

Abstract
The TGF-β signaling pathway plays a pivotal role in controlling organogenesis during fetal development. Although the role of TGF-β signaling in promoting lung alveolar epithelial growth has been determined, mesenchymal TGF-β signaling in regulating lung development has not been studied in vivo due to a lack of genetic tools for specifically manipulating gene expression in lung mesenchymal cells. Therefore, the integral roles of TGF-β signaling in regulating lung development and congenital lung diseases are not completely understood. Using a Tbx4 lung enhancer-driven Tet-On inducible Cre transgenic mouse system, we have developed a mouse model in which lung mesenchyme-specific deletion of TGF-β receptor 2 gene (Tgfbr2) is achieved. Reduced airway branching accompanied by defective airway smooth muscle growth and later peripheral cystic lesions occurred when lung mesenchymal Tgfbr2 was deleted from embryonic day 13.5 to 15.5, resulting in postnatal death due to respiratory insufficiency. Although cell proliferation in both lung epithelium and mesenchyme was reduced, epithelial differentiation was not significantly affected. Tgfbr2 downstream Smad-independent ERK1/2 may mediate these mesenchymal effects of TGF-β signaling through the GSK3β-β-catenin-Wnt canonical pathway in fetal mouse lung. Our study suggests that Tgfbr2-mediated TGF-β signaling in prenatal lung mesenchyme is essential for lung development and maturation, and defective TGF-β signaling in lung mesenchyme may be related to abnormal airway branching morphogenesis and congenital airway cystic lesions.
Keywords: airway smooth muscle cells, congenital airway cysts, lung development, lung mesenchyme, TGF-β signaling
INTRODUCTION
Prenatal lung development is a complex process. Disruption of lung growth can result in neonatal respiratory diseases and even death. In mice, lung morphogenesis begins around embryonic day (E) 9.5 when the progenitor cells from the anterior foregut endoderm grow into the splanchnic mesoderm to form the primordial of trachea and lung buds (1). Branching morphogenesis then gives rise to the bronchial trees for air conduction, which is followed by peripheral alveolar sac growth and maturation for gas exchange after birth (2). The mouse lung developmental process can be divided into five stages based on their morphology: embryonic (<E10.5), pseudoglandular (E10.5–E15.5), canalicular (E15.5–E16.5), saccular [E16.5 to postnatal day (P) 5], and alveolar stages (P5–P30) (1). The reciprocal interaction between the epithelium and mesenchyme plays a key role in controlling lung morphogenesis and maturation. Mesenchyme-derived cell lineages not only provide structural support, but also regulate growth of epithelial cells through multiple mechanisms, such as paracrine signaling (3, 4).
The transforming growth factor-β (TGF-β) pathway is an evolutionarily conserved cellular signaling system that plays an essential role in a variety of biological processes, such as cell growth, differentiation, and apoptosis (5). Abnormal TGF-β signaling is implicated in a variety of lung diseases including lung fibrosis, emphysema, and cancer (5). Active TGF-β ligands bind to a heteromeric complex of transmembrane type 1 and type 2 receptors (TGFBR1 and TGFBR2) and initiate the intracellular signaling. Both Smad2/3-dependent and Smad-independent pathways are found to mediate TGF-β signaling by modulating its downstream target gene expression. Mitogen-activated protein kinase (MAPK including ERK1/2, p38, and JNK), phosphatidylinositol-3-kinase (PI3K), and Wnt/β-catenin pathways are involved in Smad-independent TGF-β signaling (6).
Our previous studies show that abrogation of Tgfbr2 specifically in lung epithelium leads to disruption of postnatal alveolarization with a marked decrease in type I alveolar epithelial cells even though no abnormalities are observed in prenatal lung development. However, deletion of Tgfbr2 in mesoderm-derived mesenchymal cells of whole body using a Dermo1-Cre driver results in embryonic lethality around E15.5 due to multiple severe developmental defects including defective thoracic wall formation, congenital diaphragmatic hernia, and abnormal cardiac development (7). The roles of mesenchymal Tgfbr2-mediated TGF-β signaling in lung development could not be fully and specifically evaluated using this model. We have recently developed an inducible Cre expression transgenic mouse line, in which a Tbx4 lung enhancer drives rtTA expression in lung mesenchymal cells. In the presence of doxycycline, the rtTA-doxycycline complex induces TetOn-controlled Cre expression in developing lung mesenchymal cells specifically (8). Herein, we use this lung mesenchyme-specific Cre driver to study the roles of lung mesenchyme-specific Tgfbr2 signaling in lung development and pathogenesis of congenital lung diseases.
MATERIALS AND METHODS
Mice
All mice used in this study have C57Bl/6 strain background. Floxed-Tgfbr2 (Tgfbr2fx/fx) mice, in which exon 2 of the Tgfbr2 gene is flanked by loxP DNA elements, have been described in previous publications (7, 9). An inducible lung mesenchyme-specific Cre transgenic system (Tbx4-rtTA/TetO-Cre) was generated in our laboratory (8). Lung mesenchyme-specific Tgfbr2 conditional knockout mice (CKO) (Tgfbr2fx/fx/Tbx4-rtTA/TetO-Cre) were generated by crossing Tgfbr2fx/fx and Tgfbr2fx/+/Tbx4-rtTA/TetO-Cre mice, with doxycycline induction (625 mg/kg in food, TestDiet and 0.5 mg/mL in drinking water, Sigma) at different developmental stages. The genotypes of the transgenic mice were determined by genomic DNA PCR using tail tip biopsy. The primers used for identifying the floxed-Tgfbr2 allele (mScar and mEIIF) and its deletion (Tgfb2-E1 and Tgfb2-E3), TetO-Cre (Teto-1 and Cre3), and Tbx4-rtTA (rtTA-M2-3 and rtTA-M2-4) transgenes are listed in Supplemental Table S1 (see https://doi.org/10.6084/m9.figshare.13708753). Deletion of Tgfbr2 in lung tissue was verified at the mRNA and protein levels by RT-PCR and immunoblotting. Mice used in the study were housed in pathogen-free conditions, and all experimental procedures were approved by the Institutional Animal Care and Use Committee at Children’s Hospital Los Angeles.
Tissue RNA Isolation, RNA-Seq, and Real-Time PCR
E15.5 mouse lungs were dissected in RNase-free PBS and flash-frozen in liquid nitrogen. For next generation RNA-seq, total RNAs were extracted from lung tissues using the TRIzol RNA extraction reagent (Thermo Fisher Scientific, No. 15596026), and were further purified using the RNeasy Micro Kit (Qiagen, No. 74004). The RNA sample integrity was assayed using an Agilent Bioanalyzer, and the samples with RNA integrity numbers over 7.0 were used. Sequencing libraries were constructed with the addition of a common set of external RNA controls (ERCC RNA Spike-in mix, Thermo Fisher Scientific) using the Illumina TruSeq-V2 kits, and sequenced to roughly 50 million reads at 100 bp single-end by the Sequencing Core Facility at the University of Southern California. The sequences obtained were assayed for quality using the FastQC program (10), and the adapter sequences were removed using Cutadapt Python software (11). The sequences were then trimmed for quality using Trimmomatic software (12). The remaining high-quality sequences were aligned to the Gencode 23 mouse genome (GRCm38.p6) using the STAR aligner (13). Counts of sequences per gene were produced using the HTSeq-count Python software package (14). Differential gene expression between conditions was determined using the R/Bioconductor software package “edgeR” (15). Genes were considered statistically significant if they had a false discovery rate adjusted P value less than 0.05. Graphics were produced from the data using the R software “ggplot2” (16). The sequencing data was deposited to the NCBI GEO repository with accession number GSE98138.
Real-time PCR was performed using the SYBR Green Q-PCR kit on an iCycler-iQ system (Bio-Rad) as described in our previous publication (17). The relative expression of the target gene was calculated by ΔΔCT (18). The sequences of the oligonucleotide primers used are listed in Supplemental Table S1.
Histology and Immunofluorescence Staining
For histological evaluation, fetal lungs were isolated and fixed in 4% paraformaldehyde at room temperature. Tissue paraffin sections (5-μm thickness) of the left lobes of the lung samples were chosen at random and stained with hematoxylin & eosin (H&E). Immunofluorescence staining was performed following the methods published previously (19). The antibodies that were used are described in Supplemental Table S2 (see https://doi.org/10.6084/m9.figshare.13708753). The fluorescent-dye (Alexa Fluor 488, 594, or 647) conjugated secondary antibodies were purchased from Life Technologies. Fluorescence images were taken using the Zeiss LSM710 confocal microscope at the Imaging Core Facility of Children’s Hospital Los Angeles.
Cell Proliferation Assay
Timed-pregnant mice were intraperitoneally injected with a thymidine analog, 5′-ethynyl-2′-deoxyuridine (EdU, 5 mg/kg, Life Technologies) 2 h before lung specimens were harvested. Incorporated EdU was detected using Alexa Fluor 488 azide (Life Technologies), and cell nuclei were counterstained with DAPI. The ratios of EdU-labeled nuclei to total nuclei (n > 300 per field for mesenchymal cells and n > 50 per field for epithelial cells) were calculated and compared, and the independent experiments were repeated more than three times.
Protein Analysis
The expression of specific proteins in lung tissue lysate was detected by Western blot as previously described (20). Equal amounts (30 μg) of tissue lysate proteins were loaded and separated in precast SDS-PAGE gels (4%–15% gradient; Bio-Rad), and transferred to polyvinylidene difluoride membranes using Bio-Rad’s Trans-Blot Turbo System. Proteins of interest were detected using the specific primary antibodies listed in Supplemental Table S2. After washing off any unbound antibody, the membranes were incubated with horseradish peroxidase-conjugated secondary antibody, and the antibody-detected protein bands were visualized by enhanced chemiluminescence reagent (Amersham). The whole blot images are included in the Supplemental Materials (see https://doi.org/10.6084/m9.figshare.13708753).
Fetal Lung Mesenchymal Progenitor Cell Isolation, Culture, and Treatment
Lung mesenchymal progenitor cells were isolated from E15.5 lung, when significant morphological changes were just detected in Tgfbr2 CKO mice. Briefly, single-cell suspensions of lung tissue were prepared from E15.5 mice by digesting minced lung tissue with 0.2% collagenase I and 2.4 U/mL dispase II (Sigma) for 1 h at 37°C (21). After being passed through a 70-μm strainer, 1 × 105 cells were seeded onto a 100-mm plastic dish and cultured in α-MEM medium containing 20% FBS and 0.1 mM β-mercaptoethanol (β-ME). Cell colonies were formed in ∼2 wk (passage 0), and then were passed to fresh culture dishes (passage 1). The cells of passage 2, which were used in the study, were negative for expression of both epithelial (CD326) and endothelial (CD31) markers.
All cells were cultured in α-MEM medium containing 20% FBS during experiments to maintain cell growth and stemness. To determine their response to TGF-β, the cells were treated with mouse TGF-β1 (5 ng/mL) added to the aforementioned culture medium for 1 h before cell lysate preparation. To study the relationship of the cellular signaling pathways, the primary lung mesenchymal cells were pretreated with either a MAPK/ERK pathway inhibitor (20 μM U0126, Sigma) or a GSK3β inhibitor (20 μM CHIR99021, Sigma) for the indicated times. The controls were cells treated with DMSO vehicle. The cell lysates were analyzed for total and phosphorylated ERK1/2 (pERK), total and phosphorylated GSK3β protein [pGSK3β (S9) or pGSK3β (Y216)], total and nonphosphorylated β-catenin (active β-catenin). Protein band intensities were measured using ImageJ Data Acquisition Software (NIH), and phosphorylation-signal intensities were normalized to the corresponding total protein signal intensities.
Statistical Analysis
Means ± SE was used for quantitative presentation. At least six mice were included in each experimental group. All experiments were repeated at least three times, and the data represent consistent results. The statistical difference between two independent groups was assessed by two-sided Student’s t test. One-way analysis of variance (ANOVA) was used to examine differences among groups of three or more. P < 0.05 was considered statistically significant.
RESULTS
Deletion of Tgfbr2 in Embryonic Lung Mesenchyme Caused Abnormal Lung Morphogenesis and Neonatal Lethality
Lung mesenchyme-specific Tgfbr2 conditional knockout (CKO) mice were generated by crossing Tbx4-rtTA/TetO-Cre driver line with floxed-Tgfbr2 mice, with doxycycline induction from embryonic day (E) 6.5 to postnatal day (P) 1. Deletion of Tgfbr2 in fetal lung tissue was verified at the mRNA and protein levels (Supplemental Fig. S1; see https://doi.org/10.6084/m9.figshare.12543164). These Tgfbr2 CKO mice had normal body sizes at birth (Fig. 1A). However, they rapidly developed signs of respiratory distress, with gasping, retractions, and cyanosis (Fig. 1A), and died within a day. To determine the underlying cause of the neonatal respiratory failure in these mice, mouse lungs at different developmental stages from E12.5 to P1 were examined. Although no significant change in overall lung size was observed by gross view (Fig. 1B), the airway branching process was negatively impacted as early as E14 in Tgfbr2 CKO mice. This is shown by the differential distribution of branching tips in whole-mount airway epithelial staining (Fig. 1C) and by a reduction in the number of lung terminal branches in Tgfbr2 CKO lungs (Fig. 1D). Interestingly, asymmetric enlargement of the distal airways and peripheral airway cysts were found in Tgfbr2 CKO lungs at the late gestation stage (∼E18.5, Fig. 1B). The peripheral cystic lesions were further exacerbated in neonates upon respiration. Although the cysts were adjacent to the saccular structures in the peripheral part of the lung, the severity of the cystic lesions varied, which was correlated with neonatal respiratory distress and death (Fig. 1B).
Figure 1.

Abnormal lung development in mesenchyme-specific transforming growth factor-β (TGF-β) receptor 2 gene (Tgfbr2) conditional knockout (CKO) mice. A: body sizes of newborn Tgfbr2 CKO and wild-type (WT) mice are comparable. Bluish discoloration was apparent in Tgfbr2 CKO mice. B: lungs isolated from Tgfbr2 CKO and WT fetuses and neonates are compared side by side under dissecting microscope. Scale bar: 1 mm. C: whole mount anti-Cdh1 immunofluorescence staining of embryonic day (E) 14 lungs highlights airway epithelial branching structures. Different branching patterns between Tgfbr2 CKO and WT are marked by arrows. Scale bar: 0.4 mm. D: the numbers of left lung terminal branches between E14 Tgfbr2 CKO and WT lungs are compared. *P < 0.05 (n = 6, no. of mice).
The abnormal lung development in mesenchymal-specific Tgfbr2 CKO mice was further examined in H&E stained lung tissue sections under a microscope (Fig. 2A). A decrease in the number of fetal airway epithelial tubes was easily detected in E15.5 Tgfbr2 CKO lung tissue sections, which also had a reduction in mesenchymal cell density compared with the wild-type (WT) littermate controls (Fig. 2B). Although a few dilated airways in Tgfbr2 CKO lungs were seen at late gestation (E17.5–E18.5), a significant reduction of terminal air sac area was detected (Fig. 2C), suggesting that formation of primary air sacs was disrupted in the absence of mesenchymal TGF-β signaling. Lastly, large cysts with partial collapse of the lung parenchymal tissues were detected in the peripheral lungs of neonatal mice (Fig. 2, A and D). Rupturing of saccular wall structures was sometimes seen in relatively large cysts, and the epithelial cells lining the cysts were heterogeneous, ranging from columnar/cuboidal cells to flat cells depending on their relative positions to the airways (Fig. 2D).
Figure 2.

Histopathology of transforming growth factor-β (TGF-β) receptor 2 gene (Tgfbr2) conditional knockout (CKO) mouse lungs. A: the hematoxylin & eosin (H&E)-stained lung tissue sections at different developmental stages are compared between Tgfbr2 CKO and wild-type mice. B: mesenchymal cell densities between embryonic day (E) 15.5 Tgfbr2 CKO and wild-type (WT) lung tissue sections are compared. Cell nuclei were detected by DAPI staining and counted in nonepithelial area. *P < 0.05 (n = 6, no. of mice). C: the peripheral (saccular) air space at E18.5, presented as the percentage of entire tissue area, was compared between Tgfbr2 CKO and WT lung tissues. *P < 0.05 (n = 6, no. of mice). D: a H&E-stained cystic structure in postnatal day (P) 1 lung of the Tgfbr2 CKO mouse, which is lined with different epithelial cells ranging from cuboidal (arrow) to flat shapes (arrowhead).
Fetal Lung Mesenchymal TGF-β Signaling from E13.5 to E15.5 Is Critical in Controlling Airway Growth and Preventing Peripheral Cystic Lesions in Fetuses
To understand the time window during which mesenchymal TGF-β signaling is critical for lung development, we generated mesenchymal Tgfbr2 CKO mice from different gestation stages by taking advantage of our Tbx4-rtTA/TetO-Cre inducible driver with doxycycline induction starting from E13.5, E14.5, and E15.5 instead of E6.5. Lungs were harvested and analyzed at E18.5. Interestingly, mesenchymal deletion of Tgfbr2 starting from E13.5 and E14.5 still resulted in peripheral cystic lesions as confirmed by both gross view and histological examination (Fig. 3). In contrast, Tgfbr2 knockout in lung mesenchymal cells starting after E15.5 did not cause detectable defects in air sac formation and pulmonary cysts (Fig. 3), suggesting that before E15.5, lung mesenchymal TGF-β signaling is important for appropriate fetal lung development.
Figure 3.

Abnormal lung morphology in embryonic day (E) 18.5 transforming growth factor-β (TGF-β) receptor 2 gene (Tgfbr2) conditional knockout (CKO) lungs that were induced from different developmental stages. Doxycycline induction was given at different time windows as indicated. Alterations of lung morphology are shown by their gross view (A) and their histopathology (B). Arrows point to the cystic lesions. WT, wild type.
Abrogation of Lung Mesenchymal Tgfbr2-Mediated TGF-β Signaling Disrupted Proliferation and Differentiation of Both Lung Mesenchymal and Epithelial Cells
To understand the morphological abnormalities in Tgfbr2 CKO lungs, we then investigated the changes happening at the cellular level, including proliferation and differentiation. Using the EdU incorporation assay, cell proliferation was measured at E15.5 when reduced airway branching was just detectable in Tgfbr2 CKO fetuses. Compared with their wild-type littermate controls, Tgfbr2 CKO lungs had significantly reduced cell proliferation not only in lung mesenchymal cells but also in lung epithelial cells (Fig. 4, A and B), suggesting that both direct and indirect mechanisms of mesenchymal TGF-β signaling are involved in controlling overall lung cell proliferation.
Figure 4.

Blockade of transforming growth factor-β (TGF-β) receptor 2 (Tgfbr2)-mediated signaling in fetal lung mesenchymal cells resulted in altered cell proliferation and differentiation of fetal lungs. 5′-Ethynyl-2′-deoxyuridine (EdU) labeling of proliferating cells by their active DNA synthesis was detected by Alexa Fluor azide staining (red) of the lung tissue sections (A) and compared between Tgfbr2 conditional knockout (CKO) and wild-type controls (B), *P < 0.05. Epithelial cells were marked by Cdh1 immunostaining (green) and cell nuclei are counterstained with DAPI (blue). C and D: transcriptomic changes between embryonic day (E) 15.5 Tgfbr2 CKO lungs and WT controls were measured by RNA-seq. The raw data were deposited to NCBI GEO repository with the accession number GSE98138. C: gene network analysis of statistically significant genes (log2FC ≥ 1, P < 0.05) was performed using QIAGEN Ingenuity Pathway Analysis, and the information was processed using Cytoscape 3.7.1. The nodes were colored based on their P values. *Myocd gene. D: significant changes of a group of muscle cell-associated genes (red dots) and Klf4 (green dot, a transcriptional repressor for smooth muscle differentiation) were identified. E: real-time PCR to validate the changes of three smooth muscle-related genes. *P < 0.05. F: three-dimensional (3-D) airway smooth muscle structures of E14.5 left lungs with indicated genotypes were visualized by Acta2 whole mount immunostaining (red). The airway epithelia were counterstained with Cdh1 (green). G: smooth muscle cells in E15.5 lung tissue sections were also detected by Acta2 immunostaining (green), and pulmonary vasculatures were identified by their endothelial Pecam1 staining (red, arrows). H: differentiation of airway epithelial cells (including Scgb1a1-positive Club cells and Tubb4a-positive ciliated cells) and peripheral epithelial cells (including Pdpn-positive type 1 and Sftpc-positive type 2 alveolar epithelial cells) were detected in E18.5 lungs. Cell nuclei in G and H were counterstained with DAPI (blue).
We then compared the alteration of tissue transcriptomic profiles between E15.5 Tgfbr2 CKO lungs and their wild-type littermate controls by RNA-seq. Interestingly, one of the domains in the network of significantly changed genes is centered around Myocd, a key cotranscriptional activator for smooth muscle differentiation (Fig. 4C). Altered expression of a group of genes associated with muscle cell development was also detected (Fig. 4D and Supplemental Fig. S2; see https://doi.org/10.6084/m9.figshare.12543188). Changes in some of the gene expression, such as Myocd, Myh11, and Acta2, were further verified by real-time PCR (Fig. 4E). To determine the impacts of these alterations in gene expression, we examined smooth muscle cell differentiation in lung tissues using three-dimensional (3-D) whole mount tissue and two-dimensional (2-D) tissue section immunostaining (Fig. 4, F and G), we found a significant reduction of smooth muscle cells in airways, but not in pulmonary vasculatures marked by adjacent Pecam1+ vascular endothelia, in Tgfbr2 CKO lungs. This suggests that mesenchymal Tgfbr2-mediated signaling plays an important role in promoting airway smooth muscle cell growth.
Furthermore, the indirect impact on lung epithelial cell differentiation caused by mesenchymal Tgfbr2 deletion was also evaluated by co-immunostaining the molecular markers of differentiated epithelial cells, including airway secretory Club cells (Scgb1a1), airway ciliated cells (Tubb4a), and peripheral air sac epithelial cells (Sftpc for type 2 alveolar epithelial cells and Pdpn for type 1 alveolar epithelial cells). Interestingly, deletion of mesenchymal Tgfbr2 did not lead to significant changes in airway epithelial cells as seen at E18.5 (Fig. 4H). Similarly, differentiation of existing peripheral epithelial cells (Sftpc or Pdpn-positive cells) was not significantly affected although the total number of the peripheral epithelial cells was decreased due to reductions in saccular number and size. This suggests that abrogation of mesenchymal Tgfbr2-mediated signaling did not alter lung epithelial cell differentiation even though proliferation was negatively impacted.
Tgfbr2 CKO Lung Tissues Showed Reduced Activation of Both the MAPK/ERK and Wnt/β-Catenin Pathways
To understand the potential mechanisms underlying the abnormal changes in the developing lungs, particularly the cystic lesions occurring around E18.5, alterations of TGF-β and other related growth factor signaling pathways were then investigated. Interestingly, TGF-β-mediated Smad2 signaling was not significantly altered in E18.5 whole lung tissue lysate of Tgfbr2 CKO mice, whereas activation of the MAPK/ERK pathway was significantly reduced, as shown by a reduction in the phosphorylation of these proteins. It is well known that the MAPK/ERK pathway can be activated by TGF-β in a Smad-independent way (6). Furthermore, the canonical Wnt pathway was downregulated, as measured by decreased levels of active (unphosphorylated) β-catenin, reduced expression of the Wnt target gene Axin2, and reduced amounts of inactive GSK3β (phosphorylation at Ser9). Decreased Wnt pathway activation, shown by less nuclear localization of active β-catenin using immunofluorescence staining, was also seen in both lung epithelial and mesenchymal cells of E18.5 Tgfbr2 CKO mice (Fig. 5B).
Figure 5.
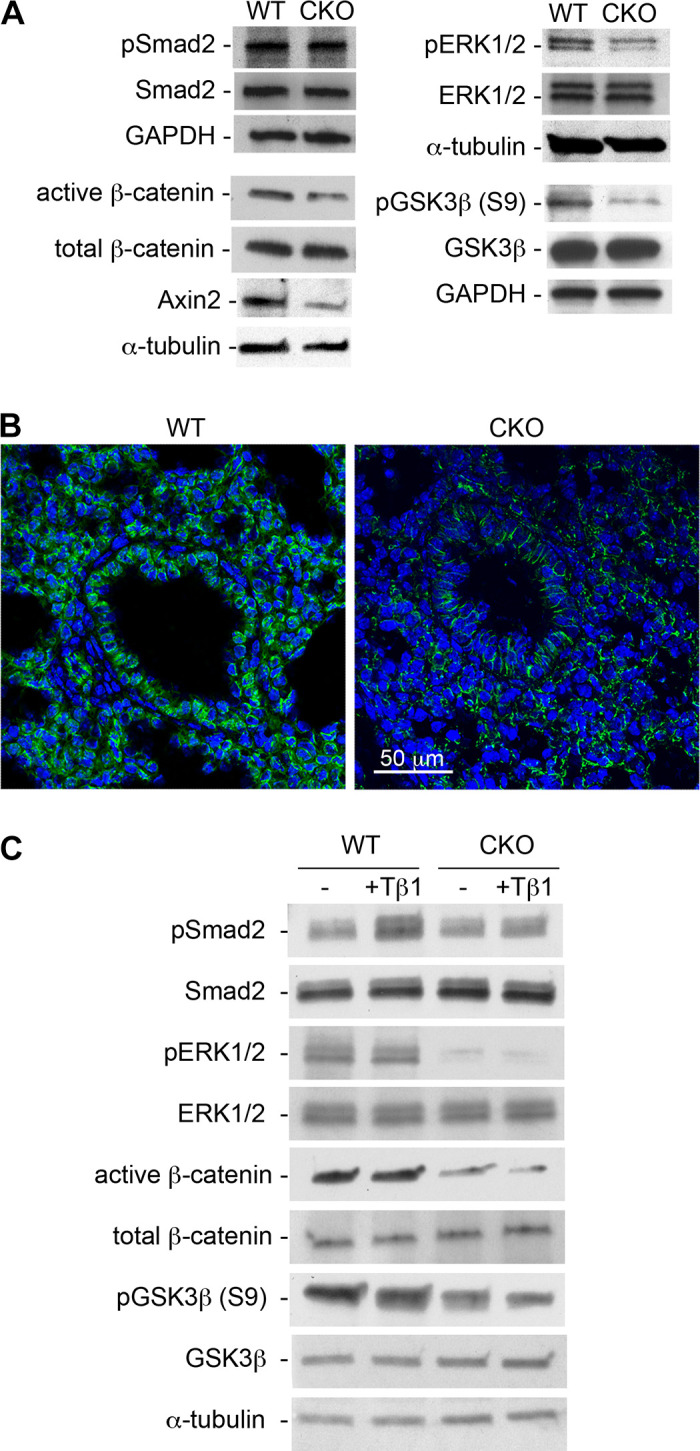
Alterations of growth factor signaling in transforming growth factor-β (TGF-β) receptor 2 gene (Tgfbr2) conditional knockout (CKO) lung tissues and cells. A: embryonic day (E) 18.5 wild-type (WT) and Tgfbr2 CKO lung tissue lysates were analyzed by immunoblot. GAPDH and α-tubulin were used as loading controls. B: active β-catenin (green) in lung tissues, as detected by immunofluorescence staining, was compared between E18.5 WT and Tgfbr2 CKO mice. Cell nuclei were counterstained with DAPI (blue). C: altered signaling activities in cultured fetal lung mesenchymal cells, which were isolated from WT and Tgfbr2 CKO lungs, were measured by immunoblot. The cells were cultured with the medium containing 20% FBS and treated with additional transforming growth factor-β1 (TGF-β1, 5 ng/mL) or vehicle control for 1 h prior to sample collection. Representative data of three independent experiments are shown.
Since lung tissue lysate consists of proteins from a variety of cell types including epithelial and mesenchymal cells, we then examined the aforementioned pathways in primary lung mesenchymal progenitor cells isolated from the fetal mouse lungs of different genotypes. To maintain the cell growth and stemness, medium containing 20% FBS was used for the cell culture throughout the experiment. The endogenous level of Smad2 activation (pSmad2) was comparable between the Tgfbr2 CKO cells and the wild-type controls (Fig. 5C). However, the Tgfbr2 CKO cells could not respond to exogenous TGF-β1 (5 ng/mL) stimulation with Smad2 activation, whereas the active pSmad2 level was drastically increased in TGF-β1-treated wild-type cells. In Smad-independent pathways, activation of ERK1/2 (pERK1/2) and β-catenin (active β-catenin) was reduced in the Tgfbr2 CKO cells (Fig. 5C), accompanied by increased activation of GSK3β as shown by a reduction in the inactive form of pGSK3β (S9). This is consistent with the earlier results from the lung tissue study. Further changes of pERK1/2, active β-catenin, and pGSK3β (S9) in response to exogenous TGF-β1 were not detected, possibly due to their high endogenous levels in 20% FBS cell culture medium.
ERK1/2 May Regulate the Wnt/β-Catenin Pathway through GSK3β in Developing Lung Mesenchymal Cells
As mentioned earlier, the ERK1/2 pathway can serve as a Smad-independent signaling pathway for mediating TGF-β regulatory effects. We then explored the relationship between ERK1/2 and Wnt signaling in cultured wild-type fetal lung mesenchymal progenitor cells as utilized above in Fig. 5C. First, we determined the consequential relationship between the ERK1/2 and Wnt pathways by treating cells with an ERK1/2-specific inhibitor U0126 (20 µM) for different durations. Significant inhibition of ERK1/2 phosphorylation (activation) was detected in the cells particularly within 1-h treatment, accompanied by reductions in inactive GSK3β [pGSK3β (S9)] and active β-catenin (Fig. 6, A and B). This suggests that reduction of ERK1/2 activation may lead to increased activation of GSK3β, resulting in hyperphosphorylation (inactivation) and degradation of β-catenin.
Figure 6.

ERK1/2 and GSKβ regulate Wnt canonical signaling in fetal lung mesenchymal cells. A: alterations of signal activation in primary fetal lung mesenchymal cells that were treated with ERK1/2 inhibitor U0126 (20 µM). Total cell lysates were analyzed by Western blot. α-Tubulin was used as a loading control. B: the relative intensities of the pERK1/2, pGSK3β(S9), and active β-catenin of the immunoblots in A were quantified using ImageJ and normalized with the total ERK1/2, GSK3β, and total β-catenin. C: Alterations of signal activation in primary fetal lung mesenchymal cells that were treated with GSK3β inhibitor CHIR99021 (20 μM). Total cell lysates were immunoblotted with antibodies as indicated. D: The relative intensities of the pERK1/2, pGSK3β(Y216), and active β-catenin of the immunoblots in C were quantified as described above. Compared with the untreated cells, changes of the band intensities above are all significant (P < 0.05, n = 3, no. of mice).
To further test whether altered GSK3β activity occurs immediately upstream of β-catenin in lung mesenchymal cells, we then treated the cells with a GSK3β-specific inhibitor CHIR99021 (20 µM). Inhibition of GSK3β was confirmed by the reduction of active form of GSK3β [pGSK3β (Y216)]. As expected, active β-catenin was increased, indicating elevated Wnt signaling pathway even though ERK1/2 phosphorylation was increasing under GSK3β inhibition (Fig. 6, C and D). The combined data suggest that there may be an ERK1/2-GSK3β-Wnt/β-catenin regulatory loop in fetal lung mesenchymal cells.
DISCUSSION
Tgfbr2-mediated TGF-β signaling plays a critical role in early embryonic and organ development. Tgfbr2 conventional knockout and Tgfbr2 pan-mesenchymal knockout results in early or mid-embryonic death, which cannot be used to study the role of lung mesenchymal TGF-β signaling in regulating fetal and neonatal lung development (7, 22). By using a lung mesenchyme-specific Tbx4-rtTA/TetO-Cre inducible driver line, we were able to delete Tgfbr2 specifically in mouse lung mesenchymal cells at different developmental stages. We found that lung mesenchymal Tgfbr2-mediated TGF-β signaling from E13.5 to E15.5 is indispensable to normal lung formation, and that abrogation of the mesenchymal TGF-β pathway results in not only hypoplastic malformation of neonatal lung, but also formation of congenital pulmonary cysts, leading to neonatal respiratory failure and death. In contrast, deletion of Tgfbr2 in fetal mouse lung epithelial cells using a Sftpc-rtTA/TetO-Cre driver does not cause significant lung malformation at birth (7). As shown in our previous publication (8), the Tbx4 lung mesenchymal enhancer is active in all mesenchymal progenitor cells of early embryonic lung, and then becomes inactive in a few committed cell lineages, such as in endothelial cells and perivascular smooth muscle cells after E11.5 and E13.5, respectively. Therefore, it is possible that the phenotypic impacts of Tgfbr2 deletion at different developmental windows may be caused by a blockade of Tgfbr2 in different compositions of mesenchymal cell types. However, airway-specific smooth muscle hyperplasia has been reported in fetal mice with global retinoid deficiency (23), and the airway-specific effect of retinoic acid is mediated through downregulation of TGF-β pathway (24). This suggests that interconnected retinoic acid and TGF-β pathways may specifically regulate airway smooth muscle cell growth during fetal lung development.
Abnormal lung development is generally caused by altered airway branching morphogenesis and/or alveolar growth, eventually affecting air conduction and/or gas exchange (2). Our study provides direct evidence that reduction of TGF-β signaling in lung mesenchymal cells could be one of the mechanisms related to prenatal lung malformation. The cell proliferation is reduced in both mesenchymal and epithelial compartments of the mesenchyme-specific Tgfbr2 knockout lungs, suggesting that a complicated mesenchymal-epithelial cross-talking mechanism underlies the pulmonary phenotypes. Future studies to elucidate the cross-talking mechanism, including paracrine signaling, cell contacts, and extracellular matrix proteins, will be very important.
In addition to the retardation of lung development mentioned above in the discussion, simultaneous development of fetal lung cysts accompanied by a defect in airway-specific smooth muscle cell growth in the mesenchymal Tgfbr2 conditional knockout mice is a novel finding. In humans, prenatal lung cystic pathology is referred to as congenital pulmonary airway malformation (CPAM), a relatively common congenital lesion (25, 26). About 25% of neonates with CPAM present with respiratory distress, infection, and pneumothorax, and the perinatal mortality of CPAM ranges from 9% to as high as 49% (27, 28). The pathology of CPAM varies depending on the timing and location of cyst formation. The peripherally located cystic lesions in our Tgfbr2 CKO mouse lungs occurred at a late gestation stage, suggesting that the lesions may originate from distal bronchiolar-saccular structures. Abnormal airway branching morphogenesis is considered as a mechanism of CPAM (29), and defects of airway smooth muscles are seen in human CPAM lungs (25). However, the pathogenic mechanisms of CPAM remain largely unknown. Different potential factors responsible for CPAM pathology are suggested by various studies (30). Using in vitro embryonic lung organ cultures, previous studies suggest that airway smooth muscle cells likely are important to embryonic lung morphogenesis by directing terminal epithelial bifurcation with physically wrapping the airway tips (31), and by maintaining positive intraluminal pressure with spontaneous peristaltic contractions (2). However, a recent in vivo study of ours suggests that sole defect of fetal mouse lung airway smooth muscle cells is not sufficient to affect the airway branching process (32). Thus, additional or multiple mechanisms are likely involved in the development of fetal airway cystic lesions in our mesenchymal Tgfbr2 conditional knockout mouse lungs. Nevertheless, our current findings suggest that altered mesenchymal TGF-β signaling may be one of the molecular mechanisms contributing to CPAM.
TGF-β activates its cellular signaling upon binding to its receptor Tgfbr1/Tgfbr2 complex, and Tgfbr1 and Tgfbr2 are expressed in both mesenchymal and epithelial cells of mouse embryonic lungs (7, 33). The intracellular TGF-β signaling can be divided into Smad-dependent and Smad-independent pathways. We have examined the signaling alterations downstream of Tgfbr2 in our mouse model. Surprisingly, no reduction in Smad2 phosphorylation was detected in the Tgfbr2 mutant lung lysate, although this does not exclude the possibility that the Smad-dependent pathway is involved in regulating lung mesenchymal development. Previous publications from us and others have reported that Smad3 conventional knockout does not result in abnormal development of prenatal lung (20, 34), suggesting that Smad3 may not play an important role in fetal lung formation. Recently, we also specifically deleted Smad2 in lung mesenchymal cells using the same Tbx4-driven Cre/lox genetic manipulation system, and no abnormalities were detected in the mesenchymal Smad2 knockout lungs up to 1 mo of age (data not included). Although this does not fully rule out the possibility that Smad2 and Smad3 complement each other in developing lung mesenchymal signaling, it does suggest that the mesenchymal Smad-independent pathway might be more important to lung organogenesis. Activation of TGF-β receptors (Tgfbr2 and Tgfbr1) is capable of recruiting the adaptor proteins Grb2/SOS and sequentially activating Ras, Raf, and ERK1/2, which further phosphorylate downstream transcription factors to regulate gene expression with or without activated Smad complexes (6). Inappropriate activation of ERK1/2-mediated TGF-β signaling has been found to be critical in several diseases. For example, abnormal activation of ERK1/2 is detected in skin samples of human systemic sclerosis, and inhibition of either TGF-β receptors or ERK1/2 suppresses the contractile activity of the skin fibroblasts (35). Upregulation of the ERK1/2 and TGF-β pathway is also implicated in kidney fibrosis (36). In our mesenchyme-specific Tgfbr2 knockout lungs, reduction of ERK1/2 phosphorylation is accompanied by decreased activation of the Wnt canonical pathway, as shown by a reduction in active β-catenin and Wnt target gene Axin2 expression. β-Catenin is negatively regulated by GSK-3β-mediated phosphorylation, whereas GSK-3β activity is controlled by protein phosphorylation, localization, and sequestration by a number of GSK-3-binding proteins (37). Phosphorylation of Ser 9 in GSK-3β results in the generation of a prephosphorylated substrate or pseudo-substrate on its N-terminal tail, which acts as a competitive inhibitor for primed substrates. On the other hand, the activity of GSK-3β is positively regulated by phosphorylation on a “T-loop” tyrosine residue (Tyr276/Tyr216). The phosphorylated Tyr 216 undergoes a conformational change that allows substrates to bind the enzyme (38). A variety of protein kinases including ERK1/2 are capable of phosphorylating GSK-3β at Ser 9 and inhibiting its activity (39). This subsequent relationship among ERK1/2, GSK3β, and β-catenin was tested in our lung mesenchymal cell culture by comparing the effects of selectively blocking ERK1/2 versus GSK-3β activity. By combining our data with the published observations described above in the discussion, we hypothesize that in developing fetal lung mesenchymal cells, blockade of Tgfbr2-mediated signaling results in increased GSK3β activation by reducing ERK1/2-mediated Ser9 phosphorylation of GSK-3β, and subsequent inactivation and degradation of β-catenin. However, alterations in other signal pathways that may cross-talk to TGF-β could not be ruled out as potential mechanisms underlying the abnormal lung phenotypes in Tgfbr2 CKO mice.
In summary, using a lung mesenchyme-specific Cre mouse line, we were able to discover novel roles of Tgfbr2-mediated signaling in regulating fetal lung development, particularly in fetal lung mesenchymal cells.
GRANTS
This work was partially supported by National Institutes of Health (NIH)/National Institute of Child Health and Human Development Grant HD090309 (to W.S.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Q.M., M.K., and W.S. conceived and designed research; Q.M., H.C., Y.L., J.C., and M.E.T. performed experiments; Q.M., H.C., Y.L., L.C., M.E.T., B.H.G., J.L., and W.S. analyzed data; Q.M., Y.L., M.E.T., B.H.G., M.K., J.L., and W.S. interpreted results of experiments; Q.M., Y.L., M.E.T., and W.S. prepared figures; Q.M., M.E.T., and W.S. drafted manuscript; Q.M., Y.L., J.C., M.E.T., and W.S. edited and revised manuscript; Q.M. and W.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Esteban Fernandez at the Cell Imaging Core of Children’s Hospital Los Angeles for helping with confocal imaging.
REFERENCES
- 1.Morrisey EE, Hogan BL. Preparing for the first breath: genetic and cellular mechanisms in lung development. Dev Cell 18: 8–23, 2010. doi: 10.1016/j.devcel.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Warburton D, El-Hashash A, Carraro G, Tiozzo C, Sala F, Rogers O, De Langhe S, Kemp PJ, Riccardi D, Torday J, Bellusci S, Shi W, Lubkin SR, Jesudason E. Lung organogenesis. Curr Top Dev Biol 90: 73–158, 2010. doi: 10.1016/s0070-2153(10)90003-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCulley D, Wienhold M, Sun X. The pulmonary mesenchyme directs lung development. Curr Opin Genet Dev 32: 98–105, 2015. doi: 10.1016/j.gde.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Warburton D, Bellusci S, De Langhe S, Del Moral PM, Fleury V, Mailleux A, Tefft D, Unbekandt M, Wang K, Shi W. Molecular mechanisms of early lung specification and branching morphogenesis. Pediatr Res 57: 26R–37R, 2005. doi: 10.1203/01.PDR.0000159570.01327.ED. [DOI] [PubMed] [Google Scholar]
- 5.Shi Y, Massague J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 113: 685–700, 2003. doi: 10.1016/S0092-8674(03)00432-X. [DOI] [PubMed] [Google Scholar]
- 6.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 425: 577–584, 2003. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 7.Chen H, Zhuang F, Liu YH, Xu B, Del Moral P, Deng W, Chai Y, Kolb M, Gauldie J, Warburton D, Moses HL, Shi W. TGF-β receptor II in epithelia versus mesenchyme plays distinct roles in the developing lung. Eur Respir J 32: 285–295, 2008. doi: 10.1183/09031936.00165407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang W, Menke DB, Jiang M, Chen H, Warburton D, Turcatel G, Lu CH, Xu W, Luo Y, Shi W. Spatial-temporal targeting of lung-specific mesenchyme by a Tbx4 enhancer. BMC Biol 11: 111, 2013. doi: 10.1186/1741-7007-11-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chytil A, Magnuson MA, Wright CV, Moses HL. Conditional inactivation of the TGF-β type II receptor using Cre:Lox. Genesis 32: 73–75, 2002. doi: 10.1002/gene.10046. [DOI] [PubMed] [Google Scholar]
- 10.Babraham Bioinformatics. FastQC: a quality control tool for high throughput sequence data, 2015. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- 11.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17: 10, 2011. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 12.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120, 2014. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21, 2013. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169, 2015. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140, 2010. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wickham H. Ggplot2: Elegant Graphics for Data Analysis. NY, USA: Springer, 2009. doi: 10.1007/978-0-387-98141-3. [DOI] [Google Scholar]
- 17.Luo Y, El Agha E, Turcatel G, Chen H, Chiu J, Warburton D, Bellusci S, Qian BP, Menke DB, Shi W. Mesenchymal adenomatous polyposis coli plays critical and diverse roles in regulating lung development. BMC Biol 13: 42, 2015. doi: 10.1186/s12915-015-0153-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi W, Chen H, Sun J, Buckley S, Zhao J, Anderson KD, Williams RG, Warburton D. TACE is required for fetal murine cardiac development and modeling. Dev Biol 261: 371–380, 2003. doi: 10.1016/S0012-1606(03)00315-4. [DOI] [PubMed] [Google Scholar]
- 19.Sun J, Chen H, Chen C, Whitsett JA, Mishina Y, Bringas P, Ma JC, Warburton D, Shi W. Prenatal lung epithelial cell-specific abrogation of Alk3-bone morphogenetic protein signaling causes neonatal respiratory distress by disrupting distal airway formation. Am J Pathol 172: 571–582, 2008. doi: 10.2353/ajpath.2008.070286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen H, Sun J, Buckley S, Chen C, Warburton D, Wang XF, Shi W. Abnormal mouse lung alveolarization caused by Smad3 deficiency is a developmental antecedent of centrilobular emphysema. Am J Physiol Lung Cell Mol Physiol 288: L683–L691, 2005. doi: 10.1152/ajplung.00298.2004. [DOI] [PubMed] [Google Scholar]
- 21.Sun L, Akiyama K, Zhang H, Yamaza T, Hou Y, Zhao S, Xu T, Le A, Shi S. Mesenchymal stem cell transplantation reverses multiorgan dysfunction in systemic lupus erythematosus mice and humans. Stem Cells 27: 1421–1432, 2009. doi: 10.1002/stem.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oshima M, Oshima H, Taketo MM. TGF-beta receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev Biol 179: 297–302, 1996. doi: 10.1006/dbio.1996.0259. [DOI] [PubMed] [Google Scholar]
- 23.Chen F, Marquez H, Kim YK, Qian J, Shao F, Fine A, Cruikshank WW, Quadro L, Cardoso WV. Prenatal retinoid deficiency leads to airway hyperresponsiveness in adult mice. J Clin Invest 124: 801–811, 2014. doi: 10.1172/JCI70291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen F, Shao F, Hinds A, Yao S, Ram-Mohan S, Norman TA, Krishnan R, Fine A. Retinoic acid signaling is essential for airway smooth muscle homeostasis. JCI Insight 3: e120398, 2018. doi: 10.1172/jci.insight.120398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang Y, Luo Y, Tang Y, Moats R, Warburton D, Zhou S, Lou J, Pryhuber GS, Shi W, Wang LL. Alteration of cystic airway mesenchyme in congenital pulmonary airway malformation. Sci Rep 9: 5296, 2019. doi: 10.1038/s41598-019-41777-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Priest JR, Williams GM, Hill DA, Dehner LP, Jaffé A. Pulmonary cysts in early childhood and the risk of malignancy. Pediatr Pulmonol 44: 14–30, 2009. doi: 10.1002/ppul.20917. [DOI] [PubMed] [Google Scholar]
- 27.Di PF, Bellia A, Inclimona G, Grasso F, Teresa M, Cassaro MN. Antenatally diagnosed congenital cystic adenomatoid malformations (CCAM): research review. J Prenat Med 6: 22–30, 2012. [PMC free article] [PubMed] [Google Scholar]
- 28.Parikh DH, Rasiah SV. Congenital lung lesions: postnatal management and outcome. Semin Pediatr Surg 24: 160–167, 2015. doi: 10.1053/j.sempedsurg.2015.01.013. [DOI] [PubMed] [Google Scholar]
- 29.Stocker JT. Cystic lung disease in infants and children. Fetal Pediatr Pathol 28: 155–184, 2009. doi: 10.1080/15513810902984095. [DOI] [PubMed] [Google Scholar]
- 30.Correia-Pinto J, Gonzaga S, Huang Y, Rottier R. Congenital lung lesions—underlying molecular mechanisms. Semin Pediatr Surg 19: 171–179, 2010. doi: 10.1053/j.sempedsurg.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 31.Kim HY, Pang MF, Varner VD, Kojima L, Miller E, Radisky DC, Nelson CM. Localized Smooth muscle differentiation is essential for epithelial bifurcation during branching morphogenesis of the mammalian lung. Dev Cell 34: 719–726, 2015. doi: 10.1016/j.devcel.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Young RE, Jones MK, Hines EA, Li R, Luo Y, Shi W, Verheyden JM, Sun X. Smooth muscle differentiation is essential for airway size, tracheal cartilage segmentation, but dispensable for epithelial branching. Dev Cell 53: 73–85.e5, 2020. doi: 10.1016/j.devcel.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seki T, Hong KH, Oh SP. Nonoverlapping expression patterns of ALK1 and ALK5 reveal distinct roles of each receptor in vascular development. Lab Invest 86: 116–129, 2006. doi: 10.1038/labinvest.3700376. [DOI] [PubMed] [Google Scholar]
- 34.Bonniaud P, Kolb M, Galt T, Robertson J, Robbins C, Stampfli M, Lavery C, Margetts PJ, Roberts AB, Gauldie J. Smad3 null mice develop airspace enlargement and are resistant to TGF-beta-mediated pulmonary fibrosis. J Immunol 173: 2099–2108, 2004. doi: 10.4049/jimmunol.173.3.2099. [DOI] [PubMed] [Google Scholar]
- 35.Chen Y, Shi-Wen X, van Beek J, Kennedy L, McLeod M, Renzoni EA, Bou-Gharios G, Wilcox-Adelman S, Goetinck PF, Eastwood M, Black CM, Abraham DJ, Leask A. Matrix contraction by dermal fibroblasts requires transforming growth factor-beta/activin-linked kinase 5, heparan sulfate-containing proteoglycans, and MEK/ERK: insights into pathological scarring in chronic fibrotic disease. Am J Pathol 167: 1699–1711, 2005. doi: 10.1016/s0002-9440(10)61252-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng X, Gao W, Dang Y, Liu X, Li Y, Peng X, Ye X. Both ERK/MAPK and TGF-Beta/Smad signaling pathways play a role in the kidney fibrosis of diabetic mice accelerated by blood glucose fluctuation. J Diabetes Res 2013: 463740, 2013. doi: 10.1155/2013/463740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fang X, Yu SX, Lu Y, Bast RC, Woodgett JR, Mills GB. Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc Natl Acad Sci USA 97: 11960–11965, 2000. doi: 10.1073/pnas.220413597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pandey MK, DeGrado TR. Glycogen synthase kinase-3 (GSK-3)-targeted therapy and imaging. Theranostics 6: 571–593, 2016. doi: 10.7150/thno.14334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ding Q, Xia W, Liu JC, Yang JY, Lee DF, Xia J, Bartholomeusz G, Li Y, Pan Y, Li Z, Bargou RC, Qin J, Lai CC, Tsai FJ, Tsai CH, Hung MC. Erk associates with and primes GSK-3β for its inactivation resulting in upregulation of β-catenin. Mol Cell 19: 159–170, 2005. doi: 10.1016/j.molcel.2005.06.009. [DOI] [PubMed] [Google Scholar]