAV-utrophin therapy rescued the muscle damage in small- and large animal models (98). For Duchenne’s muscular dystrophy caused by lack of exon 52, CRISPR-based therapy to remove exon 51 led to expression of a shortened but functional dystrophin in a pig model as well as in human induced pluripotent stem cells (hiPSC)-derived myotubes and cardiomyocytes, with partial recovery of function and increased survival (99). For visceral myopathy, similar strategies should be considered. For small insertions or deletions, CRISPR-based therapies could be used for gene correction. For heterozygous missense mutations in ACTG2 or ACTA2 that are dominant-negative, the mutant allele could be silenced using antisense oligonucleotides. To test these therapeutic strategies, we need better cell-based and animal models of visceral myopathy.
NEW MODELS TO UNDERSTAND THE PATHOPHYSIOLOGY OF VISCERAL MYOPATHIES
Many genetic changes in smooth muscle cytoskeletal proteins cause visceral myopathy. To find new treatments and cures, we need to define mechanistically how specific mutations disrupt each of these proteins and how disease-causing mutations alter cell biology, organ function, and development. Biochemical studies with purified proteins should define intra- and intermolecular interactions disrupted by mutations. In vitro cell biology methods should visualize actin cytoskeletal organization and dynamics, ideally in visceral smooth muscle-like cells. These methods can then be used to identify small molecules that impact force generation, calcium signaling, and cytoskeleton-mediated cellular functions like migration, spreading, contractility, signal transduction, apoptosis, and cell shape regulation. We need new animal models to study whole organ and whole animal physiology and to determine how mutations influence development. Animal models would also allow us to examine how inflammation, distension, and other insults affect visceral muscle function in vivo when mutations are present, and to conduct meaningful rescue experiments. Finally, we need to study visceral myopathy-inducing mutations in human cells, which might differ from non-human models. For this purpose, human induced pluripotent stem cells (hiPSCs) from people with visceral myopathy or gene edited hiPSCs could be invaluable if we had robust directed differentiation protocols to convert hiPSCs into visceral SMCs. Using patient-derived cells will allow us to identify genetic and non-genetic modifiers of disease and to conduct rescue experiments to find the best ways to improve visceral SMC function.
GRANTS
The authors were supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) of the National Institutes of Health (NIH) under Award Number F30DK118827 (to S. K. Hashmi), the Irma and Norman Braman Endowed Chair for Research in GI Motility Disorders (to R. O. Heuckeroth), the Suzi and Scott Lustgarten Center Endowment (to R. O. Heuckeroth), and The Children’s Hospital of Philadelphia Research Institute (to R. O. Heuckeroth).
DISCLAIMERS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
DISCLOSURES
R. O. Heuckeroth is a consultant for BlueRock Therapeutics and served on a Scientific Advisory Panel for Takeda. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
S.K.H., R.H.C., and R.O.H. conceived and designed research; S.K.H. prepared figures; S.K.H. and R.H.C. drafted manuscript; S.K.H., R.H.C., and R.O.H. edited and revised manuscript; S.K.H., R.H.C., and R.O.H. approved final version of manuscript.
REFERENCES
- 1.Thapar N, Saliakellis E, Benninga MA, Borrelli O, Curry J, Faure C, De GR, Gupte G, Knowles CH, Staiano A, Vandenplas Y, Di Lorenzo C. . Paediatric intestinal pseudo-obstruction: evidence and consensus-based recommendations from an ESPGHAN-Led Expert Group. J Pediatr Gastroenterol Nutr 66: 991–1019, 2018. doi: 10.1097/mpg.0000000000001982. [DOI] [PubMed] [Google Scholar]
- 2.Klar J, Raykova D, Gustafson E, Tóthová I, Ameur A, Wanders A, Dahl N. Phenotypic expansion of visceral myopathy associated with ACTG2 tandem base substitution. Eur J Hum Genet 23: 1679–1683, 2015. doi: 10.1038/ejhg.2015.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sipponen T, Karikoski R, Nuutinen H, Markkola A, Kaitila I. Three-generation familial visceral myopathy with α-actin positive inclusion bodies in intestinal smooth muscle. J Clin Gastroenterol 43: 437–443, 2009. doi: 10.1097/MCG.0b013e31817d3f84. [DOI] [PubMed] [Google Scholar]
- 4.Vargas JH, Sachs P, Ament ME. Chronic intestinal pseudo-obstruction syndrome in pediatrics. Results of a national survey by members of the North American Society of Pediatric Gastroenterology and Nutrition. J Pediatr Gastroenterol Nutr 7: 323–332, 1988. doi: 10.1097/00005176-198805000-00003. [DOI] [PubMed] [Google Scholar]
- 5.Iida H, Ohkubo H, Inamori M, Nakajima A, Sato H. Epidemiology and clinical experience of chronic intestinal pseudo-obstruction in Japan: a nationwide epidemiologic survey. J Epidemiol 23: 288–294, 2013. doi: 10.2188/jea.je20120173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muto M, Matsufuji H, Tomomasa T, Nakajima A, Kawahara H, Ida S, Ushijima K, Kubota A, Mushiake S, Taguchi T. Pediatric chronic intestinal pseudo-obstruction is a rare, serious, and intractable disease: a report of a nationwide survey in Japan. J Pediatr Surg 49: 1799–1803, 2014. doi: 10.1016/j.jpedsurg.2014.09.025. [DOI] [PubMed] [Google Scholar]
- 7.Grant D, Abu-Elmagd K, Mazariegos G, Vianna R, Langnas A, Mangus R, Farmer DG, Lacaille F, Iyer K, Fishbein T; Intestinal Transplant Association. Intestinal transplant registry report: global activity and trends. Am J Transplant 15: 210–219, 2015. doi: 10.1111/ajt.12979. [DOI] [PubMed] [Google Scholar]
- 8.Batra S, Rahman S, Rana MS, Matta S, Darbari A. Epidemiology and healthcare utilization of inpatient admissions in children with pediatric intestinal pseudo-obstruction. Neurogastroenterol Motil 32: e13781, 2020. doi: 10.1111/nmo.13781. [DOI] [PubMed] [Google Scholar]
- 9.Downes TJ, Cheruvu MS, Karunaratne TB, De Giorgio R, Farmer AD. Pathophysiology, diagnosis, and management of chronic intestinal pseudo-obstruction. J Clin Gastroenterol 52: 477–489, 2018. doi: 10.1097/MCG.0000000000001047. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalez Z, McCallum R. Small bowel dysmotility, pseudoobstruction, and functional correlation with histopathology: lessons learned. Curr Gastroenterol Rep 22: 14, 2020. doi: 10.1007/s11894-020-0748-8. [DOI] [PubMed] [Google Scholar]
- 11.Assia Batzir N, Kishor Bhagwat P, Larson A, Coban Akdemir Z, Bagłaj M, Bofferding L, et al. ; Baylor-Hopkins Center for Mendelian Genomics. Recurrent arginine substitutions in the ACTG2 gene are the primary driver of disease burden and severity in visceral myopathy. Hum Mutat 41: 641–654, 2020. doi: 10.1002/humu.23960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Billon C, Molin A, Poirsier C, Clemenson A, Dauge C, Grelet M, Sigaudy S, Patrier S, Goldenberg A, Layet V, Tantau J, Fleury C, Livard A, Diguet A, Fritih R, Verspyck E, Rendu J, Boutaud L, Tessier A, Thomas S, Razavi F, Achaiaa A, Elkhartoufi N, Hakkakian L, Magnin E, Bole-Feysot C, Masson C, Ville Y, Roth P, Prieur F, Bessieres B, Bonniere M, Attie-Bitach T. Fetal megacystis-microcolon: genetic mutational spectrum and identification of PDCL3 as a novel candidate gene. Clin Genet 98: 261–273, 2020. doi: 10.1111/cge.13801. [DOI] [PubMed] [Google Scholar]
- 13.Collins RRJ, Barth B, Megison S, Pfeifer CM, Rice LM, Harris S, Timmons CF, Rakheja D. ACTG2-associated visceral myopathy with chronic intestinal pseudoobstruction, intestinal malrotation, hypertrophic pyloric stenosis, choledochal cyst, and a novel missense mutation. Int J Surg Pathol 27: 77–83, 2019. doi: 10.1177/1066896918786586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farwell KD, Shahmirzadi L, El-Khechen D, Powis Z, Chao EC, Tippin Davis B, Baxter RM, Zeng W, Mroske C, Parra MC, Gandomi SK, Lu I, Li X, Lu H, Lu H-M, Salvador D, Ruble D, Lao M, Fischbach S, Wen J, Lee S, Elliott A, Dunlop CLM, Tang S. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med 17: 578–586, 2015. doi: 10.1038/gim.2014.154. [DOI] [PubMed] [Google Scholar]
- 15.Halim D, Hofstra RMW, Signorile L, Verdijk RM, van der Werf CS, Sribudiani Y, Brouwer RWW, van IJcken WFJ, Dahl N, Verheij JBGM, Baumann C, Kerner J, van Bever Y, Galjart N, Wijnen RMH, Tibboel D, Burns AJ, Muller F, Brooks AS, Alves MM. ACTG2 variants impair actin polymerization in sporadic megacystis microcolon intestinal hypoperistalsis syndrome. Hum Mol Genet 25: 571–583, 2016. doi: 10.1093/hmg/ddv497. [DOI] [PubMed] [Google Scholar]
- 16.Korğalı EÜ, Yavuz A, Şimşek CEÇ, Güney C, Kurtulgan HK, Başer B, Atalar MH, Özer H, Eğilmez HR. Megacystis microcolon intestinal hypoperistalsis syndrome in which a different de novo Actg2 gene mutation was detected: a case report. Fetal Pediatr Pathol 37: 109–116, 2018. doi: 10.1080/15513815.2018.1445149. [DOI] [PubMed] [Google Scholar]
- 17.Lee H, Park S, Oh J-T, Kim HM, Kim S, Lee J-S. Oral pyridostigmine-responsive visceral myopathy with ACTG2 mutations: a case series. J Pediatr Gastroenterol Nutr 68: e16–e17, 2019. doi: 10.1097/mpg.0000000000002183. [DOI] [PubMed] [Google Scholar]
- 18.Milunsky A, Baldwin C, Zhang X, Primack D, Curnow A, Milunsky J. Diagnosis of chronic intestinal pseudo-obstruction & megacystis by sequencing the ACTG2 gene. J Pediatr Gastroenterol Nutr 65: 384–387, 2017. doi: 10.1097/MPG.0000000000001608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Milunsky A, Lazier J, Baldwin C, Young C, Primack D, Milunsky JM. Prenatal diagnosis of chronic intestinal pseudo-obstruction and paternal somatic mosaicism for the ACTG2 pathogenic variant. Prenat Diagn 37: 1254–1256, 2017. doi: 10.1002/pd.5171. [DOI] [PubMed] [Google Scholar]
- 20.Monies D, Maddirevula S, Kurdi W, Alanazy MH, Alkhalidi H, Al-Owain M, Sulaiman RA, Faqeih E, Goljan E, Ibrahim N, Abdulwahab F, Hashem M, Abouelhoda M, Shaheen R, Arold ST, Alkuraya FS. Autozygosity reveals recessive mutations and novel mechanisms in dominant genes: implications in variant interpretation. Genet Med 19: 1144–1150, 2017. doi: 10.1038/gim.2017.22. [DOI] [PubMed] [Google Scholar]
- 21.Moreno CA, Metze K, Lomazi EA, Bertola DR, Barbosa RHA, Cosentino V, Sobreira N, Cavalcanti DP. Visceral myopathy: clinical and molecular survey of a cohort of seven new patients and state of the art of overlapping phenotypes. Am J Med Genet A 170: 2965–2974, 2016. doi: 10.1002/ajmg.a.37857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ravenscroft G, Pannell S, O'Grady G, Ong R, Ee HC, Faiz F, Marns L, Goel H, Kumarasinghe P, Sollis E, Sivadorai P, Wilson M, Magoffin A, Nightingale S, Freckmann M-L, Kirk EP, Sachdev R, Lemberg DA, Delatycki MB, Kamm MA, Basnayake C, Lamont PJ, Amor DJ, Jones K, Schilperoort J, Davis MR, Laing NG. Variants in ACTG2 underlie a substantial number of Australasian patients with primary chronic intestinal pseudo-obstruction. Neurogastroenterol Motil 30: e13371, 2018. doi: 10.1111/nmo.13371. [DOI] [PubMed] [Google Scholar]
- 23.Stark Z, Tan TY, Chong B, Brett GR, Yap P, Walsh M, Yeung A, Peters H, Mordaunt D, Cowie S, Amor DJ, Savarirayan R, McGillivray G, Downie L, Ekert PG, Theda C, James PA, Yaplito-Lee J, Ryan MM, Leventer RJ, Creed E, Macciocca I, Bell KM, Oshlack A, Sadedin S, Georgeson P, Anderson C, Thorne N, Gaff C, White SM; Melbourne Genomics Health Alliance. A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet Med 18: 1090–1096, 2016. doi: 10.1038/gim.2016.1. [DOI] [PubMed] [Google Scholar]
- 24.Thorson W, Diaz-Horta O, Foster J II, Spiliopoulos M, Quintero R, Farooq A, Blanton S, Tekin M. De novo ACTG2 mutations cause congenital distended bladder, microcolon, and intestinal hypoperistalsis. Hum Genet 133: 737–742, 2014. doi: 10.1007/s00439-013-1406-0. [DOI] [PubMed] [Google Scholar]
- 25.Tuzovic L, Tang S, Miller RS, Rohena L, Shahmirzadi L, Gonzalez K, Li X, LeDuc CA, Guo J, Wilson A, Mills A, Glassberg K, Rotterdam H, Sepulveda AR, Zeng W, Chung WK, Anyane-Yeboa K. New insights into the genetics of fetal megacystis: ACTG2 mutations, encoding γ-2 smooth muscle actin in megacystis microcolon intestinal hypoperistalsis syndrome (Berdon syndrome). Fetal Diagn Ther 38: 296–306, 2015. doi: 10.1159/000381638. [DOI] [PubMed] [Google Scholar]
- 26.Wangler MF, Beaudet AL. ACTG2-related disorders. In: GeneReviews, edited by Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford HC, Smith RJH, Stephens K.. Seattle, WA: University of Washington, Seattle, 2015. [Google Scholar]
- 27.Wangler MF, Gonzaga-Jauregui C, Gambin T, Penney S, Moss T, Chopra A, Probst FJ, Xia F, Yang Y, Werlin S, Eglite I, Kornejeva L, Bacino CA, Baldridge D, Neul J, Lehman EL, Larson A, Beuten J, Muzny DM, Jhangiani S, Gibbs RA, Lupski JR, Beaudet A; Baylor-Hopkins Center for Mendelian Genomics. Heterozygous de novo and inherited mutations in the smooth muscle actin (ACTG2) gene underlie megacystis-microcolon-intestinal hypoperistalsis syndrome. PLoS Genet 10: e1004258, 2014. doi: 10.1371/journal.pgen.1004258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu H, Fagnant PM, Krementsova EB, Trybus KM. Severe molecular defects exhibited by the R179H mutation in human vascular smooth muscle α-actin. J Biol Chem 291: 21729–21739, 2016. doi: 10.1074/jbc.M116.744011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dong W, Baldwin C, Choi J, Milunsky JM, Zhang J, Bilguvar K, Lifton RP, Milunsky A. Identification of a dominant MYH11 causal variant in chronic intestinal pseudo-obstruction: results of whole-exome sequencing. Clin Genet 96: 473–477, 2019. doi: 10.1111/cge.13617. [DOI] [PubMed] [Google Scholar]
- 30.Gauthier J, Ouled Amar Bencheikh B, Hamdan FF, Harrison SM, Baker LA, Couture F, Thiffault I, Ouazzani R, Samuels ME, Mitchell GA, Rouleau GA, Michaud JL, Soucy J-F. A homozygous loss-of-function variant in MYH11 in a case with megacystis-microcolon-intestinal hypoperistalsis syndrome. Eur J Hum Genet 23: 1266–1268, 2015. doi: 10.1038/ejhg.2014.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gilbert MA, Schultz-Rogers L, Rajagopalan R, Grochowski CM, Wilkins BJ, Biswas S, Conlin LK, Fiorino KN, Dhamija R, Pack MA, Klee EW, Piccoli DA, Spinner NB. Protein-elongating mutations in MYH11 are implicated in a dominantly inherited smooth muscle dysmotility syndrome with severe esophageal, gastric, and intestinal disease. Hum Mutat 41: 973–982, 2020. doi: 10.1002/humu.23986. [DOI] [PubMed] [Google Scholar]
- 32.Kloth K, Renner S, Burmester G, Steinemann D, Pabst B, Lorenz B, Simon R, Kolbe V, Hempel M, Rosenberger G. 16p13.11 microdeletion uncovers loss-of-function of a MYH11 missense variant in a patient with megacystis-microcolon-intestinal-hypoperistalsis syndrome. Clin Genet 96: 85–90, 2019. doi: 10.1111/cge.13557. [DOI] [PubMed] [Google Scholar]
- 33.Wang Q, Zhang J, Wang H, Feng Q, Luo F, Xie J. Compound heterozygous variants in MYH11 underlie autosomal recessive megacystis-microcolon-intestinal hypoperistalsis syndrome in a Chinese family. J Hum Genet 64: 1067–1073, 2019. doi: 10.1038/s10038-019-0651-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yetman AT, Starr LJ. Newly described recessive MYH11 disorder with clinical overlap of Multisystemic smooth muscle dysfunction and Megacystis microcolon hypoperistalsis syndromes. Am J Med Genet A 176: 1011–1014, 2018. doi: 10.1002/ajmg.a.38647. [DOI] [PubMed] [Google Scholar]
- 35.Halim D, Brosens E, Muller F, Wangler MF, Beaudet AL, Lupski JR, Akdemir ZHC, Doukas M, Stoop HJ, de Graaf BM, Brouwer RWW, van Ijcken WFJ, Oury J-F, Rosenblatt J, Burns AJ, Tibboel D, Hofstra RMW, Alves MM. Loss-of-function variants in MYLK cause recessive megacystis microcolon intestinal hypoperistalsis syndrome. Am J Hum Genet 101: 123–129, 2017. doi: 10.1016/j.ajhg.2017.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moreno CA, Sobreira N, Pugh E, Zhang P, Steel G, Torres FR, Cavalcanti DP. Homozygous deletion in MYL9 expands the molecular basis of megacystis-microcolon-intestinal hypoperistalsis syndrome. Eur J Hum Genet 26: 669–675, 2018. doi: 10.1038/s41431-017-0055-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kapur RP, Robertson SP, Hannibal MC, Finn LS, Morgan T, van Kogelenberg M, Loren DJ. Diffuse abnormal layering of small intestinal smooth muscle is present in patients with FLNA mutations and x-linked intestinal pseudo-obstruction. Am J Surg Pathol 34: 1528–1543, 2010. doi: 10.1097/PAS.0b013e3181f0ae47. [DOI] [PubMed] [Google Scholar]
- 38.Gunst SJ, Zhang W. Actin cytoskeletal dynamics in smooth muscle: a new paradigm for the regulation of smooth muscle contraction. Am J Physiol Cell Physiol 295: C576–C587, 2008. doi: 10.1152/ajpcell.00253.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perrin BJ, Ervasti JM. The actin gene family: function follows isoform. Cytoskeleton (Hoboken) 67: 630–634, 2010. doi: 10.1002/cm.20475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sweeney HL, Hammers DW. Muscle contraction. Cold Spring Harb Perspect Biol 10: a023200, 2018. doi: 10.1101/cshperspect.a023200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arnoldi R, Hiltbrunner A, Dugina V, Tille J-C, Chaponnier C. Smooth muscle actin isoforms: a tug of war between contraction and compliance. Eur J Cell Biol 92: 187–200, 2013. doi: 10.1016/j.ejcb.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 42.Fatigati V, Murphy RA. Actin and tropomyosin variants in smooth muscles. Dependence on tissue type. J Biol Chem 259: 14383–14388, 1984. [PubMed] [Google Scholar]
- 43.McHugh KM, Crawford K, Lessard JL. A comprehensive analysis of the developmental and tissue-specific expression of the isoactin multigene family in the rat. Dev Biol 148: 442–458, 1991. doi: 10.1016/0012-1606(91)90263-3. [DOI] [PubMed] [Google Scholar]
- 44.Lehtonen HJ, Sipponen T, Tojkander S, Karikoski R, Järvinen H, Laing NG, Lappalainen P, Aaltonen LA, Tuupanen S. Segregation of a missense variant in enteric smooth muscle actin γ-2 with autosomal dominant familial visceral myopathy. Gastroenterology 143: 1482–1491.e3, 2012. doi: 10.1053/j.gastro.2012.08.045. [DOI] [PubMed] [Google Scholar]
- 45.Matera I, Bordo D, Di Duca M, Lerone M, Santamaria G, Pongiglione M, Lezo A, Diamanti A, Spagnuolo MI, Pini Prato A, Alberti D, Mattioli G, Gandullia P, Ceccherini I. Novel ACTG2 variants disclose allelic heterogeneity and bi-allelic inheritance in pediatric chronic intestinal pseudo-obstruction. Clin Genet 99: 430–436, 2021. doi: 10.1111/cge.13895. [DOI] [PubMed] [Google Scholar]
- 46.Wei Z, Lu L, Zheng Y, Yan W, Tao Y, Xiao Y, Cai W, Wang Y. Variants in the enteric smooth muscle actin γ-2 cause pediatric intestinal pseudo-obstruction in Chinese patients. J Pediatr Gastroenterol Nutr 72: 36–42, 2021. doi: 10.1097/MPG.0000000000002897. [DOI] [PubMed] [Google Scholar]
- 47.Whittington JR, Poole AT, Dutta EH, Munn MB. A novel mutation in ACTG2 gene in mother with chronic intestinal pseudoobstruction and fetus with megacystis microcolon intestinal hypoperistalsis syndrome. Case Rep Genet 2017: 9146507, 2017. doi: 10.1155/2017/9146507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meuwissen MEC, Lequin MH, Bindels-de Heus K, Bruggenwirth HT, Knapen MFCM, Dalinghaus M, de Coo R, van Bever Y, Winkelman BHJ, Mancini GMS. ACTA2 mutation with childhood cardiovascular, autonomic and brain anomalies and severe outcome. Am J Med Genet A 161: 1376–1380, 2013. doi: 10.1002/ajmg.a.35858. [DOI] [PubMed] [Google Scholar]
- 49.Milewicz DM, Østergaard JR, Ala-Kokko LM, Khan N, Grange DK, Mendoza-Londono R, Bradley TJ, Olney AH, Adès L, Maher JF, Guo D, Buja LM, Kim D, Hyland JC, Regalado ES. De novo ACTA2 mutation causes a novel syndrome of multisystemic smooth muscle dysfunction. Am J Med Genet A 152: 2437–2443, 2010. doi: 10.1002/ajmg.a.33657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Babu GJ, Loukianov E, Loukianova T, Pyne GJ, Huke S, Osol G, Low RB, Paul RJ, Periasamy M. Loss of SM-B myosin affects muscle shortening velocity and maximal force development. Nat Cell Biol 3: 1025–1029, 2001. doi: 10.1038/ncb1101-1025. [DOI] [PubMed] [Google Scholar]
- 51.Chi M, Zhou Y, Vedamoorthyrao S, Babu GJ, Periasamy M. Ablation of smooth muscle myosin heavy chain SM2 increases smooth muscle contraction and results in postnatal death in mice. Proc Natl Acad Sci USA 105: 18614–18618, 2008. doi: 10.1073/pnas.0808162105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang Q, Babu GJ, Periasamy M, Eddinger TJ. SMB myosin heavy chain knockout enhances tonic contraction and reduces the rate of force generation in ileum and stomach antrum. Am J Physiol Cell Physiol 304: C194–C206, 2013. doi: 10.1152/ajpcell.00280.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hypolite JA, Chang S, LaBelle E, Babu GJ, Periasamy M, Wein AJ, Chacko S. Deletion of SM-B, the high ATPase isoform of myosin, upregulates the PKC-mediated signal transduction pathway in murine urinary bladder smooth muscle. Am J Physiol Renal Physiol 296: F658–F665, 2009. doi: 10.1152/ajprenal.90221.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Karagiannis P, Babu GJ, Periasamy M, Brozovich FV. The smooth muscle myosin seven amino acid heavy chain insert’s kinetic role in the crossbridge cycle for mouse bladder. J Physiol 547: 463–473, 2003. doi: 10.1113/jphysiol.2002.035717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Löfgren M, Ekblad E, Morano I, Arner A. Nonmuscle myosin motor of smooth muscle. J Gen Physiol 121: 301–310, 2003. doi: 10.1085/jgp.200208720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morano I, Chai GX, Baltas LG, Lamounier-Zepter V, Lutsch G, Kott M, Haase H, Bader M. Smooth-muscle contraction without smooth-muscle myosin. Nat Cell Biol 2: 371–375, 2000. doi: 10.1038/35014065. [DOI] [PubMed] [Google Scholar]
- 57.Kandler JL, Sklirou E, Woerner A, Walsh L, Cox E, Xue Y. Compound heterozygous loss of function variants in MYL9 in a child with megacystis-microcolon-intestinal hypoperistalsis syndrome. Mol Genet Genomic Med 8: e1516, 2020. doi: 10.1002/mgg3.1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.He W-Q, Peng Y-J, Zhang W-C, Lv N, Tang J, Chen C, Zhang C-H, Gao S, Chen H-Q, Zhi G, Feil R, Kamm KE, Stull JT, Gao X, Zhu M-S. Myosin light chain kinase is central to smooth muscle contraction and required for gastrointestinal motility in mice. Gastroenterology 135: 610–620, 2008. doi: 10.1053/j.gastro.2008.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Halim D, Wilson MP, Oliver D, Brosens E, Verheij JBGM, Han Y, Nanda V, Lyu Q, Doukas M, Stoop H, Brouwer RWW, van IJcken WFJ, Slivano OJ, Burns AJ, Christie CK, de Mesy Bentley KL, Brooks AS, Tibboel D, Xu S, Jin ZG, Djuwantono T, Yan W, Alves MM, Hofstra RMW, Miano JM. Loss of LMOD1 impairs smooth muscle cytocontractility and causes megacystis microcolon intestinal hypoperistalsis syndrome in humans and mice. Proc Natl Acad Sci USA 114: E2739–E2747, 2017. doi: 10.1073/pnas.1620507114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jenkins ZA, Macharg A, Chang C-Y, van Kogelenberg M, Morgan T, Frentz S, Wei W, Pilch J, Hannibal M, Foulds N, McGillivray G, Leventer RJ, García-Miñaúr S, Sugito S, Nightingale S, Markie DM, Dudding T, Kapur RP, Robertson SP. Differential regulation of two FLNA transcripts explains some of the phenotypic heterogeneity in the loss-of-function filaminopathies. Hum Mutat 39: 103–113, 2018. doi: 10.1002/humu.23355. [DOI] [PubMed] [Google Scholar]
- 61.Gargiulo A, Auricchio R, Barone MV, Cotugno G, Reardon W, Milla PJ, Ballabio A, Ciccodicola A, Auricchio A. Filamin A is mutated in X-linked chronic idiopathic intestinal pseudo-obstruction with central nervous system involvement. Am J Hum Genet 80: 751–758, 2007. doi: 10.1086/513321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Oda H, Sato T, Kunishima S, Nakagawa K, Izawa K, Hiejima E, Kawai T, Yasumi T, Doi H, Katamura K, Numabe H, Okamoto S, Nakase H, Hijikata A, Ohara O, Suzuki H, Morisaki H, Morisaki T, Nunoi H, Hattori S, Nishikomori R, Heike T. Exon skipping causes atypical phenotypes associated with a loss-of-function mutation in FLNA by restoring its protein function. Eur J Hum Genet 24: 408–414, 2016. doi: 10.1038/ejhg.2015.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oegema R, Hulst JM, Theuns-Valks SDM, van Unen LMA, Schot R, Mancini GMS, Schipper MEI, de Wit MCY, Sibbles BJ, de Coo IFM, Nanninga V, Hofstra RMW, Halley DJJ, Brooks AS. Novel no-stop FLNA mutation causes multi-organ involvement in males. Am J Med Genet A 161: 2376–2384, 2013. doi: 10.1002/ajmg.a.36109. [DOI] [PubMed] [Google Scholar]
- 64.Hashmi SK, Barka V, Yang C, Schneider S, Svitkina TM, Heuckeroth RO. Pseudo-obstruction-inducing ACTG2R257C alters actin organization and function. JCI Insight 5, 2020. doi: 10.1172/jci.insight.140604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gabbiani G, Schmid E, Winter S, Chaponnier C, de Ckhastonay C, Vandekerckhove J, Weber K, Franke WW. Vascular smooth muscle cells differ from other smooth muscle cells: predominance of vimentin filaments and a specific alpha-type actin. Proc Natl Acad Sci USA 78: 298–302, 1981. doi: 10.1073/pnas.78.1.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sawtell NM, Lessard JL. Cellular distribution of smooth muscle actins during mammalian embryogenesis: expression of the alpha-vascular but not the gamma-enteric isoform in differentiating striated myocytes. J Cell Biol 109: 2929–2937, 1989. doi: 10.1083/jcb.109.6.2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Milewicz DM, Trybus KM, Guo D-C, Sweeney HL, Regalado E, Kamm K, Stull JT. Altered smooth muscle cell force generation as a driver of thoracic aortic aneurysms and dissections. Arterioscler Thromb Vasc Biol 37: 26–34, 2017. [Erratum in Arterioscler Thromb Vasc Biol 37: e12, 2017]. doi: 10.1161/ATVBAHA.116.303229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Milewicz DM, Guo D-C, Tran-Fadulu V, Lafont AL, Papke CL, Inamoto S, Kwartler CS, Pannu H. Genetic basis of thoracic aortic aneurysms and dissections: focus on smooth muscle cell contractile dysfunction. Annu Rev Genomics Hum Genet 9: 283–302, 2008. doi: 10.1146/annurev.genom.8.080706.092303. [DOI] [PubMed] [Google Scholar]
- 69.Richer J, Milewicz DM, Gow R, de Nanassy J, Maharajh G, Miller E, Oppenheimer L, Weiler G, O’Connor M. R179H mutation in ACTA2 expanding the phenotype to include prune-belly sequence and skin manifestations. Am J Med Genet A 158: 664–668, 2012. doi: 10.1002/ajmg.a.35206. [DOI] [PubMed] [Google Scholar]
- 70.Zimmerman RA, Tomasek JJ, McRae J, Haaksma CJ, Schwartz RJ, Lin HK, Cowan RL, Jones AN, Kropp BP. Decreased expression of smooth muscle alpha-actin results in decreased contractile function of the mouse bladder. J Urol 172: 1667–1672, 2004. doi: 10.1097/01.ju.0000139874.48574.1b. [DOI] [PubMed] [Google Scholar]
- 71.Eddinger TJ, Meer DP. Myosin II isoforms in smooth muscle: heterogeneity and function. Am J Physiol Cell Physiol 293: C493–C508, 2007. doi: 10.1152/ajpcell.00131.2007. [DOI] [PubMed] [Google Scholar]
- 72.Capriani A, Chiavegato A, Franch R, Azzarello G, Vinante O, Sartore S. Oestrogen-dependent expression of the SM2 smooth muscle-type myosin isoform in rabbit myometrium. J Muscle Res Cell Motil 18: 413–427, 1997. doi: 10.1023/a:1018642713934. [DOI] [PubMed] [Google Scholar]
- 73.White SL, Zhou MY, Low RB, Periasamy M. Myosin heavy chain isoform expression in rat smooth muscle development. Am J Physiol Cell Physiol 275: C581–C589, 1998. doi: 10.1152/ajpcell.1998.275.2.C581. [DOI] [PubMed] [Google Scholar]
- 74.Hong F, Haldeman BD, Jackson D, Carter M, Baker JE, Cremo CR. Biochemistry of smooth muscle myosin light chain kinase. Arch Biochem Biophys 510: 135–146, 2011. doi: 10.1016/j.abb.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Boczkowska M, Rebowski G, Kremneva E, Lappalainen P, Dominguez R. How Leiomodin and Tropomodulin use a common fold for different actin assembly functions. Nat Commun 6: 8314, 2015. doi: 10.1038/ncomms9314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stossel TP, Condeelis J, Cooley L, Hartwig JH, Noegel A, Schleicher M, Shapiro SS. Filamins as integrators of cell mechanics and signalling. Nat Rev Mol Cell Biol 2: 138–145, 2001. doi: 10.1038/35052082. [DOI] [PubMed] [Google Scholar]
- 77.Mericskay M, Blanc J, Tritsch E, Moriez R, Aubert P, Neunlist M, Feil R, Li Z. Inducible mouse model of chronic intestinal pseudo-obstruction by smooth muscle-specific inactivation of the SRF gene. Gastroenterology 133: 1960–1970, 2007. doi: 10.1053/j.gastro.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 78.Park C, Lee MY, Slivano OJ, Park PJ, Ha S, Berent RM, Fuchs R, Collins NC, Yu TJ, Syn H, Park JK, Horiguchi K, Miano JM, Sanders KM, Ro S. Loss of serum response factor induces microRNA-mediated apoptosis in intestinal smooth muscle cells. Cell Death Dis 6: e2011, 2015. doi: 10.1038/cddis.2015.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee MY, Park C, Ha SE, Park PJ, Berent RM, Jorgensen BG, Corrigan RD, Grainger N, Blair PJ, Slivano OJ, Miano JM, Ward SM, Smith TK, Sanders KM, Ro S. Serum response factor regulates smooth muscle contractility via myotonic dystrophy protein kinases and L-type calcium channels. PLoS One 12: e0171262, 2017. doi: 10.1371/journal.pone.0171262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Park C, Lee MY, Park PJ, Ha SE, Berent RM, Fuchs R, Miano JM, Becker LS, Sanders KM, Ro S. Serum response factor is essential for prenatal gastrointestinal smooth muscle development and maintenance of differentiated phenotype. J Neurogastroenterol Motil 21: 589–602, 2015. doi: 10.5056/jnm15063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Huang J, Wang T, Wright AC, Yang J, Zhou S, Li L, Yang J, Small A, Parmacek MS. Myocardin is required for maintenance of vascular and visceral smooth muscle homeostasis during postnatal development. Proc Natl Acad Sci USA 112: 4447–4452, 2015. doi: 10.1073/pnas.1420363112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Houweling AC, Beaman GM, Postma AV, Gainous TB, Lichtenbelt KD, Brancati F, Lopes FM, van der Made I, Polstra AM, Robinson ML, Wright KD, Ellingford JM, Jackson AR, Overwater E, Genesio R, Romano S, Camerota L, D’Angelo E, Meijers-Heijboer EJ, Christoffels VM, McHugh KM, Black BL, Newman WG, Woolf AS, Creemers EE. Loss-of-function variants in myocardin cause congenital megabladder in humans and mice. J Clin Invest 129: 5374–5380, 2019. doi: 10.1172/JCI128545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li S, Wang D-Z, Wang Z, Richardson JA, Olson EN. The serum response factor coactivator myocardin is required for vascular smooth muscle development. Proc Natl Acad Sci USA 100: 9366–9370, 2003. doi: 10.1073/pnas.1233635100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Du KL, Chen M, Li J, Lepore JJ, Mericko P, Parmacek MS. Megakaryoblastic leukemia factor-1 transduces cytoskeletal signals and induces smooth muscle cell differentiation from undifferentiated embryonic stem cells. J Biol Chem 279: 17578–17586, 2004. doi: 10.1074/jbc.M400961200. [DOI] [PubMed] [Google Scholar]
- 85.Xie C, Guo Y, Zhu T, Zhang J, Ma PX, Chen YE. Yap1 protein regulates vascular smooth muscle cell phenotypic switch by interaction with myocardin. J Biol Chem 287: 14598–14605, 2012. doi: 10.1074/jbc.M111.329268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Scirocco A, Matarrese P, Carabotti M, Ascione B, Malorni W, Severi C. Cellular and molecular mechanisms of phenotypic switch in gastrointestinal smooth muscle. J Cell Physiol 231: 295–302, 2016. doi: 10.1002/jcp.25105. [DOI] [PubMed] [Google Scholar]
- 87.Akiho H, Ihara E, Motomura Y, Nakamura K. Cytokine-induced alterations of gastrointestinal motility in gastrointestinal disorders. World J Gastrointest Pathophysiol 2: 72–81, 2011. doi: 10.4291/wjgp.v2.i5.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Karimi A, Milewicz DM. Structure of the elastin-contractile units in the thoracic aorta and how genes that cause thoracic aortic aneurysms and dissections disrupt this structure. Can J Cardiol 32: 26–34, 2016. doi: 10.1016/j.cjca.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Puri P, Shinkai M. Megacystis microcolon intestinal hypoperistalsis syndrome. Semin Pediatr Surg 14: 58–63, 2005. doi: 10.1053/j.sempedsurg.2004.10.026. [DOI] [PubMed] [Google Scholar]
- 90.Rolle U, O'Briain S, Pearl RH, Puri P. Megacystis-microcolon-intestinal hypoperistalsis syndrome: evidence of intestinal myopathy. Pediatr Surg Int 18: 2–5, 2002. doi: 10.1007/s003830200001. [DOI] [PubMed] [Google Scholar]
- 91.Osmanagic-Myers S, Dechat T, Foisner R. Lamins at the crossroads of mechanosignaling. Genes Dev 29: 225–237, 2015. doi: 10.1101/gad.255968.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chen J, Chen H, Sanders KM, Perrino BA. Regulation of SRF/CArG-dependent gene transcription during chronic partial obstruction of murine small intestine. Neurogastroenterol Motil 20: 829–842, 2008. doi: 10.1111/j.1365-2982.2008.01149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhu B, Daoud F, Zeng S, Matic L, Hedin U, Uvelius B, Rippe C, Albinsson S, Swärd K. Antagonistic relationship between the unfolded protein response and myocardin-driven transcription in smooth muscle. J Cell Physiol 235: 7370–7382, 2020. doi: 10.1002/jcp.29637. [DOI] [PubMed] [Google Scholar]
- 94.Basford JR. The law of Laplace and its relevance to contemporary medicine and rehabilitation. Arch Phys Med Rehabil 83: 1165–1170, 2002. doi: 10.1053/apmr.2002.33985. [DOI] [PubMed] [Google Scholar]
- 95.Martin KA, Merenick BL, Ding M, Fetalvero KM, Rzucidlo EM, Kozul CD, Brown DJ, Chiu HY, Shyu M, Drapeau BL, Wagner RJ, Powell RJ. Rapamycin promotes vascular smooth muscle cell differentiation through insulin receptor substrate-1/phosphatidylinositol 3-kinase/Akt2 feedback signaling. J Biol Chem 282: 36112–36120, 2007. doi: 10.1074/jbc.M703914200. [DOI] [PubMed] [Google Scholar]
- 96.Wagner RJ, Martin KA, Powell RJ, Rzucidlo EM. Lovastatin induces VSMC differentiation through inhibition of Rheb and mTOR. Am J Physiol Cell Physiol 299: C119–C127, 2010. doi: 10.1152/ajpcell.00429.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Heo Y-A. Golodirsen: first approval. Drugs 80: 329–333, 2020. doi: 10.1007/s40265-020-01267-2. [DOI] [PubMed] [Google Scholar]
- 98.Song Y, Morales L, Malik AS, Mead AF, Greer CD, Mitchell MA, Petrov MT, Su LT, Choi ME, Rosenblum ST, Lu X, VanBelzen DJ, Krishnankutty RK, Balzer FJ, Loro E, French R, Propert KJ, Zhou S, Kozyak BW, Nghiem PP, Khurana TS, Kornegay JN, Stedman HH. Non-immunogenic utrophin gene therapy for the treatment of muscular dystrophy animal models. Nat Med 25: 1505–1511, 2019. doi: 10.1038/s41591-019-0594-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Moretti A, Fonteyne L, Giesert F, Hoppmann P, Meier AB, Bozoglu T, et al. Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy. Nat Med 26: 207–214, 2020. doi: 10.1038/s41591-019-0738-2. [DOI] [PMC free article] [PubMed] [Google Scholar]