

Abstract
Cardiovascular diseases (CVD) are the leading cause of death worldwide, and novel therapies are drastically needed to prevent or delay the onset of CVD to reduce the societal and healthcare burdens associated with these chronic diseases. One such therapy is “heat therapy,” or chronic, repeated use of hot baths or saunas. Although using heat exposure to improve health is not a new concept, it has received renewed attention in recent years as a growing number of studies have demonstrated robust and widespread beneficial effects of heat therapy on cardiovascular health. Here, we review the existing literature, with particular focus on the molecular mechanisms that underscore the cardiovascular benefits of this practice.
Keywords: heat shock proteins, hot water immersion, sauna, vascular function
INTRODUCTION
Cardiovascular diseases (CVD) remain the leading cause of morbidity and mortality in the developed world, contributing to >30% of deaths in the United States (1). Advancing age is by far the strongest risk factor for CVD, and current demographic trends predict a major increase in the number of middle-aged and older adults with attendant increases in CVD prevalence and health care costs (2). The prevalence of other traditional (modifiable) risk factors for CVD, such as obesity and hypertension, is increasing at an even greater rate (1). As such, identifying evidence-based strategies for preventing, delaying, and/or reducing the development of CVD is a high biomedical research priority (3–5).
One proposed lifestyle-based intervention is passive heat therapy, which refers to the chronic repeated use of hot baths or saunas. Although heat therapy has been used for centuries by many cultures, with common reports of improved overall well-being and quality of life (6), we have only recently begun to systematically investigate the vast physiological and health benefits of this ancient practice. Much of the research to-date has focused on the benefits of heat therapy on the cardiovascular (CV) system, and there is now evidence supporting its efficacy for reducing risk and/or severity of CVD across animal models, epidemiology, and intervention-based studies in healthy individuals and CVD patients. There has also been increasing investigation into the molecular mechanisms that may underscore the CV benefits of heat therapy. This review will briefly summarize the existing literature on the effects of passive heat therapy on the CV system but will focus on the molecular mechanisms mediating these benefits (Fig. 1). For a comprehensive review of the evidence for and against effects of heat therapy on vascular function, readers are directed to a recent review by Cheng and MacDonald (7).
Figure 1.
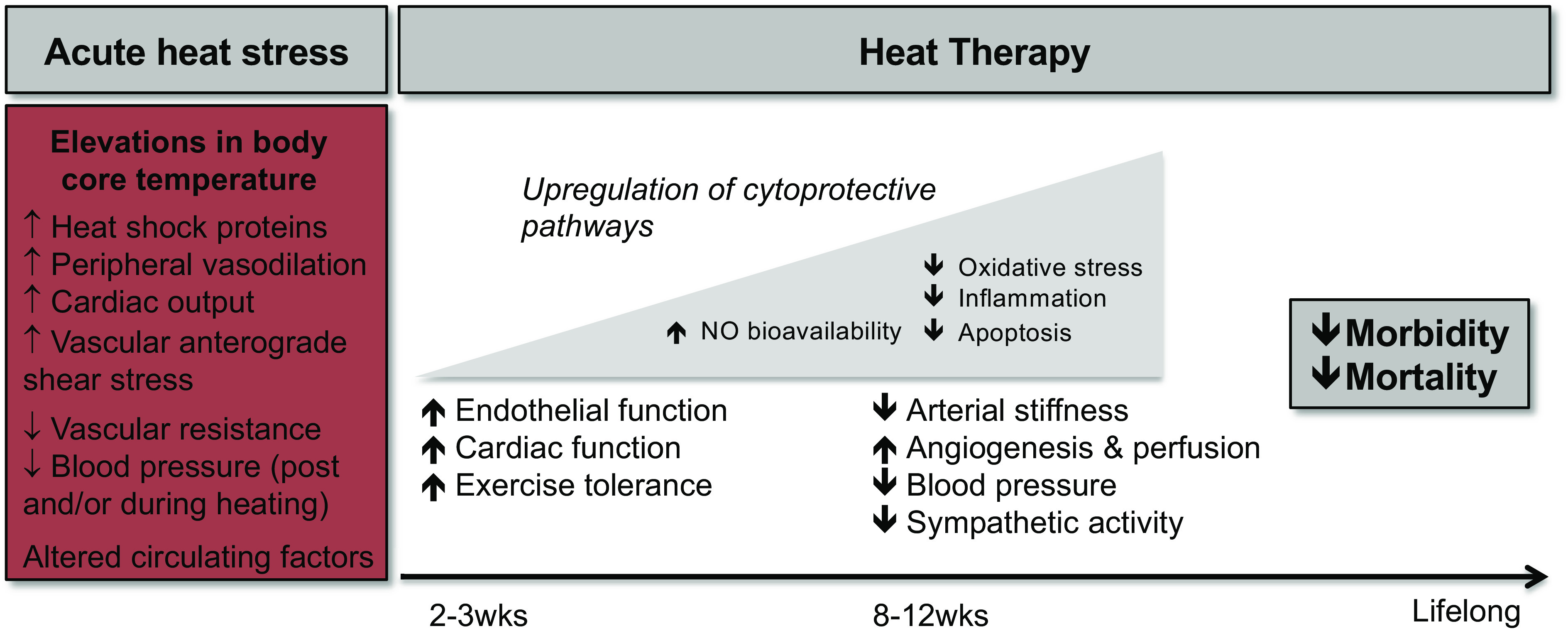
Summary of how heat stress can lead to short-term, long-term, and lifelong reductions in cardiovascular risk when repeated in the form of heat therapy.
Changes in Cardiovascular Function That Precede CVD
As discussed in later sections, heat therapy initiated later in life has been shown to ameliorate symptoms of established CVD. However, the majority of CVDs develop over decades and are preceded by age- and other risk factor-associated changes in CV function that increase susceptibility for developing CVD. Intervening to improve risk factor profile and/or overall CV function, e.g., through blood pressure (8)- or cholesterol-lowering (9) interventions, can prevent, delay, or slow the progression of CVD. Heat therapy can have profound effects on CV function and, through these effects, is therefore thought to reduce risk of developing CVD later in life. These age- and risk factor-associated changes in CV function, all of which are interrelated and drive progressive declines in one another over the life span, are summarized here.
Vascular dysfunction, characterized by vascular endothelial dysfunction and stiffening of the large elastic arteries (i.e., the aorta and carotid arteries), develops naturally with aging, is exacerbated by other risk factors (e.g., obesity, smoking, hypercholesterolemia), and is a major antecedent to clinical CVD (10–12). Endothelial dysfunction refers to an impaired ability of the vascular endothelium to produce and secrete signaling molecules important for vasodilation, vascular cell growth and coordinated proliferation/angiogenesis, and suppression of inflammation, coagulation, and the development of atherosclerotic plaques, i.e., a phenotype that is more vasoconstrictive, proinflammatory, and proatherogenic. Perhaps the most important of these signaling molecules is nitric oxide (NO), as it is upstream to most of these processes and its bioavailability is reduced with aging, with other CVD risk factors, and with most CVDs. Endothelial function is most commonly measured by endothelium-dependent dilation to either a mechanical (increased blood flow or shear stress) or chemical (acetylcholine, bradykinin, etc.) stimulus that predominantly activates endothelial NO synthase (eNOS) to produce NO and elicit vasodilation. Endothelium-dependent dilation to increased blood flow [i.e., brachial artery flow-mediated dilation (FMDBA)] is reduced in older adults, individuals with other traditional CVD risk factors, and in patients with CVD, and FMDBA is an independent predictor of future CV events and mortality (13–15).
Arterial stiffening, as measured by increased carotid-femoral (i.e., aortic) pulse wave velocity, is also an independent predictor of future CV events and mortality (16–18). Arterial stiffening is mediated by changes in the major structural proteins in the arterial wall (primarily collagen deposition and elastin degradation), as well as factors that induce an overall vasoconstrictive state, including impaired NO-mediated endothelium-dependent dilation and increased sympathetic nervous system activity. Importantly, arterial stiffening, in conjunction with endothelial dysfunction, directly contributes to the progressive increases in systolic blood pressure and a subsequent widening of pulse pressure that occur with advancing age.
Aging and other CVD risk factors (particularly hypertension) also exert direct effects on the heart, which increases susceptibility for developing CVD. Left ventricular hypertrophy, which usually occurs secondary to chronically elevated blood pressure, is characterized by detrimental changes in cardiomyocyte structure and function, as well as myocardial fibrosis and stiffening. This remodeling impairs cardiac function, particularly diastolic function (ventricular relaxation and filling), and over time may result in heart failure and increased risk of arrhythmias (19, 20). Myocardial cells from older individuals are also less tolerant of changes in calcium influx, making them more susceptible to fatal arrhythmias and less resistant to stressors, such as ischemia-reperfusion, i.e., the process that occurs with ischemic CV events. Lastly, cardiac beta-adrenergic sensitivity may also be impaired with age, affecting contractility (21).
These changes that occur to CV function over the life span are driven in large part by two molecular processes that act in a feed-forward manner to amplify one another: oxidative stress, particularly that driven by excess production of the damaging free radical superoxide, and chronic low-grade vascular inflammation. Both of these processes reduce bioavailability of NO (22, 23), mediate alterations in major arterial structural proteins (24, 25) and cardiac fibrosis (26), and directly promote the development of CVD (e.g., atherosclerosis) (19, 27, 28), among other deleterious effects. Importantly, there are several mechanisms by which passive heat therapy appears to reduce oxidative stress and inflammation, which will be discussed in later sections.
MODALITES OF PASSIVE HEATING
The goal of passive heat therapy is to increase body core temperature. Animal and some human studies have generally suggested that body core temperature should reach 38.0–38.5°C during sessions to reliably confer thermoregulatory adaptation (29). However, whether such a threshold exists for conferring cardiovascular adaptations is currently unknown. Indeed, benefits of heat therapy (albeit of lesser magnitude) have been reported even when body core temperature has not reached 38.0°C during sessions (30). As such, future work is needed to determine the ideal “dose” of heat therapy, and very likely the ideal dose will be dependent on the specific clinical condition and the desired improvements in cardiovascular risk profile.
Increases in body core temperature can be achieved with various modalities of passive heat stress. The rate at which body core temperature increases is dependent on the rate of transfer of heat from the environment to the body and the effectiveness of the body’s heat loss mechanisms, both of which vary across different modalities. In laboratory settings, the most commonly used method for passive heat stress has been water-perfused suits. Subjects wear a nylon suit that has tubes sewn into the lining, which covers their entire body with the exception of their hands, feet, and face. Water can then be circulated through the tubing at any tolerable temperature, thereby increasing skin temperature. Diathermy coils and infrared heating lamps have also been used in some studies. However, the majority of clinical studies have used saunas or hot water immersion. These modalities have the practical advantage of (generally) being available in communities, as most community and recreational centers or fitness gyms have hot tubs and/or saunas, and may be the most translational from a public health perspective.
Finnish (Dry) Sauna
There are two predominant types of saunas: traditional Finnish (dry) saunas and far infrared saunas, popularized by the Japanese (see Waon Therapy, below). Air temperature within Finnish saunas is typically maintained between 80 and 100°C. As humans are able to dissipate heat very effectively in dry air, these very high air temperatures are needed to increase body core temperature in a time-efficient manner. Some humidity can be created by pouring water over hot rocks, but these saunas still remain very dry (∼10–20% relative humidity). A targeted “rule of 200” is suggested by some in the U.S. sauna industry in which the sum of the dry sauna temperature (in Fahrenheit) and the humidity equal 200. As such, a slightly cooler sauna temperature would have a slightly higher percent humidity. In typical Finnish sauna sessions, individuals enter the sauna for 5–20 min at a time with 10- to 30-min rest periods outside the sauna in between (31, 32). They may repeat this cycle one to five more times, depending on how accustomed they are to sauna use. During such sessions, the magnitude of core temperature increase depends on the air temperature and length of stay, which can range between 70–100°C and 15–30 min (33), as well as the location of core temperature measurement. With traditional sauna use, esophageal temperature can increase to 39°C (∼1.5°C above resting) in only 10 min (34), whereas rectal temperatures are typically reported to increase by 0.2–1.0°C (33). In many cases, the increases in core temperature observed are fairly modest, and yet a multitude of long-term benefits have been reported.
Waon Therapy (Far Infrared Sauna)
A group of cardiologists in Japan conducted some of the first studies of heat therapy in humans using Waon therapy, which consists of 15 min in a 60°C far infrared sauna followed by 30 min of recovery wrapped in blankets to keep body core temperature elevated. The infrared waves generated by these saunas are able to penetrate the skin, effectively heating the body from the inside out. Thus lower air temperatures are required compared with Finnish sauna to achieve similar (or greater) increases in body core temperature. With Waon therapy, core temperature has been reported to increase by 1.0–1.2°C in 15 min (35, 36). Furthermore, maintaining core temperature at an elevated level for an extended period of time may help to potentiate greater benefits. Although this remains to be properly investigated, we speculate there may also be some hemodynamic advantages to far infrared sauna, particularly for individuals with impaired blood pressure regulation (e.g., CVD patients, those with severe spinal cord injury, and the elderly), as, given the lower air temperatures, skin temperature should not increase by as much as during hot water immersion and Finnish sauna bathing. If so, this could limit increases in cutaneous blood flow and reductions in systemic vascular resistance and blood pressure (see physiological changes with acute passive heat stress below).
Hot Water Immersion
Besides sauna, hot water immersion has been the most widely used modality of passive heat therapy. Hot water immersion is advantageous because the conduction of heat in water is ∼24 times that in air, meaning that water temperature can be considerably lower to achieve similar increases in body core temperature as achieved with sauna. Water temperature in hot baths is typically kept around 38–42°C. The rate of rise in body core temperature with hot water immersion depends on the water temperature and how deeply immersed individuals are in the water. Increases of 1.0–1.2°C in just 10 min have been reported in 41°C water with individuals immersed to the shoulder (36). Not only is heat transferred at a faster rate, but the body’s cooling mechanisms are ineffective under water: evaporation is required for sweating to dissipate heat. Thus, even as individuals become acclimated, target body core temperatures may be reached in similar amounts of time. For example, in young, sedentary subjects (37), time to reach a rectal temperature of 38.5°C via hot water immersion was unchanged across the 8-wk intervention (session 1: 24 ± 5 min vs. session 36: 27 ± 4 min; P = 0.16). However, in a subsequent study in obese women with polycystic ovary syndrome (38), the mean time to reach a rectal temperature of 38.5°C increased from 25 ± 8 min in session 1 to 37 ± 10 min for session 30 (P = 0.01), which could have been attributable to greater heat dissipation in regions not immersed in water in this population.
A key difference of hot water immersion compared with other types of passive heating is that water exerts hydrostatic pressure on the body, aiding venous return of blood back to the heart and increasing cardiac filling pressure (see Cardiovascular Hemodynamics below). Hydrostatic pressure has independent effects on CV function, such as increased cardiac output and mean arterial blood pressure (39), changes in conduit vessel diameter (40), and increased arterial compliance (41), which could contribute to long-term adaptations (42). Thus, when trying to isolate the effects of passive heating from the effects of the hydrostatic pressure, it is necessary to include a sham group that undergoes thermoneutral water immersion.
In addition to whole body hot water immersion in hot tubs and Jacuzzis, foot and leg baths have been developed and used in some studies (43, 44). Although core temperature increases to a lesser degree (∼0.4–0.5°C in 45 min), acute improvements in endothelial function (45) and exercise tolerance (44) have been observed. Foot baths may also be more accessible for some patient populations, especially those with limited physical abilities.
PHYSIOLOGICAL CHANGES WITH ACUTE PASSIVE HEAT STRESS
The acute CV effects of passive heat stress have been well characterized and are reviewed in much greater depth elsewhere (46, 47). Accordingly, here we will discuss them briefly and in the context of potentiating long-term adaptations in CV function and health. Importantly, the acute changes in CV hemodynamics that remain for a few hours following heat stress (48–50), particularly reductions in systemic vascular resistance and blood pressure (discussed below), are thought to underscore, at least in part, the chronic adaptations to heat therapy.
Sweating and Skin Blood Flow
The thermoregulatory centers in the hypothalamus respond to elevations in skin and body core temperature by first withdrawing cutaneous adrenergic sympathetic vasoconstriction and then by proportionally increasing cutaneous cholinergic sympathetic output, the latter of which stimulates the sweat glands to increase sweat output and the cutaneous microvessels to vasodilate [see Johnson et al. (51) for a comprehensive review of the mechanisms underlying these processes]. Active skin vasodilation and sweating are initiated once body core temperature reaches a temperature threshold, usually ∼0.4°C above resting core temperature (52), although this threshold may be delayed in older individuals and patient populations (53, 54). Skin temperature also modulates this threshold, in that the higher the skin temperature, the lower this threshold (55). Furthermore, directly heating the skin causes vasodilation, primarily through localized release of NO at these skin temperatures (56, 57). As such, skin blood flow may increase more in a Finnish sauna or during hot water immersion than in a far infrared sauna, even at the same body core temperature. Overall, with passive heat stress, skin blood flow can increase as much as 4.5–7.0 liters above resting in the supine posture (58), and sweat rates as high as 1.3 L/h have been reported during passive heat stress in acclimated individuals (59).
Cardiovascular Hemodynamics
For a comprehensive discussion of central cardiovascular hemodynamics associated with passive heat stress, readers are directed to the excellent review by Drs. Craig Crandall and Thad Wilson (47). Below, we focus on data relevant to the different modalities of passive heating.
Cardiac output.
Cardiac output must increase to support the increased skin blood flow and to compensate for fluid loss with sweating. The magnitude of increase will depend on the rise in body core temperature, but increases to as high as 13 L/min have been reported with passive heat stress using water-perfused suits in the supine posture (58). With hot water immersion, cardiac output increases by ∼60–140% compared with seated baseline outside of the hot tub (36, 49, 60, 61). In a recent study in young, healthy men and women, cardiac output increased from 6.6 L/min during a supine baseline to 10.4 L/min while seated in a hot tub with water at the level of the sternum, and supine cardiac output remained significantly elevated from baseline until 40 min postwater immersion (49). In another recent study, semirecumbent cardiac output increased by 3.7 L/min following 30 min of passive heating in a 42°C bath (60). With Finnish sauna, there have been varying reports from only a small increase in cardiac output (0.47 L/min) (62) up to a 75% increase (63, 64). This variability is likely attributable to the varying air temperatures, duration of sauna exposure used by community members, posture, and the effect of directly heating the skin on overall skin blood flow. Certain patient populations may also respond differently; Radtke et al. (65) observed increases in cardiac output of ∼1.5 L/min after a 10-min, 80°C sauna bath in chronic heart failure patients and healthy age-matched controls but no changes in cardiac output in coronary artery disease patients. Far infrared sauna elicits similar increases in cardiac output, with increases of 30–50% typically reported (36).
Heart rate.
Increases in heart rate during heat therapy can be substantial, to levels comparable to moderate-intensity exercise (60, 66). As changes in stroke volume are relatively modest, larger increases in heart rate are necessary to support increases in cardiac output. Heat may also have a direct chronotropic effect on the sinoatrial node (67, 68), although this will have a much lesser contribution to the increased heart rate compared with neural input. Generally, in healthy humans, heart rate increases by ∼30 beats/min for every degree increase in core temperature (52). As such, during a typical sauna session, peak heart rates of ∼80–90 beats/min are common (69–71), although heart rate has been reported to increase up to as much as 120–150 beats/min (32). When matched for core temperature changes, the rise in heart rate during hot water immersion was reported to be considerably less (49, 60).
Intracardiac pressures and stroke volume.
Changes in intracardiac pressures are dependent on the modality of passive heating. In all modalities, cutaneous vasodilation is supported by redistribution of blood volume toward the skin for thermoregulation and away from the splanchnic and renal circulations (72, 73). Despite this redistribution, with heating modalities in which venous return to the heart is not aided by hydrostatic pressure (e.g., dry sauna, far infrared sauna, and water-perfused suits): right atrial pressure decreases (often estimated from central venous pressure) (58, 72, 74), left ventricular filling pressure, as measured by pulmonary capillary wedge pressure, decreases in parallel with right atrial pressure (74, 75); and pulmonary artery pressure may either increase (76, 77) or decrease (78–80), depending on the balance between increases in right heart cardiac output and reductions in pulmonary vascular resistance. In contrast, intracardiac pressures all increase during hot water immersion as the hydrostatic compressive force of the water aids venous return to the heart (36, 81). Due to this difference, it has been proposed that sauna bathing may be safer for heart failure patients than hot water immersion, as strain on the heart is lower (36). However, the patterns observed with hot water immersion more closely mimic the hemodynamic effects of modest aerobic exercise (60). Therefore, it is possible hot water immersion could be more effective at inducing some CV adaptations, although studies comparing the two modalities are needed to determine whether this may be the case.
In young healthy individuals, stroke volume either does not change during heat stress or increases minimally (80, 82–84). This maintenance of stroke volume is achieved by augmentation of both systolic and diastolic cardiac functions (85–87) and a greater ejection fraction (36, 75, 88). In older adults, stroke volume has been reported to decrease (72, 89), although this is not a universal finding and may differ across sexes (90, 91). With hot water immersion, the compressive hydrostatic force exerted on the body submersed in water should aid venous return and allow for greater increases in or maintenance of stroke volume; however, the one study that has directly compared hemodynamic effects between sauna and hot water immersion observed no difference in stroke volume between the two heating modalities (36).
Blood pressure.
Peripheral vascular resistance generally decreases as a result of cutaneous vasodilation, although this is countered somewhat by increases in splanchnic and renal vascular resistance (72, 84). As a result, diastolic blood pressure can decrease considerably, particularly with sauna (36, 92). The hydrostatic pressure with hot water immersion may help limit decreases in vascular resistance and diastolic blood pressure (36). In healthy individuals, systolic blood pressure typically increases in the supine posture due to increases in cardiac output and heart rate (72); however, reductions in systolic blood pressure may be observed with sauna and other modalities that lack hydrostatic pressure (36, 92). In older individuals and patients, systolic blood pressure may be reduced with all modalities due to an impaired ability to offset reductions in vascular resistance and venous return to the heart via splanchnic/renal constriction (89). Blood pressure may be lower for up to 60 min following passive heat stress, which is comparable to the duration of lowered blood pressure observed following aerobic exercise (49), although this is not a universal finding (69, 70); it is possible that the duration/extent of blood pressure lowering may be dependent on the duration, type, or intensity of passive heating. Importantly, the intermittent reductions in peripheral vascular resistance and diastolic blood pressure with passive heat stress (along with other mechanisms, such as improved endothelium-dependent dilation and angiogenesis) may help facilitate long-term reductions in resting blood pressure with chronic heat therapy, as has been suggested by a number of studies and investigators (38, 45, 50, 93, 94).
Cardiovascular Contraindications for Passive Heat Stress
Most studies on the safety of passive heat stress have been conducted in Finland, where the vast majority of residents participate in sauna bathing at least once per week as a cultural family activity, from early childhood through old age. These studies have shown no adverse effects of sauna bathing across many populations [see Kukkonen-Harjula and Kauppinen (95) for a review], including patients with stable CVD, such as hypertension and heart failure (96, 97), children (98, 99), and pregnant women (100, 101). However, one caveat is that all of these studies were conducted in Finns who are well adapted to the heat. Studies conducted elsewhere in nonacclimated individuals have had mixed results. Most indicate that sauna is safe, as assessed by no obvious adverse events and CV hemodynamics (e.g., blood pressure and heart rate) remaining within safe ranges, including in healthy middle-aged and older adults (70), patients with untreated hypertension (69), and stable heart failure and coronary artery disease patients (65). However, it is still possible for nonacclimated individuals to experience adverse events. For example, nonacclimated stable coronary artery disease patients with exercise-induced myocardial ischemia also experienced myocardial ischemia during sauna bathing, although it was mostly asymptomatic and not as severe as during exercise (102). Therefore, we recommend that nonacclimated individuals be mindful when initiating a new heat therapy practice and those with elevated CVD risk should do so under the supervision of their primary care provider.
In Western medicine, heat therapy has traditionally been contraindicated in patients with CVD due to the concern that it may increase risk of cardiac arrhythmias. However, in a study monitoring 98 acute myocardial infarction patients and age-matched control subjects, only 8% experienced arrhythmias during and after sauna bathing, whereas 18% of subjects experienced arrhythmias during sub-maximal exercise (103). Furthermore, of all sudden deaths that occurred in Finland in 1970, only 1.7% occurred within 24 h after sauna bathing; this includes those that may be related to sauna and those that definitely are not, e.g., motor vehicle accidents (103). Of the nonaccidental deaths, the majority were caused by myocardial infarction related to alcohol consumption, which is known to increase risk of cardiac events independently of hyperthermia (104).
Importantly, there are some contraindications for heat therapy. Primarily, these include unstable conditions, for which exercise would also be contraindicated. Such conditions that have been studied include severe aortic stenosis, unstable angina pectoris, recent myocardial infarction, recent stroke or transient ischemic attack, and elderly individuals prone to orthostatic hypotension (96, 105).
CARDIOVASCULAR BENEFITS OF CHRONIC PASSIVE HEAT THERAPY
Although there has been much interest recently on the benefits of chronic passive heat therapy on CV health, much of the groundwork for this exciting area of research should be credited to the outstanding body of work by Dr. Michal Horowitz. Below, we present “key” studies from this field chronologically. Readers are directed elsewhere for more comprehensive recent reviews (7, 31).
Initial Studies in Animals
The first investigations into the beneficial CV adaptations of long-term passive heat therapy were conducted by Dr. Michal Horowitz and colleagues (106) in an elegant series of studies, in which they first showed that hearts from rats who received 4 wk of continuous heat exposure (housed at 34°C) were completely protected against an ischemia-reperfusion insult compared with hearts from rats housed at 24°C, determined by the percentage of infarcted tissue. They have since used this model to identify numerous protective molecular pathways that are stimulated by heat therapy, which are discussed in-depth below (see Refs.107–109 for reviews). Importantly, while some adaptations to heat can be observed quickly (e.g., lowered resting core temperature and, in humans, increased sweat rate), these authors have shown that >4 wk of continuous heat exposure are necessary to induce the full cellular-protective phenotype (109–111). This is especially important to consider when interpreting studies in humans, as very few have investigated physiological effects beyond 4 wk. Furthermore, as heat exposure is intermittent in human intervention studies, longer than 4 wk of heat therapy may be required to induce “complete” adaptations. Evidence of this is provided in a few studies in which some cardiovascular and metabolic adaptations in humans are not observed following 4–5 wk of heat therapy but are present by 8–10 wk (37, 38). That said, some adaptations may be gained more quickly, in as little as 2 wk, as discussed in the next section.
Waon Therapy in CVD Patients
The first studies to investigate CV benefits of passive heat therapy in humans were conducted by a group of cardiologists in Japan using Waon therapy. “Waon” is a Japanese word created by Dr. Chuwan Tei and is translated as “soothing and warm.” Thus Waon Therapy is “soothing warm therapy,” and much of the groundbreaking studies of heat therapy were performed by Dr. Tei and his colleagues. Most of these studies employed 3-wk interventions with daily Waon therapy sessions (i.e., a time duration shorter than that required for full adaption) and lacked long-term follow-up, but, despite these limitations, all reported profound reductions in symptoms and clinical biomarkers associated with various CVDs. For example, in left-sided heart failure patients, Waon therapy improved left-ventricular ejection fraction (35, 112, 113), increased cardiothoracic ratio (112), reduced incidence of arrhythmias (112), reduced circulating levels of atrial natriuretic peptide (a clinical biomarker of heart failure severity) (112), and reduced New York Heart Association (NYHA) functional class (114), the clinical classification system for heart failure symptom severity. In congestive heart failure patients, Waon therapy reduced mitral regurgitation and cardiothoracic ratio and increased left ventricular ejection fraction (113). In chronically occluded coronary artery-related ischemia patients, Waon therapy greatly improved myocardial perfusion (115).
Waon therapy also improves biomarkers of vascular function. FMDBA, the gold standard measured of conduit artery endothelial function (13, 14), is improved in heart failure patients (35, 116), patients with chronically occluded coronary artery-related ischemia (115), and individuals with CV risk factors (117). Systemic vascular resistance, which may contribute to chronic reductions in blood pressure, is reduced acutely following Waon therapy (36) and remains reduced chronically, measured 24 h following 2 wk of treatment (116). Autonomic function may also be improved, as measured by reduced low-frequency (sympathetic) and increased high-frequency (parasympathetic) components of heart rate variability (118).
Perhaps one of the most compelling arguments that passive heat therapy could be used as an alternative or adjunctive therapy to exercise training is the observation that just 3 wk of Waon therapy improves (35) and exercise tolerance, measured using both 6-min walk distance (35) and time-to-fatigue on a modified Bruce test (115). In the study by Sobajima et al. (115), improvements in exercise tolerance were observed in every heart failure patient studied, and 16 out of 20 patients improved in the study by Ohori et al. (35). In both studies, these improvements were correlated with improvements in FMDBA suggesting that the improvements in exercise tolerance were mediated by improved vascular function.
Only one study, as far as we are aware, has investigated the effects of longer-term Waon therapy. Following 10 wk of therapy in peripheral artery disease patients, Tei et al. (119) reported improved pain scores, walking distance, ankle-to-brachial pressure index, and resting leg skin perfusion (laser-Doppler). Waon therapy was also successful at inducing the formation of new collateral vessels in the affected legs of these patients, as shown by angiography (119). In a case study of one patient, Waon therapy also greatly improved the rate of healing of a large skin ulcer, which in turn prevented amputation of the patient’s leg (120).
Long-Term and Epidemiological Studies of Habitual Finnish Sauna Use
While extensive studies have been performed in Finland characterizing the acute effects of sauna bathing, very few have investigated long-term effects of regular sauna use and no studies have prospectively assigned individuals to treatment groups. This is attributed, in part, to the fact that saunas are available to nearly everyone in Finland (32). A 10-yr follow-up study of patients who had suffered a myocardial infarction reported that, although 60% of patients experienced symptoms of angina pectoris throughout their daily lives, only one patient experienced these symptoms associated with sauna bathing (96). In another study, the authors followed patients with essential hypertension and coronary artery disease who used sauna regularly and reported a positive effect on blood pressure at 1 and 3 yr of follow-up (121).
One of the most convincing studies to-date on the health benefits of sauna bathing was the Finnish Kuopio Ischemic Heart Disease Risk Factor Study, in which 2,315 middle-aged (42–60 yr) men who used sauna regularly were studied at enrollment, which included medical examinations and questionnaires on their sauna bathing habits, and were followed for up to 30 yr, or until time of death. In these men, both the frequency (sessions per week) and duration per session of sauna bathing predicted incidence of sudden cardiac death, fatal CVD, fatal coronary artery disease, and all-cause mortality over the follow-up period (122). The risk of CVD-related mortality was 48–50% lower in men who used sauna four to seven times per week compared with those who used it only once per week. Furthermore, the risk of developing hypertension over the follow-up period was considerably lower in individuals who used sauna either more frequently or for a longer duration per session (123). While these findings are very impressive, cautious interpretation of this study is warranted due to the lack of a true control group (the reference group still sauna bathed on average once per week), frequency and duration of sauna use only being determined at the time of enrollment, likely greater sauna use by individuals with higher income, education and discretionary time, and no inclusion of women.
Intervention-Based Studies of Heat Therapy
There is a decided lack of long-term intervention studies on the benefits of heat therapy, particularly in relation to the number of studies investigating the cardiovascular responses to an acute bout of passive heating. However, a few studies have shown promise. In young, healthy subjects, passive heat therapy interventions, which have mostly consisted of three to five sessions of hot water immersion per week for ∼8–10 wk, have improved various markers of CV function, which is impressive given that these individuals had relatively healthy function at baseline. Improvements include lowered mean arterial blood pressure (37), increases in both conduit artery (FMDBA) (30, 37, 124) and microvascular endothelial function (59, 125), reductions in arterial stiffness (37), and, interestingly, reductions in carotid artery wall thickness (37), which likely reflects reduced vascular smooth muscle hyperplasia and/or reduced vascular inflammation (126). Admittedly, some of these studies (37, 59) utilized what could be considered the upper limit of tolerability in terms of the stimulus of each heat exposure (60–90 min and a rectal temperature of 38.5–39°C), and studies that used less intense heating (shorter duration and/or lesser elevations in core temperature) observed improvements in conduit artery endothelial function of lesser magnitude (124) or that were transient (30). Collectively, these studies provide evidence that heat therapy can be effective in driving cardiovascular adaptations, but there is much we still do not know about the ideal frequency, temperature, and duration of heat therapy (7).
A few studies have investigated the potential of heat therapy interventions to improve CV function in individuals with elevated CV risk. In older adults, 30 sessions over 8–10 wk of heat therapy via hot water immersion lowered systolic blood pressure and improved endothelial function (FMDBA) (127). In obese women with polycystic ovary syndrome, 30 sessions over 8–10 wk of hot water immersion lowered systolic and diastolic blood pressure, improved endothelial function (shear rate-corrected FMDBA), reduced arterial stiffness, and reduced carotid artery wall thickness (38). The authors also observed a profound reduction in resting muscle sympathetic nerve activity (women with polycystic ovary syndrome have sympathetic overactivity, which contributes to their elevated CV risk) and protection against ischemia-reperfusion-induced impairments in endothelial function. In patients with peripheral artery disease, heat therapy via hot water immersion lowered systolic blood pressure and improved exercise tolerance, as assessed by 6-min walk distance and pain-free walk distance, but there were no significant changes in endothelial function, arterial stiffness, or ankle-brachial index (128), the primary clinical diagnostic measure for peripheral artery disease. The authors conjectured that their patients may have had too greatly advanced atherosclerosis to mitigate their vascular dysfunction within the duration of the intervention; however, the stimulus of passive heating was less than in prior studies (spa bathing at ∼39°C, 3–5 days/wk for ≤30 min sufficient to raise body temperature 1°C, followed by ≤30 min of calisthenics). Importantly, adherence to the heat therapy was much greater than adherence in the group of subjects who underwent walking and gym-based exercise. In an acute study, Pellinger et al. (44) demonstrated that acute lower leg heating improved maximal walking distance by 10–12%, whether heating duration was for 15 or 45 min. Taken together, these studies along with the work of Tei et al. (120) provide solid evidence that heat therapy may serve as a good gateway to reduce pain during subsequent exercise training in this group of patients.
MOLECULAR MECHANISMS UNDERLYING CARDIOVASCULAR BENEFITS OF HEAT THERAPY
Although the mechanisms underlying the CV benefits of heat therapy are incompletely understood, several studies were specifically targeted at elucidating the processes that underly improvements in CV biomarkers and health with passive heat therapy. From these studies, it is thought that many of the CV improvements derive from changes in shear stress within blood vessels during and following passive heating, acute and chronic upregulation of heat shock proteins (HSPs), improvements in NO bioavailability, and reductions in markers of oxidative stress and inflammation. That said, there is much that we still do not know, and more studies on the mechanisms that underpin the benefits of passive heat therapy are warranted. Importantly, with the exception of vascular shear stress, much of the work linking these pathways to improved CV function is correlative. Thus more investigation is needed to provide cause-and-effect evidence. Below, we outline the primary studies in this area from where our understanding is most complete to areas where our knowledge is incomplete.
Increases in Anterograde Vascular Shear Stress
Vascular shear stress is the mechanical frictional force exerted by the blood on the arterial walls, determined by the velocity and viscosity of the blood relative to the artery diameter. Importantly, shear stress and the adaptive effects of changes in shear stress are greatest in the conduit vessels. Although some effects of shear stress can be observed in the microvasculature, this section will focus primarily on the conduit vessels.
With perfect forward laminar flow, shear stress will be exerted in an entirely anterograde direction (i.e., in the direction of net blood flow and away from the heart). However, due to the pulsatile nature of blood flow in vivo (in conduit vessels), there are moments in the cardiac cycle where blood flow will be halted or may move in a retrograde fashion back toward the heart. The shear patterns that occur as a result of alternating anterograde and retrograde flow are referred to as oscillatory shear. Stimuli that increase blood flow typically increase anterograde shear, whereas stimuli or disease progression that result in increased peripheral vascular resistance will increase retrograde or oscillatory shear (also referred to as “disturbed flow”), as there will be greater resistance to the forward movement of blood. Many studies have investigated the effects of different shear patterns (i.e., more or less retrograde or anterograde shear) on endothelial function and adaptation. In general, high retrograde or oscillatory shear is associated with endothelial dysfunction and a proatherogenic profile, whereas high anterograde shear improves endothelial function and is antiatherogenic. The opposing effects of retrograde and anterograde shear stress on the vasculature are discussed below and summarized in Fig. 2. Passive heat stress results in increased anterograde and reduced retrograde shear stress (50, 66). Therefore, we expect that the shear-mediated vascular adaptations with heat therapy are attributable to a combination of withdrawal of the detrimental effects of retrograde/oscillatory shear stress and enhancement of the beneficial effects of increased anterograde shear stress, particularly in older adults and/or patient populations with higher retrograde/oscillatory shear stress under resting conditions compared with young, healthy adults.
Figure 2.

Summary of opposing effects of retrograde and anterograde shear stress on the vasculature. Mechanisms of vasoconstriction include greater/lesser α-adrenergic-mediated vasoconstriction and higher/lower levels of endothelin-1. NO, nitric oxide.
Detrimental effects of high retrograde/oscillatory shear stress.
There is a wealth of information showing that disturbed flow under resting conditions can promote unhealthy processes (see Refs. 129–132 for reviews). Across cell culture and rodent models, high retrograde/oscillatory shear downregulates expression eNOS (133, 134), the enzyme responsible for producing NO, and upregulates many proatherogenic factors, including adhesion molecules (135–137), proinflammatory cytokines (138), endothelin-1 (134), reactive oxygen species-producing enzymes like NADPH oxidase (133, 139), and superoxide (139). Rodents with high oscillatory shear, induced either by carotid artery partial ligation (140) or casting of the carotid artery to cause partial stenosis (138), quickly develop profound endothelial dysfunction, detrimental vascular remodeling (intimal thickening, reduced number of smooth muscle cells, and greater lipid content), and atherosclerotic plaques.
In humans, disturbed shear patterns and increases in retrograde shear are observed as a result of increased vascular tone in the downstream microvasculature, such as occurs with advanced age (141, 142), obesity (143), and hypertension (144). The extent of disturbance in flow is related to reduced NO bioavailability (145) and alpha-adrenergic mediated vasoconstriction (146). Conduit vessel endothelial dysfunction, likely secondary to greater oscillatory shear, is also present in these populations (147–149), and there is a well-established link between endothelial dysfunction and the progression of atherosclerosis (150, 151). In support of a causal link between disturbed flow and endothelial dysfunction, acutely disturbing flow in young, healthy individuals by inflating a distal blood pressure cuff to increase retrograde shear (without affecting anterograde shear) in the brachial artery reduces flow-mediated dilation (152) and causes release of the endothelial microparticles (EMPs) CD62E (E-selectin) and CD31 (153), biomarkers of proinflammatory endothelial activation and apoptosis, respectively (154). EMPs, in turn, can release C-reactive protein (155), carry regulatory microRNAs (156), reduce eNOS (157), and promote thrombosis, inflammation, and reactive oxygen species production (158). Low mean shear rates are also associated with higher levels of circulating EMPs in patient populations (159).
Beneficial effects of increases in anterograde shear stress.
On the contrary, repeated increases in anterograde shear stress are antiatherogenic and improve endothelial function. In cultured endothelial cells, physiologically relevant increases in anterograde shear stress (e.g., at levels that would be achieved in conduit vessels during exercise in humans) increase eNOS expression (160–162), the essential eNOS cofactor tetrahydrobiopterin (163), and the antioxidative enzyme superoxide dismutase (160). Anterograde shear stress can also increase phosphorylation of eNOS (i.e., activation and presumably greater NO production) through a few mechanisms, including through activating the receptor for vascular endothelial growth factor (164) and phosphoinositide 3-kinase, which in turn activates protein kinase A and then eNOS (165). Additionally, increases in anterograde shear stress can increase association of Hsp90 with eNOS (166), decrease endothelin-1 expression (167, 168), and be anti-inflammatory, reducing expression of adhesion molecules (169, 170) and protecting cells against TNFα-induced insult (170). Importantly, some of these results have been confirmed in isolated arteries, in which physiological increases in anterograde shear stress have been shown to increase eNOS and superoxide dismutase expression (171, 172), resulting in improved endothelium-dependent dilation (172).
Evidence that increases in anterograde shear stress are essential for improved vascular function with heat therapy.
In humans, Dr. Daniel Green and colleagues have performed a series of studies indicating that increases in anterograde shear stress are essential for arterial adaptation to various stimuli. These authors have utilized a model in which they assess vascular function before and after an intervention in both arms. One arm is allowed to adapt normally, whereas a blood pressure cuff is placed on the other arm and inflated to resting systolic pressure throughout the intervention sessions, such that blood flow and anterograde shear stress are prevented from increasing above resting levels. Using this model, they have shown that arterial adaptation in both conduit vessels and the microvasculature, as measured by increases in FMDBA, cutaneous microvascular endothelial function, or brachial artery diameter (i.e., arteriogenesis), is prevented in the cuffed arm following 8 wk of local arm heating (173), whole body heating (30, 125), lower limb exercise training (174), and handgrip exercise training (175). Reductions in brachial artery wall thickness with handgrip exercise training, a marker of structural remodeling, is also prevented in the cuffed arm (176). These data argue for an essential role of anterograde shear stress in arterial adaptation.
Of note, increases in local temperature seem just as important for mediating arterial adaptation to heat therapy as increases in vascular shear stress. In the same studies from Dr. Green's laboratory (125), improvements in cutaneous microvascular function following repeated whole body heating were prevented when local skin temperature was clamped to 30°C using a water bath, even though shear stress was allowed to increase. As such, it appears that the interactive effects of elevations in shear stress and temperature may be necessary for vascular adaptation.
Heat Shock Proteins
HSPs are a class of stress-sensitive proteins, named for the original observation that their expression increases rapidly and intensely at the onset of heat shock (177), although they are also expressed in response to hypoxia/ischemia (178), ultraviolet radiation (179), cytokines (180, 181), cold stress (182), exercise (183–185), peripheral injection of pyrogens to simulate fever (186, 187), local injection of neurotoxins (188), and interestingly, vascular shear stress (135, 189). Intracellular HSPs have been shown to increase in humans (measured in circulating leukocytes, skeletal muscle, and/or subcutaneous adipose tissue) following both exercise heat acclimation (e.g., 190–193) and passive heat therapy (194–197).
HSPs are believed to have two major roles in cells: 1) to protect cells from the damaging effects of heat and other stressors; and 2) to facilitate normal cell function. Upon exposure to milder/sublethal doses of stressors, HSPs confer tolerance against subsequent otherwise lethal stress (198, 199). It is believed that much of this conferred tolerance stems from the role of HSPs in processing stress-denatured proteins (200) and preventing disruption of structural proteins (201). Under nonstressed conditions, HSPs facilitate cellular function by translocating proteins to other locations within the cell, chaperoning proteins across cell membranes, stabilizing various proteins and receptors, and identifying and repairing damaged proteins. Many of these processes involve unfolding and refolding of proteins. HSPs can be found, usually associated with other proteins, in the nucleus, cytosol, mitochondria, endoplasmic reticulum, and in close proximity to the plasma membrane (202).
There are several distinct families of HSPs, grouped and named based on molecular weight and with slightly different roles within cells. The families that are most relevant to CV function are the HSP70 and HSP90 families. Roles of certain small HSPs, specifically Hsp27 and Hsp32, have also been identified. In general, proteins in the HSP70 family are important for cell growth and repair and protecting cells against damage, including processing denatured proteins (200, 203). As such, they are highly inducible with heat shock and other stressors (204). Proteins in the HSP90 family are constitutively expressed and abundant at thermoneutral temperatures but can also be further induced by heat (183). Their generalized role in cells is to associate with other proteins, thereby aiding in translocation (e.g., chaperoning newly formed proteins from the nucleus to their target location within the cell) (205), stabilization, and phosphorylation (i.e., activation) of these proteins (206, 207). Cellular (and extracellular) roles of HSPs in cellular function have been reviewed in-depth elsewhere (205, 208). Here, we focus on potential roles of “intracellular” HSPs in mediating the CV adaptations to heat therapy. Extracellular HSPs can increase during heat stress, although usually, high levels of thermal strain are needed (209). Furthermore, extracellular HSPs are typically considered to be a damage-associated molecular pattern, inducing proinflammatory signaling (210); the role (if any) of extracellular HSPs in mediating adaptations to heat acclimation/therapy is unknown.
Magnitude and time course of HSP expression.
Abundance of both Hsp70/72 and Hsp90 increases in cells and intact animals in response to heat stress (183, 192). In general, the greater the duration of heat exposure and the higher the ambient temperature, the greater the magnitude of HSP expression. For example, Harris et al. (211) observed no change in Hsp70 protein following 1 h of heat shock at 42°C in bovine aortic endothelial cells but observed an eightfold increase in Hsp70 with 45°C heat shock of the same duration. Similarly, in rats, the longer the duration of heat stress (measured following 40, 60, and 90 min) and the higher core temperature achieved (37, 39, 42, and 45°C), the greater the induction of Hsp70 and Hsp27 mRNA in brain, lung, and skin, although not in liver, tissue (212). In humans undergoing exercise heat stress, Gibson et al. (213) found that a higher rectal temperature and a longer duration of time spent with a rectal temperature ≥38.5°C was associated with a greater increase in Hsp72 mRNA in leukocytes. It is important to note that changes in mRNA expression do not necessarily infer specific changes in, or the timing of changes in, protein abundance, but still represent important signaling. Furthermore, cultured cells often require higher ambient temperatures to observe increases in HSPs compared with intact organisms or require extended periods of recovery time following heat exposure before peak expression of HSPs (205, 214). For example, Harris et al. (211) observed that 15 min of heat stress at 42°C in rats was sufficient to obtain a similar fold increase in Hsp70 in aortic tissue as what was obtained in cultured aortic cells following 1 h at 45°C.
Typically, detectibly higher levels of HSP mRNA can be observed during and immediately following heat exposure. Both Hsp70 and Hsp27 mRNA expression peak within 1 h following heat stress in rodents, returning to baseline levels by ∼6 h, with consistent responses across various tissue types, including the brain, lungs, liver, kidneys, and skin (212, 215). However, protein abundance may not peak for hours after heat stress. Furthermore, the time course of HSP induction may differ across species and tissue types. In rat skeletal muscle, Oishi et al. (216) showed that the timing for when Hsp72 protein abundance peaked following 1 h of heat stress (muscle temp reached 42°C) depended on the type of muscle fibers. In the soleus muscle of the rat, a predominantly slow-twitch muscle, Hsp72 protein content was elevated immediately following heat stress, with peak content at 4 h; whereas, in the plantaris muscle, which is predominantly fast-twitch, Hsp72 protein content was not elevated until 24–48 h following heat stress. Similar findings were observed in deep (predominantly slow-twitch) and superficial (predominantly fast-twitch) regions of the gastrocnemius (217). Two studies have measured vastus lateralis muscle HSP content in humans following acute passive heat stress, but neither observed changes in HSP content (197, 218). In fact, Hafen et al. (197) observed a slight decrease in Hsp27 phosphorylation (no change in total content). These findings may be attributable to the timing of when muscle biopsies were collected, i.e., too soon after heat stress, as Hafen et al. (196, 197) observed increases in Hsp70 and Hsp90 content after 6–10 days of daily passive heating. In exercising humans, Hsp70 content in the vastus lateralis muscle does not increase until ∼48 h after exercise and is maintained elevated above baseline levels for up to 6–7 days (219, 220); however, the exercise stimulus may produce a different pattern of expression compared with passive heat stress. Interestingly, HSP content in heat stressed human peripheral blood mononuclear cells increases much more quickly, peaking following 1 h after heat stress and returning back to baseline levels after ∼5 h (221).
Role of HSPs in cardiovascular function.
The primary signaling pathways that are altered with the progression of CVD and that have the greatest impact on CV function are the NO pathway, oxidative stress, and vascular inflammation. As described below, there is evidence that all three are improved with heat stress and/or therapy. HSPs can interact with all three, as reviewed below and summarized in Fig. 3. Thus it follows that these pathways are improved following heat therapy at least in part by upregulation of HSPs, although more investigations are needed to establish clear cause-and-effect.
Figure 3.
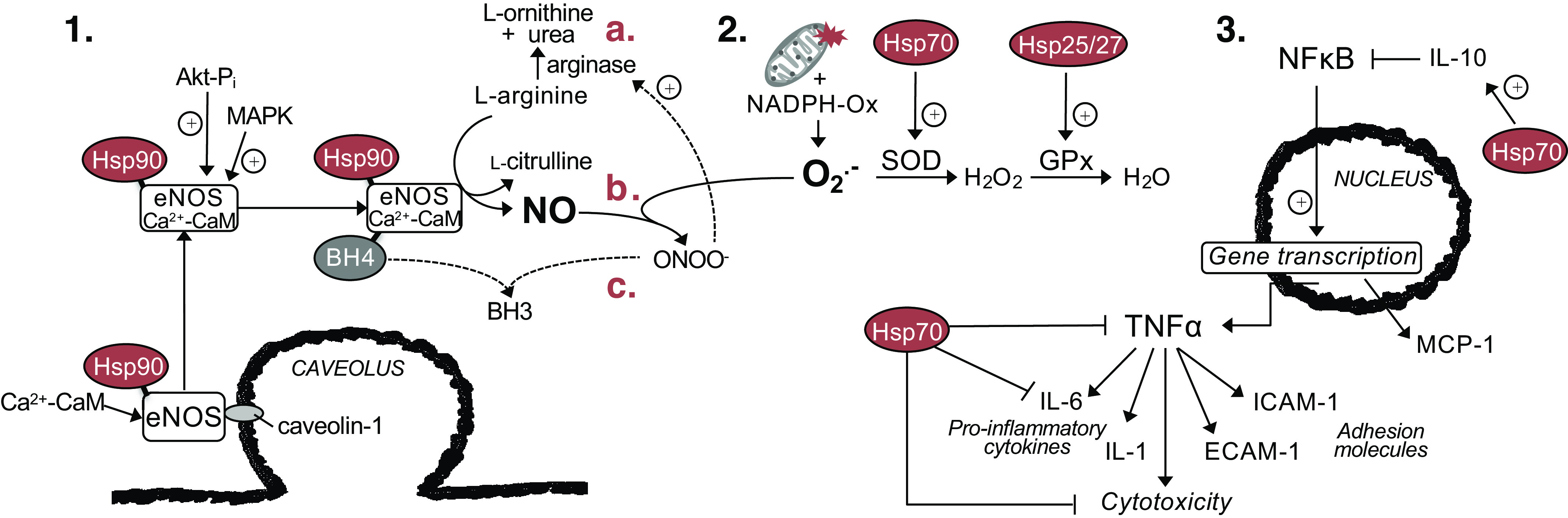
Interactions of heat shock proteins (HSPs) in endothelial cells with three primary pathways associated with vascular function: the nitric oxide (NO) pathway, oxidative stress, and inflammation. 1: Hsp90 is essential for activation of endothelial NO synthase (eNOS) by calcium-calmodulin (Ca2+-CaM) and Akt (also known as protein kinase B). 2: Hsp70 upregulates superoxide dismutase (SOD) and Hsp25/27 upregulates glutathione peroxidase (GPx), such that the damaging effects of reactive oxygen species are attenuated. These damaging effects include a) upregulation of arginase, which then decreases available l-arginine for synthesis of NO; b) scavenging of NO; and c) scavenging of tetrahydrobiopterin (BH4), and thus uncoupling of eNOS. 3: Heat stress, most likely via Hsp70, suppresses nuclear factor-kappa-B (NF-κB), a master regulator of proinflammatory gene transcription, and its downstream effects mediated by proinflammatory cytokines tumor necrosis factor (TNF)-α and interleukin (IL)-6. Hsp70 also upregulates the anti-inflammatory cytokine IL-10, which can suppress NF-κB activation. NADPH-Ox, nicotinamide adenine dinucleotide phosphate oxidase; MAPK, mitogen-activated protein kinase; ONOO-, peroxynitrite; MCP-1, monocyte chemoattractant protein-1; ICAM-1, intercellular adhesion molecule-1; ECAM-1, endothelial cell adhesion molecule-1.
Nitric oxide production.
NO has many important roles, including mediating endothelium-dependent vasodilation, angiogenesis, and suppressing the progression of atherosclerosis (222). NO is primarily produced from the conversion of l-arginine into l-citrulline and NO by the enzyme NO synthase (NOS), of which there are three primary isoforms: endothelial NOS (eNOS), neuronal NOS (nNOS), and inducible NOS (iNOS). NOS must be activated appropriately (i.e., phosphorylated in the presence of bound cofactors) in order for NO to be produced, and association with Hsp90 is essential for the activation and stabilization of both eNOS (223) and nNOS (224), which bind to the middle region (residues 259–615) of Hsp90 (225). Activation of eNOS, which is the most relevant isoform for CV function, is dependent on the calcium state of the cell. While inactive, eNOS is bound to the caveolar cell membrane by caveolin-1. Dissociation of eNOS from caveolin-1 occurs when calcium activates the messenger protein calmodulin, allowing it to bind to eNOS. This process of calmodulin-mediated dissociation of eNOS from caveolin-1 is mediated by eNOS association with Hsp90 (226, 227). Once free in the cytoplasm, eNOS can then be activated, which can be achieved by various kinases, including mitogen-activated protein kinases (MAPK) and Akt (protein kinase B). Hsp90 is also important for activation. For example, Akt phosphorylation of eNOS is dependent on Hsp90 (228).
Through these extensive roles of Hsp90, eNOS activation and therefore NO production can be enhanced by increased Hsp90 protein independent of changes in total eNOS protein. For example, Harris et al. (229) observed increased eNOS activity in mice following 10 wk of exercise training with no change in abundance of total eNOS protein. Hsp90 abundance was increased following exercise training, as well as Hsp90 association with eNOS. In contrast, when Hsp90 is pharmacologically inhibited with geldanamycin, eNOS activity in response to bradykinin, VEGF, histamine, and fluid shear stress is reduced by 50–90% (166, 225).
Oxidative stress.
Reactive oxygen species (ROS), including superoxide and hydrogen peroxide, are produced as byproducts of oxidative metabolism in the mitochondria from electron leakage at complexes I and II within the electron transport chain and by ROS-generating cytoplasmic and membrane-bound enzymes, such as nicotinamide adenine dinucleotide phosphate (NADPH) oxidases. Under normal, healthy conditions, the production of ROS is balanced by their reduction and removal from the cell by antioxidative enzymes. Furthermore, transient increases in ROS can be beneficial and may mediate cellular adaptations, e.g., aerobic exercise-induced angiogenesis (230). However, when this balance of production and removal is disrupted, either by excessive production of ROS or impaired antioxidant defenses, ROS accumulate and can have damaging effects on cells, known as “oxidative stress” (231). Oxidative stress occurs in vascular cells with normal aging, is a major mechanism of age-related endothelial dysfunction and arterial stiffening and declines in cardiac function, and is thought to be involved in the pathogenesis of many other diseases (19, 22, 23).
Superoxide is especially damaging to endothelial cells as it can readily combine with NO to produce peroxynitrite, thereby reducing the amount of bioavailable NO. Excess superoxide also reduces NO production by uncoupling eNOS, primarily achieved by the actions of peroxynitrite on tetrahydrobiopterin, one of the essential cofactors for eNOS (232). Additionally, peroxynitrite upregulates arginase activity (233), which reduces the availability of l-arginine, the substrate used by eNOS for NO production. Therefore, the actions of superoxide on the NO pathway form a positive feedback loop promoting greater and greater impairments in vascular function.
The major antioxidative enzymes in vascular cells are superoxide dismutases (SOD), which reduce superoxide into hydrogen peroxide that can be further reduced into water by catalase (234), and glutathione peroxidases, which reduce hydrogen peroxide to water (235). HSPs help to limit and/or ameliorate chronic oxidative stress, thereby improving CV function via their interactions with these antioxidative enzymes. For example, Hsp70 is essential for translocation of newly transcribed manganese SOD (MnSOD/SOD2; mitochondrial isoform) to the mitochondria and its subsequent activation (236). Activity and expression of copper/zinc SOD (Cu/ZnSOD/SOD2; intracellular isoform; measured in whole brain homogenates) is suppressed in Hsp70 knockout mice (237), suggesting that Hsp70 may be important for transcription of Cu/ZnSOD. Lastly, overexpression of Hsp25/27 in cultured mouse fibroblasts increases expression and activity of glutathione peroxidase (238).
Inflammation.
Vascular dysfunction and CVD are strongly associated with chronic systemic and vascular inflammation (28, 239, 240). Patients with CVD have elevated levels of circulating proinflammatory cytokines (241–243), and the pathogenesis of many disease states is considered to be mediated by chronic low-grade inflammation (28). A major mechanism of vascular dysfunction associated with inflammation is activation of nuclear factor-kappa-B (NF-κB) (244, 245). NF-κB is a transcription factor which, when activated, regulates the production of various proinflammatory molecules, such as TNFα, IL-6, monocyte chemoattractant protein 1, and adhesion molecules (244–247). Some of these cytokines can in turn activate NF-κB, contributing to another positive feedback loop (244).
Several studies have shown that prior heat stress suppresses NF-κB activation in response to various proinflammatory stimuli, including angiotensin-II, as shown in heart tissue in rats (248), and induced pancreatitis in rat pancreatic cells (249) and TNFα in cultured bovine aortic endothelial cells (250). Heat stress also suppresses TNFα release in response to proinflammatory stimuli in cultured macrophages (251, 252) and in intact animals (249, 253, 254). In the study by Snyder et al. (252), the authors observed a reciprocal relationship between Hsp70 and TNFα. Additionally, cells expressing elevated levels of Hsp70 are more resistant to the cytotoxic effects of TNFα (255, 256).
Heat stress has also been shown to downregulate other proinflammatory molecules, many of which are downstream to NF-κB and TNFα. For example, endotoxin-induced release of IL-1 is suppressed in heat-treated cells (251, 252). IL-1- or TNFα-mediated production of IL-6 can be suppressed by both prior heat stress and treatment with Hsp70 but not treatment with Hsp60 (257). Heat stress also suppresses TNFα-mediated expression of intracellular (ICAM-1) (258) and endothelial cell adhesion molecules (250) and pancreatitis-mediated production of ICAM-1 (89). Adhesion molecule expression is known to be partially dependent on NF-κB activation (259).
Heat stress may also increase anti-inflammatory defenses. Treatment of arthritic mice with Hsp70 increased IL-10 levels and suppressed inflammatory responses in various immune cells that are key for promoting arthritis (260); these effects of Hsp70 treatment were prevented by IL-10 knockout. As a side note, this mechanism may, at least in part, mediate reductions in symptoms reported by rheumatoid arthritis patients who use sauna regularly (261). Furthermore, IL-10 has been reported to suppress NF-κB activation (262), offering another potential mechanism by which Hsp70 elicits such profound effects on systemic inflammation.
Lastly, there is evidence that these anti-inflammatory effects of heat stress/therapy may extend to humans. Among middle-aged men enrolled in the Finnish Kuopio Ischemic Heart Disease study, there was an inverse relation between frequency of sauna use and circulating levels of C-reactive protein, a liver-derived marker of systemic inflammation, both at baseline and 11-yr follow-up (263, 264), and heat therapy interventions can reduce circulating inflammatory markers. Two weeks of hot springs bathing reduced circulating C-reactive protein, IL-6, and TNFα in chronic heart failure patients (265), and 8–10 wk of hot water immersion lowered circulating C-reactive protein in obese women with polycystic ovary syndrome (38).
Circulating Factors
Factors upregulated elsewhere (e.g., from skeletal muscle or adipocytes) in response to either acute heat stress or chronic heat therapy could enter the circulation and subsequently come in contact with vascular cells and influence cellular processes. Indeed, circulating factors have been shown to mediate effects of other types of lifestyle interventions (266–271), as assessed by exposing cells or tissues to serum/plasma from human subjects.
In cultured vascular endothelial cells, serum from young subjects who have undergone heat therapy (versus serum collected pre-heat therapy or from subjects who underwent sham/thermoneutral water immersion) increased protein abundance of the antioxidant MnSOD, reduced superoxide production, increased eNOS abundance, and increased angiogenesis (272), an established measure of endothelial cell function (93, 273). This latter effect was prevented by coincubation with the NOS inhibitor nitro-l-arginine methyl ester, indicating that it was NO-dependent and that heat therapy serum functionally increased NO bioavailability. Heat therapy serum also attenuated activation of the proinflammatory transcription factor NF-κB, ROS production, and release of proinflammatory cytokines following hypoxia-reoxygenation in endothelial cells (194), a cellular model of CV ischemic events. Importantly, all of these effects were observed without any changes in intracellular HSP abundance.
Although more studies are clearly needed, these data do suggest the circulating milieu is important for mediating, at least in part, the beneficial CV effects of heat therapy. Future research should investigate which circulating factors may confer these cellular effects, taking into account the caveat that it is more likely a combination of several factors than one or two. In addition to altered abundance of circulating metabolites, cytokines/chemokines, and/or proteins, roles of circulating microparticles and/or microvesicles should be considered. Acute passive heat stress reduces levels of circulating endothelial- and platelet-derived microparticles in young subjects (274) and individuals with spinal cord injury (275), which are known to promote endothelial oxidative stress (276) and inflammation (277) and are thought to play a pathogenic role in the progression of vascular disease (278). Microvesicles (of which exosomes are a subtype) are cell fragments that contain similar contents as their parent cells and can fuse with and/or enter other cells types to act as signaling molecules (279). As their contents are not freely circulating, they are not typically detectible by blood-based assays; thus isolating and analyzing the contents of microvesicles from humans who have undergone heat therapy could provide substantial insight into the underlying mechanisms.
Other Cellular Mechanisms
In hearts from rats that received 4 wk of continuous heat exposure, Dr. Horowitz and colleagues have identified a number of other potential mechanisms by which heat therapy may mediate CV-protective effects. They have observed that heat therapy, in addition to upregulation of HSPs (280) and antioxidative enzymes (281), upregulates genes associated with the following: 1) antiapoptosis (282); 2) maintenance of DNA and chromatin integrity (283, 284); and 3) greater reliance on glycolysis and anaerobic metabolism (285). These changes help to protect heart tissue against ischemic insults and the subsequent oxidative damage that occurs during reperfusion. Interestingly, many of these metabolic changes are believed to be mediated through hypoxia inducible factor (HIF)-1α, which is essential for complete heat acclimation (286, 287) and stabilized by Hsp72. These extensive and elegant studies provide insight that should be further investigated by others, as it remains to be determined whether these pathways are also affected in humans, with intermittent repeated (versus continuous) heat exposure, or in tissues besides the heart (e.g., the vasculature).
RESEARCH GAPS AND FUTURE DIRECTIONS
It is well accepted that repeated passive heating has many benefits in terms of improving quality of life and well-being. However, the concept that heat can be used as “therapy” to target modern diseases is just coming to the forefront of research: it is far from becoming a part of routine clinical practice (Fig. 4). Sauna use is a part of the culture in Finland, but in the rest of the world, sauna and hot tub use is viewed more as a luxury. As research on heat therapy continues, it is conceivable that heat therapy will one day be prescribed by health care providers for the prevention or treatment of disease. While this idea may be premature based on the strength of current literature, physicians are starting to prescribe exposure to nature and wilderness with similar foundations of evidence. Importantly, what is really lacking is a greater number of long-term and interventional studies utilizing the various modalities of heat therapy. In particular, there remains many unanswered questions regarding which modality may be best suited for specific clinical populations, whether there are ideal temperatures to which the body must be increased to maximize benefits in different clinical conditions, which forms of heat therapy are best tolerated by different groups, and how long the benefits of heat therapy are maintained following cessation of heat therapy, including whether a “maintenance dose” can prolong benefits with less frequent heat exposures. There also remain many questions regarding the mechanisms that underpin the health benefits of heat therapy; relatively little of the mechanistic evidence presented above has been explored yet in humans. For example, little is known about the specific roles of HSPs and circulating factors in humans, let alone the best way to measure them and their effects in vivo. More basic research on the underlying mechanisms would not only appease molecular-minded scientists but serve as a foundation by which treatments can be further explored and the benefits of heat therapy exploited. Virtually unexplored is the potential for specific drugs, interventions, and supplements to interfere with or complement heat therapy. Only one study has reported that the benefits of heat therapy appear to augment the protective effects of exercise against CVD (32). The differential and overlapping mechanisms by which exercise and heat therapy confer cardiovascular protection need to be understood. Despite the extensive literature on exercise prescription, we know very little about the prescription of heat therapy. In summary, following our ancestors who first sought out warmth for comfort and safety thousands of years ago, we are once again turning to heat to counter threats to our health, this time to defend us against modern diseases.
Figure 4.

A summary of the major gaps in the literature on the effects of heat therapy on cardiovascular (CV) function and health.
GRANTS
The authors’ work has been supported by American Heart Association Grants 14PRE20380300 and 16PRE27780085, the Eugene and Clarissa Evonuk Memorial Foundation, the Kenneth and Kenda Singer Endowment, and National Heart, Lung, and Blood Grants R01-HL-144128 and F32-HL-140875.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
V.E.B. and C.T.M. conceived and designed research; V.E.B. prepared figures; V.E.B. drafted manuscript; V.E.B. and C.T.M. edited and revised manuscript; V.E.B. and C.T.M. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors sincerely thank Emily Larson and Amy Bazzoni for assistance with preparation of the manuscript and all current and former laboratory members who have contributed to the work described herein.
REFERENCES
- 1.Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee, et al. Heart Disease and Stroke Statistics-2019 Update: A Report from the American Heart Association. Circulation 139: e56–e528, 2019. [Erratum in Circulation 141: e33, 2020] doi: 10.1161/cir.0000000000000659. [DOI] [PubMed] [Google Scholar]
- 2.Heidenreich PA, Trogdon JG, Khavjou OA, Butler J, Dracup K, Ezekowitz MD, Finkelstein EA, Hong Y, Johnston SC, Khera A, Lloyd-Jones DM, Nelson SA, Nichol G, Orenstein D, Wilson PW, Woo YJ, American Heart Association Advocacy Coordinating Committee, Stroke Council, Council on Cardiovascular Radiology and Intervention, Council on Clinical Cardiology, Council on Epidemiology and Prevention, Council on Arteriosclerosis, Thrombosis and Vascular Biology, Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation, Council on Cardiovascular Nursing, Council on the Kidney in Cardiovascular Disease, Council on Cardiovascular Surgery and Anesthesia, and Interdisciplinary Council on Quality of Care and Outcomes Research. Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation. 123: 933–944, 2011. doi: 10.1161/CIR.0b013e31820a55f5. [DOI] [PubMed] [Google Scholar]
- 3.De Backer GG. Prevention of cardiovascular disease: much more is needed. Eur J Prev Cardiol 25: 1083–1086, 2018. doi: 10.1177/2047487318770297. [DOI] [PubMed] [Google Scholar]
- 4.Seals DR, Kaplon RE, Gioscia-Ryan RA, LaRocca TJ. You’re only as old as your arteries: translational strategies for preserving vascular endothelial function with aging. Physiology (Bethesda) 29: 250–264, 2014. doi: 10.1152/physiol.00059.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.World Health Organization. Global Action Plan for the Prevention and Control of Non-Communicable Diseases. Geneva: World Health Organization, 2013. [Google Scholar]
- 6.Beever R. The effects of repeated thermal therapy on quality of life in patients with type II diabetes mellitus. J Altern Complement Med 16: 677–681, 2010. doi: 10.1089/acm.2009.0358. [DOI] [PubMed] [Google Scholar]
- 7.Cheng JL, MacDonald MJ. Effect of heat stress on vascular outcomes in humans. J Appl Physiol (1985) 126: 771–781, 2019. doi: 10.1152/japplphysiol.00682.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Williamson JD, Supiano MA, Applegate WB, Berlowitz DR, Campbell RC, Chertow GM, Fine LJ, Haley WE, Hawfield AT, Ix JH, Kitzman DW, Kostis JB, Krousel-Wood MA, Launer LJ, Oparil S, Rodriguez CJ, Roumie CL, Shorr RI, Sink KM, Wadley VG, Whelton PK, Whittle J, Woolard NF, Wright JT, Pajewski NM, SPRINT Research Group. Intensive vs standard blood pressure control and cardiovascular disease outcomes in adults aged ≥75 years: a randomized clinical trial. JAMA 315: 2673–2682, 2016. doi: 10.1001/jama.2016.7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brugts JJ, Yetgin T, Hoeks SE, Gotto AM, Shepherd J, Westendorp RG, de Craen AJ, Knopp RH, Nakamura H, Ridker P, van Domburg R, Deckers JW. The benefits of statins in people without established cardiovascular disease but with cardiovascular risk factors: meta-analysis of randomised controlled trials. BMJ 338: b2376–b2376, 2009. [PM doi: 10.1136/bmj.b2376. C [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part I: aging arteries: a “set up” for vascular disease. Circulation 107: 139–146, 2003. doi: 10.1161/01.CIR.0000048892.83521.58.doi:. [DOI] [PubMed] [Google Scholar]
- 11.Najjar SS, Scuteri A, Lakatta EG. Arterial aging: is it an immutable cardiovascular risk factor? Hypertension 46: 454–462, 2005. doi: 10.1161/01.HYP.0000177474.06749.98. [DOI] [PubMed] [Google Scholar]
- 12.Seals DR, Jablonski KL, Donato AJ. Aging and vascular endothelial function in humans. Clin Sci (Lond) 120: 357–375, 2011. doi: 10.1042/CS20100476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shechter M, Issachar A, Marai I, Koren-Morag N, Freinark D, Shahar Y, Shechter A, Feinberg MS. Long-term association of brachial artery flow-mediated vasodilation and cardiovascular events in middle-aged subjects with no apparent heart disease. Int J Cardiol 134: 52–58, 2009. doi: 10.1016/j.ijcard.2008.01.021. [DOI] [PubMed] [Google Scholar]
- 14.Yeboah J, Crouse JR, Hsu FC, Burke GL, Herrington DM. Brachial flow-mediated dilation predicts incident cardiovascular events in older adults: the Cardiovascular Health Study. Circulation 115: 2390–2397, 2007. doi: 10.1161/CIRCULATIONAHA.106.678276. [DOI] [PubMed] [Google Scholar]
- 15.Yeboah J, Folsom AR, Burke GL, Johnson C, Polak JF, Post W, Lima JA, Crouse JR, Herrington DM. Predictive value of brachial flow-mediated dilation for incident cardiovascular events in a population-based study: the multi-ethnic study of atherosclerosis. Circulation 120: 502–509, 2009. doi: 10.1161/CIRCULATIONAHA.109.864801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blacher J, Asmar R, Djane S, London GM, Safar ME. Aortic pulse wave velocity as a marker of cardiovascular risk in hypertensive patients. Hypertension 33: 1111–1117, 1999. doi: 10.1161/01.HYP.33.5.1111. [DOI] [PubMed] [Google Scholar]
- 17.Boutouyrie P, Tropeano AI, Asmar R, Gautier I, Benetos A, Lacolley P, Laurent S. Aortic stiffness is an independent predictor of primary coronary events in hypertensive patients: a longitudinal study. Hypertension 39: 10–15, 2002. doi: 10.1161/hy0102.099031. [DOI] [PubMed] [Google Scholar]
- 18.Mitchell GF, Hwang SJ, Vasan RS, Larson MG, Pencina MJ, Hamburg NM, Vita JA, Levy D, Benjamin EJ. Arterial stiffness and cardiovascular events: the Framingham Heart Study. Circulation 121: 505–511, 2010. doi: 10.1161/CIRCULATIONAHA.109.886655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lakatta EG. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part III: cellular and molecular clues to heart and arterial aging. Circulation 107: 490–497, 2003. doi: 10.1161/01.CIR.0000048894.99865.02. doi:. [DOI] [PubMed] [Google Scholar]
- 20.Nadruz W. Myocardial remodeling in hypertension. J Hum Hypertens 29: 1–6, 2015. doi: 10.1038/jhh.2014.36. [DOI] [PubMed] [Google Scholar]
- 21.Lakatta EG. Cardiovascular regulatory mechanisms in advanced age. Physiol Rev 73: 413–467, 1993. doi: 10.1152/physrev.1993.73.2.413. [DOI] [PubMed] [Google Scholar]
- 22.Bachschmid MM, Schildknecht S, Matsui R, Zee R, Haeussler D, Cohen RA, Pimental D, Loo BV. Vascular aging: chronic oxidative stress and impairment of redox signaling-consequences for vascular homeostasis and disease. Ann Med 45: 17–36, 2013. doi: 10.3109/07853890.2011.645498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van der Loo B, Labugger R, Skepper JN, Bachschmid M, Kilo J, Powell JM, Palacios-Callender M, Erusalimsky JD, Quaschning T, Malinski T, Gygi D, Ullrich V, Lüscher TF. Enhanced peroxynitrite formation is associated with vascular aging. J Exp Med 192: 1731–1744, 2000. doi: 10.1084/jem.192.12.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sell DR, Monnier VM. Molecular basis of arterial stiffening: role of glycation–a mini-review. Gerontology 58: 227–237, 2012. doi: 10.1159/000334668. [DOI] [PubMed] [Google Scholar]
- 25.Zieman SJ, Melenovsky V, Kass DA. Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler Thromb Vasc Biol 25: 932–943, 2005. doi: 10.1161/01.ATV.0000160548.78317.29. [DOI] [PubMed] [Google Scholar]
- 26.Wu QQ, Xiao Y, Yuan Y, Ma ZG, Liao HH, Liu C, Zhu JX, Yang Z, Deng W, Tang QZ. Mechanisms contributing to cardiac remodelling. Clin Sci (Lond) 131: 2319–2345, 2017. doi: 10.1042/CS20171167. [DOI] [PubMed] [Google Scholar]
- 27.Gimbrone MA, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res 118: 620–636, 2016. doi: 10.1161/CIRCRESAHA.115.306301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation 105: 1135–1143, 2002. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 29.Taylor NA. Human heat adaptation. Compr Physiol 4: 325–365, 2014. doi: 10.1002/cphy.c130022. [DOI] [PubMed] [Google Scholar]
- 30.Carter HH, Spence AL, Atkinson CL, Pugh CJ, Naylor LH, Green DJ. Repeated core temperature elevation induces conduit artery adaptation in humans. Eur J Appl Physiol 114: 859–865, 2014. doi: 10.1007/s00421-013-2817-2. [DOI] [PubMed] [Google Scholar]
- 31.Heinonen I, Laukkanen JA. Effects of heat and cold on health, with special reference to Finnish sauna bathing. Am J Physiol Regul Integr Comp Physiol 314: R629–R638, 2018. doi: 10.1152/ajpregu.00115.2017. [DOI] [PubMed] [Google Scholar]
- 32.Laukkanen JA, Laukkanen T, Kunutsor SK. Cardiovascular and other health benefits of sauna bathing: a review of the evidence. Mayo Clin Proc 93: 1111–1121, 2018. doi: 10.1016/j.mayocp.2018.04.008. [DOI] [PubMed] [Google Scholar]
- 33.Hannuksela ML, Ellahham S. Benefits and risks of sauna bathing. Am J Med 110: 118–126, 2001. doi: 10.1016/S0002-9343(00)00671-9. [DOI] [PubMed] [Google Scholar]
- 34.Kauppinen K. Sauna, shower, and ice water immersion. Physiological responses to brief exposures to heat, cool, and cold. Part II. Circulation. Arctic Med Res 48: 64–74, 1989. [PubMed] [Google Scholar]
- 35.Ohori T, Nozawa T, Ihori H, Shida T, Sobajima M, Matsuki A, Yasumura S, Inoue H. Effect of repeated sauna treatment on exercise tolerance and endothelial function in patients with chronic heart failure. Am J Cardiol 109: 100–104, 2012. doi: 10.1016/j.amjcard.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 36.Tei C, Horikiri Y, Park JC, Jeong JW, Chang KS, Toyama Y, Tanaka N. Acute hemodynamic improvement by thermal vasodilation in congestive heart failure. Circulation 91: 2582–2590, 1995. doi: 10.1161/01.CIR.91.10.2582. [DOI] [PubMed] [Google Scholar]
- 37.Brunt VE, Howard MJ, Francisco MA, Ely BR, Minson CT. Passive heat therapy improves endothelial function, arterial stiffness, and blood pressure in sedentary humans. J Physiol 594: 5329–5342, 2016. doi: 10.1113/JP272453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ely BR, Francisco MA, Halliwill JR, Bryan SD, Comrada LN, Larson EA, Brunt VE, Minson CT. Heat therapy reduces sympathetic activity and improves cardiovascular risk profile in obese women with polycystic ovary syndrome. Am J Physiol Regul Integr Comp Physiol 317: R630–R640, 2019. doi: 10.1152/ajpregu.00078.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arborelius M, Ballidin UI, Lilja B, Lundgren CE. Hemodynamic changes in man during immersion with the head above water. Aerosp Med 43: 592–598, 1972. [PubMed] [Google Scholar]
- 40.Carter HH, Spence AL, Pugh CJ, Ainslie P, Naylor LH, Green DJ. Cardiovascular responses to water immersion in humans: impact on cerebral perfusion. Am J Physiol Regul Integr Comp Physiol 306: R636–R640, 2014. doi: 10.1152/ajpregu.00516.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boussuges A. Immersion in thermoneutral water: effects on arterial compliance. Aviat Space Environ Med 77: 1183–1187, 2006. [PubMed] [Google Scholar]
- 42.Simmons EE, Bergeron ER, Florian JP. The impact of repetitive long-duration water immersion on vascular function. PLoS One 12: e0181673, 2017. doi: 10.1371/journal.pone.0181673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu Q, Zhu W, Zhu Y, Zheng L, Hughson RL. Acute effects of warm footbath on arterial stiffness in healthy young and older women. Eur J Appl Physiol 112: 1261–1268, 2012. doi: 10.1007/s00421-011-2066-1. [DOI] [PubMed] [Google Scholar]
- 44.Pellinger TK, Neighbors CB, Simmons GH. Acute lower leg heating increases exercise capacity in patients With peripheral artery disease. J Cardiovasc Nurs 34: 130–133, 2019. doi: 10.1097/JCN.0000000000000510. [DOI] [PubMed] [Google Scholar]
- 45.Romero SA, Gagnon D, Adams AN, Cramer MN, Kouda K, Crandall CG. Acute limb heating improves macro- and microvascular dilator function in the leg of aged humans. Am J Physiol Heart Circ Physiol 312: H89–H97, 2017. doi: 10.1152/ajpheart.00519.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crandall CG, González-Alonso J. Cardiovascular function in the heat-stressed human. Acta Physiol (Oxf) 199: 407–423, 2010. doi: 10.1111/j.1748-1716.2010.02119.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Crandall CG, Wilson TE. Human cardiovascular responses to passive heat stress. Compr Physiol 5: 17–43, 2015. doi: 10.1002/cphy.c140015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brunt VE, Jeckell AT, Ely BR, Howard MJ, Thijssen DH, Minson CT. Acute hot water immersion is protective against impaired vascular function following forearm ischemia-reperfusion in young healthy humans. Am J Physiol Regul Integr Comp Physiol 311: R1060–R1067, 2016. doi: 10.1152/ajpregu.00301.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Francisco MA, Colbert C, Larson EA, Sieck DC, Halliwill JR, Minson CT. Hemodynamics of post-exercise vs. post hot water immersion recovery. J Appl Physiol. In press. doi: 10.1152/japplphysiol.00260.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thomas KN, van Rij AM, Lucas SJ, Cotter JD. Lower-limb hot-water immersion acutely induces beneficial hemodynamic and cardiovascular responses in peripheral arterial disease and healthy, elderly controls. Am J Physiol Regul Integr Comp Physiol 312: R281–R291, 2017. doi: 10.1152/ajpregu.00404.2016. [DOI] [PubMed] [Google Scholar]
- 51.Johnson JM, Minson CT, Kellogg DL Jr.. Cutaneous vasodilator and vasoconstrictor mechanisms in temperature regulation. Compr Physiol 4: 33–89, 2011. doi: 10.1002/cphy.c130015h. [DOI] [PubMed] [Google Scholar]
- 52.Johnson JM, Proppe DW. Cardiovascular adjustments to heat stress. In: Handbook of Physiology: Environmental Physiology, edited by Fregly MJ, Blatteis CM. Bethesda, MD: American Physiological Society, 2011. [Google Scholar]
- 53.Armstrong CG, Kenney WL. Effects of age and acclimation on responses to passive heat exposure. J Appl Physiol 75: 2162–2167, 1993. doi: 10.1152/jappl.1993.75.5.2162. [DOI] [PubMed] [Google Scholar]
- 54.Foster KG, Ellis FP, Doré C, Exton-Smith AN, Weiner JS. Sweat responses in the aged. Age Ageing 5: 91–101, 1976. doi: 10.1093/ageing/5.2.91. [DOI] [PubMed] [Google Scholar]
- 55.Johnson JM, Park MK. Reflex control of skin blood flow by skin temperature: role of core temperature. J Appl Physiol Respir Environ Exerc Physiol 47: 1188–1193, 1979. doi: 10.1152/jappl.1979.47.6.1188. [DOI] [PubMed] [Google Scholar]
- 56.Choi PJ, Brunt VE, Fujii N, Minson CT. New approach to measure cutaneous microvascular function: an improved test of NO-mediated vasodilation by thermal hyperemia. J Appl Physiol 117: 277–283, 2014. doi: 10.1152/japplphysiol.01397.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Minson CT, Berry LT, Joyner MJ. Nitric oxide and neurally mediated regulation of skin blood flow during local heating. J Appl Physiol 91: 1619–1626, 2001. doi: 10.1152/jappl.2001.91.4.1619. [DOI] [PubMed] [Google Scholar]
- 58.Rowell LB, Brengelmann GL, Murray JA. Cardiovascular responses to sustained high skin temperature in resting man. J Appl Physiol 27: 673–680, 1969. doi: 10.1152/jappl.1969.27.5.673. [DOI] [PubMed] [Google Scholar]
- 59.Brunt VE, Eymann TM, Francisco MA, Howard MJ, Minson CT. Passive heat therapy improves cutaneous microvascular function in sedentary humans via improved nitric oxide-dependent dilation. J Appl Physiol 121: 716–723, 2016. doi: 10.1152/japplphysiol.00424.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Amin SB, Hansen AB, Mugele H, Willmer F, Gross F, Reimeir B, Cornwell WK, Simpson LL, Moore JP, Romero SA, Lawley JS. Whole body passive heating versus dynamic lower body exercise: a comparison of peripheral hemodynamic profiles. J Appl Physiol 130: 160–171, 2021. doi: 10.1152/japplphysiol.00291.2020. [DOI] [PubMed] [Google Scholar]
- 61.Larson EA, Ely BR, Francisco MA, Wright ES, Halliwill JR, Minson CT. Thermoregulatory and cardiovascular adjustments to acute passive heat exposure in low-level spinal cord injury. FASEB J 32: 722.7, 2018. doi: 10.1096/fasebj.2018.32.1_supplement.722.7 [DOI] [Google Scholar]
- 62.Kiss D, Popp W, Wagner C, Zwick H, Sertl K. Effects of the sauna on diffusing capacity, pulmonary function and cardiac output in healthy subjects. Respiration 61: 86–88, 1994. doi: 10.1159/000196312. [DOI] [PubMed] [Google Scholar]
- 63.Kauppinen K, Vuori I. Man in the sauna. Ann Clin Res 18: 173–185, 1986. [PubMed] [Google Scholar]
- 64.Vuori I. Sauna bather's circulation. Ann Clin Res 20: 249–256, 1988. [PubMed] [Google Scholar]
- 65.Radtke T, Poerschke D, Wilhelm M, Trachsel LD, Tschanz H, Matter F, Jauslin D, Saner H, Schmid JP. Acute effects of Finnish sauna and cold-water immersion on haemodynamic variables and autonomic nervous system activity in patients with heart failure. Eur J Prev Cardiolog 23: 593–601, 2016. doi: 10.1177/2047487315594506. [DOI] [PubMed] [Google Scholar]
- 66.Thomas KN, van Rij AM, Lucas SJ, Gray AR, Cotter JD. Substantive hemodynamic and thermal strain upon completing lower-limb hot-water immersion; comparisons with treadmill running. Temperature 3: 286–297, 2016. doi: 10.1080/23328940.2016.1156215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Coyle EF, González-Alonso J. Cardiovascular drift during prolonged exercise: new perspectives. Exerc Sport Sci Rev 29: 88–92, 2001. doi: 10.1097/00003677-200104000-00009. [DOI] [PubMed] [Google Scholar]
- 68.Fritzsche RG, Switzer TW, Hodgkinson BJ, Coyle EF. Stroke volume decline during prolonged exercise is influenced by the increase in heart rate. J Appl Physiol 86: 799–805, 1999. doi: 10.1152/jappl.1999.86.3.799. [DOI] [PubMed] [Google Scholar]
- 69.Gayda M, Paillard F, Sosner P, Juneau M, Garzon M, Gonzalez M, Bélanger M, Nigam A. Effects of sauna alone and postexercise sauna baths on blood pressure and hemodynamic variables in patients with untreated hypertension. J Clin Hypertens (Greenwich) 14: 553–560, 2012. doi: 10.1111/j.1751-7176.2012.00637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gravel H, Coombs GB, Behzadi P, Marcoux-Clément V, Barry H, Juneau M, Nigam A, Gagnon D. Acute effect of Finnish sauna bathing on brachial artery flow-mediated dilation and reactive hyperemia in healthy middle-aged and older adults. Physiol Rep 7: e14166, 2019. doi: 10.14814/phy2.14166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Laukkanen T, Kunutsor SK, Zaccardi F, Lee E, Willeit P, Khan H, Laukkanen JA. Acute effects of sauna bathing on cardiovascular function. J Hum Hypertens 32: 129–138, 2018. doi: 10.1038/s41371-017-0008-z. [DOI] [PubMed] [Google Scholar]
- 72.Minson CT, Wladkowski SL, Cardell AF, Pawelczyk JA, Kenney WL. Age alters the cardiovascular response to direct passive heating. J Appl Physiol 84: 1323–1332, 1998. doi: 10.1152/jappl.1998.84.4.1323. [DOI] [PubMed] [Google Scholar]
- 73.Rowell LB, Detry JR, Profant GR, Wyss C. Splanchnic vasoconstriction in hyperthermic man–role of falling blood pressure. J Appl Physiol 31: 864–869, 1971. doi: 10.1152/jappl.1971.31.6.864. [DOI] [PubMed] [Google Scholar]
- 74.Wilson TE, Tollund C, Yoshiga CC, Dawson EA, Nissen P, Secher NH, Crandall CG. Effects of heat and cold stress on central vascular pressure relationships during orthostasis in humans. J Physiol 585: 279–285, 2007. doi: 10.1113/jphysiol.2007.137901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wilson TE, Brothers RM, Tollund C, Dawson EA, Nissen P, Yoshiga CC, Jons C, Secher NH, Crandall CG. Effect of thermal stress on Frank-Starling relations in humans. J Physiol 587: 3383–3392, 2009. doi: 10.1113/jphysiol.2009.170381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.el-Sherif N, Shahwan L, Sorour AH. The effect of acute thermal stress on general and pulmonary hemodynamics in the cardiac patient. Am Heart J 79: 305–317, 1970. doi: 10.1016/0002-8703(70)90418-7. [DOI] [PubMed] [Google Scholar]
- 77.Faithfull NS, Reinhold HS, van den Berg AP, van Rhoon GC, van der Zee J, Wike-Hooley JL. Cardiovascular changes during whole body hyperthermia treatment of advanced malignancy. Eur J Appl Physiol Occup Physiol 53: 274–281, 1984. doi: 10.1007/BF00776602. [DOI] [PubMed] [Google Scholar]
- 78.Carlsten A, Gustafson A, Werko L. Hemodynamic influence of warm and dry environment in man with and without rheumatic heart disease. Acta Med Scand 169: 411–417, 1961. doi: 10.1111/j.0954-6820.1961.tb07850.x. [DOI] [PubMed] [Google Scholar]
- 79.Damato AN, Lau SH, Stein E, Haft JI, Kosowsky B, Cohen SI. Cardiovascular response to acute thermal stress (hot dry environment) in unacclimatized normal subjects. Am Heart J 76: 769–774, 1968. doi: 10.1016/0002-8703(68)90262-7. [DOI] [PubMed] [Google Scholar]
- 80.Sancetta SM, Kramer J, Husni E. The effects of dry heat on the circulation of man. I. General hemodynamics. Am Heart J 56: 212–221, 1958. doi: 10.1016/0002-8703(58)90232-1. [DOI] [PubMed] [Google Scholar]
- 81.Löllgen H, Nieding von G, Koppenhagen K, Kersting F, Just H. Hemodynamic response to graded water immersion. Klin Wochenschr 59: 623–628, 1981. doi: 10.1007/BF02593853. [DOI] [PubMed] [Google Scholar]
- 82.Koroxenidis GT, Shepherd JT, Marshall RJ. Cardiovascular response to acute heat stress. J Appl Physiol 16: 869–872, 1961. doi: 10.1152/jappl.1961.16.5.869. [DOI] [PubMed] [Google Scholar]
- 83.Murray RH. Cardiopulmonary effects of brief, intense thermal exposures. J Appl Physiol 21: 1717–1724, 1966. doi: 10.1152/jappl.1966.21.6.1717. [DOI] [PubMed] [Google Scholar]
- 84.Rowell LB, Brengelmann GL, Blackmon JR, Murray JA. Redistribution of blood flow during sustained high skin temperature in resting man. J Appl Physiol 28: 415–420, 1970. doi: 10.1152/jappl.1970.28.4.415. [DOI] [PubMed] [Google Scholar]
- 85.Brothers RM, Bhella PS, Shibata S, Wingo JE, Levine BD, Crandall CG. Cardiac systolic and diastolic function during whole body heat stress. Am J Physiol Heart Circ Physiol 296: H1150–H1156, 2009. doi: 10.1152/ajpheart.01069.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nelson MD, Haykowsky MJ, Petersen SR, DeLorey DS, Cheng-Baron J, Thompson RB. Increased left ventricular twist, untwisting rates, and suction maintain global diastolic function during passive heat stress in humans. Am J Physiol Heart Circ Physiol 298: H930–H937, 2010. doi: 10.1152/ajpheart.00987.2009. [DOI] [PubMed] [Google Scholar]
- 87.Stöhr EJ, González-Alonso J, Pearson J, Low DA, Ali L, Barker H, Shave R. Effects of graded heat stress on global left ventricular function and twist mechanics at rest and during exercise in healthy humans. Exp Physiol 96: 114–124, 2011. doi: 10.1113/expphysiol.2010.055137. [DOI] [PubMed] [Google Scholar]
- 88.Bundgaard-Nielsen M, Wilson TE, Seifert T, Secher NH, Crandall CG. Effect of volume loading on the Frank-Starling relation during reductions in central blood volume in heat-stressed humans. J Physiol 588: 3333–3339, 2010. doi: 10.1113/jphysiol.2010.191981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Minson CT, Wladkowski SL, Pawelczyk JA, Kenney WL. Age, splanchnic vasoconstriction, and heat stress during tilting. Am J Physiol Regul Integr Comp Physiol 276: R203–R212, 1999. doi: 10.1152/ajpregu.1999.276.1.r203. [DOI] [PubMed] [Google Scholar]
- 90.Gagnon D, Romero SA, Ngo H, Sarma S, Cornwell W 3rd, Poh PY, Stoller D, Levine BD, Crandall CG. Healthy aging does not compromise the augmentation of cardiac function during heat stress. J Appl Physiol 121: 885–892, 2016. doi: 10.1152/japplphysiol.00643.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Greaney JL, Stanhewicz AE, Proctor DN, Alexander LM, Kenney WL. Impairments in central cardiovascular function contribute to attenuated reflex vasodilation in aged skin. J Appl Physiol (1985) 119: 1411–1420, 2015. doi: 10.1152/japplphysiol.00729.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tei C, Horikiri Y, Park JC, Jeong JW, Chang KS, Tanaka N, Toyama Y. Effects of hot water bath or sauna on patients with congestive heart failure: acute hemodynamic improvement by thermal vasodilation. J Cardiol 24: 175–183, 1994. [PubMed] [Google Scholar]
- 93.Kuhlenhoelter AM, Kim K, Neff D, Nie Y, Blaize AN, Wong BJ, Kuang S, Stout J, Song Q, Gavin TP, Roseguini BT. Heat therapy promotes the expression of angiogenic regulators in human skeletal muscle. Am J Physiol Regul Integr Comp Physiol 311: R377–R391, 2016. doi: 10.1152/ajpregu.00134.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Thomas KN. Harnessing heat for health: a clinical application of heat stress. Temperature 4: 208–210, 2017. doi: 10.1080/23328940.2017.1317379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kukkonen-Harjula K, Kauppinen K. Health effects and risks of sauna bathing. Int J Circumpolar Health 65: 195–205, 2006. doi: 10.3402/ijch.v65i3.18102. [DOI] [PubMed] [Google Scholar]
- 96.Eisalo A, Luurila OJ. The Finnish sauna and cardiovascular diseases. Ann Clin Res 20: 267–270, 1988. [PubMed] [Google Scholar]
- 97.Keast ML, Adamo KB. The Finnish sauna bath and its use in patients with cardiovascular disease. J Cardiopulm Rehabil 20: 225–230, 2000. doi: 10.1097/00008483-200007000-00002. [DOI] [PubMed] [Google Scholar]
- 98.Jokinen E, Gregory EL, Välimäki I. The sauna and children. Ann Clin Res 20: 283–286, 1988. [PubMed] [Google Scholar]
- 99.Markkola L, Mattila KJ, Koivikko MJ. Sauna habits and related symptoms in Finnish children. Eur J Pediatr 149: 221–222, 1989. doi: 10.1007/BF01958288. [DOI] [PubMed] [Google Scholar]
- 100.Tikkanen J, Heinonen OP. Maternal hyperthermia during pregnancy and cardiovascular malformations in the offspring. Eur J Epidemiol 7: 628–635, 1991. doi: 10.1007/BF00218673. [DOI] [PubMed] [Google Scholar]
- 101.Wähä-Eskeli K, Erkkola R. The sauna and pregnancy. Ann Clin Res 20: 279–282, 1988. [PubMed] [Google Scholar]
- 102.Giannetti N, Juneau M, Arsenault A, Behr MA, Grégoire J, Tessier M, Larivée L. Sauna-induced myocardial ischemia in patients with coronary artery disease. Am J Med 107: 228–233, 1999. doi: 10.1016/S0002-9343(99)00220-X. [DOI] [PubMed] [Google Scholar]
- 103.Luurila OJ. Cardiac arrhythmias, sudden death and the Finnish sauna bath. Adv Cardiol 25: 73–81, 1978. doi: 10.1159/000402007. [DOI] [PubMed] [Google Scholar]
- 104.Singer K, Lundberg WB. Ventricular arrhythmias associated with the ingestion of alcohol. Ann Intern Med 77: 247–248, 1972. doi: 10.7326/0003-4819-77-2-247. [DOI] [PubMed] [Google Scholar]
- 105.Luurila OJ. The sauna and the heart. J Intern Med 231: 319–320, 1992. doi: 10.1111/j.1365-2796.1992.tb00938.x. [DOI] [PubMed] [Google Scholar]
- 106.Horowitz M, Shimoni Y, Parnes S, Gotsman MS, Hasin Y. Heat acclimation: cardiac performance of isolated rat heart. J Appl Physiol (1985) 60: 9–13, 1986. doi: 10.1152/jappl.1986.60.1.9. [DOI] [PubMed] [Google Scholar]
- 107.Horowitz M. Genomics and proteomics of heat acclimation. Front Biosci (Schol Ed) 2: 1068–1080, 2010. doi: 10.2741/s118. [DOI] [PubMed] [Google Scholar]
- 108.Horowitz M. Heat acclimation, epigenetics, and cytoprotection memory. Compr Physiol 4: 199–230, 2014. doi: 10.1002/cphy.c130025. [DOI] [PubMed] [Google Scholar]
- 109.Horowitz M. Epigenetics and cytoprotection with heat acclimation. J Appl Physiol (1985) 120: 702–710, 2016. doi: 10.1152/japplphysiol.00552.2015. [DOI] [PubMed] [Google Scholar]
- 110.Horowitz M, Assadi H. Heat acclimation-mediated cross-tolerance in cardioprotection. Ann N Y Acad Sci 1188: 199–206, 2010. doi: 10.1111/j.1749-6632.2009.05101.x. [DOI] [PubMed] [Google Scholar]
- 111.Horowitz M. Heat acclimation and cross-tolerance against novel stressors: genomic-physiological linkage. In: Progress in Brain Research. Amsterdam, The Netherlands: Elsevier, 2007, p. 373–392. [DOI] [PubMed] [Google Scholar]
- 112.Kihara T, Biro S, Ikeda Y, Fukudome T, Shinsato T, Masuda A, Miyata M, Hamasaki S, Otsuji Y, Minagoe S, Akiba S, Tei C. Effects of repeated sauna treatment on ventricular arrhythmias in patients with chronic heart failure. Circ J 68: 1146–1151, 2004. doi: 10.1253/circj.68.1146. [DOI] [PubMed] [Google Scholar]
- 113.Tei C, Tanaka N. Thermal vasodilation as a treatment of congestive heart failure: a novel approach. J Cardiol 27: 29–30, 1996. [PubMed] [Google Scholar]
- 114.Miyata M, Kihara T, Kubozono T, Ikeda Y, Shinsato T, Izumi T, Matsuzaki M, Yamaguchi T, Kasanuki H, Daida H, Nagayama M, Nishigami K, Hirata K, Kihara K, Tei C. Beneficial effects of Waon therapy on patients with chronic heart failure: results of a prospective multicenter study. J Cardiol 52: 79–85, 2008. doi: 10.1016/j.jjcc.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 115.Sobajima M, Nozawa T, Ihori H, Shida T, Ohori T, Suzuki T, Matsuki A, Yasumura S, Inoue H. Repeated sauna therapy improves myocardial perfusion in patients with occluded coronary artery-related ischemia. Int J Cardiol 167: 237–243, 2013. doi: 10.1016/j.ijcard.2011.12.064. [DOI] [PubMed] [Google Scholar]
- 116.Kihara T, Biro S, Imamura M, Yoshifuku S, Takasaki K, Ikeda Y, Otuji Y, Minagoe S, Toyama Y, Tei C. Repeated sauna treatment improves vascular endothelial and cardiac function in patients with chronic heart failure. J Am Coll Cardiol 39: 754–759, 2002. doi: 10.1016/S0735-1097(01)01824-1.doi:. [DOI] [PubMed] [Google Scholar]
- 117.Imamura M, Biro S, Kihara T, Yoshifuku S, Takasaki K, Otsuji Y, Minagoe S, Toyama Y, Tei C. Repeated thermal therapy improves impaired vascular endothelial function in patients with coronary risk factors. J Am Coll Cardiol 38: 1083–1088, 2001. doi: 10.1016/S0735-1097(01)01467-X. [DOI] [PubMed] [Google Scholar]
- 118.Kuwahata S, Miyata M, Fujita S, Kubozono T, Shinsato T, Ikeda Y, Hamasaki S, Kuwaki T, Tei C. Improvement of autonomic nervous activity by Waon therapy in patients with chronic heart failure. J Cardiol 57: 100–106, 2011. doi: 10.1016/j.jjcc.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 119.Tei C, Shinsato T, Miyata M, Kihara T, Hamasaki S. Waon therapy improves peripheral arterial disease. J Am Coll Cardiol 50: 2169–2171, 2007. doi: 10.1016/j.jacc.2007.08.025. [DOI] [PubMed] [Google Scholar]
- 120.Tei C, Shinsato T, Kihara T, Miyata M. Successful thermal therapy for end-stage peripheral artery disease. J Cardiol 47: 163–164, 2006. [PubMed] [Google Scholar]
- 121.Winterfeld HJ, Siewert H, Strangfeld D, Warnke H, Kruse J, Engelmann U. [Potential use of the sauna in the long-term treatment of hypertensive cardiovascular circulation disorders–a comparison with kinesiotherapy]. Schweiz Rundsch Med Prax 81: 1016–1020, 1992. [PubMed] [Google Scholar]
- 122.Laukkanen T, Khan H, Zaccardi F, Laukkanen JA. Association between sauna bathing and fatal cardiovascular and all-cause mortality events. JAMA Intern Med 175: 542–548, 2015. doi: 10.1001/jamainternmed.2014.8187. [DOI] [PubMed] [Google Scholar]
- 123.Zaccardi F, Laukkanen T, Willeit P, Kunutsor SK, Kauhanen J, Laukkanen JA. Sauna bathing and incident hypertension: a prospective cohort study. Am J Hypertens 30: 1120–1125, 2017. doi: 10.1093/ajh/hpx102. [DOI] [PubMed] [Google Scholar]
- 124.Bailey TG, Cable NT, Miller GD, Sprung VS, Low DA, Jones H. Repeated warm water immersion induces similar cerebrovascular adaptations to 8 weeks of moderate-intensity exercise training in females. Int J Sports Med 37: 757–765, 2016. doi: 10.1055/s-0042-106899. [DOI] [PubMed] [Google Scholar]
- 125.Carter HH, Spence AL, Atkinson CL, Pugh CJ, Cable NT, Thijssen DH, Naylor LH, Green DJ. Distinct effects of blood flow and temperature on cutaneous microvascular adaptation. Med Sci Sports Exerc 46: 2113–2121, 2014. doi: 10.1249/MSS.0000000000000349. [DOI] [PubMed] [Google Scholar]
- 126.Brunt VE, Howard MJ, Francisco MA, Ely BR, Minson CT. Reply from Vienna E. Brunt, Matthew J. Howard, Michael A. Francisco, Brett R. Ely and Christopher T. Minson. J Physiol 595: 3669–3670, 2017. doi: 10.1113/JP274129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Brunt VE, Rosenberg HL, Bazzoni AE, Nguyen KN, Chonchol M, Minson CT, Seals DR. Passive heat therapy lowers systolic blood pressure and improves vascular endothelial function in healthy older adults. FASEB J 33: 829.2, 2019. doI: 10.1096/fasebj.2019.33.1_supplement.829.2 [DOI] [Google Scholar]
- 128.Akerman AP, Thomas KN, van Rij AM, Body ED, Alfadhel M, Cotter JD. Heat therapy vs. supervised exercise therapy for peripheral arterial disease: a 12-wk randomized, controlled trial. Am J Physiol Heart Circ Physiol 316: H1495–H1506, 2019. doi: 10.1152/ajpheart.00151.2019. [DOI] [PubMed] [Google Scholar]
- 129.Chistiakov DA, Orekhov AN, Bobryshev YV. Effects of shear stress on endothelial cells: go with the flow. Acta Physiol (Oxf) 219: 382–408, 2017. doi: 10.1111/apha.12725. [DOI] [PubMed] [Google Scholar]
- 130.Chiu JJ, Chien S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev 91: 327–387, 2011. doi: 10.1152/physrev.00047.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gimbrone MA, García-Cardeña G. Vascular endothelium, hemodynamics, and the pathobiology of atherosclerosis. Cardiovasc Pathol 22: 9–15, 2013. doi: 10.1016/j.carpath.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhou J, Li YS, Chien S. Shear stress-initiated signaling and its regulation of endothelial function. Arterioscler Thromb Vasc Biol 34: 2191–2198, 2014. doi: 10.1161/ATVBAHA.114.303422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hwang J, Ing MH, Salazar A, Lassègue B, Griendling K, Navab M, Sevanian A, Hsiai TK. Pulsatile versus oscillatory shear stress regulates NADPH oxidase subunit expression: implication for native LDL oxidation. Circ Res 93: 1225–1232, 2003.doi: 10.1161/01.RES.0000104087.29395.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ziegler T, Bouzourène K, Harrison VJ, Brunner HR, Hayoz D. Influence of oscillatory and unidirectional flow environments on the expression of endothelin and nitric oxide synthase in cultured endothelial cells. Arterioscler Thromb Vasc Biol 18: 686–692, 1998. doi: 10.1161/01.ATV.18.5.686. [DOI] [PubMed] [Google Scholar]
- 135.Brooks AR, Lelkes PI, Rubanyi GM. Gene expression profiling of human aortic endothelial cells exposed to disturbed flow and steady laminar flow. Physiol Genomics 9: 27–41, 2002. doi: 10.1152/physiolgenomics.00075.2001. [DOI] [PubMed] [Google Scholar]
- 136.Chappell DC, Varner SE, Nerem RM, Medford RM, Alexander RW. Oscillatory shear stress stimulates adhesion molecule expression in cultured human endothelium. Circ Res 82: 532–539, 1998. doi: 10.1161/01.RES.82.5.532. [DOI] [PubMed] [Google Scholar]
- 137.O'Keeffe LM, Muir G, Piterina AV, McGloughlin T. Vascular cell adhesion molecule-1 expression in endothelial cells exposed to physiological coronary wall shear stresses. J Biomech Eng 131: 081003, 2009. doi: 10.1115/1.3148191. [DOI] [PubMed] [Google Scholar]
- 138.Cheng C, Tempel D, van Haperen R, van der Baan A, Grosveld F, Daemen MJ, Krams R, de Crom R. Atherosclerotic lesion size and vulnerability are determined by patterns of fluid shear stress. Circulation 113: 2744–2753, 2006. doi: 10.1161/CIRCULATIONAHA.105.590018. [DOI] [PubMed] [Google Scholar]
- 139.McNally JS, Davis ME, Giddens DP, Saha A, Hwang J, Dikalov S, Jo H, Harrison DG. Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am J Physiol Heart Circ Physiol 285: H2290–H2297, 2003. doi: 10.1152/ajpheart.00515.2003. [DOI] [PubMed] [Google Scholar]
- 140.Nam D, Ni CW, Rezvan A, Suo J, Budzyn K, Llanos A, Harrison D, Giddens D, Jo H. Partial carotid ligation is a model of acutely induced disturbed flow, leading to rapid endothelial dysfunction and atherosclerosis. Am J Physiol Heart Circ Physiol 297: H1535–H1534, 2009. doi: 10.1152/ajpheart.00510.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Credeur DP, Dobrosielski DA, Arce-Esquivel AA, Welsch MA. Brachial artery retrograde flow increases with age: relationship to physical function. Eur J Appl Physiol 107: 219–225, 2009. doi: 10.1007/s00421-009-1117-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Dinenno FA, Jones PP, Seals DR, Tanaka H. Limb blood flow and vascular conductance are reduced with age in healthy humans: relation to elevations in sympathetic nerve activity and declines in oxygen demand. Circulation 100: 164–170, 1999. doi: 10.1161/01.CIR.100.2.164. [DOI] [PubMed] [Google Scholar]
- 143.Cardillo C, Campia U, Iantorno M, Panza JA. Enhanced vascular activity of endogenous endothelin-1 in obese hypertensive patients. Hypertension 43: 36–40, 2004. doi: 10.1161/01.HYP.0000103868.45064.81. [DOI] [PubMed] [Google Scholar]
- 144.Taddei S, Virdis A, Ghiadoni L, Sudano I, Notari M, Salvetti A. Vasoconstriction to endogenous endothelin-1 is increased in the peripheral circulation of patients with essential hypertension. Circulation 100: 1680–1683, 1999. doi: 10.1161/01.CIR.100.16.1680. [DOI] [PubMed] [Google Scholar]
- 145.Padilla J, Simmons GH, Fadel PJ, Laughlin MH, Joyner MJ, Casey DP. Impact of aging on conduit artery retrograde and oscillatory shear at rest and during exercise: role of nitric oxide. Hypertension 57: 484–489, 2011. doi: 10.1161/HYPERTENSIONAHA.110.165365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Casey DP, Padilla J, Joyner MJ. α-adrenergic vasoconstriction contributes to the age-related increase in conduit artery retrograde and oscillatory shear. Hypertension 60: 1016–1022, 2012. doi: 10.1161/HYPERTENSIONAHA.112.200618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Cardillo C, Campia U, Kilcoyne CM, Bryant MB, Panza JA. Improved endothelium-dependent vasodilation after blockade of endothelin receptors in patients with essential hypertension. Circulation 105: 452–456, 2002. doi: 10.1161/hc0402.102989. [DOI] [PubMed] [Google Scholar]
- 148.Schinzari F, Iantorno M, Campia U, Mores N, Rovella V, Tesauro M, Di Daniele N, Cardillo C. Vasodilator responses and endothelin-dependent vasoconstriction in metabolically healthy obesity and the metabolic syndrome. Am J Physiol Endocrinol Metab 309: E787–E792, 2015. doi: 10.1152/ajpendo.00278.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Seals DR, DeSouza CA, Donato AJ, Tanaka H. Habitual exercise and arterial aging. J Appl Physiol (1985) 105: 1323–1332, 2008. doi: 10.1152/japplphysiol.90553.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zeiher AM, Drexler H, Wollschläger H, Just H. Modulation of coronary vasomotor tone in humans. Progressive endothelial dysfunction with different early stages of coronary atherosclerosis. Circulation 83: 391–401, 1991. doi: 10.1161/01.CIR.83.2.391.doi:. [DOI] [PubMed] [Google Scholar]
- 151.Zeiher AM, Drexler H, Wollschläger H, Just H. Endothelial dysfunction of the coronary microvasculature is associated with coronary blood flow regulation in patients with early atherosclerosis. Circulation 84: 1984–1992, 1991. doi: 10.1161/01.CIR.84.5.1984.doi:. [DOI] [PubMed] [Google Scholar]
- 152.Thijssen DH, Dawson EA, Tinken TM, Cable NT, Green DJ. Retrograde flow and shear rate acutely impair endothelial function in humans. Hypertension 53: 986–992, 2009. doi: 10.1161/HYPERTENSIONAHA.109.131508. [DOI] [PubMed] [Google Scholar]
- 153.Jenkins NT, Padilla J, Boyle LJ, Credeur DP, Laughlin MH, Fadel PJ. Disturbed blood flow acutely induces activation and apoptosis of the human vascular endothelium. Hypertension 61: 615–621, 2013. doi: 10.1161/HYPERTENSIONAHA.111.00561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Jimenez JJ, Jy W, Mauro LM, Soderland C, Horstman LL, Ahn YS. Endothelial cells release phenotypically and quantitatively distinct microparticles in activation and apoptosis. Thromb Res 109: 175–180, 2003. doi: 10.1016/S0049-3848(03)00064-1. [DOI] [PubMed] [Google Scholar]
- 155.Habersberger J, Strang F, Scheichl A, Htun N, Bassler N, Merivirta RM, Diehl P, Krippner G, Meikle P, Eisenhardt SU, Meredith I, Peter K. Circulating microparticles generate and transport monomeric C-reactive protein in patients with myocardial infarction. Cardiovasc Res 96: 64–72, 2012. doi: 10.1093/cvr/cvs237. [DOI] [PubMed] [Google Scholar]
- 156.Diehl P, Fricke A, Sander L, Stamm J, Bassler N, Htun N, Ziemann M, Helbing T, El-Osta A, Jowett JB, Peter K. Microparticles: major transport vehicles for distinct microRNAs in circulation. Cardiovasc Res 93: 633–644, 2012. doi: 10.1093/cvr/cvs007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Agouni A, Lagrue-Lak-Hal AH, Ducluzeau PH, Mostefai HA, Draunet-Busson C, Lefthériotis G, Heymes C, Martinez MC, Andriantsitohaina R. Endothelial dysfunction caused by circulating microparticles from patients with metabolic syndrome. Am J Pathol 173: 1210–1219, 2008. doi: 10.2353/ajpath.2008.080228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Dignat-George F, Boulanger CM. The many faces of endothelial microparticles. Arterioscler Thromb Vasc Biol 31: 27–33, 2011. doi: 10.1161/ATVBAHA.110.218123. [DOI] [PubMed] [Google Scholar]
- 159.Boulanger CM, Amabile N, Guérin AP, Pannier B, Leroyer AS, Mallat Z, Nguyen C, Tedgui A, London GM. In vivo shear stress determines circulating levels of endothelial microparticles in end-stage renal disease. Hypertension 49: 902–908, 2007. doi: 10.1161/01.HYP.0000259667.22309.df. [DOI] [PubMed] [Google Scholar]
- 160.Dimmeler S, Hermann C, Galle J, Zeiher AM. Upregulation of superoxide dismutase and nitric oxide synthase mediates the apoptosis-suppressive effects of shear stress on endothelial cells. Arterioscler Thromb Vasc Biol 19: 656–664, 1999. [PM doi: 10.1161/01.ATV.19.3.656. C]doi:. [DOI] [PubMed] [Google Scholar]
- 161.Ranjan V, Xiao Z, Diamond SL. Constitutive NOS expression in cultured endothelial cells is elevated by fluid shear stress. Am J Physiol Heart Circ Physiol 269: H550–H555, 1995. doi: 10.1152/ajpheart.1995.269.2.H550. [DOI] [PubMed] [Google Scholar]
- 162.Uematsu M, Ohara Y, Navas JP, Nishida K, Murphy TJ, Alexander RW, Nerem RM, Harrison DG. Regulation of endothelial cell nitric oxide synthase mRNA expression by shear stress. Am J Physiol Cell Physiol 269: C1371–C1378, 1995. doi: 10.1152/ajpcell.1995.269.6.C1371. [DOI] [PubMed] [Google Scholar]
- 163.Widder JD, Chen W, Li L, Dikalov S, Thöny B, Hatakeyama K, Harrison DG. Regulation of tetrahydrobiopterin biosynthesis by shear stress. Circ Res 101: 830–838, 2007. doi: 10.1161/CIRCRESAHA.107.153809. [DOI] [PubMed] [Google Scholar]
- 164.Jin ZG, Ueba H, Tanimoto T, Lungu AO, Frame MD, Berk BC. Ligand-independent activation of vascular endothelial growth factor receptor 2 by fluid shear stress regulates activation of endothelial nitric oxide synthase. Circ Res 93: 354–363, 2003. doi: 10.1161/01.RES.0000089257.94002.96. [DOI] [PubMed] [Google Scholar]
- 165.Boo YC, Hwang J, Sykes M, Michell BJ, Kemp BE, Lum H, Jo H. Shear stress stimulates phosphorylation of eNOS at Ser(635) by a protein kinase A-dependent mechanism. Am J Physiol Heart Circ Physiol 283: H1819–H1828, 2002. doi: 10.1152/ajpheart.00214.2002. [DOI] [PubMed] [Google Scholar]
- 166.Garcia-Cardena G, Fan R, Shah V, Sorrentino R, Cirino G, Papapetropoulos A, Sessa WC. Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature 392: 821–824, 1998. doi: 10.1038/33934. [DOI] [PubMed] [Google Scholar]
- 167.Kuchan MJ, Frangos JA. Shear stress regulates endothelin-1 release via protein kinase C and cGMP in cultured endothelial cells. Am J Physiol Heart Circ Physiol 264: H150–H156, 1993. doi: 10.1152/ajpheart.1993.264.1.H150. [DOI] [PubMed] [Google Scholar]
- 168.Malek A, Izumo S. Physiological fluid shear stress causes downregulation of endothelin-1 mRNA in bovine aortic endothelium. Am J Physiol Cell Physiol 263: C389–C396, 1992. doi: 10.1152/ajpcell.1992.263.2.C389. [DOI] [PubMed] [Google Scholar]
- 169.Himburg HA, Dowd SE, Friedman MH. Frequency-dependent response of the vascular endothelium to pulsatile shear stress. Am J Physiol Heart Circ Physiol 293: H645–H653, 2007. doi: 10.1152/ajpheart.01087.2006. [DOI] [PubMed] [Google Scholar]
- 170.Tsao PS, Buitrago R, Chan JR, Cooke JP. Fluid flow inhibits endothelial adhesiveness. Nitric oxide and transcriptional regulation of VCAM-1. Circulation 94: 1682–1689, 1996. doi: 10.1161/01.CIR.94.7.1682. doi:. [DOI] [PubMed] [Google Scholar]
- 171.Woodman CR, Muller JM, Rush JW, Laughlin MH, Price EM. Flow regulation of ecNOS and Cu/Zn SOD mRNA expression in porcine coronary arterioles. Am J Physiol Heart Circ Physiol 276: H1058–H1063, 1999. doi: 10.1152/ajpheart.1999.276.3.H1058. [DOI] [PubMed] [Google Scholar]
- 172.Woodman CR, Price EM, Laughlin MH. Shear stress induces eNOS mRNA expression and improves endothelium-dependent dilation in senescent soleus muscle feed arteries. J Appl Physiol (1985) 98: 940–946, 2005. doi: 10.1152/japplphysiol.00408.2004. [DOI] [PubMed] [Google Scholar]
- 173.Green DJ, Carter HH, Fitzsimons MG, Cable NT, Thijssen DH, Naylor LH. Obligatory role of hyperaemia and shear stress in microvascular adaptation to repeated heating in humans. J Physiol 588: 1571–1577, 2010. doi: 10.1113/jphysiol.2010.186965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Birk GK, Dawson EA, Atkinson C, Haynes A, Cable NT, Thijssen DH, Green DJ. Brachial artery adaptation to lower limb exercise training: role of shear stress. J Appl Physiol 112: 1653–1658, 2012. doi: 10.1152/japplphysiol.01489.2011. [DOI] [PubMed] [Google Scholar]
- 175.Tinken TM, Thijssen DH, Hopkins N, Dawson EA, Cable NT, Green DJ. Shear stress mediates endothelial adaptations to exercise training in humans. Hypertension 55: 312–318, 2010. doi: 10.1161/HYPERTENSIONAHA.109.146282. [DOI] [PubMed] [Google Scholar]
- 176.Thijssen DH, Dawson EA, van den Munckhof IC, Tinken TM, Drijver den E, Hopkins N, Cable NT, Green DJ. Exercise-mediated changes in conduit artery wall thickness in humans: role of shear stress. Am J Physiol Heart Circ Physiol 301: H241–H246, 2011. doi: 10.1152/ajpheart.00170.2011. [DOI] [PubMed] [Google Scholar]
- 177.Ritossa F. A new puffing pattern induced by temperature shock and DNP in drosophila. Experientia 18: 571–573, 1962. doi: 10.1007/BF02172188. [DOI] [Google Scholar]
- 178.Zhong N, Zhang Y, Fang QZ, Zhou ZN. Intermittent hypoxia exposure-induced heat-shock protein 70 expression increases resistance of rat heart to ischemic injury. Acta Pharmacol Sin 21: 467–472, 2000. [PubMed] [Google Scholar]
- 179.Won EJ, Han J, Lee Y, Kumar KS, Shin KH, Lee SJ, Park HG, Lee JS. In vivo effects of UV radiation on multiple endpoints and expression profiles of DNA repair and heat shock protein (Hsp) genes in the cycloid copepod Paracyclopina nana. Aquat Toxicol 165: 1–8, 2015. doi: 10.1016/j.aquatox.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 180.Freshney NW, Rawlinson L, Guesdon F, Jones E, Cowley S, Hsuan J, Saklatvala J. Interleukin-1 activates a novel protein kinase cascade that results in the phosphorylation of Hsp27. Cell 78: 1039–1049, 1994. doi: 10.1016/0092-8674(94)90278-X. C [DOI] [PubMed] [Google Scholar]
- 181.Saklatvala J, Kaur P, Guesdon F. Phosphorylation of the small heat-shock protein is regulated by interleukin 1, tumour necrosis factor, growth factors, bradykinin and ATP. Biochem J 277: 635–642, 1991. doi: 10.1042/bj2770635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Airaksinen S, Jokilehto T, Råbergh CM, Nikinmaa M. Heat- and cold-inducible regulation of HSP70 expression in zebrafish ZF4 cells. Comp Biochem Physiol B Biochem Mol Biol 136: 275–282, 2003. doi: 10.1016/S1096-4959(03)00205-7. [DOI] [PubMed] [Google Scholar]
- 183.Locke M, Noble EG, Atkinson BG. Exercising mammals synthesize stress proteins. Am J Physiol Cell Physiol 258: C723–C729, 1990. doi: 10.1152/ajpcell.1990.258.4.C723. [DOI] [PubMed] [Google Scholar]
- 184.Puntschart A, Vogt M, Widmer HR, Hoppeler H, Billeter R. Hsp70 expression in human skeletal muscle after exercise. Acta Physiol Scand 157: 411–417, 1996. doi: 10.1046/j.1365-201X.1996.512270000.x. [DOI] [PubMed] [Google Scholar]
- 185.Reichsman F, Scordilis SP, Clarkson PM, Evans WJ. Muscle protein changes following eccentric exercise in humans. Eur J Appl Physiol Occup Physiol 62: 245–250, 1991. doi: 10.1007/BF00571547. [DOI] [PubMed] [Google Scholar]
- 186.Cosgrove JW, Brown IR. Heat shock protein in mammalian brain and other organs after a physiologically relevant increase in body temperature induced by D-lysergic acid diethylamide. Proc Natl Acad Sci U S A 80: 569–573, 1983. doi: 10.1073/pnas.80.2.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Nowak TS. Effects of amphetamine on protein synthesis and energy metabolism in mouse brain: role of drug-induced hyperthermia. J Neurochem 50: 285–294, 1988. doi: 10.1111/j.1471-4159.1988.tb13262.x. [DOI] [PubMed] [Google Scholar]
- 188.Uney JB, Leigh PN, Marsden CD, Lees A, Anderton BH. Stereotaxic injection of kainic acid into the striatum of rats induces synthesis of mRNA for heat shock protein 70. FEBS Lett 235: 215–218, 1988. doi: 10.1016/0014-5793(88)81265-1. [DOI] [PubMed] [Google Scholar]
- 189.Li S, Piotrowicz RS, Levin EG, Shyy YJ, Chien S. Fluid shear stress induces the phosphorylation of small heat shock proteins in vascular endothelial cells. Am J Physiol Cell Physiol 271: C994–C1000, 1996. doi: 10.1152/ajpcell.1996.271.3.C994. [DOI] [PubMed] [Google Scholar]
- 190.Lee BJ, Mackenzie RW, Cox V, James RS, Thake CD. Human monocyte heat shock protein 72 responses to acute hypoxic exercise after 3 days of exercise heat acclimation. BioMed Research International 2015: 1–15, 2015. doi: 10.1155/2015/849809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Magalhães F de C, Amorim FT, Passos RL, Fonseca MA, Oliveira KPM, Lima MR, Guimarães JB, Ferreira-Júnior JB, Martini AR, Lima NR, Soares DD, Oliveira EM, Rodrigues LO. Heat and exercise acclimation increases intracellular levels of Hsp72 and inhibits exercise-induced increase in intracellular and plasma Hsp72 in humans. Cell Stress Chaperones 15: 885–895, 2010. doi: 10.1007/s12192-010-0197-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.McClung JP, Hasday JD, He JR, Montain SJ, Cheuvront SN, Sawka MN, Singh IS. Exercise-heat acclimation in humans alters baseline levels and ex vivo heat inducibility of HSP72 and HSP90 in peripheral blood mononuclear cells. Am J Physiol Regul Integr Comp Physiol 294: R185–R191, 2008. doi: 10.1152/ajpregu.00532.2007. [DOI] [PubMed] [Google Scholar]
- 193.Yamada PM, Amorim FT, Moseley P, Robergs R, Schneider SM. Effect of heat acclimation on heat shock protein 72 and interleukin-10 in humans. J Appl Physiol 103: 1196–1204, 2007. doi: 10.1152/japplphysiol.00242.2007. [DOI] [PubMed] [Google Scholar]
- 194.Brunt VE, Wiedenfeld-Needham K, Comrada LN, Minson CT. Passive heat therapy protects against endothelial cell hypoxia-reoxygenation via effects of elevations in temperature and circulating factors. J Physiol 596: 4831–4845, 2018. doi: 10.1113/JP276559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Ely BR, Clayton ZS, McCurdy CE, Pfeiffer J, Needham KW, Comrada LN, Minson CT. Heat therapy improves glucose tolerance and adipose tissue insulin signaling in polycystic ovary syndrome. Am J Physiol Endocrinol Metab 317: E172–E182, 2019. doi: 10.1152/ajpendo.00549.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Hafen PS, Abbott K, Bowden J, Lopiano R, Hancock CR, Hyldahl RD. Daily heat treatment maintains mitochondrial function and attenuates atrophy in human skeletal muscle subjected to immobilization. J Appl Physiol 127: 47–57, 2019. doi: 10.1152/japplphysiol.01098.2018. [DOI] [PubMed] [Google Scholar]
- 197.Hafen PS, Preece CN, Sorensen JR, Hancock CR, Hyldahl RD. Repeated exposure to heat stress induces mitochondrial adaptation in human skeletal muscle. J Appl Physiol (1985) 125: 1447–1455, 2018. doi: 10.1152/japplphysiol.00383.2018. [DOI] [PubMed] [Google Scholar]
- 198.Jones Q, Voegeli TS, Li G, Chen Y, Currie RW. Heat shock proteins protect against ischemia and inflammation through multiple mechanisms. Inflamm Allergy Drug Targets 10: 247–259, 2011. doi: 10.2174/187152811796117726. [DOI] [PubMed] [Google Scholar]
- 199.Marber MS, Mestril R, Chi SH, Sayen MR, Yellon DM, Dillmann WH. Overexpression of the rat inducible 70-kD heat stress protein in a transgenic mouse increases the resistance of the heart to ischemic injury. J Clin Invest 95: 1446–1456, 1995. doi: 10.1172/JCI117815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Mizzen LA, Welch WJ. Characterization of the thermotolerant cell. I. Effects on protein synthesis activity and the regulation of heat-shock protein 70 expression. J Cell Biol 106: 1105–1116, 1988. doi: 10.1083/jcb.106.4.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Lavoie JN, Gingras-Breton G, Tanguay RM, Landry J. Induction of Chinese hamster HSP27 gene expression in mouse cells confers resistance to heat shock. HSP27 stabilization of the microfilament organization. J Biol Chem 268: 3420–3429, 1993. doi: 10.1016/S0021-9258(18)53711-X. [DOI] [PubMed] [Google Scholar]
- 202.Chirico WJ, Waters MG, Blobel G. 70K heat shock related proteins stimulate protein translocation into microsomes. Nature 332: 805–810, 1988. doi: 10.1038/332805a0. [DOI] [PubMed] [Google Scholar]
- 203.Verner K, Schatz G. Import of an incompletely folded precursor protein into isolated mitochondria requires an energized inner membrane, but no added ATP. EMBO J 6: 2449–2456, 1987. doi: 10.1002/j.1460-2075.1987.tb02524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Velazquez JM, Lindquist S. hsp70: nuclear concentration during environmental stress and cytoplasmic storage during recovery. Cell 36: 655–662, 1984. doi: 10.1016/0092-8674(84)90345-3. [DOI] [PubMed] [Google Scholar]
- 205.Lindquist S, Craig EA. The heat-shock proteins. Annu Rev Genet 22: 631–677, 1988. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- 206.Brugge J, Yonemoto W, Darrow D. Interaction between the Rous sarcoma virus transforming protein and two cellular phosphoproteins: analysis of the turnover and distribution of this complex. Mol Cell Biol 3: 9–19, 1983. doi: 10.1128/MCB.3.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Courtneidge SA, Bishop JM. Transit of pp60v-src to the plasma membrane. Proc Natl Acad Sci U S A 79: 7117–7121, 1982. doi: 10.1073/pnas.79.23.7117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Richter K, Haslbeck M, Buchner J. The heat shock response: life on the verge of death. Mol Cell 40: 253–266, 2010. doi: 10.1016/j.molcel.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 209.Gibson OR, Dennis A, Parfitt T, Taylor L, Watt PW, Maxwell NS. Extracellular Hsp72 concentration relates to a minimum endogenous criteria during acute exercise-heat exposure. Cell Stress Chaperones 19: 389–400, 2014. doi: 10.1007/s12192-013-0468-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Asea A. Initiation of the immune response by extracellular Hsp72: chaperokine activity of Hsp72. Curr Immunol Rev 2: 209–215, 2006. doi: 10.2174/157339506778018514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Harris MB, Blackstone MA, Ju H, Venema VJ, Venema RC. Heat-induced increases in endothelial NO synthase expression and activity and endothelial NO release. Am J Physiol Heart Circ Physiol 285: H333–H340, 2003. doi: 10.1152/ajpheart.00726.2002. [DOI] [PubMed] [Google Scholar]
- 212.Blake MJ, Gershon D, Fargnoli J, Holbrook NJ. Discordant expression of heat shock protein mRNAs in tissues of heat-stressed rats. J Biol Chem 265: 15275–15279, 1990. doi: 10.1016/S0021-9258(18)77252-9. [DOI] [PubMed] [Google Scholar]
- 213.Gibson OR, Tuttle JA, Watt PW, Maxwell NS, Taylor L. Hsp72 and Hsp90α mRNA transcription is characterised by large, sustained changes in core temperature during heat acclimation. Cell Stress Chaperones. 21: 1021–1035, 2016. doi: 10.1007/s12192-016-0726-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Djurhuus R, Nossum V, Lundsett N, Hovin W, Svardal AM, Havnes MB, Fismen L, Hjelde A, Brubakk AO. Simulated diving after heat stress potentiates the induction of heat shock protein 70 and elevates glutathione in human endothelial cells. Cell Stress Chaperones 15: 405–414, 2010. doi: 10.1007/s12192-009-0156-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Nowak TS, Bond U, Schlesinger MJ. Heat shock RNA levels in brain and other tissues after hyperthermia and transient ischemia. J Neurochem 54: 451–458, 1990. doi: 10.1111/j.1471-4159.1990.tb01893.x. [DOI] [PubMed] [Google Scholar]
- 216.Oishi Y, Taniguchi K, Matsumoto H, Ishihara A, Ohira Y, Roy RR. Muscle type-specific response of HSP60, HSP72, and HSC73 during recovery after elevation of muscle temperature. J Appl Physiol 92: 1097–1103, 2002. doi: 10.1152/japplphysiol.00739.2001. [DOI] [PubMed] [Google Scholar]
- 217.Oishi Y, Taniguchi K, Matsumoto H, Ishihara A, Ohira Y, Roy RR. Differential responses of HSPs to heat stress in slow and fast regions of rat gastrocnemius muscle. Muscle Nerve 28: 587–594, 2003. doi: 10.1002/mus.10476. [DOI] [PubMed] [Google Scholar]
- 218.Morton JP, MacLaren D, Cable NT, Campbell IT, Evans L, Bongers T, Griffiths RD, Kayani AC, McArdle A, Brust B. Elevated core and muscle temperature to levels comparable to exercise do not increase heat shock protein content of skeletal muscle of physically active men. Acta Physiol 190: 319–327, 2007. doi: 10.1111/j.1748-1716.2007.01711.x. [DOI] [PubMed] [Google Scholar]
- 219.Khassaf M, Child RB, McArdle A, Brodie DA, Esanu C, Jackson MJ. Time course of responses of human skeletal muscle to oxidative stress induced by nondamaging exercise. J Appl Physiol (1985) 90: 1031–1035, 2001. doi: 10.1152/jappl.2001.90.3.1031. [DOI] [PubMed] [Google Scholar]
- 220.Morton JP, MacLaren DP, Cable NT, Bongers T, Griffiths RD, Campbell IT, Evans L, Kayani A, McArdle A, Drust B. Time course and differential responses of the major heat shock protein families in human skeletal muscle following acute nondamaging treadmill exercise. J Appl Physiol 101: 176–182, 2006. doi: 10.1152/japplphysiol.00046.2006. [DOI] [PubMed] [Google Scholar]
- 221.Deguchi Y, Negoro S, Kishimoto S. Heat-shock protein synthesis by human peripheral mononuclear cells from SLE patients. Biochem Biophys Res Commun 148: 1063–1068, 1987. doi: 10.1016/S0006-291X(87)80239-5. [DOI] [PubMed] [Google Scholar]
- 222.Vásquez-Vivar J, Kalyanaraman B, Martásek P, Hogg N, Masters BS, Karoui H, Tordo P, Pritchard KA. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci U S A 95: 9220–9225, 1998. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Pritchard KA, Ackerman AW, Gross ER, Stepp DW, Shi Y, Fontana JT, Baker JE, Sessa WC. Heat shock protein 90 mediates the balance of nitric oxide and superoxide anion from endothelial nitric-oxide synthase. J Biol Chem 276: 17621–17624, 2001. doi: 10.1074/jbc.C100084200. [DOI] [PubMed] [Google Scholar]
- 224.Song Y, Zweier JL, Xia Y. Heat-shock protein 90 augments neuronal nitric oxide synthase activity by enhancing Ca2+/calmodulin binding. Biochem J 355: 357–360, 2001. doi: 10.1042/0264-6021:3550357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Harris MB, Ju H, Venema VJ, Blackstone M, Venema RC. Role of heat shock protein 90 in bradykinin-stimulated endothelial nitric oxide release. Gen Pharmacol 35: 165–170, 2000. doi: 10.1016/S0306-3623(01)00104-5. [DOI] [PubMed] [Google Scholar]
- 226.Gratton JP, Fontana J, O'Connor DS, Garcia-Cardena G, McCabe TJ, Sessa WC. Reconstitution of an endothelial nitric-oxide synthase (eNOS), hsp90, and caveolin-1 complex in vitro. Evidence that hsp90 facilitates calmodulin stimulated displacement of eNOS from caveolin-1. J Biol Chem 275: 22268–22272, 2000. doi: 10.1074/jbc.M001644200. [DOI] [PubMed] [Google Scholar]
- 227.Takahashi S, Mendelsohn ME. Calmodulin-dependent and -independent activation of endothelial nitric-oxide synthase by heat shock protein 90. J Biol Chem 278: 9339–9344, 2003. doi: 10.1074/jbc.M212651200. [DOI] [PubMed] [Google Scholar]
- 228.Fontana J, Fulton D, Chen Y, Fairchild TA, McCabe TJ, Fujita N, Tsuruo T, Sessa WC. Domain mapping studies reveal that the M domain of hsp90 serves as a molecular scaffold to regulate Akt-dependent phosphorylation of endothelial nitric oxide synthase and NO release. Circ Res 90: 866–873, 2002. doi: 10.1161/01.RES.0000016837.26733.BE. [DOI] [PubMed] [Google Scholar]
- 229.Harris MB, Mitchell BM, Sood SG, Webb RC, Venema RC. Increased nitric oxide synthase activity and Hsp90 association in skeletal muscle following chronic exercise. Eur J Appl Physiol 104: 795–802, 2008. doi: 10.1007/s00421-008-0833-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Ushio-Fukai M, Alexander RW. Reactive oxygen species as mediators of angiogenesis signaling: role of NAD(P)H oxidase. Mol Cell Biochem 264: 85–97, 2004. doi: 10.1023/B:MCBI.0000044378.09409.b5. [DOI] [PubMed] [Google Scholar]
- 231.Kim YW, Byzova TV. Oxidative stress in angiogenesis and vascular disease. Blood 123: 625–631, 2014. doi: 10.1182/blood-2013-09-512749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Kuzkaya N, Weissmann N, Harrison DG, Dikalov S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: implications for uncoupling endothelial nitric-oxide synthase. J Biol Chem 278: 22546–22554, 2003. doi: 10.1074/jbc.M302227200. [DOI] [PubMed] [Google Scholar]
- 233.Chandra S, Romero MJ, Shatanawi A, Alkilany AM, Caldwell RB, Caldwell RW. Oxidative species increase arginase activity in endothelial cells through the RhoA/Rho kinase pathway. Br J Pharmacol 165: 506–519, 2012. doi: 10.1111/j.1476-5381.2011.01584.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol 552: 335–344, 2003. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Lubos E, Loscalzo J, Handy DE. Glutathione peroxidase-1 in health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal 15: 1957–1997, 2011. doi: 10.1089/ars.2010.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Afolayan AJ, Alexander M, Holme RL, Michalkiewicz T, Rana U, Teng RJ, Zemanovic S, Sahoo D, Pritchard KA, Konduri GG. Domain mapping of heat shock protein 70 reveals that glutamic acid 446 and arginine 447 are critical for regulating superoxide dismutase 2 function. J Biol Chem 292: 2369–2378, 2017. doi: 10.1074/jbc.M116.756122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Choi S, Park KA, Lee HJ, Park MS, Lee JH, Park KC, Kim M, Lee SH, Seo JS, Yoon BW. Expression of Cu/Zn SOD protein is suppressed in hsp 70.1 knockout mice. J Biochem Mol Biol 38: 111–114, 2005. doi: 10.5483/bmbrep.2005.38.1.111. [DOI] [PubMed] [Google Scholar]
- 238.Baek SH, Min JN, Park EM, Han MY, Lee YS, Lee YJ, Park YM. Role of small heat shock protein HSP25 in radioresistance and glutathione-redox cycle. J Cell Physiol 183: 100–107, 2000. doi: 10.1002/(SICI)1097-4652(200004)183:1<100::AID-JCP12>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 239.Cahill PA, Redmond EM. Vascular endothelium–gatekeeper of vessel health. Atherosclerosis 248: 97–109, 2016. doi: 10.1016/j.atherosclerosis.2016.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Ruparelia N, Chai JT, Fisher EA, Choudhury RP. Inflammatory processes in cardiovascular disease: a route to targeted therapies. Nat Rev Cardiol 14: 133–144, 2017. [Erratum in Nat Rev Cardiol 14: 314, 2017]. doi: 10.1038/nrcardio.2016.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Brevetti G, Silvestro A, Di Giacomo S, Bucur R, Di Donato A, Schiano V, Scopacasa F. Endothelial dysfunction in peripheral arterial disease is related to increase in plasma markers of inflammation and severity of peripheral circulatory impairment but not to classic risk factors and atherosclerotic burden. J Vasc Surg 38: 374–379, 2003. doi: 10.1016/S0741-5214(03)00124-1. [DOI] [PubMed] [Google Scholar]
- 242.Castro AR, Silva SO, Soares SC. The use of high sensitivity c-reactive protein in cardiovascular disease detection. J Pharm Pharm Sci 21: 496–503, 2018. doi: 10.18433/jpps29872. [DOI] [PubMed] [Google Scholar]
- 243.Fichtlscherer S, Rosenberger G, Walter DH, Breuer S, Dimmeler S, Zeiher AM. Elevated C-reactive protein levels and impaired endothelial vasoreactivity in patients with coronary artery disease. Circulation 102: 1000–1006, 2000. doi: 10.1161/01.CIR.102.9.1000. [DOI] [PubMed] [Google Scholar]
- 244.de Winther MP, Kanters E, Kraal G, Hofker MH. Nuclear factor kappaB signaling in atherogenesis. Arterioscler Thromb Vasc Biol 25: 904–914, 2005. doi: 10.1161/01.ATV.0000160340.72641.87. [DOI] [PubMed] [Google Scholar]
- 245.Guzik TJ, Harrison DG. Endothelial NF-kappaB as a mediator of kidney damage: the missing link between systemic vascular and renal disease? Circ Res 101: 227–229, 2007. doi: 10.1161/CIRCRESAHA.107.158295. [DOI] [PubMed] [Google Scholar]
- 246.Neish AS, Williams AJ, Palmer HJ, Whitley MZ, Collins T. Functional analysis of the human vascular cell adhesion molecule 1 promoter. J Exp Med 176: 1583–1593, 1992. doi: 10.1084/jem.176.6.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Osborn L. Leukocyte adhesion to endothelium in inflammation. Cell 62: 3–6, 1990. doi: 10.1016/0092-8674(90)90230-C.doi:. [DOI] [PubMed] [Google Scholar]
- 248.Chen Y, Arrigo AP, Currie RW. Heat shock treatment suppresses angiotensin II-induced activation of NF-kappaB pathway and heart inflammation: a role for IKK depletion by heat shock? Am J Physiol Heart Circ Physiol 287: H1104–14, 2004. doi: 10.1152/ajpheart.00102.2004. [DOI] [PubMed] [Google Scholar]
- 249.Frossard JL, Pastor CM, Hadengue A. Effect of hyperthermia on NF-kappaB binding activity in cerulein-induced acute pancreatitis. Am J Physiol Gastrointest Liver Physiol 280: G1157–G1162, 2001. doi: 10.1152/ajpgi.2001.280.6.G1157. [DOI] [PubMed] [Google Scholar]
- 250.Nakabe N, Kokura S, Shimozawa M, Katada K, Sakamoto N, Ishikawa T, Handa O, Takagi T, Naito Y, Yoshida N, Yoshikawa T. Hyperthermia attenuates TNF-alpha-induced up regulation of endothelial cell adhesion molecules in human arterial endothelial cells. Int J Hyperthermia 23: 217–224, 2007. doi: 10.1080/02656730601143295. [DOI] [PubMed] [Google Scholar]
- 251.Ensor JE, Wiener SM, McCrea KA, Viscardi RM, Crawford EK, Hasday JD. Differential effects of hyperthermia on macrophage interleukin-6 and tumor necrosis factor-alpha expression. Am J Physiol Cell Physiol 266: C967–C974, 1994. doi: 10.1152/ajpcell.1994.266.4.C967. [DOI] [PubMed] [Google Scholar]
- 252.Snyder YM, Guthrie L, Evans GF, Zuckerman SH. Transcriptional inhibition of endotoxin-induced monokine synthesis following heat shock in murine peritoneal macrophages. J Leukoc Biol 51: 181–187, 1992. doi: 10.1002/jlb.51.2.181. [DOI] [PubMed] [Google Scholar]
- 253.Kluger MJ, Rudolph K, Soszynski D, Conn CA, Leon LR, Kozak W, Wallen ES, Moseley PL. Effect of heat stress on LPS-induced fever and tumor necrosis factor. Am J Physiol Regul Integr Comp Physiol 273: R858–R863, 1997. doi: 10.1152/ajpregu.1997.273.3.R858. [DOI] [PubMed] [Google Scholar]
- 254.Rakonczay Z, Takács T, Mándi Y, Iványi B, Varga S, Pápai G, Boros I, Lonovics J. Water immersion pretreatment decreases pro-inflammatory cytokine production in cholecystokinin-octapeptide-induced acute pancreatitis in rats: possible role of HSP72. Int J Hyperthermia 17: 520–535, 2001. doi: 10.1080/02656730110081785. [DOI] [PubMed] [Google Scholar]
- 255.Jäättelä M, Wissing D. Heat-shock proteins protect cells from monocyte cytotoxicity: possible mechanism of self-protection. J Exp Med 177: 231–236, 1993. doi: 10.1084/jem.177.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Landry J, Chrétien P, Lambert H, Hickey E, Weber LA. Heat shock resistance conferred by expression of the human HSP27 gene in rodent cells. J Cell Biol 109: 7–15, 1989. doi: 10.1083/jcb.109.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Kim I, Shin HM, Baek W. Heat-shock response is associated with decreased production of interleukin-6 in murine aortic vascular smooth muscle cells. Naunyn Schmiedebergs Arch Pharmacol 371: 27–33, 2005. doi: 10.1007/s00210-004-1007-5. [DOI] [PubMed] [Google Scholar]
- 258.Kohn G, Wong HR, Bshesh K, Zhao B, Vasi N, Denenberg A, Morris C, Stark J, Shanley TP. Heat shock inhibits tnf-induced ICAM-1 expression in human endothelial cells via I kappa kinase inhibition. Shock 17: 91–97, 2002. doi: 10.1097/00024382-200202000-00002. [DOI] [PubMed] [Google Scholar]
- 259.Gao L, Wang F, Wang B, Gong B, Zhang J, Zhang X, Zhao J. Cilostazol protects diabetic rats from vascular inflammation via nuclear factor-kappa B-dependent down-regulation of vascular cell adhesion molecule-1 expression. J Pharmacol Exp Ther 318: 53–58, 2006. doi: 10.1124/jpet.106.101444. [DOI] [PubMed] [Google Scholar]
- 260.Wieten L, Berlo SE, Brink Ten CB, van Kooten PJ, Singh M, van der Zee R, Glant TT, Broere F, van Eden W. IL-10 is critically involved in mycobacterial HSP70 induced suppression of proteoglycan-induced arthritis. PLoS One 4: e4186, 2009. doi: 10.1371/journal.pone.0004186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Isomäki H. The sauna and rheumatic diseases. Ann Clin Res 20: 271–275, 1988. [PubMed] [Google Scholar]
- 262.Wang P, Wu P, Siegel MI, Egan RW, Billah MM. Interleukin (IL)-10 inhibits nuclear factor kappa B (NF kappa B) activation in human monocytes. IL-10 and IL-4 suppress cytokine synthesis by different mechanisms. J Biol Chem 270: 9558–9563, 1995. doi: 10.1074/jbc.270.16.9558. [DOI] [PubMed] [Google Scholar]
- 263.Kunutsor SK, Laukkanen T, Laukkanen JA. Longitudinal associations of sauna bathing with inflammation and oxidative stress: the KIHD prospective cohort study. Ann Med 50: 437–442, 2018. doi: 10.1080/07853890.2018.1489143. [DOI] [PubMed] [Google Scholar]
- 264.Laukkanen JA, Laukkanen T. Sauna bathing and systemic inflammation. Eur J Epidemiol 33: 351–353, 2018. doi: 10.1007/s10654-017-0335-y. [DOI] [PubMed] [Google Scholar]
- 265.Oyama JI, Kudo Y, Maeda T, Node K, Makino N. Hyperthermia by bathing in a hot spring improves cardiovascular functions and reduces the production of inflammatory cytokines in patients with chronic heart failure. Heart Vessels 28: 173–178, 2013. doi: 10.1007/s00380-011-0220-7. [DOI] [PubMed] [Google Scholar]
- 266.Omodei D, Licastro D, Salvatore F, Crosby SD, Fontana L. Serum from humans on long-term calorie restriction enhances stress resistance in cell culture. Aging (Albany NY) 5: 599–606, 2013. doi: 10.18632/aging.100584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Soliman S, Aronson WJ, Barnard RJ. Analyzing serum-stimulated prostate cancer cell lines after low-fat, high-fiber diet and exercise intervention. Evid Based Complement Alternat Med 2011: 529053–529057, 2011. doi: 10.1093/ecam/nep031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Tymchuk CN, Barnard RJ, Heber D, Aronson WJ. Evidence of an inhibitory effect of diet and exercise on prostate cancer cell growth. J Urol 166: 1185–1189, 2001. doi: 10.1016/S0022-5347(05)65943-5. [DOI] [PubMed] [Google Scholar]
- 269.Tymchuk CN, Barnard RJ, Ngo TH, Aronson WJ. Role of testosterone, estradiol, and insulin in diet- and exercise-induced reductions in serum-stimulated prostate cancer cell growth in vitro. Nutr Cancer 42: 112–116, 2002. doi: 10.1207/S15327914NC421_15. [DOI] [PubMed] [Google Scholar]
- 270.Weber NC, Riedemann I, Smit KF, Zitta K, van de Vondervoort D, Zuurbier CJ, Hollmann MW, Preckel B, Albrecht M. Plasma from human volunteers subjected to remote ischemic preconditioning protects human endothelial cells from hypoxia–induced cell damage. Basic Res Cardiol 110: 17, 2015. doi: 10.1007/s00395-015-0474-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Zitta K, Meybohm P, Bein B, Heinrich C, Renner J, Cremer J, Steinfath M, Scholz J, Albrecht M. Serum from patients undergoing remote ischemic preconditioning protects cultured human intestinal cells from hypoxia-induced damage: involvement of matrixmetalloproteinase-2 and -9. Mol Med 18: 29–37, 2012. doi: 10.2119/molmed.2011.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Brunt VE, Weidenfeld-Needham KM, Comrada LN, Francisco MA, Eymann TM, Minson CT. Serum from young, sedentary adults who underwent passive heat therapy improves endothelial cell angiogenesis via improved nitric oxide bioavailability. Temperature 6: 169–178, 2019. doi: 10.1080/23328940.2019.1614851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Akasaki Y, Miyata M, Eto H, Shirasawa T, Hamada N, Ikeda Y, Biro S, Otsuji Y, Tei C. Repeated thermal therapy up-regulates endothelial nitric oxide synthase and augments angiogenesis in a mouse model of hindlimb ischemia. Circ J 70: 463–470, 2006. doi: 10.1253/circj.70.463. [DOI] [PubMed] [Google Scholar]
- 274.Bain AR, Ainslie PN, Bammert TD, Hijmans JG, Sekhon M, Hoiland RL, Flück D, Donnelly J, DeSouza CA. Passive heat stress reduces circulating endothelial and platelet microparticles. Exp Physiol 102: 663–669, 2017. doi: 10.1113/EP086336. [DOI] [PubMed] [Google Scholar]
- 275.Coombs GB, Barak OF, Phillips AA, Mijacika T, Sarafis ZK, Lee AH, Squair JW, Bammert TD, DeSouza NM, Gagnon D, Krassioukov AV, Dujic Z, DeSouza CA, Ainslie PN. Acute heat stress reduces biomarkers of endothelial activation but not macro- or microvascular dysfunction in cervical spinal cord injury. Am J Physiol Heart Circ Physiol 316: H722–H733, 2019. doi: 10.1152/ajpheart.00693.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Burger D, Montezano AC, Nishigaki N, He Y, Carter A, Touyz RM. Endothelial microparticle formation by angiotensin II is mediated via Ang II receptor type I/NADPH oxidase/Rho kinase pathways targeted to lipid rafts. Arterioscler Thromb Vasc Biol 31: 1898–1907, 2011. doi: 10.1161/ATVBAHA.110.222703. [DOI] [PubMed] [Google Scholar]
- 277.Melki I, Tessandier N, Zufferey A, Boilard E. Platelet microvesicles in health and disease. Platelets 28: 214–221, 2017. doi: 10.1080/09537104.2016.1265924. [DOI] [PubMed] [Google Scholar]
- 278.Barteneva NS, Fasler-Kan E, Bernimoulin M, Stern JN, Ponomarev ED, Duckett L, Vorobjev IA. Circulating microparticles: square the circle. BMC Cell Biol 14: 23, 2013. doi: 10.1186/1471-2121-14-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Lovren F, Verma S. Evolving role of microparticles in the pathophysiology of endothelial dysfunction. Clin Chem 59: 1166–1174, 2013. doi: 10.1373/clinchem.2012.199711. [DOI] [PubMed] [Google Scholar]
- 280.Horowitz M, Maloyan A, Shlaier J. HSP 70 kDa dynamics in animals undergoing heat stress superimposed on heat acclimation. Ann N Y Acad Sci 813: 617–619, 1997. doi: 10.1111/j.1749-6632.1997.tb51755.x. [DOI] [PubMed] [Google Scholar]
- 281.Beit-Yannai E, Trembovler V, Horowitz M, Lazarovici P, Kohen R, Shohami E. Neuroprotection against oxidative stress by serum from heat acclimated rats. Neurosci Lett 254: 89–92, 1998. doi: 10.1016/S0304-3940(98)00670-3. [DOI] [PubMed] [Google Scholar]
- 282.Assayag M, Gerstenblith G, Stern MD, Horowitz M. Long- but not short-term heat acclimation produces an apoptosis-resistant cardiac phenotype: a lesson from heat stress and ischemic/reperfusion insults. Cell Stress Chaperones 15: 651–664, 2010. doi: 10.1007/s12192-010-0178-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Gaffney DK, Lundquist M, Warters RL, Rosley R. Effects of modifying topoisomerase II levels on cellular recovery from radiation damage. Radiat Res 154: 461–466, 2000. doi: 10.1667/0033-7587(2000)154[0461:EOMTIL]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 284.Kültz D, Chakravarty D. Maintenance of genomic integrity in mammalian kidney cells exposed to hyperosmotic stress. Comp Biochem Physiol A Mol Integr Physiol 130: 421–428, 2001. doi: 10.1016/S1095-6433(01)00440-8. [DOI] [PubMed] [Google Scholar]
- 285.Eynan M, Knubuvetz T, Meiri U, Navon G, Gerstenblith G, Bromberg Z, Hasin Y, Horowitz M. Heat acclimation-induced elevated glycogen, glycolysis, and low thyroxine improve heart ischemic tolerance. J Appl Physiol 93: 2095–2104, 2002. doi: 10.1152/japplphysiol.00304.2002. [DOI] [PubMed] [Google Scholar]
- 286.Maloyan A, Eli-Berchoer L, Semenza GL, Gerstenblith G, Stern MD, Horowitz M. HIF-1alpha-targeted pathways are activated by heat acclimation and contribute to acclimation-ischemic cross-tolerance in the heart. Physiol Genomics 23: 79–88, 2005. doi: 10.1152/physiolgenomics.00279.2004. [DOI] [PubMed] [Google Scholar]
- 287.Treinin M, Shliar J, Jiang H, Powell-Coffman JA, Bromberg Z, Horowitz M. HIF-1 is required for heat acclimation in the nematode Caenorhabditis elegans. Physiol Genomics 14: 17–24, 2003. doi: 10.1152/physiolgenomics.00179.2002. [DOI] [PubMed] [Google Scholar]