


Keywords: exercise, ketogenic diet, mitochondria, peroxisomal, skeletal muscle
Abstract
Ketogenic diets (KDs) are reported to improve body weight, fat mass, and exercise performance in humans. Unfortunately, most rodent studies have used a low-protein KD, which does not recapitulate diets used by humans. Since skeletal muscle plays a critical role in responding to macronutrient perturbations induced by diet and exercise, the purpose of this study was to test if a normal-protein KD (NPKD) impacts shifts in skeletal muscle substrate oxidative capacity in response to exercise training (ExTr). A high fat, carbohydrate-deficient NPKD (16.1% protein, 83.9% fat, 0% carbohydrate) was given to C57BL/6J male mice for 6 wk, whereas controls (Con) received a low-fat diet with similar protein (15.9% protein, 11.9% fat, 72.2% carbohydrate). After 3 wk on the diet, mice began treadmill training 5 days/wk, 60 min/day for 3 wks. The NPKD increased body weight and fat mass, whereas ExTr negated a continued rise in adiposity. ExTr increased intramuscular glycogen, whereas the NPKD increased intramuscular triglycerides. Neither the NPKD nor ExTr alone altered mitochondrial content; however, in combination, the NPKD-ExTr group showed increases in PGC-1α and markers of mitochondrial fission/fusion. Pyruvate oxidative capacity was unchanged by either intervention, whereas ExTr increased leucine oxidation in NPKD-fed mice. Lipid metabolism pathways had the most notable changes as the NPKD and ExTr interventions both enhanced mitochondrial and peroxisomal lipid oxidation and many adaptations were additive or synergistic. Overall, these results suggest that a combination of a NPKD and ExTr induces additive and/or synergistic adaptations in skeletal muscle oxidative capacity.
NEW & NOTEWORTHY A ketogenic diet with normal protein content (NPKD) increases body weight and fat mass, increases intramuscular triglyceride storage, and upregulates pathways related to protein metabolism. In combination with exercise training, a NPKD induces additive and/or synergistic activation of AMPK, PGC-1α, mitochondrial fission/fusion genes, mitochondrial fatty acid oxidation, and peroxisomal adaptations in skeletal muscle. Collectively, results from this study provide mechanistic insight into adaptations in skeletal muscle relevant to keto-adaptation.
INTRODUCTION
In 1974, ketolytic enzyme activity was found to be increased in three different types of muscle after a treadmill running program, sparking interest in ketone-based energy metabolism in skeletal muscle (1). Athletes often seek nutritional interventions to optimize sports performance and it has become increasingly popular to adopt a ketogenic diet (KD) or consume exogenous ketone esters in an attempt to achieve their goals (2–5). A core concept of KDs is that carbohydrate content is substantially limited, and this drives nutritional ketosis that is typically defined as blood ketone levels between 0.5–3.0 mM (3, 6). Whether KDs actually improve exercise performance remains contentious; however, a concept of keto-adaptation that is consistently reported is that athletes that consume a carbohydrate-restricted KD adapt to have high rates of fat oxidation during exercise that limits reliance on glucose (5, 7).
Interactions between a low-carbohydrate, lipid-rich KD and exercise have been extensively studied and include several reports describing shifts of in vivo substrate utilization (5, 7–18); however, the combined effects of a KD and exercise training on substrate oxidative capacity in skeletal muscle are less well known. Exercise training is routinely reported to increase mitochondrial content/function and substrate oxidative capacity in rodents and humans (19–21). Likewise, KDs are often described to alter enzymes involved in fatty acid oxidation, the citric acid cycle, and oxidative phosphorylation (22–25). Unfortunately, most studies testing the effects of KDs on physiological responses in rodents use a protein-restricted (∼5% kcal) version of the diet (26–30), which does not recapitulate the macronutrient composition of KDs most often consumed by humans that typically contain 15%–30% protein (7, 18, 31, 32). The purpose of this investigation was to address this matter by testing the hypothesis that the combination of treadmill exercise training while consuming a KD with normal protein content (NPKD) would induce additive and/or synergistic adaptations in peroxisomal lipid metabolism, as well as mitochondrial pathways that dictate carbohydrate, amino acid, and lipid metabolism in skeletal muscle of lean, healthy C57BL/6J male mice.
MATERIALS AND METHODS
Animals
C57BL/6J male mice were ordered from Jackson Laboratories (Stock No. 000664; Bar Harbor, ME) at 12 wk of age and studied at 20 wk of age. Mice were group-housed at room temperature under a 12:12-h light-dark cycle and allowed ad libitum access to food and water throughout the study. The Pennington Biomedical Research Center has an American Association for the Accreditation of Laboratory Animal Care-approved Comparative Biology Core facility and veterinary staff that monitor animal health via a sentinel program and daily inspection. All studies were approved by the Institutional Animal Care and Use Committee.
Study Design
Figure 1A illustrates the study design. Upon arrival mice were provided a low-fat chow diet (Purina 5001). Two weeks later (14 wk of age), all mice were tested for body weight and composition with a Bruker Minispec LF50 Time-Domain NMR and results were used to assign mice into groups that ensured similar body composition before beginning the dietary intervention. Mice (n = 20) in the control (Con) group were switched to a low-fat diet (TestDiet No. 5TJS, St. Louis, MO) containing 15.9% calories from protein, 11.9% from fat, and 72.2% from carbohydrate. Alternatively, mice in the experimental group (n = 19) were provided an isocaloric protein-matched high-fat diet with no carbohydrate (NPKD; TestDiet No. 5TJQ, St. Louis, MO), which provided 16.1% calories from protein, 83.9% from fat, and 0% from carbohydrate. Matched diets using the same sources of protein (casein) and lipid (lard, milk fat, and vegetable shortening) were used in this study; however, carbohydrate (sucrose and maltodextrin) was only present in the control diet. Mice were introduced to their experimental diet during a 3-day lead-in period where mice had access to both Purina 5001 and their experimental diet. At week 0, mice received only their appropriate experimental diet (Con or NPKD) and this was sustained for 6 wk. After 2.5 wk on the diet, all mice (n = 39) were habituated on an Exer 3/6 treadmill (Columbus Instruments; Columbus, OH) for 3 days. The treadmill was set at 10° incline and the habituation protocol consisted of 5 min stages at 0, 5, and 10 m/min followed by 2 min at 15 m/min. All mice responded well to the habituation protocol, so they were randomly assigned to the following groups: control (low fat) sedentary (Con-Sed, n = 11), control (low fat) exercise training (Con-ExTr, n = 9), NPKD sedentary (NPKD-Sed, n = 10), or NPKD exercise training (NPKD-ExTr, n = 9). The 3-wk training protocol was initiated after 3 wk on the dietary intervention (Fig. 1A). At the end of week 6, muscle tissues were collected 24 h after the last exercise bout and food was removed 2 h before harvest.
Figure 1.

Schematic of study design and body composition. Graphical depiction of the study design and timeline (A). Mice received either a control diet (Con) or a normal protein, ketogenic diet (NPKD) and were randomly assigned to control sedentary (Con-Sed, n = 11 mice), control exercise training (Con-ExTr, n = 9 mice), NPKD sedentary (NPKD-Sed, n = 10 mice), or NPKD exercise training (NPKD-ExTr, n = 9 mice) groups. Circulating ketone (β-hydroxybutyrate) levels (B), exercise training workload (C), body weight (D), fat mass (F), and lean mass (H) are reported, as well as changes from baseline in body weight (E), fat mass (G), and lean mass (I). Circulating ketone data were analyzed using a two-way repeated-measure ANOVA with a Bonferroni post hoc test, while body composition data were analyzed using a three-way repeated measures ANOVA with a Bonferroni post hoc test and are presented as means ± SE. #P < 0.05 main effect of diet; $main effect for exercise; Bonferroni post hoc analysis revealed significant differences (identified with an asterisk) at the following P values *P < 0.05; **P < 0.01; ***P < 0.001. Changes in body composition between Pre and week 3 are shown as ^P < 0.05, ^^P < 0.01, ^^^P < 0.001. Changes in body composition from week 3–week 6 are shown as †P < 0.05, ††P < 0.01, †††P < 0.001.
Treadmill Exercise Training Protocol
Mice in the ExTr groups ran on a treadmill for 3 wk, 5 days/wk, 1 h/day and the stimulus was designed to yield a moderate-to-high exercise intensity. As shown in Table 1, speed was increased in a step-wise fashion throughout the individual exercise bouts to ensure mice reached the appropriate intensity. To account for adaptations in aerobic fitness, exercise intensity was increased weekly and a Lactate Plus Meter (Nova Biomedical, Waltham, MA) was used to test blood lactate before, and at the end of, an exercise bout. Aversive stimuli (light tapping with a brush, light electrical shock) were used to motivate mice to run to ensure they completed each exercise bout. Mice were monitored continuously throughout each exercise bout and the interventions were well-tolerated as none exhibited signs of exhaustion (no righting reflex and/or acceptance of light electrical shock for >5 s) or injury (awkward gait, foot injury, etc.). Mice were immediately returned to their cages after each exercise bout and granted ad libitum access to food and water.
Table 1.
Exercise training protocol for week 4–6
|
Week 4 |
Week 5 |
Week 6 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Stage | Speed, m/min | Duration, min | Distance, m | Stage | Speed, m/min | Duration, min | Distance, m | Stage | Speed, m/min | Duration, min | Distance, m |
| 1 | 10 | 10 | 100 | 1 | 12 | 10 | 120 | 1 | 14 | 10 | 140 |
| 2 | 12 | 10 | 120 | 2 | 14 | 10 | 140 | 2 | 16 | 10 | 160 |
| 3 | 14 | 10 | 140 | 3 | 16 | 10 | 160 | 3 | 18 | 10 | 180 |
| 4 | 16 | 10 | 160 | 4 | 18 | 10 | 180 | 4 | 20 | 15 | 300 |
| 5 | 18 | 10 | 180 | 5 | 20 | 10 | 200 | 5 | 22 | 15 | 330 |
| 6 | 20 | 10 | 200 | 6 | 22 | 10 | 220 | ||||
| TOTAL | 10° incline | 60 | 900 | TOTAL | 10° incline | 60 | 1,020 | TOTAL | 10° incline | 60 | 1,110 |
After 3 wk of dietary interventions, mice were trained for 3 wk (week 4–6 of the study) on a treadmill set at 10° incline and each session started with a 5-min warmup (W), followed by a 60-min exercise session where a progressive increase in speed was used to increase intensity.
Tissue Harvest
Tissue collection occurred during the light cycle between 9:00 and 11:00 AM and mice were anesthetized via intraperitoneal (ip) injection of ketamine/xylazine/acepromazine (16 mg/mL ketamine, 0.8 mg/mL xylazine, and 0.32 mg/mL acepromazine delivered at a dose of 0.125 mL/20 g body wt). Muscle samples were collected and either 1) used fresh for substrate oxidation assays, or 2) snap-frozen in liquid nitrogen and stored at −80°C until subsequent analyses could be performed.
Blood Ketones
Blood ketones (β-hydroxybutyrate) (Nova Ketone Monitor; Nova Biomedical, Waltham, MA) were measured at the end of week 6 via tail vein in conscious mice in the fed state at the end of the dark cycle (7:00 AM). Food was then removed and measurements were taken 2–4 h later (between 9:00 and 11:00 AM) during the light cycle to test the effects of a mild fast on circulating ketones.
Muscle Substrate Storage
Muscle glycogen and intramuscular triglycerides (TAGs) were measured in quadriceps muscle.
Muscle glycogen.
Frozen powdered tissue was weighed (∼15–30 mg) and used to extract and measure glycogen content using a commercially available kit (Abcam ab65620, Cambridge, MA) according to the manufacturer’s instructions. Results are normalized to tissue weight.
Muscle triglycerides.
Frozen powdered tissue was weighed (∼25–30 mg) and homogenized two times for 15 s in 300 µL ice-cold 5% NP-40 using a handheld homogenizer (VWR, Radnor, PA). Tissue homogenates were slowly heated to 95°C in a water bath for 5 min until the solution became white and then cooled at room temperature for ∼15 min. This heating-cooling procedure was performed twice to solubilize tissue TAGs. Homogenates were centrifuged (14,000 g, 2 min, room temperature) and supernatants were collected to determine intramuscular TAG content. A commercially available TAG assay kit (SIGMA Cat. No. TR0100) was used to measure glycerol in these extracts as an indirect measure of total TAGs. This kit allows for determination of free glycerol, total TAGs, and true TAGs (total TAG minus free glycerol): all data are reported as true TAG and are normalized to tissue weight.
Gene Expression
RNA was isolated from powdered muscle (∼25 mg) using an RNeasy kit (Qiagen, Germantown, MD) with proteinase K digestion and on-column DNase treatment, as per the manufacturer’s instructions. RNA concentration and quality were tested using a Nanodrop ND-1000 spectrophotometer (Thermo Scientific, Wilmington, DE) and integrity was confirmed using an Agilent 2100 bioanalyzer (Santa Clara, CA). Samples containing RNA concentrations above 50 ng/μL (average ∼188), 260/280 ratios above 1.9 (average ∼2.1), and RNA integrity numbers above 5.0 (average ∼6.9) were used to prepare cDNA. Briefly, 1 µg of RNA was used to develop cDNA using an iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). Gene expression was measured by real-time PCR on an ABI 7900HT Sequence Detection System (Life Technologies, Carlsbad, CA) using iTaq Universal SYBR Green Supermix (Biorad; Cat. No. 172-5124). Primer pairs were designed using Primer-BLAST software and sequences have been previously reported (33–35) and are included in Supplemental Table S1 (all Supplemental material is available at https://doi.org/10.6084/m9.figshare.13335932). mRNA expression was determined using the comparative CT method (ΔΔCT) with an endogenous control (cyclophilin) and converted to a linear function by using a base 2 antilog transformation (2−ΔΔCT).
Tissue Protein Analysis
Tissues lysates were prepared using T-PER buffer (Thermo Fisher No. 78510) containing protease inhibitor cocktail and phosphatase inhibitors from Sigma-Aldrich (St. Louis, MO). Western blot analyses were performed using standard PAGE-SDS as described previously (36). Primary antibodies were as follows: acetyl-CoA carboxylase (ACC; No. 3676), phospho (p)-ACC (No. 11818), AMP-activated protein kinase alpha (AMPKα; No. 5831), p-AMPKα (No. 2535), peroxin 5 (Pex5; No. 83020), mammalian target of rapamycin (mTOR; No. 2983), p-mTOR (No. 5536), ribosomal protein s6 (RPs6; No. 2217), p-RPs6 (No. 2211), eukaryotic translation initiation factor 4E (4EBP1; No. 9644), p-4EBP1 (No. 2855), Unc-51 like autophagy activating kinase 1 (ULK1; No. 8054), p-ULK1 (No. 6888), autophagy related 7 (Atg7; No. 8558), autophagy light chain 3 A/B (LC3 A/B; No. 12741), and adipose triglyceride lipase (ATGL; No. 2439) from Cell Signaling (Danvers, MA); Total oxidative phosphorylation (OXPHOS) cocktail (ab110413), Pex19 (ab137072), and peroxisomal membrane protein, 70 kDa (PMP70) (ab3421) from Abcam (Cambridge, MA); Pex14 (ABC-142) from EMD Millipore (Burlington, MA); peroxisomal acyl-coenzyme A oxidase 1 (ACOX1; sc-517306) and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α; sc-517380) from Santa Cruz Biotechnology (Dallas, TX); and carnitine palmitoyltransferase 1b (Cpt1b; 22170-1-AP) from ProteinTech (Rosemont, IL). Horseradish peroxidase-linked secondary antibodies (anti-mouse IgG Cat. No. NXA931 and anti-rabbit IgG Cat. No. NA934V) were purchased from GE Healthcare (Piscataway, NJ) and proteins were detected using ECL chemistry. Reversible protein stain-MemCode (MemC; Thermo Fisher No. 24580) was used to confirm equal protein transfer and quantitation of these bands served as a loading control. A ChemiDoc imaging system and Image Lab software (Bio-Rad; Hercules, CA) were used to image bands and quantitate band intensity. Western blot results are expressed as fold change compared with the Con-Sed group after normalization to MemC lane intensity. Supplemental Table S2 contains references to manuscripts that reported validation of antibodies used against ACC, AMPKα, ULK1, and Atg7 via gene deletion models. We provide knockout confirmation of antibodies against Cpt1b, Pex5, and PGC1α in Supplemental Fig. S1. All other antibodies yielded single bands at the reported molecular weight of the protein of interest.
Substrate Oxidation
Substrate oxidation rates were measured in mixed gastrocnemius homogenates using established methods that yield 75%–80% intact mitochondria (37–39). Briefly, homogenates were incubated in the presence of [14C]radiolabeled substrates (American Radiolabeled Chemicals, St. Louis, MO) and complete oxidation was measured as the liberation of 14CO2 from the following substrates: [1-14C]pyruvate (1 mM) was used to measure pyruvate dehydrogenase (PDH) activity, [2-14C]pyruvate (1 mM) was used to measure pyruvate oxidation, [U-14C]leucine (100 µM) was used to measure oxidation of the branched-chain amino acid leucine, [1-14C]palmitate (200 µM) was used to measure fatty acid oxidation, and [1-14C]lignocerate (25 µM) was used to specifically measure peroxisomal fatty acid oxidation. Incomplete palmitate oxidation was measured as the production of 14C-acid soluble metabolites (ASMs), whereas ASMs were not analyzed for other substrates tested. Palmitate was bound to fatty acid-free bovine serum albumin (BSA) at a molar ratio of 4.7:1 (Palmitate:BSA), whereas lignocerate was solubilized in α-cyclodextrin (10 mg/mL final concentration).
Data Analysis
Bloodwork measured before and after fasting was analyzed using a two-way repeated measure ANOVA (diet × exercise training) with a Bonferroni post hoc analysis. Body composition measurements were analyzed using a three-way repeated measure ANOVA (diet × exercise × time) with a Bonferroni post hoc analysis. All other data were analyzed using a two-way ANOVA with a Bonferroni post hoc analysis. GraphPad Prism software was used for two-way ANOVA statistical analyses and IBM SPSS v. 27 was used for the three-way repeated measure ANOVA. A P ≤ 0.05 was established a priori as representing a statistically significant difference.
RESULTS
Schematic of Study Design and Body Composition
The overall study design is depicted in Fig. 1A. As shown in Fig. 1B, provision of a NPKD for 6 wk induced a mild level of nutritional ketosis that was similar to a short-term fast (2–4 h) in mice fed the Con diet. Animals on both diets performed a similar amount of work throughout the 3-wk ExTr protocol, although values tended to be slightly higher in NPKD-fed mice (Fig. 1C) due to slightly higher body weight. As previously reported (35), this ExTr protocol induced a vigorous-intensity stimulus as average blood lactate levels immediately postexercise approached the lactate threshold (4–5 mM) as lactate values rose from 1.1 ± 0.1 mM (resting) to 4.2 ± 0.4 mM (postexercise) in the Con-ExTr group and from 1.0 ± 0.3 mM (resting) to 4.6 ± 0.5 mM (postexercise) in the NPKD-ExTr group. Animals placed on the lipid-rich NPKD gained significantly more body weight than those fed a low-fat Con diet during the first 3 wk of the dietary interventions, whereas weight gain from weeks 3–6 of the diet were similar between Con and NPKD groups (Fig. 1, D and E). Likewise, mice fed the NPKD gained greater amounts of fat mass (Fig. 1, F and G) and lean mass (Fig. 1, H and I) during the initial 3 wk of the dietary intervention than those receiving the Con diet; however, differences between these dietary groups were not observed from weeks 3–6 (i.e., no main effect of diet). Main effects of ExTr were detected in body weight (Fig. 1E), fat mass (Fig. 1G), and lean mass (Fig. 1I), suggesting all were lowered by ExTr. Looking more closely, both Con-Sed and NPKD-Sed groups gained significant amounts of fat mass from weeks 3–6, whereas neither Con-ExTr nor NPKD-ExTr groups gained fat mass; therefore, ExTr effectively prevented further gains in adiposity in both groups.
Mitochondrial Adaptations
To understand the interactive effects of the NPKD and ExTr on cellular energy sensing pathways, Western blot analyses were performed to measure total and phosphorylated forms of AMPKα (Fig. 2, A and B). No significant differences between groups were detected in total or phospho-AMPKα; however, the pAMPKα/AMPKα ratio was significantly higher in NPKD-ExTr than Con-ExTr mice. Next, we examined factors related to mitochondrial biogenesis including PGC-1α and the mitochondrial transcription factor A (Tfam). Results indicate that the ExTr stimulus increased PGC-1α and Tfam gene expression (Fig. 2C), as well as PGC-1α protein (Fig. 2D), only in mice fed the NPKD, whereas this was not observed in the Con-ExTr mice. PGC-1α plays an important role in remodeling pathways involved in mitochondrial biogenesis and dynamics, so we measured complexes of the oxidative phosphorylation (OxPhos) system (Fig. 2E), as well as genes involved in mitochondrial fission and fusion (Fig. 2F). The general expression pattern of OxPhos proteins was similar to the results from PGC-1α and Tfam; however, none of these effects reached statistical significance (Fig. 2E). Alternatively, robust changes were detected in mitochondrial fission and fusion (Fig. 2F), which also mirrored the patterns of PGC-1α and Tfam. Overall, these results suggest there is additive and/or synergistic activation by the NPKD and ExTr on major regulators of energy sensing pathways (i.e., AMPK and PGC-1α) and mitochondrial remodeling (i.e., fission and fusion) in skeletal muscle.
Figure 2.
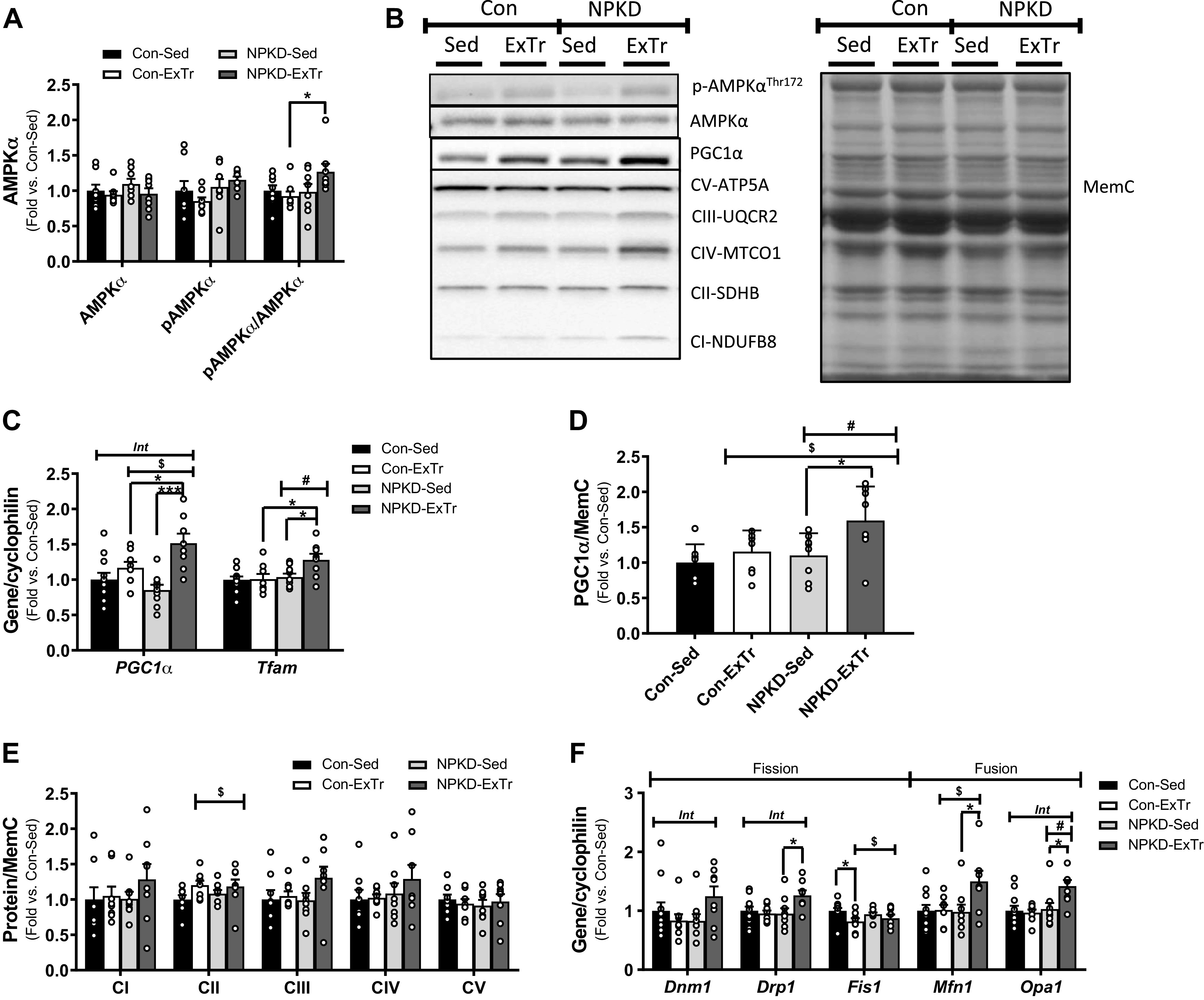
Mitochondrial adaptations. Quadriceps lysates were used to measure protein content of AMPKα (total and phospho-; A), PGC-1α (D), and subunits involved in Complex I–V of the OxPhos system (E). Western blots were run using n = 7 or 9/group and representative Western blot images are shown in (B). Genes involved in mitochondrial biogenesis (C) and mitochondrial fission and fusion (F) were also tested. Control sedentary (Con-Sed; n = 11 mice), control exercise training (Con-ExTr; n = 9 mice), normal protein, ketogenic diet sedentary (NPKD-Sed; n = 10 mice), and NPKD exercise training (NPKD-ExTr; n = 9 mice) were used for gene expression analyses. Data were analyzed using a two-way ANOVA with a Bonferroni post hoc analysis and are presented as means ± SE relative to the Con-Sed group. Quantitated Western blots images and gene expression are normalized to total protein stain using MemCode (MemC) and cyclophilin (PPIB), respectively. #P < 0.05 main effect of diet; $main effect for exercise; Bonferroni post hoc test revealed significant differences (identified with an asterisk) at the following P values *P < 0.05; ***P < 0.001. Int, interaction effect.
Glucose Metabolism
Since the NPKD was devoid of carbohydrate, which is critical for exercise performance, we next tested adaptations in pathways related to intramuscular glucose utilization. Intramuscular glycogen reserves did not change in response to the NPKD; however, a main effect of the 3-wk ExTr program was observed which indicated glycogen content increased in response to ExTr, regardless of diet (Fig. 3A). We next tested whether the capacity to utilize carbohydrate as metabolic fuel was altered. Despite changes in intramuscular glycogen, no changes in PDH activity (Fig. 3B) or pyruvate oxidation (Fig. 3C) capacity were observed in skeletal muscle. Next, we examined pyruvate dehydrogenase kinase 4 (Pdk4), which is a lipid-responsive gene that modulates the substrate switch to favor fatty acid utilization. Pdk4 gene expression was elevated in NPKD groups compared with control-fed mice; however, ExTr had no effect in either dietary condition (Fig. 3D). Together, our data suggest that 1) mice increase intramuscular glycogen in response to ExTr and 2) neither ExTr nor NPKDs alter maximal oxidative capacity to utilize pyruvate in mice.
Figure 3.

Glucose metabolism. Intramuscular glycogen content (A) was measured in samples collected at the tissue harvest (2–4 h food pull). Pyruvate dehydrogenase (PDH) activity (B) and pyruvate oxidation (C) were measured in fresh homogenates. Expression of the Pdk4 gene (D) was assessed by RT-PCR. Control sedentary (Con-Sed; n = 11 mice), control exercise training (Con-ExTr; n = 9 mice), normal protein, ketogenic diet sedentary (NPKD-Sed; n = 10 mice), and NPKD exercise training (NPKD-ExTr; n = 9 mice) were used for all analyses. Data were analyzed using a two-way ANOVA with a Bonferroni post hoc analysis and are presented as means ± SE. Gene expression is normalized to cyclophilin (PPIB). #P < 0.05 main effect of diet; $main effect for exercise; Bonferroni post hoc test revealed significant differences (identified with an asterisk) at the following P values *P < 0.05; ***P < 0.001; ****P < 0.0001.
Amino Acid Metabolism and mTOR Signaling
Protein content was matched in both diets used in this study; however, since the NPKD was devoid of carbohydrate, we tested adaptations in amino acid metabolism. Our data show a main effect of ExTr to increase oxidative capacity to utilize leucine as an alternative fuel; however, post hoc analysis indicated this only occurred in NPKD-fed mice that underwent ExTr (Fig. 4A). Changes in leucine oxidation could not be ascribed to regulation of key genes involved in branched-chain amino acid metabolism (Fig. 4B). The amino acid sensing mTOR signaling pathway was also tested due to its role in regulating protein synthesis and autophagy. When looking at key proteins related to protein synthesis, the NPKD group had higher total and pmTOR (Ser 2448) (Fig. 4D), pRPs6 (Ser235/236), and pRPs6/RPs6 ratio (Fig. 4E), and p4EBP1 (Thr37/46) and p4EBP1/4EBP1 ratio (Fig. 4F) compared with the Con diet. Interestingly, ExTr reduced the phosphorylation status of RPs6 compared with Sed controls (Fig. 4E) but did not alter 4EBP1. The ExTr and NPKD interventions had less potent effects on indices of autophagy. The NPKD modestly increased pULK1 (Ser757) and the pULK1 (Ser757)/ULK1 ratio (Fig. 4G); however, neither treatment had any effects on Atg7 (Fig. 4H). Conversely, ExTr moderately increased LC3A/B-II content, but this was not sufficient to alter the ratio of LC3A/B-II/I (Fig. 4I). Collectively, these results indicate that skeletal muscle adapts to the NPKD in a manner that appears to slightly favor protein synthesis over autophagy.
Figure 4.
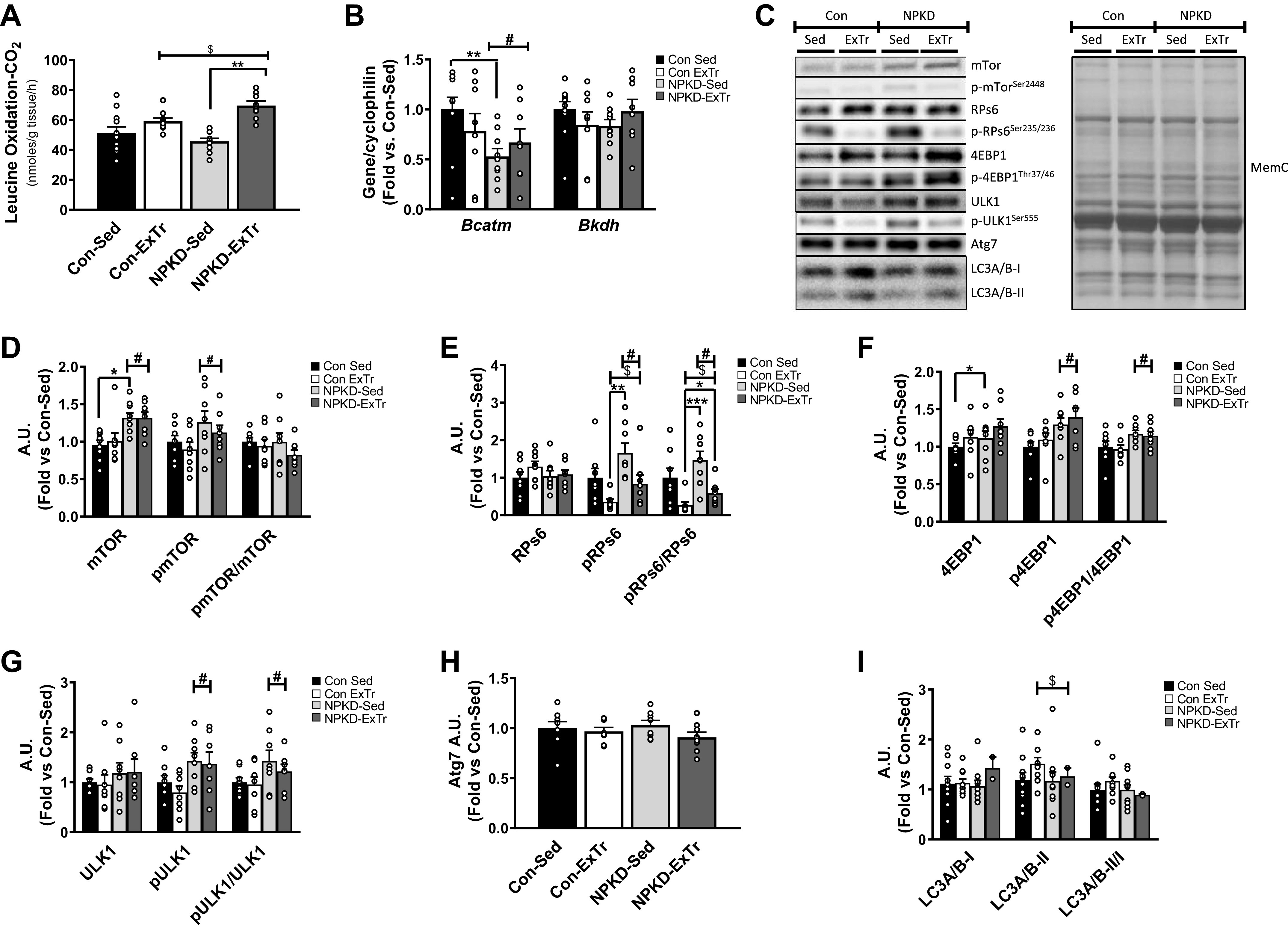
Amino acid metabolism and mammalian target of rapamycin (mTOR) signaling. Branched-chain amino acid metabolism (BCAA) was studied by assessing leucine oxidation (A), as well as expression of the Bcatm and Bkdh genes (B). mTOR signaling was assessed by measuring protein content of mTOR (D), ribosomal protein s6 (RPs6) (E), eukaryotic translation initiation factor 4E (4EBP1) (F), Unc-51 like autophagy activating kinase 1 (ULK1) (G), autophagy related 7 (Atg7) (H), and autophagy light chain 3 A/B (LC3A/B) (I). Protein blots were normalized to total protein stain MemCode (MemC) and expressed relative to the Control sedentary (Con-Sed) animals. Representative protein blots and protein stain are provided (C). Data were analyzed using a two-way ANOVA with a Bonferroni post hoc test and are presented as means ± SE. Con-Sed (n = 11 mice), control exercise training (Con-ExTr; n = 9 mice), normal protein, ketogenic diet sedentary (NPKD-Sed; n = 10 mice), and NPKD exercise training (NPKD-ExTr; n = 9 mice) were used for leucine oxidation and gene expression analyses, while n = 7–9/group were used for Western blots. Data were analyzed using a two-way ANOVA with a Bonferroni post hoc analysis and are presented as means ± SE. #P < 0.05 main effect of diet; $main effect for exercise. Bonferroni post hoc test revealed significant differences (identified with an asterisk) at the following P values *P < 0.05; **P < 0.01; ***P < 0.0001.
Fatty Acid Metabolism
Since the NPKD provided ∼84% calories from fat with no carbohydrate, we next tested adaptations in pathways related to fatty acid metabolism. Intramuscular TAGs showed a main effect for diet, suggesting the lipid-rich NPKD diet increased TAGs (Fig. 5A); however, ExTr had little effect on this parameter. Next, we tested genes involved in lipid storage. Acetyl-CoA carboxylase (ACC) produces malonyl-CoA, which inhibits fatty acid oxidation, thus favoring lipid storage. The NPKD decreased Acc1 gene expression (Fig. 5B); however, this was not observed at the level of protein as the NPKD increased both total and phosphorylated (inactive) ACC (Fig. 5C). Examination of genes that encode proteins that interact with lipid droplets revealed an interaction effect for Atgl gene expression, which showed that neither diet nor exercise alone altered Atgl levels; however, the combination of NPKD plus ExTr induced a robust increase (Fig. 5B). This effect was mirrored at the level of ATGL protein expression (Fig. 5D). Testing members of the perilipin (Plin) family revealed that ExTr induced an inhibitory adaptation in Plin2 in mice fed the Con diet, but the NPKD negated this effect (Fig. 5B). An interaction effect was observed for Plin3 showing ExTr increased Plin3 specifically in mice fed the NPKD; however, it is notable that NPKD-Sed mice generally appeared to have a lower baseline level than Con-Sed mice (Fig. 5B). The Plin5 gene profile was similar to that of intramuscular TAGs as expression was increased by the NPKD, but ExTr had no effect (Fig. 5B). We next measured palmitate oxidative capacity and results showed NPKD and ExTr had additive effects to increase both complete (CO2) and incomplete (ASM) oxidation (Fig. 5E). The NPKD increased incomplete oxidation to a slightly greater extent than complete oxidation, resulting in an higher ASM:CO2 ratio than mice fed the Con diet; however, ExTr did not alter this ratio (Fig. 5F). These adaptations are partially consistent with observations showing the NPKD increased Cpt1b gene (Fig. 5G) and protein (Fig. 5H) expression, which is a key factor that modulates mitochondrial lipid entry; however, ExTr did not alter Cpt1b. Collectively, these findings confirmed that skeletal muscle responds to a lipid-rich NPKD diet by increasing the capacity to both store and utilize lipid, whereas ExTr exerted additive effects to further increase lipid oxidative capacity.
Figure 5.
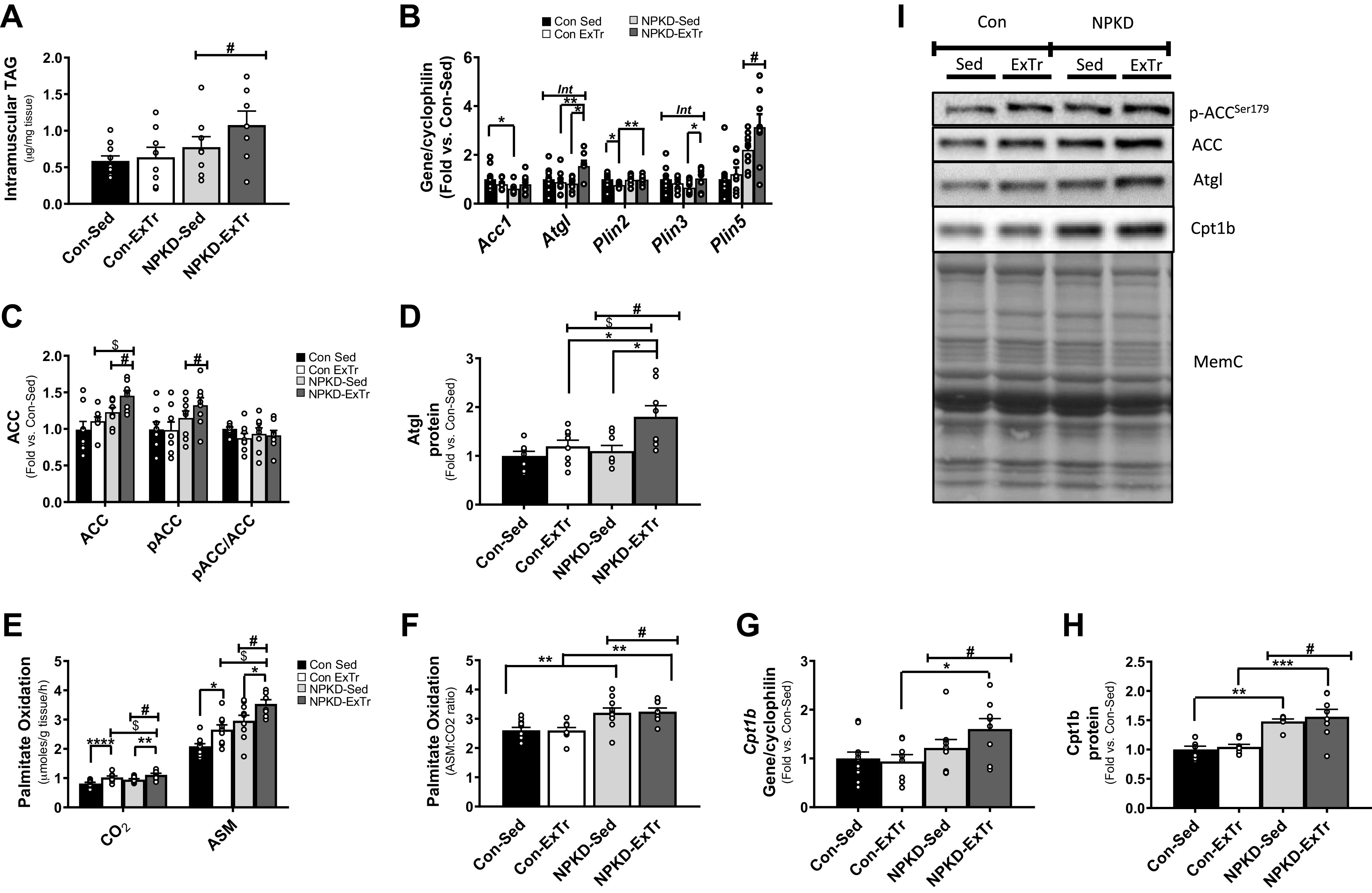
Fatty acid metabolism. Parameters related to muscle lipid storage were tested by measuring intramuscular triglyceride (TAG) content (A), as well as genes (B) and proteins (C, D, and I) related to lipid storage and utilization. Factors related to tissue lipid oxidation were examined by measuring complete (CO2) and incomplete (acid soluble metabolite; ASM) palmitate oxidation (E), ratio of incomplete-to-complete oxidation (F), as well as Cpt1b gene (G) and protein (H and I) expression. Control sedentary (Con-Sed; n = 11 mice), control exercise training (Con-ExTr; n = 9 mice), normal protein, ketogenic diet sedentary (NPKD-Sed; n = 10 mice), and NPKD exercise training (NPKD-ExTr; n = 9 mice) were used for palmitate oxidation, intramuscular triglyceride, and gene expression analyses, while n = 7 or 9/group were used for Western blots. Data were analyzed using a two-way ANOVA with a Bonferroni post hoc test and are presented as means ± SE. Quantitated RT-PCR data were normalized to cyclophilin (PPIB), while protein blots were normalized to total protein stain MemCode (MemC). #P < 0.05 main effect of diet; $main effect for exercise; Bonferroni post hoc test revealed significant differences (identified with an asterisk) at the following P values *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Int, interaction effect.
Peroxisomal Adaptations
Peroxisomes are ubiquitous organelles that play an irreplaceable role in lipid metabolism; therefore, we assessed peroxisomal adaptations in response to a NPKD and ExTr. Since fatty acids with a chain length greater than 20 carbons can only be oxidized by peroxisomes, oxidation of lignocerate (C24:0) was used to assess peroxisomal function. The NPKD increased lignocerate oxidation in sedentary animals (Fig. 6A). ExTr increased lignocerate oxidation in mice fed the Con diet, but did not have an effect in mice fed the NPKD (Fig. 6A). Interestingly, patterns for lignocerate oxidation were not matched by changes in peroxisomal proteins or genes. Pex14 and Pex19 protein exhibited main effects of the NPKD (increased), whereas only Pex14 showed a main effect of ExTr (increased) (Fig. 6B). Roughly, half of the peroxisomal genes tested exhibited significant differences in this study (Fig. 6C) and included key components of peroxisomal biogenesis (Pex11α and Pex19), lipid uptake (PMP70), β-oxidation (Acox1), and antioxidant defense (catalase). With this in mind, it is interesting to note that ExTr alone did not alter any peroxisomal genes in mice fed the low-fat Con diet. Alternatively, mice fed the lipid-rich NPKD not only exhibited increased expression of the aforementioned peroxisomal genes, but ExTr appeared to further potentiate upregulation of these genes in the NPKD group (Fig. 6C). Overall, these results suggest that 1) a NPKD and ExTr alone increase peroxisomal fat oxidation, and 2) the combination of a NPKD along with 3-wk ExTr can further induce remodeling of peroxisomal genes in mouse skeletal muscle.
Figure 6.
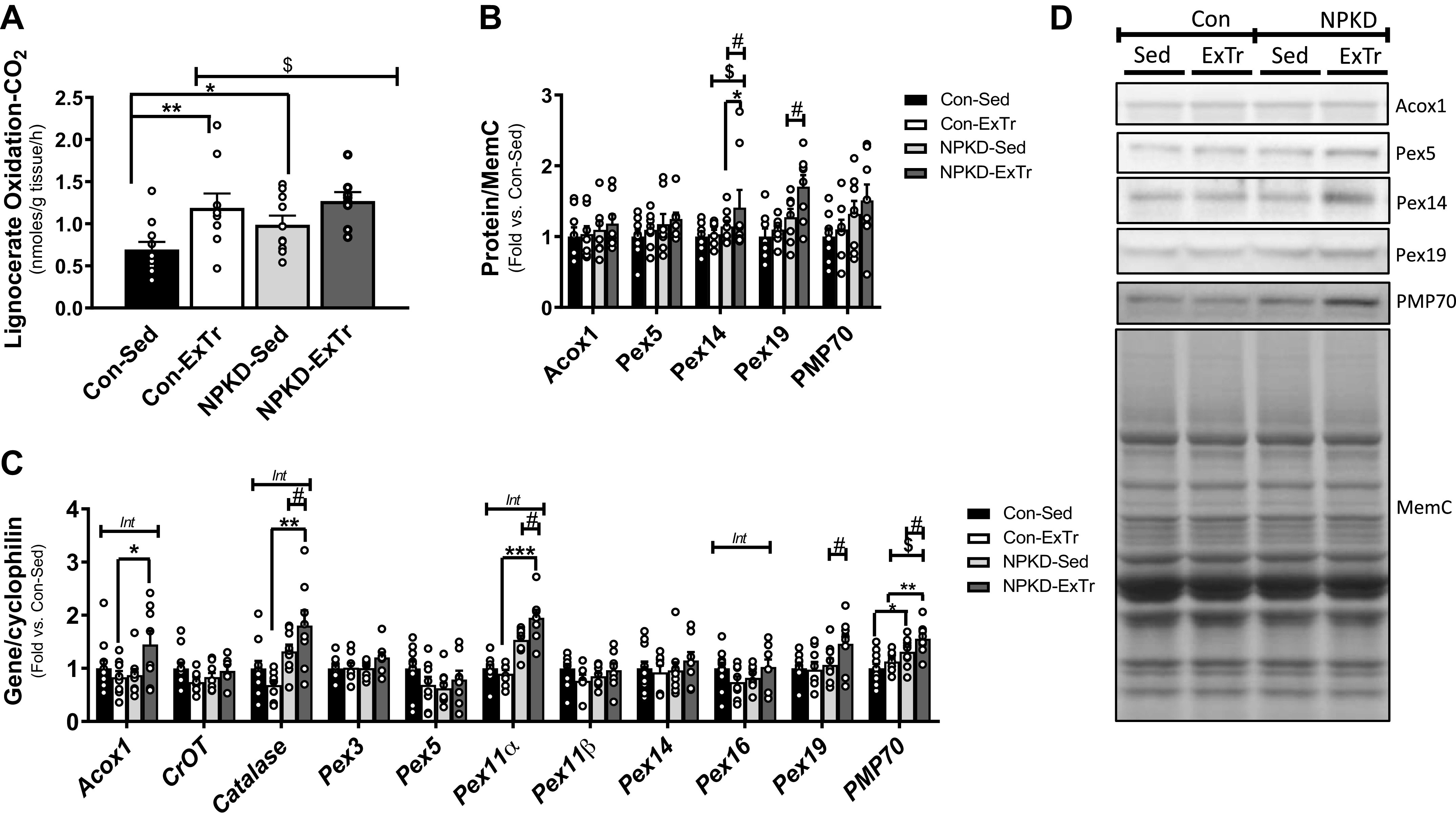
Peroxisomal adaptations. Peroxisomal fatty acid oxidation was tested by measuring oxidation of the very long-chain fatty acid (C24:0), lignocerate (A) in mixed gastrocnemius muscle homogenates. Peroxisomal proteins (B and D) and genes (C) were also measured. Protein images were normalized to total protein using MemCode (MemC), while gene expression was normalized to cyclophilin (PPIB). Control sedentary (Con-Sed; n = 11 mice), control exercise training (Con-ExTr; n = 9 mice), normal protein, ketogenic diet sedentary (NPKD-Sed; n = 10 mice), and NPKD exercise training (NPKD-ExTr; n = 9 mice) were used for lignocerate oxidation and gene expression analyses, while n = 7 or 9/group were used for Western blots. Data were analyzed using a two-way ANOVA with a Bonferroni post hoc test and are presented as means ± SE. #P < 0.05 main effect of diet; $main effect for exercise; Bonferroni post hoc test revealed significant differences (identified with an asterisk) at the following P values *P < 0.05; **P < 0.01; ***P < 0.001. Int, interaction effect.
DISCUSSION
Ketogenic diets have been found to enhance energy expenditure, while improving body weight, fat mass, lean mass, lipid profiles, glycemic index, and glucose homeostasis in overweight subjects with metabolic disease (6, 26, 40–42). These reports, however, are nuanced as a review of studies using human subjects found that while KDs having high protein content reduce body weight, studies matching protein content with the control diet have largely failed to show an advantage of KDs to lower fat mass (6). In stark contrast, studies in rodents indicate that a protein-restricted KD is optimal for lowering body weight and fat mass (26–30). A likely resolution to this apparent schism between species is that while high-protein KDs induce nutritional ketosis in humans with circulating ketones reaching 0.5–3.0 mM (3, 6), they do not do so in rodents (43). To achieve similar ketone levels in rodents, a protein-restricted KD is required (43). The importance of raising ketones to improve body composition is perhaps best illustrated by studies that show provision of exogenous ketone esters is sufficient to reduce body weight and fat mass without significantly altering macronutrient content of the diet (3, 44–46). As such, it appears that the beneficial changes in body composition in response to KDs likely depend on the degree of nutritional ketosis reached. Although the NPKD in the present study was chosen to more closely resemble the protein content of KDs consumed by humans than the protein-restricted KD most commonly used in rodent studies, the failure to induce robust nutritional ketosis may have negated effects to improve body composition often reported in response to KDs.
We previously reported that low-intensity treadmill running induced acute shifts in skeletal muscle substrate oxidative pathways in lean, healthy C57BL/6J male mice; however, training at this intensity for 6 wk failed to induce robust adaptations in substrate oxidative capacity (33). Using the same mouse model, the present study employed a more vigorous exercise stimulus that is reported to induce shifts in mitochondrial content/function and substrate oxidative capacity (19–21). Comparing mice fed low-fat control diets from both studies shows that in contrast to low-intensity exercise, training at vigorous-intensity did induce adaptations in skeletal muscle as evidenced by increased intramuscular glycogen, as well as elevated mitochondrial and peroxisomal fatty acid oxidation. Despite these adaptations, Con-ExTr mice did not exhibit other exercise training adaptations commonly reported such as increases in PGC-1α, mitochondrial content, markers of mitochondrial fission/fusion, intramuscular TAGs, or peroxisomal content. Interestingly, however, nearly all these parameters showed additive or synergistic adaptations in NPKD-ExTr mice. With this in mind, it is important to note two key matters that are common to rodents and humans: 1) lipid-rich diets enhance fatty acid oxidative capacity (22, 37, 47, 48), and 2) the postexercise recovery period is a time during which substantial metabolic remodeling of tissue can occur and consuming carbohydrates before, during, or within 2 h after exercise can negate remodeling of mitochondria and fatty acid oxidative capacity (21, 49, 50). Since mice were immediately allowed access to food after each exercise bout, it is tempting to speculate that the carbohydrate-deficient, lipid-rich NPKD may have helped optimize adaptations in lipid metabolism pathways in response to ExTr, whereas abundant carbohydrates in the Con-ExTr group may have minimized these adaptations. Alternatively, ketones 1) continue to rise after exercise cessation and are sustained for several hours during the postexercise recovery period (51–53) and 2) can impact cell signaling and metabolic processes that impact metabolic remodeling of tissues (3). We previously reported that circulating ketones were elevated immediately postexercise in this study, but the effect was not necessarily more robust in the NPKD-ExTr group than the Con-ExTr group (12). Unfortunately, we did not continue to monitor them during the postexercise recovery period. Since mice in this study were immediately allowed access to food after each exercise bout, ketone levels were likely rapidly reduced in the Con-ExTr group by the carbohydrate-rich diet but were sustained at higher levels in the NPKD-ExTr group during this critical adaptive phase. In addition, although the increase in circulating ketones induced by the NPKD was modest and did not reach the reported threshold of nutritional ketosis (0.5–3.0 mM), a recent study reported that treatment of cells with low amounts of exogenous ketones (0.5 mM) more effectively increased mitochondrial respiration in cultured cells than moderate-to-high doses (54); thus, it is possible that ketones near the low end of the spectrum of nutritional ketosis may be more beneficial. Collectively, it seems logical and likely that most adaptations in the present study were driven by the elevated lipid content of the NPKD; however, we cannot completely discount the possibility that the level of ketosis reached contributed to the adaptations reported.
Exercise-induced activation of AMPK (55–58) and PGC-1α (59, 60) induces metabolic remodeling in human and rodent skeletal muscle. In the present study, little activation of these factors was observed in Con-ExTr mice; however, both were significantly elevated in NPKD-ExTr mice. Activation of these factors likely explains the increase in genes related to mitochondrial fission/fusion, as well as heightened fatty acid oxidative capacity, since both mechanisms stimulate these pathways (61–64). Importantly, previous studies report increased AMPK activity in skeletal muscle from mice fed a protein-restricted KD (29), but this was not observed in NPKD-Sed mice. As such, results within the present study show additive or synergistic activation of these pathways in NPKD-ExTr mice that is not confounded by protein-restriction. These adaptations were likely primarily driven by the high lipid content of the NPKD, rather than ketone bodies per se because 1) the NPKD only modestly increased ketones, and 2) previous studies show ingestion of ketone esters directly does not alter similar mitochondrial factors in mouse skeletal muscle (44). Interestingly, previous studies that have employed a similar design to the present study reported that KDs increase genes involved in lipid utilization (65), as well as activity and coupling of complexes of the mitochondrial oxidative phosphorylation system (13); however, these studies did not report interaction effects with exercise training. Alternatively, although traditional high-fat diets (HFD) often increase mitochondrial content, we report that the lipid-rich NPKD alone did not increase OxPhos proteins. Interestingly, evidence suggests mitochondrial adaptations to a KD may not be highly consistent as the same group has reported decreased (14), little change (13), or increased (66) mitochondrial content and/or function using a similar diet across all studies. Key differences between a traditional HFD and a lipid-rich KD are that the KD 1) has minimal carbohydrate compared to a HFD, 2) typically induces nutritional ketosis, and 3) does not induce insulin resistance. As such, whether mitochondrial biogenesis 1) is facilitated by carbohydrates in the HFD, 2) is limited by ketones, or 3) requires a more substantial metabolic stress (e.g., insulin resistance) to occur warrants further study. Overall, more research is required to elucidate whether metabolic changes are driven primarily by the high lipid content of the diet, are a result of severe carbohydrate restriction, or whether ketones directly regulate these responses before we can establish if KDs potentiate remodeling of metabolic pathways in skeletal muscle in response to exercise.
In addition to its role in regulating mitochondrial content and function, we previously found that PGC-1α induces peroxisomal biogenesis in human myotubes (36). We also reported that acute low-intensity exercise induces a rapid, short-term increase in peroxisomal lipid oxidation (33). In addition, peroxisomal oxidative capacity increases in response to lipid-rich environments in rodent and human muscle (38, 67). Data herein extend upon these findings as they are the first to report that vigorous-intensity exercise training increases peroxisomal oxidation in mice fed a low-fat diet; however, while being fed a lipid-rich NPKD, exercise training further induced several peroxisomal genes and proteins in skeletal muscle. Since peroxisomes are one of two organelles equipped with a β-oxidation system, they play an important role in lipid metabolism in mammalian cells (68). Indeed, peroxisomes provide an alternative route for lipid catabolism that allows them to rescue mitochondrial fatty acid oxidation when mitochondrial lipid entry is impaired (38, 39); however, this is not likely to be the primary function of peroxisomes during exercise. Rather, since lipolysis of endogenous lipid stores during exercise delivers far more fatty acids to the skeletal muscle than is required to meet energy demand (21, 69, 70), peroxisomes likely act as a safety valve to limit the mitochondrial lipid burden in exercising muscle. In theory, limiting lipid excess in mitochondria would allow them to maintain optimal substrate metabolism to meet the increasing energy demand during exercise; however, this remains speculative and requires further investigation.
Skeletal muscle is a major site for glucose uptake and utilization under various conditions, including interventions of diet and exercise. Since carbohydrates are far less abundant than fatty acids, glucose availability is particularly important to prevent exhaustion during exercise. With this in mind, it is important to recall that the NPKD in this study was devoid of carbohydrate, yet mice consuming this diet maintained normal glycogen content in skeletal muscle. We previously reported that mice fed this carbohydrate-deficient NPKD had slightly lower hepatic glycogen levels; however, they maintained normal baseline blood glucose and, more importantly, exhibited similar increases in glucose levels immediately after exercise (35). Anecdotally, mice fed the NPKD also did not exhibit any signs of impaired exercise tolerance during individual exercise sessions. Collectively, these results suggest that despite consuming a carbohydrate-deficient diet, mice receiving the NPKD were not limited in their ability to exercise by glucose supply. These findings are consistent with the concept of keto-adaptation, where the body adapts to a KD by preferentially using fatty acids while sparing glycogen (7, 21).
Skeletal muscle mass is highly affected by diet and exercise as these factors can affect mTOR signaling and its downstream effects on protein synthesis and autophagy. To date, the effect of KDs on muscle mass and mTOR signaling are equivocal. Some suggest KDs may contribute to skeletal muscle mass maintenance or growth through upregulation of mTOR signaling, whereas others indicate KDs may inhibit mTOR signaling and lower skeletal muscle mass (71–73). Unfortunately, most rodent research looking at the effects of KDs have done so using a protein-restricted KD that does not recapitulate the macronutrient composition of KDs consumed by humans. Many tissues rely on amino acid consumption to support energetic demands, and branched-chain amino acids activate mTOR complex 1 activity and contribute to organ hypertrophy (74, 75). Since the mTOR pathway is directly linked to protein metabolism and is highly responsive to changes in amino acid levels, the applicability of previous findings from rodent studies using protein-restricted KDs toward humans is unclear. As such, we tested responses in mTOR signaling using a NPKD to address this matter and show that the NPKD activated mTOR-related proteins that drive protein synthesis. This is partially consistent with body composition assessments as the NPKD increased lean mass during the first 3 wk of the diet but this effect was largely lost from week 3–6, thus resulting in maintenance of lean mass across the 6-wk dietary intervention. Also, mTOR is an important regulator of autophagy (76, 77). Here, we show that the NPKD increased ULK1 phosphorylation at Ser757, a site known to inhibit autophagy; however, no other autophagy-related proteins were affected by diet. When taken together these findings suggest that a NPKD may differentially impact mTOR-related signaling pathways in a manner that favors protein synthesis over autophagy. Additional studies using more stringent assessments of protein synthesis and catabolism are needed to better understand how the provision of a NPKD may affect overall skeletal muscle mass and integrity.
Reports suggesting that athletes use KDs to improve sport performance has driven great interest in this field; however, whether these diets truly induce ergogenic effects remain quite contentious. Much of the initial research suggested KDs and/or exogenous ketone ester supplementation improve performance (2–5); however, recent reports challenge this notion as both have been found to impair performance (8, 9, 78). Advocates for the KD cite that improved performance is due to keto-adaptation which increases fatty acid utilization and promotes glucose sparing (5, 7). Alternatively, an argument can be made for the opposing view as well since 1) mitochondria do not efficiently utilize ketone bodies (79), 2) ketones are not readily used during exercise (80), and 3) increased reliance on fatty acids during exercise can reduce exercise economy (8). Unfortunately, the present study cannot provide further clarity to this controversy since we did not include performance assessments, but further studies certainly need to be done before a consensus can be reached.
The NPKD increased the ASM:CO2 ratio for palmitate oxidation indicating the diet increased incomplete oxidation to a slightly greater extent than complete oxidation. This is entirely predictable since environments of lipid excess, such as that provided by the NPKD, are known to increase incomplete lipid metabolism (38, 81, 82). There are, however, two points that are worth noting. First, previous studies that show increased incomplete lipid metabolism typically report that this is associated with metabolic disease (glucose intolerance, insulin resistance, diabetes, etc) (38, 81, 82); however, the increase in incomplete oxidation in the present study likely occurred in the absence of metabolic disease because KDs typically improve glucose homeostasis and the NPKD used in this study did not alter baseline glycemia or insulin levels (12). Second, exercise training did not normalize the ASM:CO2 ratio in NPKD-fed mice. This observation likely points toward context-dependent interpretation of measures of incomplete lipid metabolism. Studies suggest that limiting incomplete lipid oxidation is likely beneficial for those with metabolic disease (34, 39, 81, 82); however, results herein suggest that a heightened ratio of incomplete oxidation in the absence of metabolic disease may not need correction.
Due to tissue limitations, substrate storage (glycogen and triglycerides) was tested in quadriceps, whereas all other tissue-level outcomes (genes, proteins, and substrate oxidation) were measured in gastrocnemius. Both are mixed muscles, yet fiber-type differences are often reported between these muscle groups; however, we do not believe that this confounds the interpretation of the results presented in this manuscript. First, outcomes tested using the gastrocnemius are very consistent with, rather than in contrast to, substrate storage results from the quadriceps. Second, despite evidence indicating that intramuscular glycogen and triglyceride exhibit fiber-type differences, studies that have directly compared intramuscular glycogen (83, 84), triglycerides (85, 86), and mitochondrial respiration (87) between quadriceps and gastrocnemius show minimal differences. As such, we are confident that results obtained from the gastrocnemius and quadriceps complement each other and the interpretation is not confounded by potential fiber-type differences.
There are limitations to this study. First, as discussed previously (35), C57BL/6J mice used in this study have an inherent deficiency in nicotinamide nucleotide dehydrogenase that may compromise exercise adaptations (88, 89). Second, all animals were exercised at the same absolute intensity, but NPKD-ExTr mice experienced a slightly higher (not statistically significant) workload (Fig. 1C) than Con-ExTr mice because they were slightly heavier; therefore, it is possible that some of the adaptations in NPKD-ExTr mice were linked to them exercising at a slightly higher relative intensity. Third, a NPKD was chosen as it more closely resembles the protein content of KDs consumed by humans than the protein-restricted KD most often used in rodent research; however, it only induced a mild increase in circulating ketones, so it does not completely reproduce the physiological response to KDs in humans. It could be argued that mice may not be the optimal model for studying responses to KDs; however, excluding use of mouse models to study the effects of ketogenic diets in future studies is not likely realistic since mice are the most frequently used model in metabolic research and are often critical for mechanistic studies due to the availability of genetic models. As such, it may be best if future studies in this area that use mice are designed to mirror the macronutrient composition of KDs to those consumed by humans and provide the mice with supplemental ketone esters (diet or drinking water) to best replicate both the dietary composition and nutritional ketosis observed in humans. Fourth, food intake/energy consumption were not measured, which makes it difficult to discern whether differences in caloric intake between all groups contributed to some of the differences noted. Finally, only male mice were used in this study, so it is important to determine whether females exhibit a similar response.
Overall, we show that feeding a lipid-rich, carbohydrate-deficient NPKD to young, lean, healthy C57BL/6J male mice actually increases body weight and fat mass, but exercise training is capable of preventing this weight gain. Moreover, using a 6-wk dietary intervention and implementation of a 3-wk treadmill exercise training protocol, we showed that interactions of a NPKD and ExTr result in additive or synergistic modulation of key factors involved in energy sensing (AMPK and PGC-1α), mitochondrial dynamics (fission and fusion), as well as mitochondrial and peroxisomal fatty acid utilization in skeletal muscle. Finally, since the NPKD used in this study only modestly increased circulating ketones, it allows us to begin to delineate that adaptations in the aforementioned pathways are likely primarily driven by the lipid-content of the diet, whereas improvements in body composition that are often reported in response to a KD may be driven primarily by ketone bodies themselves; however, this requires further study.
GRANTS
This work was supported in part by National Institutes of Health (NIH)-COBRE (NIH 3P30-GM118430) and NIH-NORC (NIH 2P30-DK072476) center grants, as well as NIH 1R01DK103860 (to R. C. Noland). F. R. Goldsmith and S. E. Fuller were supported by a T32 fellowship (AT004094). M. A. Linden is supported by T32 fellowship DK064584.
DISCLAIMERS
The results are presented clearly, honestly, and without fabrication, falsification, or inappropriate data manipulation.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
T-Y.H., S.E.F., F.R.G., and R.C.N. conceived and designed research; T-Y.H., M.A.L., S.E.F., F.R.G., J.S., H.M.B., M.C.S., N.M.E., J.M.B., and R.C.N. performed experiments; T-Y.H., M.A.L., S.E.F., F.R.G., J.S., and R.C.N. analyzed data; T-Y.H., M.A.L., and R.C.N. interpreted results of experiments; T-Y.H., M.A.L., and R.C.N. prepared figures; T-Y.H., M.A.L., and R.C.N. drafted manuscript; T-Y.H., M.A.L., and R.C.N. edited and revised manuscript; T-Y.H., M.A.L., and R.C.N. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the Comparative Biology and Animal Metabolism and Behavior Core, as well as the Genomics Core at PBRC for expert technical assistance and support.
REFERENCES
- 1.Winder WW, Baldwin KM, Holloszy JO. Enzymes involved in ketone utilization in different types of muscle: adaptation to exercise. Eur J Biochem 47: 461–467, 1974. doi: 10.1111/j.1432-1033.1974.tb03713.x. [DOI] [PubMed] [Google Scholar]
- 2.Cox PJ, Kirk T, Ashmore T, Willerton K, Evans R, Smith A, Murray AJ, Stubbs B, West J, McLure SW, King MT, Dodd MS, Holloway C, Neubauer S, Drawer S, Veech RL, Griffin JL, Clarke K. Nutritional ketosis alters fuel preference and thereby endurance performance in athletes. Cell Metab 24: 256–268, 2016. doi: 10.1016/j.cmet.2016.07.010. [DOI] [PubMed] [Google Scholar]
- 3.Evans M, Cogan KE, Egan B. Metabolism of ketone bodies during exercise and training: physiological basis for exogenous supplementation. J Physiol 595: 2857–2871, 2017. doi: 10.1113/JP273185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pinckaers PJ, Churchward-Venne TA, Bailey D, van Loon LJ. Ketone bodies and exercise performance: the next magic bullet or merely hype? Sports Med 47: 383–391, 2017. doi: 10.1007/s40279-016-0577-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Volek JS, Freidenreich DJ, Saenz C, Kunces LJ, Creighton BC, Bartley JM, Davitt PM, Munoz CX, Anderson JM, Maresh CM, Lee EC, Schuenke MD, Aerni G, Kraemer WJ, Phinney SD. Metabolic characteristics of keto-adapted ultra-endurance runners. Metabolism 65: 100–110, 2016. doi: 10.1016/j.metabol.2015.10.028. [DOI] [PubMed] [Google Scholar]
- 6.Aragon AA, Schoenfeld BJ, Wildman R, Kleiner S, VanDusseldorp T, Taylor L, Earnest CP, Arciero PJ, Wilborn C, Kalman DS, Stout JR, Willoughby DS, Campbell B, Arent SM, Bannock L, Smith-Ryan AE, Antonio J. International society of sports nutrition position stand: diets and body composition. J Int Soc Sports Nutr 14: 16, 2017. doi: 10.1186/s12970-017-0174-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Volek JS, Noakes T, Phinney SD. Rethinking fat as a fuel for endurance exercise. Eur J Sport Sci 15: 13–20, 2015. doi: 10.1080/17461391.2014.959564. [DOI] [PubMed] [Google Scholar]
- 8.Burke LM, Ross ML, Garvican-Lewis LA, Welvaert M, Heikura IA, Forbes SG, Mirtschin JG, Cato LE, Strobel N, Sharma AP, Hawley JA. Low carbohydrate, high fat diet impairs exercise economy and negates the performance benefit from intensified training in elite race walkers. J Physiol 595: 2785–2807, 2017. doi: 10.1113/JP273230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burke LM, Whitfield J, Heikura IA, Ross MLR, Tee N, Forbes SF, Hall R, McKay AKA, Wallett AM, Sharma AP. Adaptation to a low carbohydrate high fat diet is rapid but impairs endurance exercise metabolism and performance despite enhanced glycogen availability. J Physiol 599: 771–790, 2021. doi: 10.1113/JP280221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gollnick PD, Piehl K, Saubert CW, Armstrong RB, Saltin B. Diet, exercise, and glycogen changes in human muscle fibers. J Appl Physiol (1985) 33: 421–425, 1972. doi: 10.1152/jappl.1972.33.4.421. [DOI] [PubMed] [Google Scholar]
- 11.Huang Q, Ma S, Tominaga T, Suzuki K, Liu C. An 8-week, low carbohydrate, high fat, ketogenic diet enhanced exhaustive exercise capacity in mice part 2: effect on fatigue recovery, post-exercise biomarkers and anti-oxidation capacity. Nutrients 10: 1339, 2018. doi: 10.3390/nu10101339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang TY, Goldsmith FR, Fuller SE, Simon J, Batdorf HM, Scott MC, Essajee NM, Brown JM, Burk DH, Morrison CD, Burke SJ, Collier JJ, Noland RC. Response of liver metabolic pathways to ketogenic diet and exercise are not additive. Med Sci Sports Exerc 52: 37–48, 2020. doi: 10.1249/MSS.0000000000002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hyatt HW, Kephart WC, Holland AM, Mumford P, Mobley CB, Lowery RP, Roberts MD, Wilson JM, Kavazis AN. A ketogenic diet in rodents elicits improved mitochondrial adaptations in response to resistance exercise training compared to an isocaloric western diet. Front Physiol 7: 533, 2016. doi: 10.3389/fphys.2016.00533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kephart WC, Mumford PW, Mao X, Romero MA, Hyatt HW, Zhang Y, Mobley CB, Quindry JC, Young KC, Beck DT, Martin JS, McCullough DJ, D'Agostino DP, Lowery RP, Wilson JM, Kavazis AN, Roberts MD. The 1-week and 8-month effects of a ketogenic diet or ketone salt supplementation on multi-organ markers of oxidative stress and mitochondrial function in rats. Nutrients 9: 1019, 2017. doi: 10.3390/nu9091019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma S, Huang Q, Yada K, Liu C, Suzuki K. An 8-week ketogenic low carbohydrate, high fat diet enhanced exhaustive exercise capacity in mice. Nutrients 10: 673, 2018. doi: 10.3390/nu10060673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pilegaard H, Osada T, Andersen LT, Helge JW, Saltin B, Neufer PD. Substrate availability and transcriptional regulation of metabolic genes in human skeletal muscle during recovery from exercise. Metabolism 54: 1048–1055, 2005. doi: 10.1016/j.metabol.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 17.Urbain P, Strom L, Morawski L, Wehrle A, Deibert P, Bertz H. Impact of a 6-week non-energy-restricted ketogenic diet on physical fitness, body composition and biochemical parameters in healthy adults. Nutr Metab (Lond) 14, 2017. doi: 10.1186/s12986-017-0175-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zajac A, Poprzecki S, Maszczyk A, Czuba M, Michalczyk M, Zydek G. The effects of a ketogenic diet on exercise metabolism and physical performance in off-road cyclists. Nutrients 6: 2493–2508, 2014. doi: 10.3390/nu6072493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drake JC, Wilson RJ, Yan Z. Molecular mechanisms for mitochondrial adaptation to exercise training in skeletal muscle. FASEB J 30: 13–22, 2016. doi: 10.1096/fj.15-276337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Egan B, Zierath JR. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab 17: 162–184, 2013. doi: 10.1016/j.cmet.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 21.Noland RC. Exercise and regulation of lipid metabolism. Prog Mol Biol Transl Sci 135: 39–74, 2015. doi: 10.1016/bs.pmbts.2015.06.017. [DOI] [PubMed] [Google Scholar]
- 22.Garcia-Roves P, Huss JM, Han DH, Hancock CR, Iglesias-Gutierrez E, Chen M, Holloszy JO. Raising plasma fatty acid concentration induces increased biogenesis of mitochondria in skeletal muscle. Proc Natl Acad Sci USA 104: 10709–10713, 2007. doi: 10.1073/pnas.0704024104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McAinch AJ, Lee JS, Bruce CR, Tunstall RJ, Hawley JA, Cameron-Smith D. Dietary regulation of fat oxidative gene expression in different skeletal muscle fiber types. Obes Res 11: 1471–1479, 2003. doi: 10.1038/oby.2003.197. [DOI] [PubMed] [Google Scholar]
- 24.Miller VJ, Villamena FA, Volek JS. Nutritional ketosis and mitohormesis: potential implications for mitochondrial function and human health. J Nutr Metab 2018: 1–27, 2018. doi: 10.1155/2018/5157645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simi B, Sempore B, Mayet MH, Favier RJ. Additive effects of training and high-fat diet on energy metabolism during exercise. J Appl Physiol (1985) 71: 197–203, 1991. doi: 10.1152/jappl.1991.71.1.197. [DOI] [PubMed] [Google Scholar]
- 26.Badman MK, Kennedy AR, Adams AC, Pissios P, Maratos-Flier E. A very low carbohydrate ketogenic diet improves glucose tolerance in ob/ob mice independently of weight loss. Am J Physiol Endocrinol Metab 297: E1197–E1204, 2009. doi: 10.1152/ajpendo.00357.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ellenbroek JH, van Dijck L, Tons HA, Rabelink TJ, Carlotti F, Ballieux BE, de Koning EJ. Long-term ketogenic diet causes glucose intolerance and reduced beta- and alpha-cell mass but no weight loss in mice. Am J Physiol Endocrinol Metab 306: E552–E558, 2014. doi: 10.1152/ajpendo.00453.2013. [DOI] [PubMed] [Google Scholar]
- 28.Hutfles LJ, Wilkins HM, Koppel SJ, Weidling IW, Selfridge JE, Tan E, Thyfault JP, Slawson C, Fenton AW, Zhu H, Swerdlow RH. A bioenergetics systems evaluation of ketogenic diet liver effects. Appl Physiol Nutr Metab 42: 955–962, 2017. doi: 10.1139/apnm-2017-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kennedy AR, Pissios P, Otu H, Roberson R, Xue B, Asakura K, Furukawa N, Marino FE, Liu FF, Kahn BB, Libermann TA, Maratos-Flier EA. high-fat, ketogenic diet induces a unique metabolic state in mice. Am J Physiol Endocrinol Metab 292: E1724–E1739, 2007. doi: 10.1152/ajpendo.00717.2006. [DOI] [PubMed] [Google Scholar]
- 30.Laeger T, Henagan TM, Albarado DC, Redman LM, Bray GA, Noland RC, Munzberg H, Hutson SM, Gettys TW, Schwartz MW, Morrison CD. FGF21 is an endocrine signal of protein restriction. J Clin Invest 124: 3913–3922, 2014. doi: 10.1172/JCI74915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brouns F. Overweight and diabetes prevention: is a low-carbohydrate-high-fat diet recommendable? Eur J Nutr 57: 1301–1312, 2018. doi: 10.1007/s00394-018-1636-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Christodoulides C, Dyson P, Sprecher D, Tsintzas K, Karpe F. Circulating fibroblast growth factor 21 is induced by peroxisome proliferator-activated receptor agonists but not ketosis in man. J Clin Endocrinol Metab 94: 3594–3601, 2009. doi: 10.1210/jc.2009-0111. [DOI] [PubMed] [Google Scholar]
- 33.Fuller SE, Huang TY, Simon J, Batdorf HM, Essajee NM, Scott MC, Waskom CM, Brown JM, Burke SJ, Collier JJ, Noland RC. Low-intensity exercise induces acute shifts in liver and skeletal muscle substrate metabolism but not chronic adaptations in tissue oxidative capacity. J Appl Physiol (1985) 127: 143–156, 2019. doi: 10.1152/japplphysiol.00820.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ghosh S, Wicks SE, Vandanmagsar B, Mendoza TM, Bayless DS, Salbaum JM, Dearth SP, Campagna SR, Mynatt RL, Noland RC. Extensive metabolic remodeling after limiting mitochondrial lipid burden is consistent with an improved metabolic health profile. J Biol Chem 294: 12313–12327, 2019. doi: 10.1074/jbc.RA118.006074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang TY, Goldsmith FR, Fuller SE, Simon J, Batdorf HM, Scott MC, Essajee NM, Brown JM, Burk DH, Morrison CD, Burke SJ, Collier JJ, Noland RC. Response of liver metabolic pathways to ketogenic diet and exercise are not additive. Med Sci Sports Exerc 52: 37–48, 2020. doi: 10.1249/MSS.0000000000002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang TY, Zheng D, Houmard JA, Brault JJ, Hickner RC, Cortright RN. Overexpression of PGC-1alpha increases peroxisomal activity and mitochondrial fatty acid oxidation in human primary myotubes. Am J Physiol Endocrinol Metab 312: E253–E263, 2017. doi: 10.1152/ajpendo.00331.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Noland RC, Thyfault JP, Henes ST, Whitfield BR, Woodlief TL, Evans JR, Lust JA, Britton SL, Koch LG, Dudek RW, Dohm GL, Cortright RN, Lust RM. Artificial selection for high-capacity endurance running is protective against high-fat diet-induced insulin resistance. Am J Physiol Endocrinol Metab 293: E31–E41, 2007. doi: 10.1152/ajpendo.00500.2006. [DOI] [PubMed] [Google Scholar]
- 38.Noland RC, Woodlief TL, Whitfield BR, Manning SM, Evans JR, Dudek RW, Lust RM, Cortright RN. Peroxisomal-mitochondrial oxidation in a rodent model of obesity-associated insulin resistance. Am J Physiol Endocrinol Metab 293: E986–E1001, 2007. doi: 10.1152/ajpendo.00399.2006. [DOI] [PubMed] [Google Scholar]
- 39.Wicks SE, Vandanmagsar B, Haynie KR, Fuller SE, Warfel JD, Stephens JM, Wang M, Han X, Zhang J, Noland RC, Mynatt RL. Impaired mitochondrial fat oxidation induces adaptive remodeling of muscle metabolism. Proc Natl Acad Sci USA 112: E3300–E3309, 2015. doi: 10.1073/pnas.1418560112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Castellana M, Conte E, Cignarelli A, Perrini S, Giustina A, Giovanella L, Giorgino F, Trimboli P. Efficacy and safety of very low calorie ketogenic diet (VLCKD) in patients with overweight and obesity: a systematic review and meta-analysis. Rev Endocr Metab Disord 21: 5–16, 2020. doi: 10.1007/s11154-019-09514-y. [DOI] [PubMed] [Google Scholar]
- 41.Mobbs CV, Mastaitis J, Isoda F, Poplawski M. Treatment of diabetes and diabetic complications with a ketogenic diet. J Child Neurol 28: 1009–1014, 2013. doi: 10.1177/0883073813487596. [DOI] [PubMed] [Google Scholar]
- 42.Walton CM, Perry K, Hart RH, Berry SL, Bikman BT. Improvement in glycemic and lipid profiles in type 2 diabetics with a 90-day ketogenic diet. J Diabetes Res 2019: 1–6, 2019. doi: 10.1155/2019/8681959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bielohuby M, Menhofer D, Kirchner H, Stoehr BJ, Muller TD, Stock P, Hempel M, Stemmer K, Pfluger PT, Kienzle E, Christ B, Tschop MH, Bidlingmaier M. Induction of ketosis in rats fed low-carbohydrate, high-fat diets depends on the relative abundance of dietary fat and protein. Am J Physiol Endocrinol Metab 300: E65–E76, 2011. doi: 10.1152/ajpendo.00478.2010. [DOI] [PubMed] [Google Scholar]
- 44.Davis RAH, Deemer SE, Bergeron JM, Little JT, Warren JL, Fisher G, Smith DL , Jr., Fontaine KR, Dickinson SL, Allison DB, Plaisance EP. Dietary R,S-1,3-butanediol diacetoacetate reduces body weight and adiposity in obese mice fed a high-fat diet. FASEB J 33: 2409–2421, 2019. doi: 10.1096/fj.201800821RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deemer SE, Davis RAH, Gower BA, Koutnik AP, Poff AM, Dickinson SL, Allison DB, D'Agostino DP, Plaisance EP. Concentration-dependent effects of a dietary ketone ester on components of energy balance in mice. Front Nutr 6: 56, 2019. doi: 10.3389/fnut.2019.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murray AJ, Knight NS, Cole MA, Cochlin LE, Carter E, Tchabanenko K, Pichulik T, Gulston MK, Atherton HJ, Schroeder MA, Deacon RM, Kashiwaya Y, King MT, Pawlosky R, Rawlins JN, Tyler DJ, Griffin JL, Robertson J, Veech RL, Clarke K. Novel ketone diet enhances physical and cognitive performance. FASEB J 30: 4021–4032, 2016. doi: 10.1096/fj.201600773R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hancock CR, Han DH, Chen M, Terada S, Yasuda T, Wright DC, Holloszy JO. High-fat diets cause insulin resistance despite an increase in muscle mitochondria. Proc Natl Acad Sci USA 105: 7815–7820, 2008. doi: 10.1073/pnas.0802057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Turner N, Bruce CR, Beale SM, Hoehn KL, So T, Rolph MS, Cooney GJ. Excess lipid availability increases mitochondrial fatty acid oxidative capacity in muscle: evidence against a role for reduced fatty acid oxidation in lipid-induced insulin resistance in rodents. Diabetes 56: 2085–2092, 2007. doi: 10.2337/db07-0093. [DOI] [PubMed] [Google Scholar]
- 49.Gonzalez JT, Stevenson EJ. New perspectives on nutritional interventions to augment lipid utilisation during exercise. Br J Nutr 107: 339–349, 2012. doi: 10.1017/S0007114511006684. [DOI] [PubMed] [Google Scholar]
- 50.Kiens B, Alsted TJ, Jeppesen J. Factors regulating fat oxidation in human skeletal muscle. Obes Rev 12: 852–858, 2011. doi: 10.1111/j.1467-789X.2011.00898.x. [DOI] [PubMed] [Google Scholar]
- 51.Kim MJ, Hong SH, Cho W, Park DH, Lee EB, Song Y, Choe YS, Lee JH, Jang Y, Lee W, Jeon JY. Breath acetone measurement-based prediction of exercise-induced energy and substrate expenditure. Sensors (Basel) 20: 6878, 2020. doi: 10.3390/s20236878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Matoulek M, Svobodova S, Vetrovska R, Stranska Z, Svacina S. Post-exercise changes of beta hydroxybutyrate as a predictor of weight changes. Physiol Res 63: S321–S325, 2014. doi: 10.33549/physiolres.932815. [DOI] [PubMed] [Google Scholar]
- 53.Walsh NP, Blannin AK, Clark AM, Cook L, Robson PJ, Gleeson M. The effects of high-intensity intermittent exercise on the plasma concentrations of glutamine and organic acids. Eur J Appl Physiol Occup Physiol 77: 434–438, 1998. doi: 10.1007/s004210050356. [DOI] [PubMed] [Google Scholar]
- 54.Mey JT, Erickson ML, Axelrod CL, King WT, Flask CA, McCullough AJ, Kirwan JP. beta-Hydroxybutyrate is reduced in humans with obesity-related NAFLD and displays a dose-dependent effect on skeletal muscle mitochondrial respiration in vitro. Am J Physiol Endocrinol Metab 319: E187–E195, 2020. doi: 10.1152/ajpendo.00058.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen ZP, Stephens TJ, Murthy S, Canny BJ, Hargreaves M, Witters LA, Kemp BE, McConell GK. Effect of exercise intensity on skeletal muscle AMPK signaling in humans. Diabetes 52: 2205–2212, 2003. doi: 10.2337/diabetes.52.9.2205. [DOI] [PubMed] [Google Scholar]
- 56.Jorgensen SB, Treebak JT, Viollet B, Schjerling P, Vaulont S, Wojtaszewski JF, Richter EA. Role of AMPKalpha2 in basal, training-, and AICAR-induced GLUT4, hexokinase II, and mitochondrial protein expression in mouse muscle. Am J Physiol Endocrinol Metab 292: E331–E339, 2007. doi: 10.1152/ajpendo.00243.2006. [DOI] [PubMed] [Google Scholar]
- 57.Winder WW, Hardie DG. Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. Am J Physiol Endocrinol Metab 270: E299–E304, 1996. doi: 10.1152/ajpendo.1996.270.2.E299. [DOI] [PubMed] [Google Scholar]
- 58.Wojtaszewski JF, Nielsen P, Hansen BF, Richter EA, Kiens B. Isoform-specific and exercise intensity-dependent activation of 5′-AMP-activated protein kinase in human skeletal muscle. J Physiol 528: 221–226, 2000. doi: 10.1111/j.1469-7793.2000.t01-1-00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci USA 104: 12017–12022, 2007. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leick L, Fentz J, Bienso RS, Knudsen JG, Jeppesen J, Kiens B, Wojtaszewski JF, Pilegaard H. PGC-1{alpha} is required for AICAR-induced expression of GLUT4 and mitochondrial proteins in mouse skeletal muscle. Am J Physiol Endocrinol Metab 299: E456–E465, 2010. doi: 10.1152/ajpendo.00648.2009. [DOI] [PubMed] [Google Scholar]
- 61.Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab 17: 491–506, 2013. doi: 10.1016/j.cmet.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Parker BA, Walton CM, Carr ST, Andrus JL, Cheung ECK, Duplisea MJ, Wilson EK, Draney C, Lathen DR, Kenner KB, Thomson DM, Tessem JS, Bikman BT. β-hydroxybutyrate elicits favorable mitochondrial changes in skeletal muscle. Int J Mol Sci 19: 2247, 2018. doi: 10.3390/ijms19082247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pilegaard H, Ordway GA, Saltin B, Neufer PD. Transcriptional regulation of gene expression in human skeletal muscle during recovery from exercise. Am J Physiol Endocrinol Metab 279: E806–E814, 2000. doi: 10.1152/ajpendo.2000.279.4.E806. [DOI] [PubMed] [Google Scholar]
- 64.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98: 115–124, 1999. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 65.Shimizu K, Saito H, Sumi K, Sakamoto Y, Tachi Y, Iida K. Short-term and long-term ketogenic diet therapy and the addition of exercise have differential impacts on metabolic gene expression in the mouse energy-consuming organs heart and skeletal muscle. Nutr Res 60: 77–86, 2018. doi: 10.1016/j.nutres.2018.09.004. [DOI] [PubMed] [Google Scholar]
- 66.Parry HA, Kephart WC, Mumford PW, Romero MA, Mobley CB, Zhang Y, Roberts MD, Kavazis AN. Ketogenic diet increases mitochondria volume in the liver and skeletal muscle without altering oxidative stress markers in rats. Heliyon 4: e00975, 2018. doi: 10.1016/j.heliyon.2018.e00975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang TY, Zheng D, Hickner RC, Brault JJ, Cortright RN. Peroxisomal gene and protein expression increase in response to a high-lipid challenge in human skeletal muscle. Metabolism 98: 53–61, 2019. doi: 10.1016/j.metabol.2019.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lazarow PB. The role of peroxisomes in mammalian cellular metabolism. J Inherit Metab Dis 10: 11–22, 1987. doi: 10.1007/BF01812843. [DOI] [PubMed] [Google Scholar]
- 69.Romijn JA, Coyle EF, Sidossis LS, Gastaldelli A, Horowitz JF, Endert E, Wolfe RR. Regulation of endogenous fat and carbohydrate metabolism in relation to exercise intensity and duration. Am J Physiol Endocrinol Metab 265: E380–E391, 1993. doi: 10.1152/ajpendo.1993.265.3.E380. [DOI] [PubMed] [Google Scholar]
- 70.Turcotte LP, Richter EA, Kiens B. Increased plasma FFA uptake and oxidation during prolonged exercise in trained vs. untrained humans. Am J Physiol Endocrinol Metab 262: E791–E799, 1992. doi: 10.1152/ajpendo.1992.262.6.E791. [DOI] [PubMed] [Google Scholar]
- 71.McDaniel SS, Rensing NR, Thio LL, Yamada KA, Wong M. The ketogenic diet inhibits the mammalian target of rapamycin (mTOR) pathway. Epilepsia 52: e7–e11, 2011. doi: 10.1111/j.1528-1167.2011.02981.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nakao R, Abe T, Yamamoto S, Oishi K. Ketogenic diet induces skeletal muscle atrophy via reducing muscle protein synthesis and possibly activating proteolysis in mice. Sci Rep 9: 19652, 2019. doi: 10.1038/s41598-019-56166-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ochaba J, Powers AF, Tremble KA, Greenlee S, Post NM, Matson JE, MacLeod AR, Guo S, Aghajan M. A novel and translational role for autophagy in antisense oligonucleotide trafficking and activity. Nucleic Acids Res 47: 11284–11303, 2019. doi: 10.1093/nar/gkz901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Neinast MD, Jang C, Hui S, Murashige DS, Chu Q, Morscher RJ, Li X, Zhan L, White E, Anthony TG, Rabinowitz JD, Arany Z. Quantitative analysis of the whole-body metabolic fate of branched-chain amino acids. Cell Metab 29: 417–429.e4, 2019. doi: 10.1016/j.cmet.2018.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Neishabouri SH, Hutson SM, Davoodi J. Chronic activation of mTOR complex 1 by branched chain amino acids and organ hypertrophy. Amino Acids 47: 1167–1182, 2015. doi: 10.1007/s00726-015-1944-y. [DOI] [PubMed] [Google Scholar]
- 76.Inokuchi-Shimizu S, Park EJ, Roh YS, Yang L, Zhang B, Song J, Liang S, Pimienta M, Taniguchi K, Wu X, Asahina K, Lagakos W, Mackey MR, Akira S, Ellisman MH, Sears DD, Olefsky JM, Karin M, Brenner DA, Seki E. TAK1-mediated autophagy and fatty acid oxidation prevent hepatosteatosis and tumorigenesis. J Clin Invest 124: 3566–3578, 2014. doi: 10.1172/JCI74068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci 122: 3589–3594, 2009. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Helge JW. A high carbohydrate diet remains the evidence based choice for elite athletes to optimise performance. J Physiol 595: 2775–2775, 2017. doi: 10.1113/JP273830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Petrick HL, Brunetta HS, Pignanelli C, Nunes EA, Loon LJC, Burr JF, Holloway GP. In vitro ketone-supported mitochondrial respiration is minimal when other substrates are readily available in cardiac and skeletal muscle. J Physiol 598: 4869–4885, 2020. doi: 10.1113/JP280032. [DOI] [PubMed] [Google Scholar]
- 80.Dearlove DJ, Harrison OK, Hodson L, Jefferson A, Clarke K, Cox PJ. The effect of blood ketone concentration and exercise intensity on exogenous ketone oxidation rates in athletes. Med Sci Sports Exerc 53: 505–516, 2021. doi: 10.1249/MSS.0000000000002502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, Bain J, Stevens R, Dyck JR, Newgard CB, Lopaschuk GD, Muoio DM. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab 7: 45–56, 2008. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 82.Noland RC, Koves TR, Seiler SE, Lum H, Lust RM, Ilkayeva O, Stevens RD, Hegardt FG, Muoio DM. Carnitine insufficiency caused by aging and overnutrition compromises mitochondrial performance and metabolic control. J Biol Chem 284: 22840–22852, 2009. doi: 10.1074/jbc.M109.032888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Patel S, Macaulay K, Woodgett JR. Tissue-specific analysis of glycogen synthase kinase-3alpha (GSK-3alpha) in glucose metabolism: effect of strain variation. PLoS One 6: e15845, 2011. doi: 10.1371/journal.pone.0015845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sun B, Zhang H, Bird A, Li S, Young SP, Koeberl DD. Impaired clearance of accumulated lysosomal glycogen in advanced Pompe disease despite high-level vector-mediated transgene expression. J Gene Med 11: 913–920, 2009. doi: 10.1002/jgm.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Guo Z. Triglyceride content in skeletal muscle: variability and the source. Anal Biochem 296: 1–8, 2001. doi: 10.1006/abio.2001.5233. [DOI] [PubMed] [Google Scholar]
- 86.Hagstrom-Toft E, Qvisth V, Nennesmo I, Ryden M, Bolinder H, Enoksson S, Bolinder J, Arner P. Marked heterogeneity of human skeletal muscle lipolysis at rest. Diabetes 51: 3376–3383, 2002. doi: 10.2337/diabetes.51.12.3376. [DOI] [PubMed] [Google Scholar]
- 87.Jacobs RA, Diaz V, Meinild AK, Gassmann M, Lundby C. The C57Bl/6 mouse serves as a suitable model of human skeletal muscle mitochondrial function. Exp Physiol 98: 908–921, 2013. doi: 10.1113/expphysiol.2012.070037. [DOI] [PubMed] [Google Scholar]
- 88.Fisher-Wellman KH, Ryan TE, Smith CD, Gilliam LA, Lin CT, Reese LR, Torres MJ, Neufer PD. A direct comparison of metabolic responses to high-fat diet in C57BL/6J and C57BL/6NJ mice. Diabetes 65: 3249–3261, 2016. doi: 10.2337/db16-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Massett MP, Berk BC. Strain-dependent differences in responses to exercise training in inbred and hybrid mice. Am J Physiol Regul Integr Comp Physiol 288: R1006–R1013, 2005. doi: 10.1152/ajpregu.00476.2004. [DOI] [PubMed] [Google Scholar]