

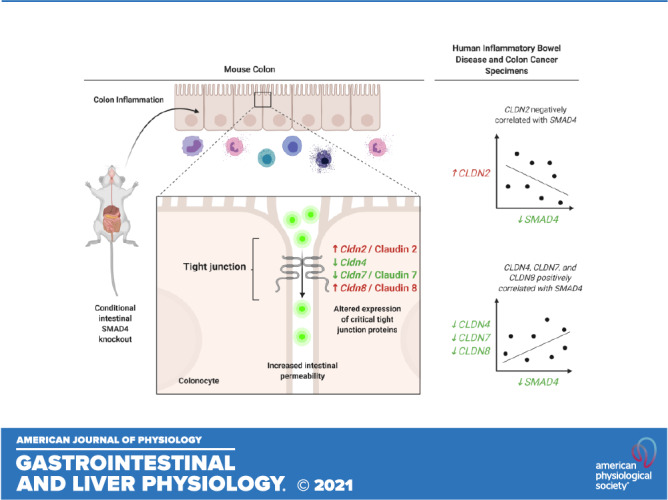
Keywords: claudin, inflammatory bowel disease, tight junction, transforming growth factor β
Abstract
Defective barrier function is a predisposing factor in inflammatory bowel disease (IBD) and colitis-associated cancer (CAC). Although TGFβ signaling defects have been associated with IBD and CAC, few studies have examined the relationship between TGFβ and intestinal barrier function. Here, we examine the role of TGFβ signaling via SMAD4 in modulation of colon barrier function. The Smad4 gene was conditionally deleted in the intestines of adult mice and intestinal permeability assessed using an in vivo 4 kDa FITC-Dextran (FD4) permeability assay. Mouse colon was isolated for gene expression (RNA-sequencing), Western blot, and immunofluorescence analysis. In vitro colon organoid culture was utilized to assess junction-related gene expression by qPCR and transepithelial resistance (TER). In silico analyses of human IBD and colon cancer databases were performed. Mice lacking intestinal expression of Smad4 demonstrate increased colonic permeability to FD4 without gross mucosal damage. mRNA/protein expression analyses demonstrate significant increases in Cldn2/Claudin 2 and Cldn8/Claudin 8, and decreases in Cldn3, Cldn4, and Cldn7/Claudin 7 with intestinal SMAD4 loss in vivo without changes in Claudin protein localization. TGFβ1/BMP2 treatment of polarized SMAD4+ colonoids increases TER. Cldn2, Cldn4, Cldn7, and Cldn8 are regulated by canonical TGFβ signaling, and TGFβ-dependent regulation of these genes is dependent on nascent RNA transcription (Cldn2, Cldn4, Cldn8) but not nascent protein translation (Cldn4, Cldn8). Human IBD/colon cancer specimens demonstrate decreased SMAD4, CLDN4, CLDN7, and CLDN8 and increased CLDN2 compared with healthy controls. Canonical TGFβ signaling modulates the expression of tight junction proteins and barrier function in mouse colon.
NEW & NOTEWORTHY We demonstrate that canonical TGFβ family signaling modulates the expression of critical tight junction proteins in colon epithelial cells, and that expression of these tight junction proteins is associated with maintenance of colon epithelial barrier function in mice.
BACKGROUND
Although the pathophysiology of inflammatory bowel disease (IBD) and colitis-associated cancer (CAC) is multifactorial, there is increasing evidence that defective intestinal barrier function plays a role in the pathogenesis of IBD and CAC (1–8). At the same time, the transforming growth factor-beta (TGFβ) signaling pathway is known to be frequently altered in IBD (1, 9–12), sporadic colorectal cancer (CRC) (13), and CAC (1, 14). Although the TGFβ signaling pathway has been implicated in the modulation of endothelial and epithelial barrier function previously, the impact of this signaling on maintaining or, conversely, degrading barrier integrity is widely variable between tissue types and contexts. For example, TGFβ is a potent inhibitor of barrier integrity in pulmonary endothelium (15–18) and esophageal epithelium (19). On the other hand, TGFβ signaling has been demonstrated to increase transepithelial resistance (TER) and preserve barrier function in two-dimensional immortalized jejunal and colon cancer cell lines (20–27). However, it remains unclear whether canonical TGFβ signaling affects colon barrier integrity in vivo or whether TGFβ signaling defects could lead to impaired intestinal barrier function and/or IBD and CAC.
Importantly, the TGFβ signaling pathway has both canonical and noncanonical components. In the canonical pathway, binding of the TGFβ family of extracellular ligands [including TGFβs, bone morphogenetic proteins (BMPs), activins, and nodals] to cell surface receptors activates intracellular, receptor-associated SMAD (R-SMAD) proteins (28). TGFβ-1, -2, and -3 bind to TGFβ-type serine/threonine kinase receptors, causing phosphorylation of R-SMADs-2 and -3 (SMAD2/3). BMPs (including BMPs 2–15) bind to BMP-type receptors and cause phosphorylation of R-SMADs-1, -5, and -9 (SMAD1/5/9). Once phosphorylated, R-SMADs from either side of the pathway must interact with the common SMAD, SMAD4, to translocate to the nucleus and regulate transcription. The canonical signaling activity downstream of all TGFβ ligands and receptors is therefore dependent on SMAD4, and loss of SMAD4 abrogates all canonical signaling by TGFβ family members (29). Importantly, the TGFβ signaling pathway is also postulated to have noncanonical (SMAD-independent) components wherein receptors interact directly with and activate non-SMAD protein kinases (30, 31).
Previous work from our group demonstrated that loss of canonical TGFβ signaling through conditional intestine-specific deletion of SMAD4 results in a profound increase in colon epithelial inflammatory gene expression, a concomitant increase in colon immune cell infiltration, and significant susceptibility to CAC development (14). Although the canonical TGFβ signaling pathway was observed to directly modulate the expression of selected inflammatory genes in a cell-autonomous manner, expression of numerous other inflammatory genes that were significantly altered with intestinal SMAD4 loss in vivo were not significantly changed with SMAD4 loss or TGFβ pathway stimulation in cultured colon epithelial cells (14). These findings led us to postulate that there may be other mechanisms triggered by loss of epithelial TGFβ signaling in vivo—aside from direct regulation of inflammatory gene expression—that contribute to inflammation and CAC susceptibility. One such possibility is a role for TGFβ in preservation of intestinal barrier function.
We hypothesized that TGFβ signaling via SMAD4 preserves colon mucosal barrier integrity through direct modulation of critical junctional protein expression. Through the following experiments, we demonstrate that loss of intestinal SMAD4 expression is associated with impaired colon mucosal barrier function in vivo, and that TGFβ signaling via SMAD4 modulates the expression of critical junctional proteins. Furthermore, we show that altered expression levels of both SMAD4 and critical junction-related genes are observed in human IBD and colon cancer specimens and that SMAD4 expression is significantly correlated with junction-related gene expression, suggesting that alterations in critical junctional proteins may play an important role in intestinal barrier dysfunction and/or inflammation due to SMAD4 loss or in the pathogenesis of human IBD, CRC, and/or CAC.
METHODS
Mouse Model
Animal work was performed with approval from the Vanderbilt University Institutional Animal Care and Use Committee and followed ARRIVE guidelines. Mouse alleles Lrig1CreERT2 and Smad4fl/fl have been previously published (14, 32–34) and were bred into the C57BL/6J background for at least 10 generations. Controls were sibling littermates and cage mates, and male/female mice were split evenly between experimental arms. Mice were given tamoxifen (2 mg in 0.1 mL corn oil) intraperitoneally two times on alternating days after 8 wk of age to ensure that Smad4 gene deletion occurred during adulthood and not during development. After tamoxifen treatment, bedding was mixed among cages within an experiment once per week.
Mice with Lrig1CreERT2 Smad4fl/fl genotype that received tamoxifen injections and who were confirmed by immunohistochemistry (IHC) to have undergone recombination with loss of SMAD4 protein in the intestinal crypts are referred to as Smad4ΔLrig1. Smad4ΔLrig1 mice demonstrate loss of SMAD4 protein in 90% or more of colon crypts (14). SMAD4-expressing control mice (mice with Smad4fl/fl genotype + tamoxifen injection) are referred to as Smad4fl/fl, SMAD4+, or simply “control” mice for simplicity.
RNA-Sequencing Data Analysis
Previously published RNA-sequencing (RNA-seq) datasets generated in our laboratory (14) were utilized for in silico analysis of differentially expressed junctional protein-encoding genes. Data files are publicly available on the National Institute of Health Gene Expression Omnibus (GEO) database (35, 36), accession number GSE100082.
Tissue Preparation and Imaging
For hematoxylin and eosin (H&E), IHC, and immunofluorescence (IF), mouse colons were fixed in 4% paraformaldehyde and embedded in paraffin or preserved fresh in optimal cutting temperature (OCT) compound as previously described (37–39). For fluorescence in situ hybridization (FISH) and mucous staining, colons were fixed in poloxamer solution as previously published (40). All antibodies have been previously validated and published (14, 41–46), and antibody catalogue numbers and dilutions for all staining are listed in Table 1.
Table 1.
Antibodies, qPCR primers, and fluorescence in situ hybridization probes utilized
| Antibodies | ||||
|---|---|---|---|---|
| Target Protein | Company | Product Number | Dilution | References |
| SMAD4 | Abcam | 40759 | 1:1,000 (IHC [FFPE/Cryo])1:500 (WB) | (14) |
| Claudin 2 | Invitrogen | 51-6100 | 1:125 (IF [Cryo])1:1,000 (WB) | (41) |
| Claudin 3 | Invitrogen | 34-1700 | 1:150 (IF [FFPE])1:500 (WB) | (45) |
| Claudin 7 | Invitrogen | 34-9100 | 1:250 (IF [Cryo])1:20,000 (WB) | (45) |
| Claudin 8 | Abcam | 183738 | 1:1000 (IF [FFPE])1:1000 (WB) | (42, 43, 46) |
| FITC | Abcam | ab6656 | 1:1000 (IF [Cryo]) | (44) |
|
qPCR Primers | ||
|---|---|---|
| Target Gene | Forward (5′ to 3′) | Reverse (5′ to 3′) |
| Cldn2 | tgaacacggaccactgaaag | ttagcaggaagctgggtcag |
| Cldn3 | gtggccactgcagctactt | gtttcatggtttgcctgtctc |
| Cldn4 | ttttgtggtcaccgactttg | tgtagtcccatagacgccatc |
| Cldn7 | tgtcttgtggagggcttga | caagcatggccattgaaa |
| Cldn8 | gggcctggggataaaagag | aatccttaagctgtttttaggcaat |
| Smad7 | acccccatcaccttagtcg | gaaaatccattgggtatctgga |
| Pmm1 (Reference Gene) | gggtggctctgactactctaagat | acacgtagtcaaacttctcaatgact |
| Fluorescence In Situ Hybridization (FISH) Probes | |
|---|---|
| Eub338 | 5′-Cy3-GCTGCCTCCCGTAGGAGT-3′ |
| Non-Eub (Negative Control) | 5′-Cy3-CGACGGAGGGCATCCTCA-3′ |
Cryo, cryopreserved (fresh frozen) tissue; FFPE, formalin-fixed and paraffin-embedded; IF, immunofluorescence; IHC, immunohistochemistry; WB, Western blot.
Bright-field images were captured on an Axioskop 40 microscope using Axiovision software (Carl Zeiss Microimaging, Thornwood, NY) through the Vanderbilt University Digital Histology Shared Resource. Fluorescent images were captured with the Zeiss LSM 510 Meta Inverted Confocal Microscope through the Vanderbilt University Cell Imaging Shared Resource.
In Vivo Permeability Assay
Mice (eight Smad4ΔLrig1 and eight control) were administered 484 mg/kg body weight of 4 kDa fluorescein isothiocyanate-Dextran (FITC-D, Sigma Aldrich, 46944) in phosphate-buffered saline (PBS) by oral gavage. Four hours later, mice were anesthetized in isoflurane until unresponsive. Once unresponsive, a midline abdominal incision was made, and the inferior vena cava was exposed. One mL of blood was drawn from the vena cava using a 22-G needle and blood was immediately put in Eppendorf tubes containing 15 µL 1 M EDTA and placed on ice. Eppendorf tubes were spun down at 3,000 RPM for 20 min. Briefly, 200 µL of plasma was transferred into a new Eppendorf tube and diluted 1:1 with PBS. Diluted plasma of 120 µL was transferred into a fluorimeter tray in triplicate. Samples were subsequently analyzed by spectrophotofluorometer (Promega, San Luis Obispo, CA) with excitation/emission wavelengths of 485 nm/530 nm (FITC). Signal intensity across the three measurements per mouse was averaged, and plasma Dextran concentration was then calculated for each mouse using a standard curve. Of note, blood from one Smad4ΔLrig1 mouse clotted during collection and thus we were unable to isolate plasma for fluorescent analysis from this mouse. This left the final experiment cohort for this element of the experiment to be 15 mice (Smad4ΔLrig1 mice, n = 7; Control mice, n = 8).
Following blood collection, mice were euthanized by cervical dislocation and the colon was removed. Mouse colon specimens were preserved in OCT and were cut and stained with a horseradish peroxidase (HRP)-conjugated anti-FITC antibody (Table 1). Signal was amplified using a Cy3-Tyramide amplification kit (Perkin Elmer, Boston, MA) diluted 1:1,000 for 10 min at room temperature and counterstained with TOTO-3 (1:3,000) in 50 mM HEPES, pH 7.7. Of note, all eight mice from each arm, regardless of ability to measure plasma FITC concentration, had their colons preserved in OCT and were included in this element of the experiment (total n = 16; n = 8 in each cohort).
Fluorescent In Situ Hybridization
Custom oligonucleotides were generated by the Vanderbilt University Molecular Cell Biology Resource Core in partnership with Sigma/Genosys (Woodlands, TX). The pan-bacterial probe (Eub338) and negative control probe (Non-Eub) sequences are listed in Table 1.
Five Smad4ΔLrig1 and five littermate control mice were used for this analysis. All female mice (3 Smad4ΔLrig1 and 2 control mice) were housed together while all male mice (2 Smad4ΔLrig1 and 3 control mice) were housed together. Bedding was mixed weekly between the two cages. Two centimeters of distal colon was collected and poloxamer-preserved (40) 4 wk after tamoxifen injection. Poloxamer-preserved colon sections were cut and placed in a hybridization oven at 50°C for 10 min to melt paraffin and then deparaffinized in Histoclear (National Diagnostics) and rehydrated by ethanol gradient before being placed in 20 mM Tris buffer. Bacterial probes were diluted to 2 µM in prewarmed hybridization buffer (20 mM Tris·HCl [pH 8.0] + 0.9 M NaCl + 0.01% sodium dodecyl sulfate). Approximately 150 µL probe solution was then placed on each slide until the sample was completely covered and slides were incubated for 1.5 h at 46°C in humidity chamber. Slides were then washed in fluorescence in situ hybridization (FISH) wash buffer (225 mM NaCl + 20 mM Tris + 5 mM EDTA) for 5 min, three times followed by Hoechst 33342 (1:10,000 diluted in PBS) for 5 min. Slides were then washed again in FISH wash buffer for 5 min, twice, and the slides were cover slipped with prolong gold and allowed to cure in the dark for 48 h before sealing.
16S Targeted Sequencing of Mouse Colon Microbiome
The same ten mice used in the FISH experiment described above were used for this analysis. Stool pellets were collected from mice before dissection and colon mucosal scrapings were taken from 5 cm of middle and distal colon (proximal to the 2 cm used for poloxamer preservation). Following collection, microbiota samples were processed and analyzed by the ZymoBIOMICS Service: Targeted Metagenomic Sequencing (Zymo Research, Irvine, CA). DNA was extracted using the ZymoBIOMICS-96 MagBead DNA Kit (Zymo Research, Irvine, CA). DNA samples were prepared for targeted sequencing using the Quick-16STM NGS Library Prep Kit (Zymo Research, Irvine, CA). DNA library was sequenced on Illumina MiSeq with a v3 reagent kit (600 cycles). The sequencing was performed with >10% PhiX spike-in. Unique amplicon sequences were inferred from raw reads and chimeric sequences removed using the Dada2 pipeline (47). Taxonomy assignment and composition visualization, alpha-diversity, and beta-diversity analyses were performed using Uclust from Qiime v.1.9.1 (48). Taxonomy was assigned with the Zymo Research Database.
Mucin Staining
The ten sections of poloxamer-preserved (40) distal colon (as in Fluorescent In Situ Hybridization) were cut, deparaffinized, and rehydrated with an ethanol gradient. Slides were placed in 3% acetic acid for 3 min followed by Alcian blue solution (5 g Alcian blue-8GX, 500 mL acetic acid 3%, pH 2.5) for 30 min at room temperature. Slides were then rinsed in 3% acetic acid and washed in running tap water for 10 min before being dehydrated, counterstained with eosin, and cover slipped.
Western Blotting
Five Smad4ΔLrig1 and five littermate control mice were dissected and their colon isolated. Genotypes were split evenly between male/female mice, and mice of the same sex were housed together with bedding mixed between cages weekly. Colon crypts were isolated by removing and flushing the colon, opening it longitudinally, rinsing, and incubating at 4°C in 1.5 mmol/L EDTA in PBS followed by shaking for 1 min. Following EDTA chelation, protein lysates were generated, Western blots (WB) performed as published (49), and density quantified using ImageJ software (50). Antibodies are listed in Table 1.
Colon Organoid (“Colonoid”) Experiments
Mouse colonoids were generated and cultured as previously described (14). Colonoids were suspended and plated in 50-μL beads of growth factor reduced Matrigel (GFR; Corning, Tewksbury, MA). Complete colonoid medium was composed of 40% basal medium [advanced DMEM/F12 (Gibco) supplemented with penicillin/streptomycin (Gibco), N2 (Gibco), B27 (Gibco), Glutamax (Gibco), HEPES (Sigma Aldrich), 50 ng/mL epidermal growth factor (R&D Systems)], 40% Wnt3a-conditioned medium, 10% R-Spondin-conditioned medium, and 10% Noggin-conditioned medium. Colonoids were grown at 37°C in 5% CO2, media were changed every 2–3 days, and colonoids were passaged every 5–7 days. Colonoids at density of 70–100 per well were treated 3 days after passage with TGFβ1 (3 ng/mL), BMP2 (100 ng/mL), TGFβ1/BMP2 (3 ng/mL and 100 ng/mL, respectively), actinomycin D (ActD) (5 μg/mL), cycloheximide (CHX, 100 μM), and/or vehicle control at designated time points for quantitative reverse-transcription polymerase chain reaction (qPCR) and RNA-seq experiments. Vehicle for TGFβ1 and BMP2 was 4 mM HCl + 0.1% BSA in PBS. Vehicle for ActD and CHX was dimethyl sulfoxide (DMSO). Wells treated with BMP2 had the BMP-inhibitor, Noggin, withheld from the media.
Quantitative Reverse-Transcription Polymerase Chain Reaction
RNA was extracted from colonoids from at least three experimental replicates and purified as described (51). Samples were run using a standard SYBR Green qPCR protocol (52). All samples were run in triplicate with a negative control on a CFX96 Thermal Cycler (Bio-Rad, Hercules, CA). A well-known TGFβ/SMAD-response gene (53), Smad7, was used for a positive control to confirm TGFβ pathway stimulation in all experimental replicates. mRNA levels were normalized to the level of Pmm1. All qPCR primer sequences are listed in Table 1. Each point on each qPCR graph represents a single experimental replicate (each done on a separate day), and each experimental replicate reflects the mean value of three technical replicates.
In Vitro Measures of Permeability
Colonoids were grown on transwell membranes and TER measurements were taken on colonoids as previously described (54). Briefly, colonoids were collected and dissociated using 0.25% Trypsin. Following dissociation, cells were washed and resuspended in complete media + ROCK Inhibitor (Y27632, R&D Systems, 1:1,000) and plated on collagen (Collagen I, Gibco)-coated 0.4-µm transwell filters (Corning) with 75,000 cells plated per transwell. After one day, ROCK inhibitor was removed from culture media. TER was measured by Ohm meter daily after plating. Following polarization on days 2–3 after plating, media were removed from the apical chamber to create an air-liquid interface (ALI) and to induce three-dimensional differentiation, and transwells were treated with 3 ng/mL TGFβ1 and 100 ng/mL BMP2 or equivalent volume vehicle control in the basolateral chamber. TER was subsequently measured at 24- and 48-h following treatment.
Graphs provided represent the mean and standard deviation of three biological replicates, each representing separate experiments performed on different days. Each experiment included 3–6 wells per arm, depending on cell number availability.
In Silico Analysis of Microarray Data and the Cancer Genome Atlas (TCGA)
We queried a previously published (7) database consisting of transcriptomic data from patients with IBD and healthy controls generated from endoscopic biopsy samples and analyzed by Microarray [data accessible at NCBI GEO database (35, 36), Accession No. GSE75214]. This database included colon mucosal biopsies from 74 patients with active UC (UCa), 23 patients with inactive UC (UCi), 8 patients with active Crohn’s disease (CDa), and 11 healthy controls (HC). Data are represented in Log2 scale.
We additionally queried The Cancer Genome Atlas (TCGA) database (https://www.cancer.gov/tcga) utilizing the Firehose web browser from the Broad Institute (https://gdac.broadinstitute.org/). mRNA-seq expression data for 282 colon cancer (CC) specimens and 41 healthy control colon (HCc) specimens were available and downloaded for in silico comparison of gene expression. Data are represented in Log2 scale.
Statistical Analysis
Results from in vivo assays including FITC-D permeability assays and Western blots were compared using nonparametric Mann–Whitney test. Results from in vivo and in vitro RNA-seq assays were compared as described (14). For the 16S targeted sequencing microbiome analysis, species detection levels and Simpson Diversity scores (55) were compared using the nonparametric Wilcoxon test. In vitro colonoid qPCR assays were compared using nonparametric Kruskal–Wallis test and post hoc Welch’s t test. In vitro colonoid TER assays were compared using repeated-measured ANOVA/mixed-effects model. In silico analysis of gene expression in human biopsy samples were compared using the nonparametric Kruskal–Wallis test with post hoc Welch’s t test (IBD specimens), nonparametric Mann–Whitney test (TCGA specimens), or nonparametric Spearman’s correlation (IBD and TCGA specimens), as appropriate. Statistical analyses were performed using GraphPad Prism 9 Software (San Diego, CA). Throughout the manuscript, statistical significance is designated as: ns (P ≥ 0.05), * (P < 0.05), ** (P < 0.01), or *** (P < 0.001).
RESULTS
Loss of SMAD4 Expression within the Intestinal Epithelium is Associated with Increased Intestinal Permeability in Vivo
We previously demonstrated that Lrig1CreERT2 Smad4fl/fl (Smad4ΔLrig1) adult mice lose expression of SMAD4 protein in >90% of their colonic crypts by one month after tamoxifen treatment and that Smad4ΔLrig1 mice demonstrate increased colon epithelial inflammatory signaling and increased immune cell infiltration into the subepithelial colonic stroma compared with their SMAD4+ (either vehicle-treated Lrig1CreERT2 Smad4fl/fl or tamoxifen-treated Smad4fl/fl mice) counterparts (14). We sought to determine whether an alteration in intestinal permeability in Smad4ΔLrig1 mice existed and could at least partially explain the observed inflammatory phenotype in mice with intestinal SMAD4 deletion. To assess this, we performed an in vivo FITC-Dextran permeability assay (56), whereby mice were given 4 kDa FITC-Dextran (FD4) by oral gavage and then blood was extracted by inferior vena cava puncture 4 h later for measurement of plasma FD4 concentration (Fig. 1A). This analysis demonstrated a 2.5-fold increase in gastrointestinal permeability to FD4 in Smad4ΔLrig1 versus SMAD4+ mice (Fig. 1B). Colon frozen sections from these mice were stained with an anti-FITC antibody. This demonstrated increased translocation of the FD4 molecule across the colon epithelium in Smad4ΔLrig1 mice compared with control (Fig. 1C), supporting the notion that a colon mucosal barrier defect exists in mice with conditional intestinal Smad4 deletion.
Figure 1.

Intestinal Smad4 loss associated with impaired barrier function in mice. A: seven Smad4ΔLrig1 mice and eight control mice were administered 4 kDa fluorescein isothiocyanate-Dextran (FITC-D, FD4) by oral gavage. Blood was collected and colon was fresh frozen 4 h later. B: Smad4ΔLrig1 mice demonstrated a 2.5-fold increase in plasma concentration of FD4 compared with control mice, suggesting an intestinal barrier defect (P = 0.029 by Mann–Whitney test). C: FITC molecules in preserved colon sections from eight Smad4ΔLrig1 mice and eight control mice were detected using an anti-FITC antibody (red), demonstrating more FITC molecules per crypt in Smad4ΔLrig1 mice compared with control (0.187 vs. 0.908, P = 0.007 by Mann–Whitney test). Statistical significance is designated as: *P < 0.05, **P < 0.01. Image created with BioRender.com.
Three distinct pathways of transepithelial permeability have been described. The “pore” and “leak” pathways both govern tight junction-mediated flux via the paracellular route without associated evidence of gross epithelial damage/ulceration and are distinguished by their size and charge selectivity (57–62). A third tight junction-independent “damage” pathway is characterized by gross tissue damage, ulceration, and epithelial cell death (62, 63). Although the “pore” pathway is typically permeable only to very small molecules (diameter <4–6 Å), both the “leak” and “damage” pathways allow for passage of larger uncharged solutes, including FD4 (diameter 28 Å). Although the “leak” pathway allows passage of molecules with diameters up to 100 Å, the “damage” pathway allows unrestricted movement of macromolecules and even bacterial organisms. Given that the aforementioned experiments demonstrated a 2.5-fold increase in permeability to FD4, it is unlikely that the permeability defect observed is due solely to a defect in the “pore” pathway [which would show no increase in permeability to a FD4 (60)] or the “damage” pathway [which would show a much larger increase in permeability to FD4 (64, 65)].
To validate our hypothesis that intestinal SMAD4 loss is associated with a barrier defect via the tight junction-dependent “leak” pathway rather than a more extensive “damage” phenotype, we examined the colon mucosa of Smad4ΔLrig1 mice both grossly and histologically and saw no evidence of widespread mucosal damage, ulceration, or tissue breakdown compared with control mice (Fig. 2A). To further explore whether intestinal SMAD4 loss is associated with a widespread epithelial “damage” phenotype wherein luminal bacteria are able to cross the epithelial barrier and invade the colon wall, we performed FISH for bacterial organisms on poloxamer-preserved sections of distal colon. FISH was performed using the pan-bacterial probe, Eub338, which demonstrated no difference in bacterial translocation across the colon epithelium of Smad4ΔLrig1 compared with SMAD4+ mice (Fig. 2B).
Figure 2.

Colons from Smad4ΔLrig1 mice show no evidence of gross mucosal damage. A: representative hematoxylin and eosin (H&E) stains from Smad4ΔLrig1 and control mice showing epithelial integrity. B: fluorescence in situ hybridization (FISH) staining with the pan-bacterial probe, Eub338 (red). Nuclei in light blue. Bacterial species indicated by solid arrowhead. Autofluorescent red blood cells indicated by open arrowhead. Border between lumen and epithelium demarcated with dashed line. C: Alcian blue (pH 2.5) stain for mucins (blue).
Loss of Intestinal SMAD4 Expression is Not Associated with Major Changes in Mucin Accumulation or Microbiome Composition
As mucins play a very important role in barrier function and mucosal protection in the gastrointestinal tract and colon (5), major alterations in mucin expression, accumulation, or localization have the potential to be a confounding variable in the inflammatory and permeability phenotypes we’ve observed with intestinal SMAD4 loss in vivo. To examine this possibility, we performed an Alcian blue (pH 2.5) stain, which is known to stain all sulfated and carboxylated acid mucopolysaccharides and sialomucins (glycoproteins) (66) and thus represents a reliable indicator of general mucin accumulation and distribution, on poloxamer-preserved colon sections from mice with and without intestinal SMAD4 expression. We observed no change in mucin localization or accumulation in Smad4ΔLrig1 compared with SMAD4+ mice (Fig. 2C).
As the microbiome is also known to regulate gastrointestinal inflammation and pathology (67, 68), we examined matched stool pellets and colonic mucosal scrapings with 16S targeted sequencing to characterize the microbiome in mice with intestinal SMAD4 loss. 16S sequencing data for all samples (stool pellets and mucosal scrapings) at all taxonomic levels (phylum, class, order, family, genus, and species) are available in Supplemental Table S1 (see https://doi.org/10.5281/zenodo.4628541). Comparison of species detection levels demonstrated that, when comparing the microbiome composition between five Smad4ΔLrig1 and five SMAD4+ control mice, there was significant mouse-to-mouse variability with some similarities between mice of the same sex that were housed together (Fig. 3, A and B). In addition, when unsupervised hierarchical clustering was performed, minimal clustering between mouse genotypes occurred for either the stool pellets or colon mucosal scrapings, and mice of the same sex/housed together appeared to cluster more closely together (Fig. 3, C and D). Importantly, over the 232 unique bacterial species identified in all 20 samples, 25 species were present in a significantly different proportion (increased or decreased) in Smad4ΔLrig1 compared with SMAD4+ control mice in either the stool pellet or colon mucosal scraping samples (Table 2). Of those 25 species, 22 species were significantly different in the stool pellets of Smad4ΔLrig1 relative to SMAD4+ control mice, 7 were significantly different in the colon mucosal scrapings of Smad4ΔLrig1 relative to control, and 4 species were significantly different in both stool pellets and colon mucosal scrapings from Smad4ΔLrig1 mice relative to control. All four of these species were detected at significantly lower levels in Smad4ΔLrig1 mice compared with control and all four species belong to the phylum Firmicutes. These four species were the following: Firmicutes Bacilli Lactobacillales Streptococcaceae Streptococcus danieliae; Firmicutes Clostridia Clostridiales Lachnospiraceae sp33625; Firmicutes Clostridia Clostridiales Ruminococcaceae sp35382-sp35403-sp35432; and Firmicutes Clostridia Clostridiales Lachnospiraceae sp32758. Of these four species, all were detected in Smad4ΔLrig1 mice at levels at least twofold lower than control mice in both stool pellets and mucosal scrapings.
Figure 3.
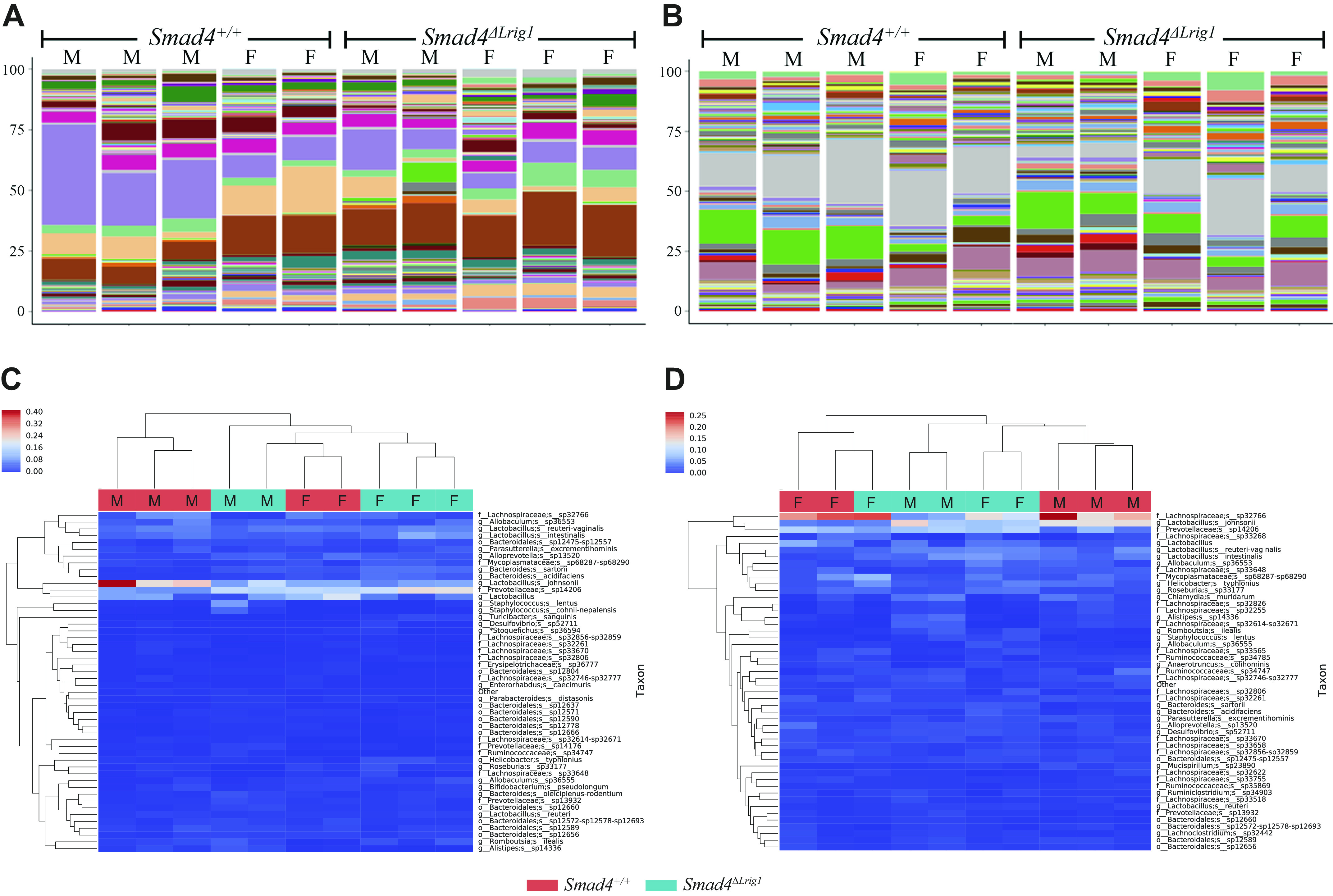
Colonic microbiome composition in five Smad4ΔLrig1 and five control mice. Composition bar plots from stool pellets (A) and colon mucosal scrapings (B) of five Smad4ΔLrig1 and five control mice. Each color represents a unique bacterial species. Heatmaps with unsupervised hierarchical clustering of stool pellets (C) and colon mucosal scrapings (D) from five Smad4ΔLrig1 and five control mice. All mice were littermates. “F” indicates female mice who were housed together, and “M” indicates male mice who were housed together (bedding mixed between the one male and one female cage weekly).
Table 2.
Bacterial species that are significantly increased or decreased as a proportion of all detected bacteria in Smad4ΔLrig1 vs. control mice
| Stool Pellets |
Colon Mucosal Scrapings |
|||||
|---|---|---|---|---|---|---|
| Species Name | % Detected Species (SMAD4+) | % Detected Species (Smad4ΔLrig1) | P Value | % Detected Species (SMAD4+) | % Detected Species (Smad4ΔLrig1) | P Value |
| Firmicutes; Clostridia; Clostridiales; Peptostreptococcaceae; Romboutsia; ilealis | 0.8379 | 2.0481 | 0.095 | 0.1991 | 1.0541 | 0.016 |
| Bacteroidetes; Bacteroidia; Bacteroidales; Bacteroidaceae; Bacteroides; oleiciplenus-rodentium | 0.5212 | 1.2221 | 0.032 | 0.2307 | 0.6080 | 0.095 |
| Firmicutes; Clostridia; Clostridiales; Family XIII; NA; sp31521 | 0.0135 | 0.0462 | 0.036 | 0.0076 | 0.0188 | 0.265 |
| Bacteroidetes; Bacteroidia; Bacteroidales; Porphyromonadaceae; Parabacteroides; sp13265 | 0.1112 | 0.2483 | 0.016 | 0.0485 | 0.1046 | 0.151 |
| Bacteroidetes; Bacteroidia; Bacteroidales; Bacteroidaceae; Bacteroides; sartorii | 1.3250 | 3.8431 | 0.016 | 0.6562 | 1.3833 | 0.095 |
| Bacteroidetes; Bacteroidia; Bacteroidales; NA; NA; sp12520 | 0.1155 | 0.2571 | 0.008 | 0.0662 | 0.1035 | 0.056 |
| Bacteroidetes; Bacteroidia; Bacteroidales; Prevotellaceae; NA; sp14206 | 10.8367 | 18.3292 | 0.032 | 6.0529 | 8.0463 | 0.222 |
| Firmicutes; Bacilli; Lactobacillales; Lactobacillaceae; Lactobacillus; johnsonii | 21.3527 | 9.8224 | 0.032 | 9.6831 | 8.9486 | 1.000 |
| Firmicutes; Clostridia; Clostridiales; Lachnospiraceae; NA; sp32746-sp32777 | 0.4938 | 0.2613 | 0.008 | 1.2803 | 1.0871 | 0.691 |
| Firmicutes; Clostridia; Clostridiales; Lachnospiraceae; NA; sp32655 | 0.1455 | 0.0452 | 0.016 | 0.2828 | 0.2337 | 0.691 |
| Firmicutes; Clostridia; Clostridiales; Lachnospiraceae; Roseburia; sp33136 | 0.0388 | 0.0050 | 0.011 | 0.1075 | 0.0883 | 0.548 |
| Actinobacteria; Coriobacteriia; Coriobacteriales; Coriobacteriaceae; Enterorhabdus; caecimuris | 0.3641 | 0.2244 | 0.008 | 0.4050 | 0.2743 | 0.095 |
| Firmicutes; Clostridia; Clostridiales; Lachnospiraceae; NA; sp33577 | 0.1223 | 0.0576 | 0.095 | 0.3545 | 0.2342 | 0.016 |
| Firmicutes; Clostridia; Clostridiales; Lachnospiraceae; Tyzzerella; sp33289-sp33291 | 0.0167 | 0.0000 | 0.025 | 0.0289 | 0.0186 | 0.672 |
| Firmicutes; Clostridia; Clostridiales; Lachnospiraceae; NA; sp33451-sp33593 | 0.1123 | 0.0221 | 0.036 | 0.2692 | 0.1586 | 0.151 |
| Bacteroidetes; Bacteroidia; Bacteroidales; NA; NA; sp12597-sp12621 | 0.2880 | 0.2173 | 0.421 | 0.2329 | 0.1312 | 0.008 |
| Firmicutes; Erysipelotrichia; Erysipelotrichales; Erysipelotrichaceae; NA; sp36777 | 0.4539 | 0.1368 | 0.032 | 0.3133 | 0.1547 | 0.222 |
| Actinobacteria; Coriobacteriia; Coriobacteriales; Coriobacteriaceae; Paraeggerthella; hongkongensis | 0.0872 | 0.0235 | 0.008 | 0.1238 | 0.0595 | 0.095 |
| Firmicutes; Clostridia; Clostridiales; Lachnospiraceae; Roseburia; sp33168 | 0.2563 | 0.0244 | 0.012 | 0.5081 | 0.2373 | 0.095 |
| Firmicutes; Clostridia; Clostridiales; Lachnospiraceae; NA; sp32623 | 0.0563 | 0.0040 | 0.045 | 0.0862 | 0.0321 | 0.094 |
| Firmicutes; Bacilli; Lactobacillales; Streptococcaceae; Streptococcus; danieliae | 0.0394 | 0.0053 | 0.045 | 0.0392 | 0.0098 | 0.020 |
| Firmicutes; Clostridia; Clostridiales; Lachnospiraceae; NA; sp33625 | 0.0490 | 0.0000 | 0.008 | 0.1880 | 0.0426 | 0.020 |
| Firmicutes; Clostridia; Clostridiales; Ruminococcaceae; NA; sp35382-sp35403-sp35432 | 0.0966 | 0.0105 | 0.045 | 0.0643 | 0.0120 | 0.045 |
| Firmicutes; Clostridia; Clostridiales; Lachnospiraceae; NA; sp32758 | 0.0698 | 0.0000 | 0.026 | 0.1215 | 0.0044 | 0.045 |
| Firmicutes; Erysipelotrichia; Erysipelotrichales; Erysipelotrichaceae; *Stoquefichus; sp36592-sp36804 | 0.0178 | 0.0028 | 0.045 | 0.0047 | 0.000 | 0.424 |
Bolded species are bacterial species that are significantly altered (P < 0.05) in both stool pellet and colon mucosal scraping samples. All species listed are from the kingdom Bacteria. Species name are listed as Phylum; Class; Order; Family; Genus; Species. NA = not applicable. Data represent five Smad4ΔLrig1 and five control mice. Genotypes were split evenly between male and female male. All mice were littermates. All female mice were housed together while all male mice were housed together. Bedding was mixed between male/female cages weekly.
When microbiome alterations were examined at the family level, small but statistically significant differences in Bacteroidetes Bacteroidia Bacteroidales Bacteroidaceae (stool pellet fold change −1.6, P = 0.03; mucosal scraping fold change −1.05, P = 0.01) and Firmicutes Bacilli Lactobacillales Streptococcaceae (stool pellet fold change 0.87, P = 0.04; mucosal scraping fold change 0.75, P = 0.02) were observed in Smad4ΔLrig1 mice relative to control. At the genus level, small but significant differences in Bacteroidetes Bacteroidia Bacteroidales Bacteroidaceae Bacteroides (stool pellet fold change −1.6, P = 0.03; mucosal scraping fold change −1.05, P = 0.01) and Firmicutes Bacilli Lactobacillales Streptococcaceae Streptococcus (stool pellet fold change 0.87, P = 0.05; mucosal scraping fold change 0.75, P = 0.02) were again detected.
We additionally examined microbial diversity between samples. Comparison of the Simpson Diversity Index (55) demonstrated an increase in diversity in stool pellets from Smad4ΔLrig1 mice compared with control mice at the family (0.84 vs. 0.76, P = 0.02) and genus (0.85 vs. 0.77, P = 0.02) levels but not at the species level (0.93 vs. 0.90, P = 0.15). There was no significant difference in Simpson Diversity Index score in colon mucosal scraping samples between Smad4ΔLrig1 and control mice at the family (0.82 vs. 0.76, P = 0.10), genus (0.85 vs. 0.80, P = 0.10), or species (0.95 vs. 0.93, P = 0.10) levels.
Although some statistically significant differences in microbiome composition and diversity were detected, the implications for their influence on inflammatory, tumorigenic, and barrier phenotypes seen in Smad4ΔLrig1 mice are unclear.
These data relating to histological architecture, mucin distribution, bacterial translocation, and microbiota composition/diversity collectively suggest that while mice with intestine-specific deletion of SMAD4 exhibit increased intestinal permeability, neither a global mucosal “damage” pathway nor major changes in mucin accumulation nor microbiome composition are likely to singularly explain this phenotype. Rather, a tight junction-dependent “leak” pathway is most likely to explain the barrier defect detected in Smad4ΔLrig1 mice.
Loss of Smad4 Expression within Intestinal Epithelium is Associated with Altered Gene Expression Levels for Multiple Junctional Proteins in Vivo
To begin understanding how the regulation of intestinal barrier function is altered with SMAD4 loss in vivo, colon epithelial cells from Smad4ΔLrig1 and SMAD4+ mice were analyzed by RNA-seq (14). An exhaustive survey of differential junction-related gene expression identified at least 30 genes related to barrier function (including junctional complexes, cell adhesion, and mucin production) that were significantly altered in Smad4ΔLrig1 compared with SMAD4+control mice (Table 3, left columns).
Table 3.
Junctional protein-related genes are altered with loss of epithelial Smad4 expression in vivo and with TGFβ pathway stimulation in vitro
| SMAD4 KO In Vivo |
TGFβ1/BMP2 Treatment In Vitro |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Gene Symbol (Protein Name) | Fold Change | Log2 Fold Change | P Value | FDR | Fold Change | Log2 Fold Change | P Value | FDR | |
| Tight Junction | Tjp1 (ZO-1) | 1.64 | 0.71 | <0.001 | <0.001 | 1.71 | 0.78 | <0.001 | <0.001 |
| Tjp2 (ZO-2) | 1.30 | 0.38 | 0.005 | 0.016 | 1.85 | 0.89 | <0.001 | <0.001 | |
| Ocln (Occludin) | 0.85 | −0.24 | 0.109 | 0.198 | 1.35 | 0.43 | 0.023 | 0.051 | |
| Cldn2 (Claudin 2) | 1.22 | 0.28 | 0.045 | 0.098 | 0.06 | −4.14 | <0.001 | <0.001 | |
| Cldn3 (Claudin 3) | 0.56 | −0.85 | <0.001 | <0.001 | 1.29 | 0.37 | 0.040 | 0.081 | |
| Cldn4 (Claudin 4) | 0.64 | −0.64 | 0.002 | 0.007 | 5.79 | 2.53 | <0.001 | <0.001 | |
| Cldn7 (Claudin 7) | 0.82 | −0.29 | 0.041 | 0.092 | 1.49 | 0.58 | 0.002 | 0.004 | |
| Cldn8 (Claudin 8) | 2.13 | 1.09 | <0.001 | <0.001 | 0.06 | −4.10 | <0.001 | <0.001 | |
| Cldn10 (Claudin 10) | 10.45 | 3.39 | <0.001 | <0.001 | |||||
| Cldn12 (Claudin 12) | 1.11 | 0.15 | 0.323 | 0.454 | 0.81 | −0.31 | 0.112 | 0.188 | |
| Cldn14 (Claudin 14) | 1.04 | 0.06 | 0.821 | 0.882 | 0.12 | −3.02 | <0.001 | <0.001 | |
| Cldn15 (Claudin 15) | 0.53 | −0.91 | <0.001 | <0.001 | 0.06 | −4.09 | <0.001 | <0.001 | |
| Cldn23 (Claudin 23) | 1.03 | 0.04 | 0.796 | 0.865 | 3.66 | 1.87 | <0.001 | <0.001 | |
| Cldn25 (Claudin 25) | 0.91 | −0.13 | 0.316 | 0.447 | 1.45 | 0.54 | 0.013 | 0.033 | |
| Marveld2 (Tricellulin) | 1.04 | 0.06 | 0.642 | 0.747 | 1.51 | 0.59 | 0.009 | 0.023 | |
| Adherens Junction | Cdh1 (E-Cadherin) | 1.27 | 0.35 | 0.012 | 0.033 | 2.54 | 1.35 | <0.001 | <0.001 |
| Ctnna1 (α-1-Catenin) | 1.15 | 0.20 | 0.142 | 0.242 | 2.31 | 1.21 | <0.001 | <0.001 | |
| Ctnnb1 (β-Catenin) | 0.99 | −0.01 | 0.929 | 0.958 | 2.11 | 1.08 | <0.001 | <0.001 | |
| Ctnnd1 (P120 Catenin) | 1.47 | 0.56 | <0.001 | <0.001 | 3.20 | 1.68 | <0.001 | <0.001 | |
| Jup (Plakoglobulin) | 0.93 | −0.11 | 0.495 | 0.622 | 3.39 | 1.76 | <0.001 | <0.001 | |
| Pkp1 (Plakophilin 1) | 1.03 | 0.04 | 0.771 | 0.845 | 2.70 | 1.43 | <0.001 | <0.001 | |
| Pkp2 (Plakophilin 2) | 1.36 | 0.44 | <0.001 | 0.002 | 1.65 | 0.72 | <0.001 | 0.001 | |
| Pkp3 (Plakophilin 3) | 0.66 | −0.59 | <0.001 | <0.001 | 2.85 | 1.51 | <0.001 | <0.001 | |
| Vcl (Vinculin) | 1.27 | 0.34 | 0.012 | 0.032 | 1.79 | 0.84 | <0.001 | <0.001 | |
| Actn1 (Actinin Alpha 1) | 1.74 | 0.80 | <0.001 | <0.001 | 2.93 | 1.55 | <0.001 | <0.001 | |
| Actn4 (Actinin Alpha 4) | 1.09 | 0.13 | 0.405 | 0.539 | 2.50 | 1.32 | <0.001 | <0.001 | |
| Desmosome | Dsc2 (Desmocollin 2) | 1.40 | 0.49 | 0.001 | 0.005 | 2.89 | 1.53 | <0.001 | <0.001 |
| Dsg2 (Desmoglein 2) | 1.51 | 0.59 | <0.001 | <0.001 | 2.25 | 1.17 | <0.001 | <0.001 | |
| Dsg3 (Desmoglein 3) | 9.13 | 3.19 | <0.001 | <0.001 | |||||
| Gap Junction | Gjb4 (Connexin 30.3) | 14.1 | 3.82 | <0.001 | <0.001 | ||||
| Gjb3 (Connexin 31) | 0.75 | −0.42 | 0.003 | 0.010 | 3.20 | 1.68 | <0.001 | <0.001 | |
| Gjb5 (Connexin 31.1) | 9.19 | 3.20 | <0.001 | <0.001 | |||||
| Gjb1 (Connexin 32) | 0.80 | −0.32 | 0.010 | 0.028 | 0.35 | −1.51 | <0.001 | <0.001 | |
| Gja5 (Connexin 40) | 29.0 | 4.86 | <0.001 | <0.001 | |||||
| Gja1 (Connexin 43) | 2.19 | 1.13 | 0.012 | 0.032 | 0.46 | −1.12 | 0.002 | 0.005 | |
| Cell Adhesion Molecules | Ceacam1 (Carcinoembryonic Antigen-related Cell Adhesion Molecule 1) | 1.54 | 0.63 | <0.001 | <0.001 | 2.33 | 1.22 | <0.001 | <0.001 |
| Ceacam10 (Carcinoembryonic Antigen-related Cell Adhesion Molecule 10) | 1.35 | 0.43 | 0.007 | 0.022 | 0.17 | −2.57 | <0.001 | <0.001 | |
| Ceacam18 (Carcinoembryonic Antigen-related Cell Adhesion Molecule 18) | 0.58 | −0.79 | <0.001 | <0.001 | 2.12 | 1.09 | 0.030 | 0.070 | |
| Ceacam20 (Carcinoembryonic Antigen/CEA) | 1.47 | 0.56 | <0.001 | 0.001 | 2.01 | 1.01 | <0.001 | 0.002 | |
| Mucins | Muc1 (Mucin 1) | 2.36 | 1.24 | <0.001 | <0.001 | 0.49 | −1.02 | 0.066 | 0.123 |
| Muc2 (Mucin 2) | 0.76 | −0.39 | 0.015 | 0.041 | 0.20 | −2.34 | <0.001 | <0.001 | |
| Muc3 (Mucin 3) | 0.98 | −0.03 | 0.864 | 0.913 | |||||
| Muc4 (Mucin 4) | 2.01 | 1.01 | <0.001 | <0.001 | 2.19 | 1.13 | <0.001 | <0.001 | |
| Muc13 (Mucin 13) | 1.31 | 0.39 | 0.008 | 0.023 | 0.65 | −0.62 | 0.001 | 0.002 | |
| Muc20 (Mucin 20) | 2.07 | 1.05 | 0.006 | 0.019 | |||||
Comparison of two RNA-sequencing (RNA-seq) datasets. Left columns titled “SMAD4 KO in vivo” represent RNA-seq data comparing gene expression in the colon epithelium of three Smad4ΔLrig1 and three control mice. Right columns titled “TGFβ1/BMP2 Treatment in vitro” represent RNA-seq data comparing gene expression in wild-type colonoids treated with TGFβ1 + BMP2 or vehicle × 24 h (three biological replicates). Rows bolded represent genes that are significantly changed (P < 0.05) in opposite directions with SMAD4 knockout in vivo and TGFβ/BMP treatment in vitro, suggesting a possible role for direct regulation by canonical TGFβ signaling. BMP, bone morphogenetic protein; FDR, false discover rate; KO, knockout; TGFβ, transforming growth factor-β.
TGFβ Pathway Stimulation Results in Junctional Protein Gene Expression Changes in Vitro
Using an unbiased approach to determine whether the barrier function-related genes altered in Smad4ΔLrig1 mice are due to a cell-intrinsic process of TGFβ-dependent gene expression versus other downstream functions of SMAD4 in vivo, a complimentary RNA-seq experiment was conducted in vitro. Importantly, because many transformed and immortalized two-dimensional cell lines either fail to polarize or exhibit abnormal expression of tight junction proteins in vitro, these experiments were performed using three-dimensional colon organoids (hereafter referred to as “colonoids”) which are known to form tight junctions (69). Colonoids generated from wild-type mice were incubated in vitro with TGFβ1 and BMP2 or vehicle for 24 h and RNA-seq analysis was again performed (14). An exhaustive survey of the RNA-seq results for barrier function-related genes was performed (Table 3, right columns). This analysis revealed 40 differentially expressed barrier function-related genes.
A comparison of the differentially expressed genes due to SMAD4 loss in vivo and TGFβ pathway stimulation in vitro revealed 11 candidate genes that could potentially be directly regulated by canonical TGFβ family signaling as evidenced by significant (P < 0.05) but opposite gene expression changes in the two described experimental systems. These 11 genes included Cldn2, Cldn3, Cldn4, Cldn7, Cldn8, Pkp3, Gjb3, Gja1, Ceacam10, Ceacam18, and Muc13 (bolded in Table 3).
Five of the aforementioned genes are of particular interest in the context of our observed tight junction-dependent barrier defect: Cldn2 (encoding Claudin 2), Cldn3 (Claudin 3), Cldn4 (Claudin 4), Cldn7 (Claudin 7), and Cldn8 (Claudin 8). The Claudin proteins are an integral part of the tight junction complex (70), and altered Claudin protein levels are associated with tight junction disassembly and impaired barrier integrity (60, 63). Because intestine-specific SMAD4 loss appears to be related to a tight junction-dependent “leak” pathway phenotype in vivo, we chose to focus our subsequent experiments primarily on the above implicated Claudins.
Loss of SMAD4 within Intestinal Epithelium of Mice is Associated with Altered Levels of Claudin Protein but Normal Cellular Localization
To determine if alterations in Cldn mRNA levels were reflected in altered protein levels, we isolated the colon epithelium from five Smad4ΔLrig1 and five SMAD4+ control mice and created protein lysates which we used to perform Western blots (WB). WB analysis demonstrated an increase in Claudin 2 and Claudin 8 protein levels in the colon epithelium of Smad4ΔLrig1 mice compared with control. Conversely, a significant decrease in Claudin 7 protein was observed in the colon epithelium of mice with intestinal deletion of SMAD4, consistent with the changes in RNA expression observed by RNA-seq. Claudin 3 protein levels were not significantly changed in Smad4ΔLrig1 mice compared with control (Fig. 4, A and B).
Figure 4.


Loss of intestinal Smad4 expression is associated with altered levels, but not localization, of several Claudin proteins. A: Western blots (WB) from protein lysates generated from colon epithelium of five Smad4ΔLrig1 and five control mice. B: quantification of relative protein density by WB. Protein density was compared between mouse genotypes by Mann–Whitney test. C: immunohistochemistry for SMAD4 protein (brown) in SMAD4+ control and Smad4ΔLrig1 mice. D: immunofluorescence staining for indicated Claudin proteins. Claudin proteins in red, nuclei in green. Statistical significance is designated as nsP ≥ 0.05, *P < 0.05, **P < 0.01.
To determine if there were any alterations in junctional protein localization in Smad4ΔLrig1 mice compared with control, we performed immunostaining of mouse colons with and without SMAD4 expression. Immunostaining confirmed >90% SMAD4 loss in the colons of Smad4ΔLrig1 mice (Fig. 4C). For both Smad4ΔLrig1 and SMAD4+ mice, immunofluorescence staining demonstrated Claudin 2 subapical localization largely concentrated in cells at the crypt base (Fig. 4D). Claudins 3 and 7 were localized to the subapical and basolateral membranes and were uniformly present along the crypt axis in both Smad4ΔLrig1 and SMAD4+ mice. Similar to Claudins 3 and 7, Claudin 8 was expressed subapically and basolaterally, however unlike Claudins 3 and 7, Claudin 8 was predominantly localized in the middle and tops of crypts with very low levels observed at the crypt base. Protein localization for each of the aforementioned Claudin proteins was consistent with previously published reports (71–73).
These data suggest that epithelial TGFβ signaling via SMAD4 modulates the levels, but not the localization, of Claudin 2, Claudin 7, and Claudin 8 protein and that epithelial TGFβ signaling does not impact Claudin 3 production or localization in vivo. Unfortunately, we were unable to reliably detect Claudin 4 by immunostaining or Western Blot and thus were unable to assess Claudin 4 production or localization in vivo.
TGFβ and BMP Signaling via SMAD4 Regulate Cldn Gene Expression in a Cell-Autonomous Manner
To determine if TGFβ signaling is modulating the expression of critical junctional proteins in a cell-autonomous manner, a series of experiments were performed in vitro. Wild-type colonoids were treated with TGFβ1, BMP2, both, or vehicle for 24 h and RNA was subsequently isolated for qPCR. Induction of Smad7 expression served as a control for ligand activity. This largely validated our RNA-seq findings and demonstrated that stimulation of the TGFβ signaling pathway with either TGFβ1, BMP2, or both resulted in differential expression of tight junction-related genes. Although Cldn2 and Cldn8 expression were significantly decreased with TGFβ pathway stimulation by either TGFβ1, BMP2, or both, Cldn4 and Cldn7 expression were significantly increased with TGFβ1, BMP2, or both. Expression levels of Cldn3, on the other hand, did not change significantly with TGFβ pathway stimulation by qPCR (Fig. 5A).
Figure 5.

Transforming growth factor-β (TGFβ)/bone morphogenetic protein (BMP) signaling via SMAD4 modulates the expression of Cldn2, Cldn4, Cldn7, and Cldn8 in a cell-autonomous manner. A: wild-type colonoids treated with TGFβ1, BMP2, both, or vehicle for 24 h. B: SMAD4 knockout and control colonoids treated with TGFβ1/BMP2 (T/B) or vehicle for 24 h. Fold change indicates the relative level of mRNA compared with vehicle-treated wild-type controls for indicated genes. Each data point represents a single biological replicate, with biological replicates treated and harvested on different days. Experimental arms were compared using nonparametric Kruskal–Wallis test with post hoc Welch’s t test. Smad7 is used as a positive control to confirm ligand activity. Statistical significance is designated as *P < 0.05, **P < 0.01, ***P < 0.001.
Colonoids generated from Lrig1CreERT2;Smad4fl/fl mice (that did not receive tamoxifen in vivo) were additionally treated with 4OH-tamoxifen or vehicle in vitro to generate SMAD4 knockout or SMAD4 positive control colonoids, respectively. Knockout and control colonoids were then stimulated with either TGFβ1/BMP2 co-treatment or vehicle control for 24 h before RNA isolation. This experiment revealed that in the setting of TGFβ pathway stimulation, SMAD4 knockout in vitro resulted in increased Cldn2 and Cldn8 expression and decreased Cldn4 and Cldn7 expression. In addition, in the absence of SMAD4 expression, TGFβ pathway stimulation failed to elicit significant changes in Claudin gene expression, confirming that the above-described changes in Claudin gene expression due to TGFβ/BMP treatment are dependent on the SMAD-dependent (canonical) signaling pathway rather than the SMAD-independent (noncanonical) TGFβ pathways (Fig. 5B). Of note, gene expression levels of Cldn3 were not significantly changed with SMAD4 knockout in vitro. Collectively, these results suggest that TGFβ signaling via SMAD4 modulates Cldn2, Cldn4, Cldn7, and Cldn8 gene expression in mouse colon epithelial cells in a cell-autonomous manner, and that the decreased Cldn3 expression observed with SMAD4 loss in vivo may not be due to a cell-intrinsic process.
TGFβ Pathway Signaling Regulates Transepithelial Resistance in a Cell-Autonomous Manner
To determine if canonical TGFβ signaling has a direct effect on colonocyte barrier function in a cell autonomous manner, SMAD4-null and SMAD4+ control colonoids were plated onto collagen-coated transwells and allowed to polarize. Following polarization [TER ≥ 500 ohms (Ω)] on days 2–3 after plating (Fig. 6A), ALI was initiated (Fig. 6B). Simultaneous to ALI initiation, transwells were treated with TGFβ/BMP co-treatment or vehicle. TGFβ/BMP co-treatment resulted in a significant increase in TER in SMAD4+ colonoids compared with vehicle-treated controls at both 24- (fold change 1.86 vs. 1.31) and 48-h (fold change 1.95 vs. 1.09) (P < 0.001, Fig. 6C). Importantly, SMAD4-null colonoids were unresponsive to TGFβ pathway stimulation and demonstrated no significant difference in TER compared with vehicle-treated control colonoids at 24- (fold change 1.35 vs. 1.26) or 48-h (fold change 1.22 vs. 1.16) (P = 0.641, Fig. 6D). These data suggest that TGFβ pathway stimulation directly increases TER in mouse colonocytes in a manner dependent on SMAD4.
Figure 6.

Transforming growth factor-β (TGFβ)/bone morphogenetic protein (BMP) signaling via SMAD4 modulates barrier function/transepithelial resistance in a cell-autonomous manner. Representative images of hematoxylin and eosin (H&E) stain demonstrating polarized monolayers of colon epithelial cells grown on collagen-coated transwell membranes before (A) and after (B) initiation of air-liquid interface (ALI). C: normalized transepithelial resistance (TER) for wild-type (SMAD4+) colonoids on collagen-coated transwells in an ALI system. TGFβ/BMP co-treatment in the basolateral chamber was associated with a significant increase in TER compared with vehicle-treated controls at 24- and 48-h (P < 0.001 by repeated-measured ANOVA/mixed effects model). D: normalized TER for SMAD4 knockout colonoids grown on collagen-coated transwells in an ALI system. TGFβ/BMP co-treatment in the basolateral chamber failed to elicit a change in TER compared with vehicle-treated controls at 24- or 48-h. For both C and D, data represent the mean and standard deviation of three biological replicates, with biological replicates being plated, treated, and measured on separate days. Experimental arms were compared using repeat measures ANOVA/Mixed effects model. TER measurements for each well were normalized to the final pretreatment TER for that well (Day 0 on the X-axis).
TGFβ Signaling Modulates Junctional Protein Gene Expression by Disparate Mechanisms
To begin to understand the mechanism by which canonical TGFβ signaling pathway is modulating the expression of these critical junctional proteins, a time course experiment was performed. Wild-type colonoids underwent vehicle or TGFβ1/BMP2 co-treatment for 0-, 2-, 4-, or 16-h. TGFβ pathway stimulation significantly decreased the expression of Cldn2 as early as 4 h posttreatment with a further decrease in expression at 16 h posttreatment compared with vehicle-treated control colonoids (Fig. 7A). On the other hand, Cldn4 and Cldn8 were not significantly altered at 0-, 2-, or 4-h post-treatment, but were significantly altered (Cldn4 increased and Cldn8 decreased) by 16 h posttreatment compared with vehicle-treated control colonoids. Cldn7 expression levels, however, were not significantly changed at 0-, 2-, 4-, or 16-h although they were significantly changed with TGFβ1/BMP2 co-treatment and SMAD4 knockout at 24 h in the previously described experiments (Fig. 5). These data suggest that more than one mechanism may be implicated in the modulation of Cldn gene expression by canonical TGFβ signaling.
Figure 7.

Transforming growth factor-β (TGFβ)/bone morphogenetic protein (BMP) signaling via SMAD4 modulates the expression of Cldn genes by disparate mechanisms. A: wild-type colonoids treated with TGFβ1/BMP2 (gray bars/open boxes) or vehicle (white bars/closed circles) for the indicated periods of time. B: wild-type colonoids treated with TGFβ1/BMP2 (T/B) vs. vehicle (white bars, left), T/B vs. vehicle in the presence of actinomycin D (ActD) (light gray bars, middle), or T/B vs. vehicle in the presence of cyclohexamide (CHX) (dark gray bars, right) for 6.5 h. Fold change indicates the relative level of mRNA for indicated genes compared with vehicle-treated control. Each data point represents a single biological replicate, with biological replicates treated and harvested on different days. Experimental arms were compared using nonparametric Kruskal–Wallis test with post hoc Welch’s t test. Smad7 is used as a positive control to confirm ligand activity. Statistical significance is designated as *P < 0.05, **P < 0.01.
TGFβ Signaling Modulates Cldn2, Cldn4, Cldn7, and Cldn8 Gene Expression in a Manner That is Dependent on Nascent RNA Transcription
To determine if changes in Cldn gene expression levels were due to altered mRNA stability or to altered transcription, we blocked transcription using actinomycin D (ActD) (74) in wild-type colonoids with and without TGFβ1/BMP2 stimulation. We found that ActD was 100% lethal to our colonoids by 24 h but colonoids retained at least 50% viability in the presence of ActD for 6.5 h. As a positive control, ActD prevented the induction of Smad7, a known direct target of SMAD-mediated transcription (53). Treatment of wild-type colonoids with TGFβ1/BMP2, +/−ActD, or their respective vehicles for 6.5 h demonstrated that, for Cldn2, Cldn4, and Cldn8, TGFβ1/BMP2-dependent changes in gene expression levels were blocked by ActD co-treatment, suggesting that TGFβ pathway-dependent changes in Cldn2, Cldn4, and Cldn8 gene expression levels are dependent on a mechanism which includes nascent transcription (Fig. 7B). Because Cldn7 levels were not significantly changed at 6.5 h with TGFβ pathway stimulation in the absence of ActD, it is impossible to tell from this experiment if TGFβ-mediated regulation of Cldn7 requires nascent transcription.
TGFβ Signaling Modulates the Transcription of Cldn4 and Cldn8 Genes in a Manner That is Independent of Nascent Protein Translation
To determine if these TGFβ-dependent changes in Cldn expression are dependent on novel protein synthesis, we blocked protein translation using cycloheximide (CHX) (75) in wild-type colonoids with and without TGFβ1/BMP2 stimulation. Similar to the ActD experiment described in TGFβ Signaling Modulates Cldn2, Cldn4, Cldn7, and Cldn8 Gene Expression in a Manner That is Dependent on Nascent RNA Transcription, the 6.5-h timepoint was used for this experiment as the colonoids retained at least 50% viability in the presence of CHX at 6.5 h and colonoid viability dropped precipitously after that point. CHX did not alter the ability of TGFβ1/BMP2 to induce Smad7 expression, as expected (Fig. 7B). Similarly, in the setting of CHX, TGFβ1/BMP2 were still able to increase Cldn4 expression and decrease Cldn8 expression, suggesting that TGFβ-dependent changes in Cldn4 and Cldn8 expression occur in a manner independent of nascent protein translation.
Interestingly, CHX treatment dramatically decreased Cldn2 levels regardless of the presence or absence of TGFβ pathway stimulation, suggesting that Cldn2 mRNA expression is dependent on nascent protein synthesis. Given this finding, it is not possible to determine from this experiment whether TGFβ pathway-dependent regulation of Cldn2 expression relies on nascent protein synthesis. As in the ActD experiment, Cldn7 levels were not significantly changed at 6.5 h with TGFβ pathway stimulation even in the absence of CHX, making it impossible to determine if TGFβ-dependent Cldn7 expression relies on nascent protein production.
Altered Expression of SMAD4 and Claudin Genes Are Associated with Human IBD and Colon Cancer
To correlate the earlier-described findings in mice with human disease, we examined the relationship between TGFβ pathway signaling and CLDN expression in human specimens by querying a previously published database consisting of transcriptomic data generated from colonoscopic biopsy samples and analyzed by Microarray (GSE75214) (7). This database included colon mucosal biopsies from 74 patients with active UC (UCa), 23 patients with inactive UC (UCi), 8 patients with active Crohn’s disease (CDa), and 11 healthy controls (HC). This analysis demonstrated that SMAD4 expression was significantly decreased in the colon of UCa and CDa specimens compared with HC specimens, similar to previously published reports (14, 76). In addition, there were significant perturbations in CLDN expression in IBD colon specimens compared with HC specimens (Fig. 8A). CLDN2 expression was significantly increased in UCa specimens compared with HC specimens whereas CLDN4, CLDN7, and CLDN8 were significantly decreased in UCa specimens compared with HC specimens. These alterations in claudin gene expression in IBD patient samples are consistent with previously published reports (7, 77–81). Importantly, a significant inverse correlation exists between SMAD4 expression and CLDN2 expression, whereas a positive correlation was observed between CLDN4, CLDN7, and CLDN8 expression and SMAD4 expression in the colons of patients with IBD (Fig. 8B).
Figure 8.

SMAD4 and CLDN genes are dysregulated in human inflammatory bowel disease (IBD) and colon cancer. A: in silico analysis of human microarray database (accession number GSE75214). Samples represent human colon biopsy samples from patients with active ulcerative colitis (UCa, n = 74), inactive ulcerative colitis (UCi, n = 23), active Crohn’s disease (CDa, n = 8), and healthy controls (HC, n = 11). Gene expression in human biopsy samples were compared between groups using the nonparametric Kruskal–Wallis test with post hoc Welch’s t test. B: correlation between SMAD4 expression and indicated CLDN gene expression in human colon biopsy specimens from GSE75214. Spearman’s correlation was used to measure correlation between SMAD4 and CLDN gene expression. C: in silico analysis of RNA-sequencing data from The Cancer Genome Atlas (TCGA) database. Samples represent mRNA expression in colon cancer (CC, n = 283) or healthy control colon (HCc, n = 41) specimens. Nonparametric Mann–Whitney test was used to compare gene expression levels between groups. D: correlation between SMAD4 expression and indicated CLDN gene expression in human colon specimens from TCGA. Spearman’s correlation was used to measure correlation between SMAD4 and CLDN gene expression. For A–D, gene expression levels are represented on a Log2 scale. Statistical significance is designated as **P < 0.01, ***P < 0.001.
We additionally queried the TCGA database to determine whether this relationship between SMAD4 and CLDN gene expression was observed in sporadic colon cancers in addition to IBD. The TCGA database includes gene expression data generated from RNA-seq analysis of 282 primary colon cancers (CC) and 41 healthy control colon (HCc) (Fig. 8C). By TCGA analysis, SMAD4 expression was significantly decreased in CC specimens compared with HCc specimens, consistent with prior reports of decreased SMAD4 expression in CRC (14, 82, 83). In addition, CLDN2 expression is significantly increased in CC specimens compared with HCc specimens whereas expression levels of CLDN4, CLDN7, and CLDN8 are significantly lower in CC specimens compared with HCc. Changes in claudin gene expression levels observed here by TCGA analysis are largely consistent with prior published reports (84–88). Importantly, CLDN2 and SMAD4 expression are inversely correlated whereas CLDN7 and CLDN8 expression are both positively correlated with SMAD4 expression in CC and HCc specimens. Interestingly, CLDN4 expression is not significantly correlated with SMAD4 expression in human CC and HCc specimens in the TCGA database (Fig. 8D).
DISCUSSION
Although the pathophysiology of IBD and CAC is complex with multiple genetic, environmental, and dietary factors likely contributing (89), increased mucosal permeability and impaired barrier function have been observed in first-degree relatives of patients with Crohn’s disease (90, 91) and have also been noted to precede inflammatory symptoms in patients with IBD (92), suggesting that a primary intestinal barrier defect may be a contributing factor in some cases (3–8). At the same time, TGFβ signaling defects have been associated with IBD (1, 9–12, 14, 93). However, the relationship between intestinal TGFβ signaling, barrier function, and IBD has not been deeply explored. Both TGFβ signaling in general as well as its regulation of barrier function specifically has been shown to be highly tissue- and context-dependent. While in the pulmonary endothelium and in the esophagus TGFβ signaling profoundly disrupts barrier integrity and decreases TER (15–19), in immortalized two-dimensional jejunal cell lines and in colon cancer cell lines in vitro, TGFβ treatment increases TER (20–25, 27). However, until now, the impact of canonical TGFβ signaling for maintenance of barrier function in nonmalignant colon epithelium in vitro or in colon epithelium in vivo had yet to be explored.
Our data indicate that loss of canonical TGFβ signaling in the intestinal epithelium of mice through conditional and tissue-specific deletion of SMAD4 leads to increased colon mucosal permeability to a 4 kDa dextran molecule (diameter 28 Å). However, these mice show no gross or histologic evidence of mucosal ulceration or damage, and no increased translocation of luminal bacteria across the colon epithelium, suggesting that the permeability defect due to epithelial SMAD4 loss likely represents a tight junction-dependent “leak” phenotype rather than a more extensive mucosal “damage” phenotype (60). Although we did observe minor changes in mucin-related gene expression with intestinal SMAD4 loss in vivo (including increased expression of Muc1, Muc13, and Muc20 and decreased expression of Muc2), only Muc13 was significantly and reciprocally altered with TGFβ pathway stimulation in vitro. In addition, staining for mucin accumulation demonstrated no obvious changes in mucin quantity or distribution, making a primary TGFβ-dependent mucin barrier defect unlikely to be a major driver of barrier dysfunction. Although paracellular transport via the tight junction is mediated by two functionally distinct pathways [the “pore” and “leak” pathways (60)], increased permeability to macromolecule 4 kDa dextran is suggestive of a “leak” pathway-dependent process (57–62) whereby passage of larger ions and molecules up to 100 Å are able to pass without charge selectivity. In addition, although some statistically significant differences in microbiome composition/diversity were detected by 16S sequencing, the implications for their influence on inflammatory, tumorigenic, or barrier phenotypes seen in Smad4ΔLrig1 mice are unclear.
RNA-seq analysis of Smad4ΔLrig1 mice compared with control revealed alterations in a number of genes related to barrier function. Although alterations in any of these genes could contribute to the barrier dysfunction observed here or to the inflammatory phenotype previously described (14), we chose to focus our attention on five Claudin genes (Cldn2, Cldn3, Cldn4, Cldn7, and Cldn8) for two main reasons. First, in addition to demonstrating altered expression levels in colon epithelium in vivo with SMAD4 loss, these five genes additionally demonstrated reciprocal changes in mouse colonoids with TGFβ pathway stimulation in vitro, suggesting that these genes could be directly modulated by TGFβ signaling as opposed to being a downstream effect of other pathways regulated by SMAD4 loss in vivo (such as increased inflammation). Second, among the 11 genes that were reciprocally changed with both SMAD4 loss in vivo and TGFβ pathway stimulation in vitro, these five Claudin genes are known to transcribe proteins with critical roles in tight junction function and are therefore likely to regulate the observed barrier phenotype (70, 72, 73, 78–81, 94–96).
We also demonstrated that at least some of these observed changes in tight junction-related genes observed in vivo are the result of colonocyte-intrinsic TGFβ signaling via SMAD4. Although TGFβ pathway stimulation resulted in decreased Cldn2 and Cldn8 expression and increased Cldn4 and Cldn7 expression in wild-type colonoids, this TGFβ-responsiveness was lost with SMAD4 deletion in vitro, confirming that these Claudin genes are regulated by the SMAD-dependent (canonical) TGFβ signaling pathway in mouse colon epithelium and that this regulation occurs in a cell-autonomous manner. Importantly, we also demonstrated the TGFβ pathway stimulation of wild-type colonoids grown on transwell membranes in vitro results in increased TER and that this effect is SMAD4-dependent. Collectively, these data suggest an epithelium-intrinsic role for canonical TGFβ signaling in colonic barrier function.
Although we have yet to fully understand the mechanism for TGFβ-dependent modulation of Claudin gene expression, our data do allow us to make several conclusions regarding mechanism of action. First, from the in vitro SMAD4 knockout experiment described in Fig. 5, we know that Cldn2, Cldn4, Cldn7, and Cldn8 are regulated by TGFβ through the canonical (SMAD-dependent) pathway. This differs from what has been published previously with regard to TGFβ regulation of barrier function, the majority of which implicates noncanonical pathways (15, 16, 20, 21, 97–99). Second, the impact of TGFβ signaling on the expression of Cldn2, Cldn4, and Cldn8 is blunted with blockade of RNA transcription, demonstrating that TGFβ modulation of Cldn2, Cldn4, and Cldn8 are dependent on nascent RNA transcription. Third, TGFβ pathway regulation of Cldn4 and Cldn8 appears to be independent of nascent protein translation, as evidenced by the fact that CHX co-treatment did not abrogate the ability of TGFβ pathway stimulation to alter their expression levels. An additional point of interest is that Qi et al. (100) examined SMAD4-DNA binding in small intestinal stem cells through chromatin immunoprecipitation sequencing (ChIP-seq) and reported in their supplemental data that SMAD4 binding sites existed in the promoter/enhancer regions of multiple junctional protein-related genes including Cldn3, Cldn4, Cldn7, Cldn12, Cldn14, Cldn22, Cldn23, and Cldn25. Given the tissue- and context-dependent nature of SMAD complex binding and Claudin gene expression, beyond the scope of the current manuscript, similar chromatin-binding experiments will be required to determine whether SMAD binding to the same sites is evident in colon epithelium. Nonetheless, collectively, these data suggest the possibility that the canonical TGFβ signaling pathway modulates the expression of a subset of Claudin genes through direct SMAD complex binding to their respective promoter/enhancer regions.
Importantly, we found that human biopsy specimens from colon cancers and patients with IBD have significantly altered expression levels of SMAD4 as well as CLDN2, CLDN4, CLDN7, and CLDN8, consistent with prior reports (7, 14, 76–80, 82–88). In addition, we found that SMAD4 levels are significantly correlated with CLDN2, CLDN4, CLDN7, and CLDN8 levels in human IBD and/or CRC. Interestingly, although CLDN2, CLDN4, and CLDN7 are directionally altered in ways that are consistent with the changes observed with intestinal SMAD4 loss in mice, CLDN8 expression levels are altered in the opposite direction. Although we found Cldn8 to be significantly decreased in mouse colonoids with TGFβ pathway stimulation in vitro and to be significantly increased with SMAD4 loss in mice, CLDN8 is significantly decreased and positively correlated with SMAD4 expression in both human IBD and colon cancer specimens. The reason for this disparity between our observations in mice and those observed in human disease are not entirely understood. One possibility is that other important pathways that are frequently altered in human IBD and colon cancers besides the TGFβ pathway [including APC/WNT, KRAS, BRAF, TP53 pathways and others (101)] also regulate CLDN8 expression, masking the SMAD-dependent effects in these human disease specimens. Alternatively, it is possible that the TGFβ signaling pathway via SMAD4 in humans has a different effect on CLDN8 expression than it does on the homologous gene in mice. Such a hypothesis is supported by a recent publication by Toyonaga et al. (26), which demonstrated that TGFβ pathway stimulation via the ALK1 receptor increased CLDN8 expression in cultured human intestinal epithelial cells. Further studies are required to understand the relationship more fully between canonical TGFβ signaling, CLDN8 expression, and human IBD and colon cancer.
The implications of our findings are wide ranging. First, TGFβ signaling is known to be frequently disrupted in IBD, CRC, and CAC through TGFβ receptor mutations, loss of SMAD protein expression, and other pathway alterations. In addition, barrier defects are known to contribute to intestinal inflammation and IBD, and our findings suggest that TGFβ-dependent barrier regulation may be a contributing factor in IBD pathogenesis. Second, Claudin proteins, while best known for roles in governing paracellular permeability, also have additional roles in cell proliferation, transformation, carcinogenesis, and metastasis (85, 102–105). The alterations we see in Cldn2, Cldn4, Cldn7, and Cldn8 in these experiments very well could have implications for CRC and CAC development in mouse models and in human cancers beyond their direct role in maintaining barrier function. Third, with the increased prevalence of TGFβ inhibitors in therapeutics and clinical trials (106), it is important to understand that these therapies may ultimately have an impact on intestinal barrier function/junctional protein expression which may have clinical implications.
Several elements of the relationship between TGFβ signaling and colon epithelial barrier function remain to be discovered. First, the precise mechanisms by which these Claudin genes are regulated by TGFβ signaling are yet to be resolved. Although our data suggest that the TGFβ-dependent regulation of the Claudin genes is dependent on the canonical signaling pathway, is reliant on nascent transcription and, in some cases, is independent of novel protein translation, more work needs to be done to definitively determine whether these genes are regulated by direct SMAD complex binding to promoter/enhancer regions or whether other cell-intrinsic processes are governing these changes. Second, although our data support the idea that TGFβ via SMAD4 signaling directly modulates the expression of critical junctional proteins in vitro, whether a primary barrier defect due to intestinal SMAD4 deletion contributes to the inflammatory and CAC phenotypes that we previously discovered in Smad4ΔLrig1 mice remains unexplored. Future in vivo experiments targeting specific junctional protein alterations will help to determine whether abrogating some of the barrier-specific effects of SMAD4 loss can reverse the observed barrier defect and/or inflammatory phenotype previously described. Third, how SMAD4-dependent regulation of inflammation in vivo impinges on barrier function remains unexplored. Cytokines, including TNFα and IFNγ, have been previously shown to impact the expression of Claudins in multiple tissue types (41, 107–109), and intestinal SMAD4 loss has previously been demonstrated to significantly impact inflammatory gene expression and leukocyte recruitment (14). It is possible, even likely, that in addition to TGFβ-dependent barrier regulation, direct regulation of inflammatory gene expression by SMAD4 may have an additive downstream effect on Claudin gene expression and/or barrier function in vivo, and this remains to be explored. Finally, we chose to focus this paper primarily on the tight junction-related genes given the “leak” phenotype observed in vivo with intestinal SMAD4 loss, however it is highly possible that some of the other barrier function-related genes (including Pkp3, Gjb3, Gja1, Ceacam10, Ceacam18, and Muc13) are also playing an important role in TGFβ-dependent maintenance of barrier function, and these genes and their protein products deserve closer examination in the future.
In summary, we discovered that loss of epithelial canonical TGFβ signaling via intestine-specific SMAD4 loss is associated with a barrier defect in mouse colon that is consistent with a deficiency in the tight junction-mediated “leak” pathway. Further, we conclude from our observations that the canonical TGFβ signaling pathway regulates the expression of several tight junction-related genes as well as colonic epithelial cell TER in a cell-autonomous manner. Further investigation is necessary to fully understand the mechanism of TGFβ regulation of Claudin gene expression as well as the clinical implications of these findings.
GRANTS
This research was funded in part by National Cancer Institute Grants (F32 CA236309; T32 CA106183; R01 CA235016), the Burroughs Wellcome Fund Physician-Scientist Institutional Award No. 1018894/Vanderbilt Supporting Careers in Research for Interventional Physicians and Surgeons (SCRIPS), the Vanderbilt Ingram Cancer Center (VICC) Grant (P30 CA068485), the Vanderbilt Digestive Diseases Research Center Grant (P30 DK058404), and the Department of Veterans Affairs Career Development Award (IK2BX004648).
DISCLAIMERS
The results published here are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga, and the authors recognize the contribution of the specimen donors and research groups.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
P.M.S, Y.A.C., N.O.M., I.K., A.L.M., and R.D.B. conceived and designed research; P.M.S., Y.A.C., D.N.H., J.Z., C.J.W., J.A.H., and K.B.L. performed experiments; P.M.S., N.O.M., D.N.H., J.Z., C.J.W., J.A.H., J.Y., Q.L., I.K., A.L.M., and R.D.B., analyzed data; P.M.S., Y.A.C., N.O.M., D.N.H., J.Z., C.J.W., K.B.L., J.Y., Q.L., I.K., A.L.M., and R.D.B. interpreted results of experiments; P.M.S. prepared figures; P.M.S. drafted manuscript; P.M.S., Y.A.C., N.O.M., D.N.H., J.Y., Q.L., I.K., A.L.M., and R.D.B. edited and revised manuscript; P.M.S., Y.A.C., N.O.M., D.N.H., J.Z., C.J.W., J.A.H., K.B.L., J.Y., Q.L., I.K., A.L.M., and R.D.B. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors acknowledge the multiple Vanderbilt University and Vanderbilt University Medical Center core services for making this work possible, including: Vanderbilt University Cell Imaging Shared Resource (VU-CISR), Vanderbilt Technologies for Advanced Genomics (VANTAGE), Vanderbilt University Medical Center Translational Pathology Shared Resource (TPSR), the Vanderbilt University Molecular Cell Biology Resource Core, and the Vanderbilt University Medical Division of Animal Care (DAC). The authors used BioRender.com to generate the graphical abstract and Fig. 1A.
REFERENCES
- 1.Allaire JM, Darsigny M, Marcoux SS, Roy SAB, Schmouth J-F, Umans L, Zwijsen A, Boudreau F, Perreault N. Loss of Smad5 leads to the disassembly of the apical junctional complex and increased susceptibility to experimental colitis. Am J Physiol Gastrointest Liver Physiol 300: G586–G597, 2011. doi: 10.1152/ajpgi.00041.2010. [DOI] [PubMed] [Google Scholar]
- 2.Laukoetter MG, Nava P, Lee WY, Severson EA, Capaldo CT, Babbin BA, Williams IR, Koval M, Peatman E, Campbell JA, Dermody TS, Nusrat A, Parkos CA. JAM-A regulates permeability and inflammation in the intestine in vivo. J Exp Med 204: 3067–3076, 2007. doi: 10.1084/jem.20071416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martini E, Krug SM, Siegmund B, Neurath MF, Becker C. Mend your fences the epithelial barrier and its relationship with mucosal immunity in inflammatory bowel disease. Cell Mol Gastroenterol Hepatol 4: 33–46, 2017. doi: 10.1016/j.jcmgh.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McGuckin MA, Eri R, Simms LA, Florin THJ, Radford-Smith G. Intestinal barrier dysfunction in inflammatory bowel diseases. Inflamm Bowel Dis 15: 100–113, 2009. doi: 10.1002/ibd.20539. [DOI] [PubMed] [Google Scholar]
- 5.Sluis M. D, Koning BAED, Bruijn A, Velcich A, Meijerink JPP, Goudoever JBV, Büller HA, Dekker J, Seuningen IV, Renes IB, Einerhand AWC. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 131: 117–129, 2006. doi: 10.1053/j.gastro.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 6.Söderholm JD, Olaison G, Lindberg E, Hannestad U, Vindels A, Tysk C, Järnerot G, Sjödahl R. Different intestinal permeability patterns in relatives and spouses of patients with Crohn’s disease: an inherited defect in mucosal defence? Gut 44: 96, 1999. doi: 10.1136/gut.44.1.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vancamelbeke M, Vanuytsel T, Farré R, Verstockt S, Ferrante M, Assche GV, Rutgeerts P, Schuit F, Vermeire S, Arijs I, Cleynen I. Genetic and transcriptomic bases of intestinal epithelial barrier dysfunction in inflammatory bowel disease. Inflamm Bowel Dis 23: 1718–1729, 2017. doi: 10.1097/MIB.0000000000001246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wehkamp J, Koslowski M, Wang G, Stange EF. Barrier dysfunction due to distinct defensin deficiencies in small intestinal and colonic Crohn’s disease. Mucosal Immunol 1: S67–S74, 2008. doi: 10.1038/mi.2008.48. [DOI] [PubMed] [Google Scholar]
- 9.Babyatsky MW, Rossiter G, Podolsky DK. Expression of transforming growth factors alpha and beta in colonic mucosa in inflammatory bowel disease. Gastroenterology 110: 975–984, 1996. doi: 10.1053/gast.1996.v110.pm8613031. [DOI] [PubMed] [Google Scholar]
- 10.Monteleone G, Kumberova A, Croft NM, McKenzie C, Steer HW, MacDonald TT. Blocking Smad7 restores TGF-β1 signaling in chronic inflammatory bowel disease. J Clin Invest 108: 601–609, 2001. doi: 10.1172/JCI12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Monteleone G, Neurath MF, Ardizzone S, Sabatino AD, Fantini MC, Castiglione F, Scribano ML, Armuzzi A, Caprioli F, Sturniolo GC, Rogai F, Vecchi M, Atreya R, Bossa F, Onali S, Fichera M, Corazza GR, Biancone L, Savarino V, Pica R, Orlando A, Pallone F. Mongersen, an oral SMAD7 antisense oligonucleotide, and Crohn’s disease. New Engl J Med 372: 1104–1113, 2015. doi: 10.1056/NEJMoa1407250. [DOI] [PubMed] [Google Scholar]
- 12.Sedda S, Marafini I, Dinallo V, Fusco DD, Monteleone G. The TGF-β/Smad system in IBD pathogenesis. Inflamm Bowel Dis 21: 2921–2925, 2015. doi: 10.1097/MIB.0000000000000542. [DOI] [PubMed] [Google Scholar]
- 13.Alazzouzi H, Alhopuro P, Salovaara R, Sammalkorpi H, Jarvinen H, Mecklin J-P, Hemminki A, Schwartz S, Aaltonen LA, Arango D. SMAD4 as a prognostic marker in colorectal cancer. Clin Cancer Res 11: 2606–2611, 2005. doi: 10.1158/1078-0432.CCR-04-1458. [DOI] [PubMed] [Google Scholar]
- 14.Means AL, Freeman TJ, Zhu J, Woodbury LG, Marincola-Smith P, Wu C, Meyer AR, Weaver CJ, Padmanabhan C, An H, Zi J, Wessinger BC, Chaturvedi R, Brown TD, Deane NG, Coffey RJ, Wilson KT, Smith JJ, Sawyers CL, Goldenring JR, Novitskiy SV, Washington MK, Shi C, Beauchamp RD. Epithelial Smad4 deletion upregulates inflammation and promotes inflammation-associated cancer. Cell Mol Gastroenterol Hepatol 6: 257–276, 2018. doi: 10.1016/j.jcmgh.2018.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Birukova AA, Birukov KG, Adyshev D, Usatyuk P, Natarajan V, Garcia JGN, Verin AD. Involvement of microtubules and Rho pathway in TGF-β1-induced lung vascular barrier dysfunction. J Cell Physiol 204: 934–947, 2005. doi: 10.1002/jcp.20359. [DOI] [PubMed] [Google Scholar]
- 16.Goldberg PL, MacNaughtten DE, Clements RT, Minnear FL, Vincent PA. p38 MAPK activation by TGF-β1 increases MLC phosphorylation and endothelial monolayer permeability. Am J Physiol Lung Cell Mol Physiol 282: L146–L154, 2002. doi: 10.1152/ajplung.2002.282.1.l146. [DOI] [PubMed] [Google Scholar]
- 17.Hurst V, Goldberg PL, Minnear FL, Hiemark RL, Vincent PA. Rearrangement of adherens junctions by transforming growth factor-β1: role of contraction. Am J Physiol Lung Cell Mol Physiol 276: L582–L595, 1999. doi: 10.1152/ajplung.1999.276.4.L582. [DOI] [PubMed] [Google Scholar]
- 18.Pittet J-F, Griffiths MJD, Geiser T, Kaminski N, Dalton SL, Huang X, Brown LAS, Gotwals PJ, Koteliansky VE, Matthay MA, Sheppard D. TGF-β is a critical mediator of acute lung injury. J Clin Invest 107: 1537–1544, 2001. doi: 10.1172/JCI11963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nguyen N, Fernando SD, Biette KA, Hammer JA, Capocelli KE, Kitzenberg DA, Glover LE, Colgan SP, Furuta GT, Masterson JC. TGF-β1 alters esophageal epithelial barrier function by attenuation of claudin-7 in eosinophilic esophagitis. Mucosal Immunol 11: 415–426, 2018. doi: 10.1038/mi.2017.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hering NA, Andres S, Fromm A, Tol Ea van, Amasheh M, Mankertz J, Fromm M, Schulzke JD. Transforming growth factor-β, a whey protein component, strengthens the intestinal barrier by upregulating claudin-4 in HT-29/B6 cells. J Nutr 141: 783–789, 2011. doi: 10.3945/jn.110.137588. [DOI] [PubMed] [Google Scholar]
- 21.Howe KL, Reardon C, Wang A, Nazli A, McKay DM. Transforming growth factor-β regulation of epithelial tight junction proteins enhances barrier function and blocks enterohemorrhagic Escherichia coli O157:H7-induced increased permeability. Am J Pathol 167: 1587–1597, 2005. doi: 10.1016/S0002-9440(10)61243-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKay D, Singh P. Superantigen activation of immune cells evokes epithelial (T84) transport and barrier abnormalities via IFN-gamma and TNF-alpha: inhibition of increased permeability, but not diminished secretory responses, by TGF-beta2. J Immunol 159: 2382–2390, 1997. [PubMed] [Google Scholar]
- 23.Planchon S, Fiocchi C, Takafuji V, Roche JK. Transforming growth factor-β1 preserves epithelial barrier function: identification of receptors, biochemical intermediates, and cytokine antagonists. J Cell Physiol 181: 55–66, 1999. doi:. [DOI] [PubMed] [Google Scholar]
- 24.Planchon S, Martins C, Guerrant R, Roche J. Regulation of intestinal epithelial barrier function by TGF-beta1. Evidence for its role in abrogating the effect of a T cell cytokine. J Immunol 153: 5730–5739, 1994. [PubMed] [Google Scholar]
- 25.Roche JK, Martins CAP, Cosme R, Fayer R, Guerrant RL. Transforming growth factor β1 ameliorates intestinal epithelial barrier disruption by Cryptosporidium parvum in vitro in the absence of mucosal T lymphocytes. Infect Immun 68: 5635–5644, 2000. doi: 10.1128/IAI.68.10.5635-5644.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Toyonaga T, Steinbach EC, Keith BP, Barrow JB, Schaner MR, Wolber EA, Beasley C, Huling J, Wang Y, Allbritton NL, Chaumont N, Sadiq TS, Koruda MJ, Jain A, Long MD, Barnes EL, Herfarth HH, Isaacs KL, Hansen JJ, Shanahan MT, Rahbar R, Furey TS, Sethupathy P, Sheikh SZ. Decreased colonic activin receptor-like kinase 1 disrupts epithelial barrier integrity in patients with Crohn’s disease. Cell Mol Gastroenterol Hepatol 10: 779–796, 2020. doi: 10.1016/j.jcmgh.2020.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiao K, Cao S, Jiao L, Song Z, Lu J, Hu C. TGF-β1 protects intestinal integrity and influences Smads and MAPK signal pathways in IPEC-J2 after TNF-α challenge. Innate Immun 23: 276–284, 2017. doi: 10.1177/1753425917690815. [DOI] [PubMed] [Google Scholar]
- 28.Shi Y, Massagué J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 113: 685–700, 2003. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 29.Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol 13: 616–630, 2012. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moustakas A, Heldin C-H. Non-Smad TGF-β signals. J Cell Sci 118: 3573–3584, 2005. doi: 10.1242/jcs.02554. [DOI] [PubMed] [Google Scholar]
- 31.Zhang YE. Non-Smad pathways in TGF-β signaling. Cell Res 19: 128–139, 2009. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bardeesy N, Cheng K, Berger JH, Chu GC, Pahler J, Olson P, Hezel AF, Horner J, Lauwers GY, Hanahan D, DePinho RA. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Gene Dev 20: 3130–3146, 2006. doi: 10.1101/gad.1478706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Means AL, Xu Y, Zhao A, Ray KC, Gu G. A CK19CreERT knockin mouse line allows for conditional DNA recombination in epithelial cells in multiple endodermal organs. Genesis 46: 318–323, 2008. doi: 10.1002/dvg.20397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Powell AE, Wang Y, Li Y, Poulin EJ, Means AL, Washington MK, Higginbotham JN, Juchheim A, Prasad N, Levy SE, Guo Y, Shyr Y, Aronow BJ, Haigis KM, Franklin JL, Coffey RJ. The pan-ErbB negative regulator lrig1 is an intestinal stem cell marker that functions as a tumor suppressor. Cell 149: 146–158, 2012. doi: 10.1016/j.cell.2012.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, Yefanov A, Lee H, Zhang N, Robertson CL, Serova N, Davis S, Soboleva A. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res 41: D991–D995, 2013. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edgar R, Domrachev M, Lash AE. Gene expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30: 207–210, 2002. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blaine SA, Ray KC, Anunobi R, Gannon MA, Washington MK, Means AL. Adult pancreatic acinar cells give rise to ducts but not endocrine cells in response to growth factor signaling. Development 137: 2289–2296, 2010. doi: 10.1242/dev.048421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Means AL, Ray KC, Singh AB, Washington MK, Whitehead RH, Harris RC, Wright CVE, Coffey RJ, Leach SD. Overexpression of heparin-binding EGF-like growth factor in mouse pancreas results in fibrosis and epithelial metaplasia. Gastroenterology 124: 1020–1036, 2003. doi: 10.1053/gast.2003.50150. [DOI] [PubMed] [Google Scholar]
- 39.Ray KC, Moss ME, Franklin JL, Weaver CJ, Higginbotham J, Song Y, Revetta FL, Blaine SA, Bridges LR, Guess KE, Coffey RJ, Crawford HC, Washington MK, Means AL. Heparin-binding epidermal growth factor-like growth factor eliminates constraints on activated Kras to promote rapid onset of pancreatic neoplasia. Oncogene 33: 823–831, 2014. doi: 10.1038/onc.2013.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Macedonia MC, Drewes JL, Markham NO, Simmons AJ, Roland JT, Vega PN, Scurrah CR, Coffey RJ, Shrubsole MJ, Sears CL, Lau KS. Clinically adaptable polymer enables simultaneous spatial analysis of colonic tissues and biofilms. NPJ Biofilms Microbiomes 6: 33, 2020. doi: 10.1038/s41522-020-00143-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Amoozadeh Y, Dan Q, Xiao J, Waheed F, Szászi K. Tumor necrosis factor-α induces a biphasic change in claudin-2 expression in tubular epithelial cells: role in barrier functions. Am J Physiol Cell Physiol 309: C38–C50, 2015. doi: 10.1152/ajpcell.00388.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ashikari D, Takayama K, Obinata D, Takahashi S, Inoue S. CLDN8, an androgen-regulated gene, promotes prostate cancer cell proliferation and migration. Cancer Sci 108: 1386–1393, 2017. doi: 10.1111/cas.13269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheng B, Rong A, Zhou Q, Li W. CLDN8 promotes colorectal cancer cell proliferation, migration, and invasion by activating MAPK/ERK signaling. Cancer Manage Res 11: 3741–3751, 2019. doi: 10.2147/CMAR.S189558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Healing E, Charlier CF, Meira LB, Elliott RM. A panel of colorimetric assays to measure enzymatic activity in the base excision DNA repair pathway. Nucleic Acids Res 47: gkz171, 2019. doi: 10.1093/nar/gkz171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Otani T, Nguyen TP, Tokuda S, Sugihara K, Sugawara T, Furuse K, Miura T, Ebnet K, Furuse M. Claudins and JAM-A coordinately regulate tight junction formation and epithelial polarity Claudins and JAM-A in tight junction formation. J Cell Biol 218: 3372–3396, 2019. doi: 10.1083/jcb.201812157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou S, Piao X, Wang C, Wang R, Song Z. Identification of claudin-1, -3, -7 and -8 as prognostic markers in human laryngeal carcinoma. Mol Med Rep 20: 393–400, 2019. doi: 10.3892/mmr.2019.10265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13: 581–583, 2016. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336, 2010. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shiou S-R, Datta PK, Dhawan P, Law BK, Yingling JM, Dixon DA, Beauchamp RD. Smad4-dependent regulation of urokinase plasminogen activator secretion and RNA stability associated with invasiveness by autocrine and paracrine transforming growth factor-β. J Biol Chem 281: 33971–33981, 2006. doi: 10.1074/jbc.M607010200. [DOI] [PubMed] [Google Scholar]
- 50.Schneider CA, Rasband WS, Eliceiri KW. NIH image to ImageJ: 25 years of image analysis. Nat Methods 10: 847–856, 2012. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Freeman TJ, Smith JJ, Chen X, Washington MK, Roland JT, Means AL, Eschrich SA, Yeatman TJ, Deane NG, Beauchamp RD. Smad4-mediated signaling inhibits intestinal neoplasia by inhibiting expression of β-catenin. Gastroenterology 142: 562–571.e2, 2012. doi: 10.1053/j.gastro.2011.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Green MR, Sambrook J. Quantification of RNA by real-time reverse transcription-polymerase chain reaction (RT-PCR). Cold Spring Harb Protoc 2018: 2018. doi: 10.1101/pdb.prot095042.[30275077] [DOI] [PubMed] [Google Scholar]
- 53.Zhao J, Crowe D, Castillo C, Wuenschell C, Chai Y, Warburton D. Smad7 is a TGF-β-inducible attenuator of Smad2/3-mediated inhibition of embryonic lung morphogenesis. Mech Dev 93: 71–81, 2000. doi: 10.1016/s0925-4773(00)00281-1. [DOI] [PubMed] [Google Scholar]
- 54.Choksi YA, Reddy VK, Singh K, Barrett CW, Short SP, Parang B, Keating CE, Thompson JJ, Verriere TG, Brown RE, Piazuelo MB, Bader DM, Washington MK, Mittal MK, Brand T, Gobert AP, Coburn LA, Wilson KT, Williams CS. BVES is required for maintenance of colonic epithelial integrity in experimental colitis by modifying intestinal permeability. Mucosal Immunol 11: 1363–1374, 2018. doi: 10.1038/s41385-018-0043-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Simpson EH. Measurement of diversity. Nature 163: 688–688, 1949. doi: 10.1038/163688a0. [DOI] [Google Scholar]
- 56.Volynets V, Reichold A, Bárdos G, Rings A, Bleich A, Bischoff SC. Assessment of the intestinal barrier with five different permeability tests in healthy C57BL/6J and BALB/cJ mice. Digest Dis Sci 61: 737–746, 2016. doi: 10.1007/s10620-015-3935-y. [DOI] [PubMed] [Google Scholar]
- 57.Anderson JM, Itallie CMV. Physiology and function of the tight junction. Cold Spring Harb Perspect Biol 1: a002584, 2009. doi: 10.1101/cshperspect.a002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Itallie CMV, Holmes J, Bridges A, Gookin JL, Coccaro MR, Proctor W, Colegio OR, Anderson JM. The density of small tight junction pores varies among cell types and is increased by expression of claudin-2. J Cell Sci 121: 298–305, 2008. doi: 10.1242/jcs.021485. [DOI] [PubMed] [Google Scholar]
- 59.Itallie CV, Fanning A, Bridges A, Anderson J. ZO-1 stabilizes the tight junction solute barrier through coupling to the perijunctional cytoskeleton. Mol Biol Cell 20: 3930–3940, 2009. doi: 10.1091/mbc.e09-04-0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shen L, Weber CR, Raleigh DR, Yu D, Turner JR. Tight junction pore and leak pathways: a dynamic duo. Annu Rev Physiol 73: 283–309, 2011. doi: 10.1146/annurev-physiol-012110-142150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weber CR, Raleigh DR, Su L, Shen L, Sullivan EA, Wang Y, Turner JR. Epithelial myosin light chain kinase activation induces mucosal interleukin-13 expression to alter tight junction ion selectivity. J Biol Chem 285: 12037–12046, 2010. doi: 10.1074/jbc.M109.064808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zuo L, Kuo W-T, Turner JR. Tight junctions as targets and effectors of mucosal immune homeostasis. Cell Mol Gastroenterol Hepatol 10: 327–340, 2020. doi: 10.1016/j.jcmgh.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.France MM, Turner JR. The mucosal barrier at a glance. J Cell Sci 130: 307–314, 2017. doi: 10.1242/jcs.193482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim Y, Volpert G, Shin K, Kim S, Shin S, Lee Y, Sung SH, Lee Y, Ahn J, Pewzner-Jung Y, Park W, Futerman AH, Park J. Ablation of ceramide synthase 2 exacerbates dextran sodium sulphate-induced colitis in mice due to increased intestinal permeability. J Cell Mol Med 21: 3565–3578, 2017. doi: 10.1111/jcmm.13267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Panpetch W, Chancharoenthana W, Bootdee K, Nilgate S, Finkelman M, Tumwasorn S, Leelahavanichkul A. Lactobacillus rhamnosus L34 attenuates gut translocation-induced bacterial sepsis in murine models of leaky gut. Infect Immun 86: e00700-17, 2018. doi: 10.1128/IAI.00700-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Myers RB, Fredenburgh JL, Grizzle WE. Carbohydrates. In: Theory and Practice of Histological Techniques (6th ed.), edited by Suvarna K, Layton C, Bancroft J.. Philadelphia, USA: Elsevier, 2018, p. 161–186. [Google Scholar]
- 67.Glassner KL, Abraham BP, Quigley EMM. The microbiome and inflammatory bowel disease. J Allergy Clin Immun 145: 16–27, 2020. doi: 10.1016/j.jaci.2019.11.003. [DOI] [PubMed] [Google Scholar]
- 68.Song M, Chan AT, Sun J. Influence of the gut microbiome, diet, and environment on risk of colorectal cancer. Gastroenterology 158: 322–340, 2019. doi: 10.1053/j.gastro.2019.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McClintock SD, Attili D, Dame MK, Richter A, Silvestri SS, Berner MM, Bohm MS, Karpoff K, McCarthy CL, Spence JR, Varani J, Aslam MN. Differentiation of human colon tissue in culture: effects of calcium on trans-epithelial electrical resistance and tissue cohesive properties. PLoS One 15: e0222058, 2020. doi: 10.1371/journal.pone.0222058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tsukita S, Tanaka H, Tamura A. The claudins: from tight junctions to biological systems. Trends Biochem Sci 44: 141–152, 2018. doi: 10.1016/j.tibs.2018.09.008. [DOI] [PubMed] [Google Scholar]
- 71.Darsigny M, Babeu J-P, Dupuis A-A, Furth EE, Seidman EG, Lévy É, Verdu EF, Gendron F-P, Boudreau F. Loss of hepatocyte-nuclear-factor-4α affects colonic ion transport and causes chronic inflammation resembling inflammatory bowel disease in mice. Plos One 4: e7609, 2009. doi: 10.1371/journal.pone.0007609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Raju P, Shashikanth N, Tsai P-Y, Pongkorpsakol P, Chanez-Parades S, Steinhagen PR, Kuo W-T, Singh G, Tsukita S, Turner JR. Inactivation of paracellular cation-selective claudin-2 channels attenuates immune-mediated experimental colitis in mice. J Clin Invest 130: 5197–5208, 2020. doi: 10.1172/JCI138697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tanaka H, Takechi M, Kiyonari H, Shioi G, Tamura A, Tsukita S. Intestinal deletion of Claudin-7 enhances paracellular organic solute flux and initiates colonic inflammation in mice. Gut 64: 1529, 2015. doi: 10.1136/gutjnl-2014-308419. [DOI] [PubMed] [Google Scholar]
- 74.Sobell HM. Actinomycin and DNA transcription. Proc Natl Acad Sci USA 82: 5328–5331, 1985. doi: 10.1073/pnas.82.16.5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schneider-Poetsch T, Ju J, Eyler DE, Dang Y, Bhat S, Merrick WC, Green R, Shen B, Liu JO. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol 6: 209–217, 2010. doi: 10.1038/nchembio.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Klausen P, Karstensen JG, Coskun M, Săftoiu A, Vilmann P, Cowland JB, Riis LB. SMAD4 protein expression is downregulated in ileal epithelial cells from patients with Crohn’s disease with significant inverse correlation to disease activity. Gastroenterol Res Pract 2018: 1–8, 2018. doi: 10.1155/2018/9307848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lameris AL, Huybers S, Kaukinen K, Mäkelä TH, Bindels RJ, Hoenderop JG, Nevalainen PI. Expression profiling of claudins in the human gastrointestinal tract in health and during inflammatory bowel disease. Scand J Gastroenterol 48: 58–69, 2012. doi: 10.3109/00365521.2012.741616. [DOI] [PubMed] [Google Scholar]
- 78.Luettig J, Rosenthal R, Barmeyer C, Schulzke J. Claudin-2 as a mediator of leaky gut barrier during intestinal inflammation. Tissue Barriers 3: e977176, 2015. doi: 10.4161/21688370.2014.977176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Prasad S, Mingrino R, Kaukinen K, Hayes KL, Powell RM, MacDonald TT, Collins JE. Inflammatory processes have differential effects on claudins 2, 3 and 4 in colonic epithelial cells. Lab Invest 85: 1139–1162, 2005. doi: 10.1038/labinvest.3700316. [DOI] [PubMed] [Google Scholar]
- 80.Randall K, Henderson N, Reens J, Eckersley S, Nyström A-C, South MC, Balendran CA, Böttcher G, Hughes G, Price SA. Claudin-2 expression levels in ulcerative colitis: development and validation of an in-situ hybridisation assay for therapeutic studies. PLoS One 11: e0162076, 2016. doi: 10.1371/journal.pone.0162076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zeissig S, Bürgel N, Günzel D, Richter J, Mankertz J, Wahnschaffe U, Kroesen AJ, Zeitz M, Fromm M, Schulzke J-D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 56: 61, 2007. doi: 10.1136/gut.2006.094375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wasserman I, Lee LH, Ogino S, Marco MR, Wu C, Chen X, et al. SMAD4 loss in colorectal cancer patients correlates with recurrence, loss of immune infiltrate, and chemoresistance. Clin Cancer Res 25: 1948–1956, 2018. doi: 10.1158/1078-0432.CCR-18-1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yan P, Klingbiel D, Saridaki Z, Ceppa P, Curto M, McKee TA, Roth A, Tejpar S, Delorenzi M, Bosman FT, Fiocca R. Reduced expression of SMAD4 is associated with poor survival in colon cancer. Clin Cancer Res 22: 3037–3047, 2016. doi: 10.1158/1078-0432.CCR-15-0939. [DOI] [PubMed] [Google Scholar]
- 84.Bornholdt J, Friis S, Godiksen S, Poulsen SS, Santoni-Rugiu E, Bisgaard HC, Lothe IM, Ikdahl T, Tveit KM, Johnson E, Kure EH, Vogel LK. The level of claudin-7 is reduced as an early event in colorectal carcinogenesis. BMC Cancer 11: 65, 2011. doi: 10.1186/1471-2407-11-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dhawan P, Ahmad R, Chaturvedi R, Smith JJ, Midha R, Mittal MK, Krishnan M, Chen X, Eschrich S, Yeatman TJ, Harris RC, Washington MK, Wilson KT, Beauchamp RD, Singh AB. Claudin-2 expression increases tumorigenicity of colon cancer cells: role of epidermal growth factor receptor activation. Oncogene 30: 3234–3247, 2011. doi: 10.1038/onc.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ueda J, Semba S, Chiba H, Sawada N, Seo Y, Kasuga M, Yokozaki H. Heterogeneous expression of claudin-4 in human colorectal cancer: decreased claudin-4 expression at the invasive front correlates cancer invasion and metastasis. Pathobiology 74: 32–41, 2007. doi: 10.1159/000101049. [DOI] [PubMed] [Google Scholar]
- 87.Hahn-Strömberg V, Ahmad A, Befekadu R, Nilsson TK. Expression of claudin 1, claudin 4, and claudin 7 in colorectal cancer and its relation with CLDN DNA methylation patterns. Tumor Biol 39: 1–10, 2017. doi: 10.1177/1010428317697569. [DOI] [PubMed] [Google Scholar]
- 88.Xu C, Wang X, Li W, Wang K, Ding L. Expression and clinical significance of claudin-7 in patients with colorectal cancer. Technol Cancer Res Treat 17: 1–10, 2018. doi: 10.1177/1533033818817774. [DOI] [Google Scholar]
- 89.Guan Q. A comprehensive review and update on the pathogenesis of inflammatory bowel disease. J Immunol Res 2019: 1–16, 2019. doi: 10.1155/2019/7247238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hollander D, Vadheim C, Brittholz E, Petersen G, Delahunty T, Rotter J. Increased intestinal permeability in patients with Crohn’s disease and their relatives. A possible etiologic factor. Ann Intern Med 105: 883–885, 1986. doi: 10.7326/0003-4819-105-6-883. [DOI] [PubMed] [Google Scholar]
- 91.Katz KD, Hollander D, Vadheim CM, McElree C, Delahunty T, Dadufalza VD, Krugliak P, Rotter JI. Intestinal permeability in patients with Crohn’s disease and their healthy relatives. Gastroenterology 97: 927–931, 1989. doi: 10.1016/0016-5085(89)91499-6. [DOI] [PubMed] [Google Scholar]
- 92.Wyatt J, Vogelsang H, Hubl W, Waldhoer T, Lochs H. Intestinal permeability and the prediction of relapse in Crohn’s disease. Lancet 341: 1437–1439, 1993. doi: 10.1016/0140-6736(93)90882-H. [DOI] [PubMed] [Google Scholar]
- 93.Smith PM, Means AL, Beauchamp RD. Immunomodulatory effects of TGF-β family signaling within intestinal epithelial cells and carcinomas. Gastrointest Disord (Basel) 1: 290–300, 2019. doi: 10.3390/gidisord1020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Günzel D, Yu ASL. Claudins and the modulation of tight junction permeability. Physiol Rev 93: 525–569, 2013. doi: 10.1152/physrev.00019.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rosenthal R, Milatz S, Krug SM, Oelrich B, Schulzke J-D, Amasheh S, Günzel D, Fromm M. Claudin-2, a component of the tight junction, forms a paracellular water channel. J Cell Sci 123: 1913–1921, 2010. doi: 10.1242/jcs.060665. [DOI] [PubMed] [Google Scholar]
- 96.Xu C, Wang K, Ding Y-H, Li W-J, Ding L. Claudin-7 gene knockout causes destruction of intestinal structure and animal death in mice. World J Gastroenterol 25: 584–599, 2019. doi: 10.3748/wjg.v25.i5.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Clements RT, Minnear FL, Singer HA, Keller RS, Vincent PA. RhoA and Rho-kinase dependent and independent signals mediate TGF-β-induced pulmonary endothelial cytoskeletal reorganization and permeability. Am J Physiol Lung Cell Mol Physiol 288: L294–L306, 2005. doi: 10.1152/ajplung.00213.2004. [DOI] [PubMed] [Google Scholar]
- 98.Howe K, Gauldie J, McKay DM. TGF-β effects on epithelial ion transport and barrier: reduced Cl− secretion blocked by a p38 MAPK inhibitor. Am J Physiol Cell Physiol 283: C1667–C1674, 2002. doi: 10.1152/ajpcell.00414.2001. [DOI] [PubMed] [Google Scholar]
- 99.Lu Q, Harrington EO, Jackson H, Morin N, Shannon C, Rounds S. Transforming growth factor-β1-induced endothelial barrier dysfunction involves Smad2-dependent p38 activation and subsequent RhoA activation. J Appl Physiol 101: 375–384, 2006.. doi: 10.1152/japplphysiol.01515.2005. [DOI] [PubMed] [Google Scholar]
- 100.Qi Z, Li Y, Zhao B, Xu C, Liu Y, Li H, Zhang B, Wang X, Yang X, Xie W, Li B, Han J-DJ, Chen Y-G. BMP restricts stemness of intestinal Lgr5+ stem cells by directly suppressing their signature genes. Nat Commun 8: 13824, 2017. doi: 10.1038/ncomms13824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Koveitypour Z, Panahi F, Vakilian M, Peymani M, Forootan FS, Esfahani MHN, Ghaedi K. Signaling pathways involved in colorectal cancer progression. Cell Biosci 9: 97, 2019. doi: 10.1186/s13578-019-0361-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bhat AA, Pope JL, Smith JJ, Ahmad R, Chen X, Washington MK, Beauchamp RD, Singh AB, Dhawan P. Claudin-7 expression induces mesenchymal to epithelial transformation (MET) to inhibit colon tumorigenesis. Oncogene 34: 4570–4580, 2015. doi: 10.1038/onc.2014.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dhawan P, Singh AB, Deane NG, No Y, Shiou S-R, Schmidt C, Neff J, Washington MK, Beauchamp RD. Claudin-1 regulates cellular transformation and metastatic behavior in colon cancer. J Clin Invest 115: 1765–1776, 2005. doi: 10.1172/jci24543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gowrikumar S, Ahmad R, Uppada SB, Washington MK, Shi C, Singh AB, Dhawan P. Upregulated claudin-1 expression promotes colitis-associated cancer by promoting β-catenin phosphorylation and activation in Notch/p-AKT-dependent manner. Oncogene 38: 5321–5337, 2019. doi: 10.1038/s41388-019-0795-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pope JL, Ahmad R, Bhat AA, Washington MK, Singh AB, Dhawan P. Claudin-1 overexpression in intestinal epithelial cells enhances susceptibility to adenamatous polyposis coli-mediated colon tumorigenesis. Mol Cancer 13: 167, 2014. doi: 10.1186/1476-4598-13-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ciardiello D, Elez E, Tabernero J, Seoane J. Clinical development of therapies targeting TGFβ: current knowledge and future perspectives. Ann Oncol 31: 1336–1349, 2020. doi: 10.1016/j.annonc.2020.07.009. [DOI] [PubMed] [Google Scholar]
- 107.Capaldo CT, Nusrat A. Cytokine regulation of tight junctions. Biochim Biophys Acta 1788: 864–871, 2009. doi: 10.1016/j.bbamem.2008.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mazzon E, Cuzzocrea S. Role of TNF-α in lung tight junction alteration in mouse model of acute lung inflammation. Respir Res 8: 75, 2007. doi: 10.1186/1465-9921-8-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Watson CJ, Hoare CJ, Garrod DR, Carlson GL, Warhurst G. Interferon-γ selectively increases epithelial permeability to large molecules by activating different populations of paracellular pores. J Cell Sci 118: 5221–5230, 2005. doi: 10.1242/jcs.02630. [DOI] [PubMed] [Google Scholar]